

Abstract
Purpose of review
The process of aging involves biological changes that increases susceptibility for disease. In the aging lung disease IPF, GWAS studies identified genes associated with risk for disease. Recently, several of these genes were also found to be involved in risk for COPD or lung cancer. This review describes GWAS-derived risk genes for IPF that overlap with risk genes for lung cancer or COPD.
Recent findings
Risk genes that overlap between aging lung diseases, include FAM13A, DSP and TERT. Most interestingly, disease predisposing alleles for IPF are opposite to those for COPD or lung cancer. Studies show that the alleles are associated with differential gene expression and with physiological traits in the general population. The opposite allelic effect sizes suggest the presence of trade-offs in the aging lung. For TERT, the trade-off involves cellular senescence versus proliferation and repair. For FAM13A and DSP, trade-offs may involve protection from noxious gases or tissue integrity.
Summary
The overlap in risk genes in aging lung diseases provides evidence that processes associated with FAM13A, DSP and TERT are important for healthy aging. The opposite effect size of the disease risk alleles may represent trade-offs, for which a model involving an apicobasal gene expression gradient is presented.
Keywords: aging, chronic obstructive pulmonary disease, genome-wide association studies, idiopathic pulmonary fibrosis, lung cancer, trade-off
INTRODUCTION
It is well accepted that the influence of genetic variation on disease development is large. In the last decades, tremendous progress was made in identifying spurious deleterious alleles of high penetrance that strongly contribute to disease development. This has provided unique opportunities, not only for disease diagnostics, but also for development of therapies that aim to biologically compensate or nullify the mutational effect. However, most diseases are not caused by deleterious alleles but are associated with common polymorphisms in our DNA. These polymorphisms have a minor effect on gene function and in ideal circumstances have neutral impact on sustaining healthy life. However, under changing circumstances and in certain combinations, their impact may be altered and particular alleles may contribute to disease development.
The results of over a decade's worth of genome-wide association studies (GWAS) have taught us a lot. On the down-side, the effect size of common polymorphisms is most often extremely low, which precludes translation to the clinic. However, on the up-side, patterns between diseases are starting to arise [1]. Disease-associated genes overlap among many diseases, and these patterns indicate that similar pathobiological processes underlie widely varying clinical entities [2–6,7▪▪]. Recent GWAS results provide evidence that one such group of diseases with overlapping genes consists of pulmonary aging diseases: idiopathic pulmonary fibrosis (IPF), chronic obstructive pulmonary disease (COPD) and lung cancer [7▪▪,8▪▪]. With increasing age, the lung changes to the extent that alleles that initially had neutral impact, may increase risk for disease development in aged tissue. Most interestingly though is that the implicated genes in aging pulmonary diseases do overlap, but the alleles do not: opposing alleles at the same locus confer risk for very different clinical entities. This review focuses on overlapping genes and opposite risk alleles in the aging lung diseases IPF, COPD and lung cancer.
Box 1.

no caption available
AGING AND LUNG DISEASE
As we get older, the chance of developing a lung disease increases. Older persons, aged at least 65 years, report high rates of respiratory symptoms, which commonly associate with COPD, IPF and lung cancer [9▪]. Overall, there is an almost five-fold increase in incidence of IPF and COPD related solely to age [10], and two-thirds of new lung cancer cases are diagnosed in patients over the age of 65 [11].
COPD is prevalent and a significant cause of mortality in the elderly population. The key element in the diagnosis of COPD is the presence of a persistently reduced ratio of forced expiratory volume in one second/forced vital capacity (FEV1/FVC), which is also a characteristic of a naturally aged lung [12]. Increase in alveolar size with emphysematous changes in the upper lobes of the lungs are commonly present in more severe COPD. An increase in alveolar size is also a feature of a naturally aging lung [13], however, in COPD the process is associated with inflammation and alveolar wall destruction [12].
IPF is a rare fibrotic lung disease and typically characterized by symmetric bibasilar peripheral fibrogenesis of the lung [14]. IPF survival is 2–3 years and worsens with increasing age [14]. Progressive decline in lung function parameters such as diffusing capacity of the lungs for carbon monoxide (DLCO) is characteristic for IPF. However, in contrast to the decrease in DLCO that is seen in the naturally aging lung, the decrease in IPF is associated with ongoing interstitial fibrogenesis. Naturally aging lung may also contain increased thickening of alveolar septa, without inflammation or fibrosis [13]. A characteristic of IPF lung biopsy is spatial heterogeneity, showing normal lung areas interspersed among areas of fibrosis [15]. Interestingly, these normal areas commonly show thickening of alveolar septa, which may be a sign of aging.
Malignant transformation of cells in lung cancer is caused by accumulated DNA damage. Usually, smoking is the main source of the damage, however, natural biological processes do also contribute. Several forms of lung cancer exist and all are positively associated with age [16]. Median survival in lung cancer is extremely low, and depends on cancer type and stage. Age is a major risk factor with 72% of deaths in lung cancer occurring in patients over 65 years of age [11].
AGING LUNG DISEASES MAY OVERLAP
COPD is an independent risk factor for lung cancer. Several epidemiologic studies and lung cancer screening trials have shown a two- to four-fold increase in lung cancer risk in patients with COPD in comparison with non-COPD smokers [12]. This risk is highest when airflow obstruction and emphysema coexist in a patient [17]. Squamous cell carcinoma in particular is more commonly seen in patients with COPD, and tumor localization strongly associates with areas with the highest degree of emphysema [17].
The risk of developing lung cancer in patients with IPF is approximately seven times higher than that of the general population [18]. IPF patients with lung cancer seem to be slightly older and more commonly male. They most often develop squamous cell carcinoma in the fibrotic areas in the lower lung periphery. Patients with co-occuring lung cancer and IPF have worse survival than with only lung cancer or only IPF [19,20].
IPF and COPD features may coexist, a condition known as combined pulmonary fibrosis and emphysema (CPFE). CPFE is characterized by both upper-lobe emphysema and lower-lobe fibrosis [21]. Patients with CPFE are at increased risk of developing lung cancer than with IPF only, and tumors are commonly localized in fibrotic areas [22▪▪].
GENETIC DISEASE
Although aging is a risk factor for IPF, COPD and lung cancer, the majority of the aging population does not develop any of these diseases. This is also true for smokers, and indicates that genes play an important role in disease development.
Germline mutations associated with COPD account for approximately 5% of cases, almost all smokers. The most frequent cause is the presence of autosomal recessive mutations in SERPINA1[23] which associates with lower lobe emphysema. However, a few cases with dominant mutations in TERT and upper lobe emphysema have also been described [24,25].
The most common consequence of TERT mutations, however, is IPF [26]. In IPF, approximately 20% of patients have a familial form of the disease. Familial IPF is most often caused by mutations in telomere related genes [27–29] or by mutations in surfactant processing genes. A remarkable overlap between aging lung diseases and mutations in surfactant processing genes exists. Carriers of autosomal recessive [30] or dominant surfactant processing mutations [31], can have coexisting emphysematous or large cystic changes [32–34] or may develop lung cancer [35–37], all depending on which gene harbors the mutation.
In lung cancer, somatic mutations are the mainstay and these mutations are also found in tumors of patients with preexisting COPD [38] or IPF [39,40]. Twin siblings of affected persons had a 7-fold increased risk for lung cancer, with no difference in risk between monozygous and dizygous twins, underlining the importance of the environment in development of lung cancer [41].
GENOME-WIDE ASSOCIATION STUDIES
The majority of elderly lung patients do not carry highly penetrant mutations. In these patients, small constitutional genetic differences, may become of consequence during aging and a history of noxious exposure.
Observations that GWAS risk loci for COPD and lung cancer overlap have been numerous [42–44]. However, recently it was found that several risk genes for COPD or lung cancer were also involved in IPF. A recent GWAS showed that the genes FAM13A and DSP that associate with COPD overlap with IPF although with opposite risk alleles [7▪▪]. Furthermore, meta-analysis of cancerous diseases showed that the gene TERT confers risk for lung cancer [8▪▪], and for IPF [45,46] although again, opposite risk alleles are involved [3].
Although the risk alleles in FAM13A, DSP and TERT are intronic, genetic and physiological consequences of allele carriership have been described. Figure 1 and Table 1 summarize shared risk genes, alleles, associated phenotypes and expression.
FIGURE 1.
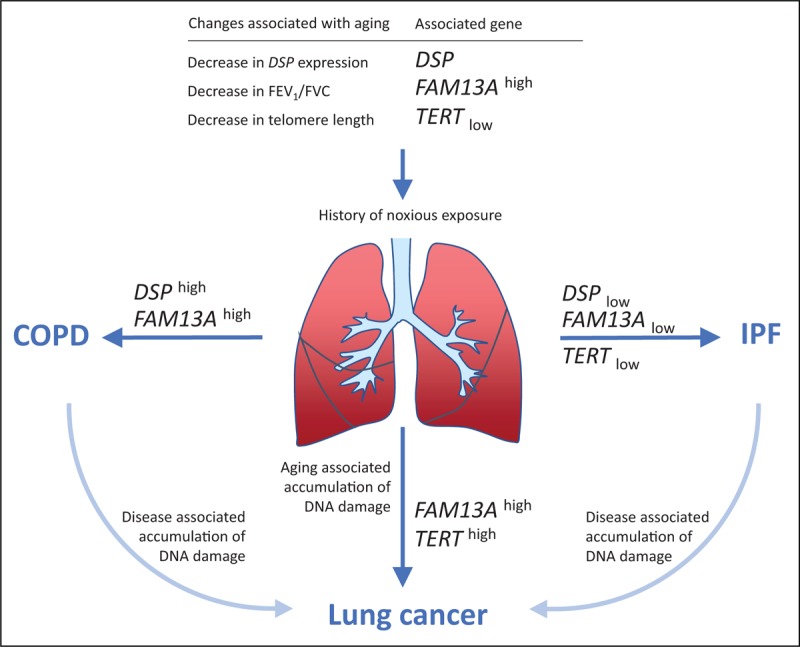
Genes associated with development of aging lung diseases idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease and lung cancer. Disease-predisposing genes overlap but alleles have opposite effect size. Aging and a history of noxious exposure changes genetic requirements for maintenance of a healthy lung. In the aged lung, subtle differences in gene expression conferred by risk alleles in DSP, FAM13A and TERT, can influence biological processes and increase risk for specific aging-associated lung diseases. Presence of disease contributes to tissue aging and increase the risk for secondary lung cancer. GENENAMEhigh, allele associated with increased gene expression; GENENAMElow, allele associated with decreased gene expression.
Table 1.
Characteristics of shared disease loci in idiopathic pulmonary fibrosis, lung cancer and chronic obstructive pulmonary disease
| Risk allele | Expression | |||||||||||
| Gene | Locus | Ancestral allele a | Variant allele | Global MAF a | COPD | Lung cancer | IPF | General population | Risk allele | High | Low | Ref |
| FAM13A | rs2609255 | T | G | G = 0.35 | T (7) | G (7,46,47) | Lower FEV1/FVC | T d (48) | T | G | [49] | |
| rs7671167 | T | C | C = 0.48 | T (42,43) | T (43) | Cb | Lower FEV1/FVC c | T (50) | T | C | [51] | |
| DSP | rs2076295 | G | T | G = 0.43 | T (7) | G (7,52) | T | G | [52▪] | |||
| TERT | rs2736100 | A | C | C = 0.48 | C (53) | A (45,46,54) | Short leukocyte telomere length | A (55) | C | A | [56▪] | |
COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; MAF, minor allele frequency; Ref, reference.
aThe minor allele differed per study population, therefore, the ancestral allele and MAF were derived from https://www.ncbi.nlm.nih.gov/projects/SNP.
bJoint analysis United States Patent Application 20160060701 at http://www.freepatentsonline.com/y2016/0060701.html.
cCases and controls.
dThe referenced study presents results for rs2609264; however, there is complete/tight linkage disequilibrium (r2) between rs2609255 and rs2609264: r2 = 0.97 in Asian and r2 = 1 in European population computed at http://archive.broadinstitute.org/mpg/snap/ldsearchpw.php.
FAMILY WITH SEQUENCE SIMILARITY 13 MEMBER A
Alleles localized at chromosome 4q22, in the gene Family with sequence similarity 13 member A (FAM13A) are among the strongest risk factors for aging lung diseases. The same risk allele is independently associated with COPD [42,57] and lung cancer [43]. Moreover, variants in the gene are independently associated with lung function in the general population [42,48,58,59]. In COPD, the allele is not only associated with risk for disease, but also with phenotypes of disease, such as reduced FEV1/FVC ratio and presence of chronic bronchitis [50,60–62]. In IPF the same allele also associates with worse lung function (low DLCO) and with worse survival [47▪]. However, surprisingly, the risk for IPF disease development is conferred by the opposite allele [46].
For several FAM13A polymorphisms a quantitative effect on gene expression has been demonstrated [59,63]. Risk alleles for COPD, and lung cancer associate with increased gene expression of FAM13A[49,59,64,65]. The risk allele for IPF is the opposite and associates with decreased gene expression.
So far FAM13A was only shown to contribute to development of disease in the elderly, however its contribution to changes in lung function was also found in pediatric cohorts [66]. In human fetal lung, expression of FAM13A is influenced by polymorphisms, and expression levels increase with fetal lung age [67] but an essential role in lung development seems unlikely because Fam13a-deficient mice showed no gross defects in major organs and had normal lung function [68▪▪].
The mechanisms by which FAM13A contributes to disease is not understood. FAM13A has a diverse role in signal transduction that seems to be highly dependent on context [68▪▪,69–71]. In lung cancer, FAM13A was identified as a key regulator of tumor growth and progression [69]. In human lungs, FAM13A is expressed in airway epithelial and mucosal cells, club cells, alveolar type II epithelial cells and macrophages [68▪▪,70] and can be induced by hypoxia [72]. In COPD and IPF lung tissue, FAM13A expression was not influenced by allele carriage and was not significantly different from controls [46,49]. However, increased protein levels of FAM13A were detected in very severe COPD lungs whenever compared with healthy ex-smokers [68▪▪]. Furthermore, only Fam13a wild type mice can develop emphysema, whereas Fam13a-deficient mice are protected from emphysema development, even after 6 months of exposure to cigarette smoke [68▪▪]. This suggests that FAM13A expression may be essential for emphysema development, in line with the finding that higher expressing alleles associate with development of COPD.
DESMOPLAKIN
The desmoplakin (DSP) gene at 6p24 harbors one of the top risk alleles in IPF GWAS [46,73], and the opposite allele was recently found to confer risk for COPD [7▪▪].
In both control and IPF lung samples, the DSP risk locus associated with differential expression of DSP. Lower expression levels associated with the risk allele for IPF [52▪], higher expression levels associated with the opposite allele. Interestingly, the expression of DSP decreased with age in control lung samples [52▪]. However, in case of disease, the expression of DSP increases and levels in IPF lung samples are higher than in controls [52▪].
Desmoplakin is a critical component of desmosomes that are important for cell–cell adhesion. Desmosomes have also been shown to influence cell proliferation, differentiation, migration and apoptosis [74]. Staining of fibrotic and normal human lung tissue localized DSP to airway epithelia and epithelial cells lining cystic areas of the fibrotic lung [52▪]. Staining in normal alveolar tissue was not detected, but one must keep in mind that DSP may be present at the intersections of alveolar type I cells, which may not be visible by immunohistochemistry.
DSP is essential for development, mutations in DSP cause Mendelian disorders primarily affecting the skin and heart. Dsp-deficient mice are not viable [75], and mice heterozygous for cardiac-restricted deficiency of DSP have reduced survival and develop arrhythmogenic right ventricular cardiomyopathy, including fibrosis in the myocardium [76].
Desmosomes are found in tissue that experience intense mechanical stress or shear stress, hence the association with cardiomyopathy [77]. Induction of loss of desmoplakin in cardiomyocytes causes upregulation of profibrotic genes [78]. A possible cause of IPF was suggested to involve increased tractional stress, because IPF is typically characterized by fibrogenesis at bibasilar peripheral lung regions wherever mechanical stress is the highest [79]. This process was further suggested to be accelerated by dysfunctional surfactant fluid through admixture with MUC5B protein [80]. The IPF predisposing allele in DSP associates with decreased expression of desmoplakin, which may decrease structural integrity at sites of highest tractional stress and subsequently trigger fibrogenesis.
Interestingly, loss of DSP is also considered an early step in carcinogenesis. Reduction of DSP can be caused by an epigenetic mechanism and reduced levels are present in primary lung tumors independent of tumor grade, tumor stage and lymph node status [81]. Further cancer cell experiments showed that overexpression of DSP led to significant reduction of lung cancer cell proliferation and anchorage-independent growth [81].
TELOMERASE REVERSE TRANSCRIPTASE
Telomerase reverse transcriptase (TERT) at 5p15 encodes an enzyme essential for telomere length maintenance. The TERT allele that increases risk for IPF [45,46,54] is the opposite of the risk allele for lung cancer [3,8▪▪,53]. The risk allele for IPF associates with lower expression of the gene [56▪] and with shorter leukocyte telomere length in the general population [55]. The allele for lung cancer is the opposite and associated with higher expression and longer telomeres. Moreover, longer leukocyte telomere length associates with an increased risk of developing lung cancer [82].
Germline mutations in TERT that cause IPF or (rarely) emphysema lead to haploinsufficiency of telomerase and accelerated shortening of telomeres upon cell division [83]. Critically short telomeres signal senescence or apoptosis [84]. Mouse models deficient for normal telomere function develop pulmonary emphysema when exposed to cigarette smoke [24], or pulmonary fibrosis when exposed to bleomycin [85].
Telomere shortening or dysfunction in alveolar type II cells seems critical for fibrogenesis. Telomere shortening was observed in alveolar epithelial cells from patients with COPD [86] and IPF [87▪▪]. Recently, shortest telomeres were found in alveolar type II cells in fibrotic areas of IPF lung in comparison with nonfibrotic areas [87▪▪]. Furthermore, selective shortening of telomeres in alveolar type II cells in mice resulted in age-dependent lung remodeling and fibrosis [88▪▪].
Progressive telomere shortening from cell division, also known as replicative aging, provides a barrier for tumor progression [89]. However, in lung tumors, somatic mutations in the promoter region of TERT contribute to increased transcription and cellular immortality [90]. Tumorigenesis is dependent on sequential accumulation of mutations, which are crucial for malignant transformation of the cell [91]. Carriage of the allele for longer telomeres may allow for more cell divisions and increase the chance of accumulating critical oncogenic mutations. Furthermore, longer telomere length associates with higher intrinsic epigenetic age acceleration [92]. Telomerase is thought to regulate the balance between ageing and cancer. Indeed, Tert overexpression in mice increased tumor development, but in a model of tumor-resistant mice, Tert overexpression showed to be beneficial and increased longevity [89].
Reduction in leukocyte telomere length was proposed as a biomarker of aging [93]. Patients with COPD have accelerated shortening of leukocyte telomere length, but this is not related to clinical parameters [94]. Patients with IPF, however, not only have very short telomeres, the decrease in blood and lung is also associated with worse survival [87▪▪,95]. Furthermore, the leukocyte telomere length in IPF is significantly shorter than in other lung diseases with comparable disease burden [96]. It can, therefore, be concluded that only in IPF – and not in COPD or lung cancer – the genetic constitution of the patient is the cause of the short telomeres.
TRADE-OFFS IN THE AGING LUNG
The identification of genetic correlations between diseases can provide useful pathological insights [1]. In the aging lung, polymorphisms in FAM13A, DSP and TERT connect three lung diseases: IPF, COPD and lung cancer. Clinically, these three diseases are also connected by shared risk factors: noxious particles and gases (i.e. from smoking), and aging.
Aging involves changes in cellular levels of gene transcript and proteins that are partly regulated by the presence of genetic variants. The risk alleles in FAM13A, DSP and TERT for IPF were opposite to those for COPD or lung cancer. Furthermore, the polymorphisms were shown to influence gene expression levels: risk alleles for IPF associate with low expression and risk alleles for COPD and lung cancer associate with high expression.
The opposing alleles in TERT, FAM13A and DSP probably represent trade-offs in an aging organism. A trade-off exists whenever a benefit in one context entails a cost in another [97]. A textbook example of trade-off is the sickle cell causing mutation HbS, which also protects against malaria. The trade-off associated with the TERT variant is easiest to understand: high expression of TERT is associated with the capacity of rejuvenation and repair but confers an increased risk of developing cancer. Trade-offs for FAM13A and DSP may involve protection from noxious gases or tissue integrity.
Trade-offs associated with optimal expression levels may be caused by involvement of different cell types or different lung areas. Hypothetically, low FAM13A levels in airway cells may be optimal to decrease risk for COPD but may be too low in alveolar cells to prevent IPF. In the context of lung localization, a gradient of optimal expression levels may exist. In IPF, fibrogenesis follows an apicobasal gradient with basal and peripheral predominance of fibrosis [14]. The cause of this gradient is unknown, but may be associated with regional differences in lung mechanical stress or perfusion. Its presence, however, suggests that a trade-off between the upper and lower parts of the lung may exist. The low-expressing alleles – associated with IPF – may be too low at basal lung areas. Moreover, the high expressing alleles – associated with COPD – may be too high for the apical lung regions (Fig. 2). Further research is required to understand which trade-offs are present in the aging lung.
FIGURE 2.
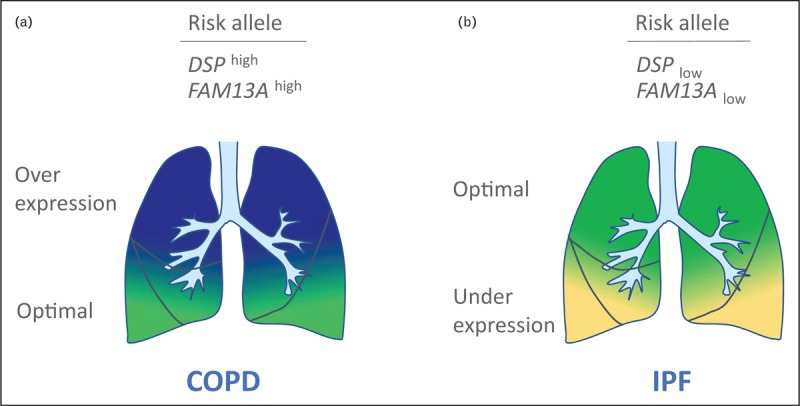
Trade-off gene expression gradient in the aging lung: a model for genotype dependent risk for development of COPD and IPF. As a consequence of aging, optimal expression levels may differ between lung areas. A model is presented for optimal expression levels that follow an apicobasal gradient. Optimal gene expression levels for healthy aging lung are low in the apical zones and high in the basal zones. (a) Carriers of COPD risk alleles that confer high-gene expression have optimal levels (green) in the basal lung, whereas levels for the middle and apical zones are too high (dark blue). This increases the risk for development of COPD. (b) Carriers of IPF risk alleles that confer low-gene expression have optimal levels (green) in the apical and middle lung, whereas levels are too low for the basal zones (light yellow). This increases the risk for development of IPF. COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis.
Given the presence of opposite risk alleles and the additive effect of each allele on expression, it appears that in the aging lung, heterozygotes are at an advantage. In fact, in humans a significant association between increased genome-wide heterozygosity and survival is present [98]. Lung diseases contribute significantly to mortality; thus it is possible that individuals heterozygous for the studied polymorphisms in TERT, DSP and FAM13A are at an advance whenever becoming of age. Allele frequencies are driven by evolutionary processes, maintaining the alleles best fitted for survival in the context of reproduction. Alleles are, therefore, not optimized for aging. The alleles in TERT, DSP and FAM13A that influence susceptibility for aging lung diseases IPF, lung cancer and COPD are well tolerated in early life, they influence phenotypic traits but do not associate with disease. It is conceivable that alleles influencing expression of these genes to a higher degree may not be well tolerated and will affect health at a prereproductive age. Some experimental studies on over-expression and under-expression of the genes showed deleterious consequences, supporting this hypothesis. This has to be kept in mind whenever trying to translate findings on risk alleles into therapies that interfere with expression levels of genes associated with aging lung diseases.
CONCLUSION
In conclusion, IPF risk genes, FAM13A, DSP and TERT are shared with COPD or lung cancer. This underlines the importance of these genes in the development of lung disease in the aging population. Risk alleles were shown to have opposite effect size and opposite influence on expression levels (Fig. 1 and Table 1). A trade-off model is presented (Fig. 2) demonstrating how opposing alleles may influence disease risk. Further studies are required to understand how these genes contribute to health and disease in the aging lung.
Acknowledgements
The author would like to thank Hendrik-Jan Megens, Joanne van der Vis and Jan Grutters for valuable discussion on the subject.
Financial support and sponsorship
This work was supported by ZonMW TopZorg St Antonius Lung grantnr 842002001.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Pasaniuc B, Price AL. Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet 2017; 18:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Moorsel CHM, Christiani DC. Genetic susceptibility to sarcoidosis, a chronic inflammatory disorder. Am J Respir Crit Care Med 2012; 186:816–818. [DOI] [PubMed] [Google Scholar]
- 3.Snetselaar R, van Oosterhout MFM, Grutters JC, Van Moorsel CHM. TERT polymorphism rs2736100: a balancing act between cancer and noncancer disease, a meta-analysis. Front Med 2018; 5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lareau CA, DeWeese CF, Adrianto I, et al. Polygenic risk assessment reveals pleiotropy between sarcoidosis and inflammatory disorders in the context of genetic ancestry. Genes Immun 2017; 18:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smeland OB, Wang Y, Lo M-T, et al. Identification of genetic loci shared between schizophrenia and the Big Five personality traits. Sci Rep 2017; 7:2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witoelar A, Jansen IE, Wang Y, et al. International Parkinson's Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) Investigators. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol 2017; 74:780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7▪▪.Hobbs BD, de Jong K, Lamontagne M, et al. COPDGene Investigators; ECLIPSE Investigators; LifeLines Investigators; SPIROMICS Research Group; International COPD Genetics Network Investigators; UK BiLEVE Investigators; International COPD Genetics Consortium. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet 2017; 49:426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence for overlapping risk genes with opposite effect size in COPD and IPF.
- 8▪▪.Haycock PC, Burgess S, et al. Telomeres Mendelian Randomization Collaboration. Association between telomere length and risk of cancer and non-neoplastic diseases: a Mendelian Randomization Study. JAMA Oncol 2017; 3:636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that alleles associated with increased telomere length confer risk for many types of cancer, including lung cancer, and opposite alleles increase risk for pulmonary fibrosis.
- 9▪.Vaz Fragoso CA. Epidemiology of lung disease in older persons. Clin Geriatr Med 2017; 33:491–501. [DOI] [PubMed] [Google Scholar]; Review summarizing epidemiological data on lung disease in the elderly, including COPD, IPF and lung cancer.
- 10.Ascher K, Elliot SJ, Rubio GA, Glassberg MK. Lung diseases of the elderly: cellular mechanisms. Clin Geriatr Med 2017; 33:473–490. [DOI] [PubMed] [Google Scholar]
- 11.Barta JA, Zinner RG, Unger M. Lung cancer in the older patient. Clin Geriatr Med 2017; 33:563–577. [DOI] [PubMed] [Google Scholar]
- 12.Cortopassi F, Gurung P, Pinto-Plata V. Chronic obstructive pulmonary disease in elderly patients. Clin Geriatr Med 2017; 33:539–552. [DOI] [PubMed] [Google Scholar]
- 13.Verbeken EK, Cauberghs M, Mertens I, et al. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest 1992; 101:793–799. [DOI] [PubMed] [Google Scholar]
- 14.Raghu G, Collard HR, Egan JJ, et al. An officIAL ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith M, Dalurzo M, Panse P, et al. Usual interstitial pneumonia-pattern fibrosis in surgical lung biopsies. Clinical, radiological and histopathological clues to aetiology. J Clin Pathol 2013; 66:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol 2013; 30:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai J, Yang P, Cox A, Jiang G. Lung cancer and chronic obstructive pulmonary disease: from a clinical perspective. Oncotarget 2017; 8:18513–18524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5:72–84. [DOI] [PubMed] [Google Scholar]
- 19.Lee T, Park JY, Lee HY, et al. Lung cancer in patients with idiopathic pulmonary fibrosis: clinical characteristics and impact on survival. Respir Med 2014; 108:1549–1555. [DOI] [PubMed] [Google Scholar]
- 20.Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J 2015; 46:1113–1130. [DOI] [PubMed] [Google Scholar]
- 21.Cottin V, Nunes H, Brillet P-Y, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005; 26:586–593. [DOI] [PubMed] [Google Scholar]
- 22▪▪.Koo HJ, Do K-H, Lee JB, et al. Lung cancer in combined pulmonary fibrosis and emphysema: a systematic review and meta-analysis. PLoS One 2016; 11:e0161437. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article summarizes knowledge regarding patients with lung cancer, IPF and emphysema and shows that survival in patients is worse then any one or two diseases combined.
- 23.Brode SK, Ling SC, Chapman KR. Alpha-1 antitrypsin deficiency: a commonly overlooked cause of lung disease. CMAJ 2012; 184:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alder JK, Guo N, Kembou F, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med 2011; 184:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanley SE, Chen JJL, Podlevsky JD, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest 2015; 125:563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res 2012; 730:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai J, Cai H, Zhuang Y, et al. Telomerase gene mutations and telomere length shortening in patients with idiopathic pulmonary fibrosis in a Chinese population. Respirology 2015; 20:122–128. [DOI] [PubMed] [Google Scholar]
- 28.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 2007; 104:7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res 2012; 13:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campo I, Zorzetto M, Mariani F, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res 2014; 15:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas AQ, Lane K, Phillips J, et al. Heterozygosity for a surfactant protein c gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165:1322–1328. [DOI] [PubMed] [Google Scholar]
- 32.van Moorsel CHM, van Oosterhout MFM, Barlo NP, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch Cohort. Am J Respir Crit Care Med 2010; 182:1419–1425. [DOI] [PubMed] [Google Scholar]
- 33.Cottin V, Reix P, Khouatra C, et al. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 2011; 66:918–919. [DOI] [PubMed] [Google Scholar]
- 34.Epaud R, Delestrain C, Louha M, et al. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur Respir J 2014; 43:638–641. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 2009; 84:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Moorsel CHM, ten Klooster L, van Oosterhout MFM, et al. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2015; 192:1249–1252. [DOI] [PubMed] [Google Scholar]
- 37.Nathan N, Giraud V, Picard C, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet 2016; 25:1457–1467. [DOI] [PubMed] [Google Scholar]
- 38.Xiao D, Li F, Pan H, et al. Integrative analysis of genomic sequencing data reveals higher prevalence of LRP1B mutations in lung adenocarcinoma patients with COPD. Sci Rep 2017; 7:2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guyard A, Danel C, Théou-Anton N, et al. Morphologic and molecular study of lung cancers associated with idiopathic pulmonary fibrosis and other pulmonary fibroses. Respir Res 2017; 18:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang JA, Kim D, Chun S-M, et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J Pathol 2018; 244:25–35. [DOI] [PubMed] [Google Scholar]
- 41.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer — analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343:78–85. [DOI] [PubMed] [Google Scholar]
- 42.Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet 2010; 42:200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young RP, Hopkins RJ, Hay BA, et al. FAM13A locus in COPD is independently associated with lung cancer - evidence of a molecular genetic link between COPD and lung cancer. Appl Clin Genet 2011; 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziółkowska-Suchanek I, Mosor M, Gabryel P, et al. Susceptibility loci in lung cancer and COPD: association of IREB2 and FAM13A with pulmonary diseases. Sci Rep 2015; 5:13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet 2008; 45:654–656. [DOI] [PubMed] [Google Scholar]
- 46.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013; 45:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47▪.Hirano C, Ohshimo S, Horimasu Y, et al. FAM13A polymorphism as a prognostic factor in patients with idiopathic pulmonary fibrosis. Respir Med 2017; 123:105–109. [DOI] [PubMed] [Google Scholar]; Analysis of a Japanese cohort of patients with IPF shows that both DLCO and survival is better in carriers of the IPF disease predisposing allele in FAM13A.
- 48.Kim WJ, Lee MK, Shin C, et al. Genome-wide association studies identify locus on 6p21 influencing lung function in the Korean population. Respirology 2014; 19:360–368. [DOI] [PubMed] [Google Scholar]
- 49.Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep 2017; 7:44232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardin M, Zielinski J, Wan ES, et al. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am J Respir Cell Mol Biol 2012; 47:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obeidat M, Hao K, Bossé Y, et al. Molecular mechanisms underlying variations in lung function: a systems genetics analysis. Lancet Respir Med 2015; 3:782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52▪.Mathai SK, Pedersen BS, Smith K, et al. Desmoplakin variants are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016; 193:1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]; Analysis of DSP expression in lung biopsies shows that risk alleles are associated with decreased expression in IPF.
- 53.Nie W, Zang Y, Chen J, Xiu Q. TERT rs2736100 polymorphism contributes to lung cancer risk: a meta-analysis including 49,869 cases and 73,464 controls. Tumour Biol 2014; 35:5569–5574. [DOI] [PubMed] [Google Scholar]
- 54.Wei R, Li C, Zhang M, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res 2014; 163:494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Codd V, Nelson CP, Albrecht E, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 2013; 45:422–427. 427.e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56▪.Ge M, Shi M, An C, et al. Functional evaluation of TERT-CLPTM1L genetic variants associated with susceptibility of papillary thyroid carcinoma. Sci Rep 2016; 6:26037. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, it was shown that the TERT SNPs that associate with risk for aging lung diseases influence gene expression levels.
- 57.Cho MH, McDonald M-LN, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med 2014; 2:214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet 2010; 42:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Plaat DA, de Jong K, Lahousse L, et al. Genome-wide association study on the FEV1/FVC ratio in never-smokers identifies HHIP and FAM13A. J Allergy Clin Immunol 2017; 139:533–540. [DOI] [PubMed] [Google Scholar]
- 60.Pillai SG, Kong X, Edwards LD, et al. ECLIPSE and ICGN Investigators. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 182:1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang B, Liang B, Yang J, et al. Association of FAM13A polymorphisms with COPD and COPD-related phenotypes in Han Chinese. Clin Biochem 2013; 46:1683–1688. [DOI] [PubMed] [Google Scholar]
- 62.Lee JH, Cho MH, Hersh CP, et al. COPDGene and ECLIPSE Investigators. Genetic susceptibility for chronic bronchitis in chronic obstructive pulmonary disease. Respir Res 2014; 15:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamontagne M, Couture C, Postma DS, et al. Refining susceptibility loci of chronic obstructive pulmonary disease with lung eqtls. PLoS One 2013; 8:e70220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim WJ, Lim MN, Hong Y, et al. Association of lung function genes with chronic obstructive pulmonary disease. Lung 2014; 192:473–480. [DOI] [PubMed] [Google Scholar]
- 65.Wain LV, Shrine N, Artigas MS, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 2017; 49:416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corvol H, Hodges CA, Drumm ML, Guillot L. Moving beyond genetics: is FAM13A a major biological contributor in lung physiology and chronic lung diseases? J Med Genet 2014; 51:646–649. [DOI] [PubMed] [Google Scholar]
- 67.Miller S, Melén E, Merid SK, et al. Genes associated with polymorphic variants predicting lung function are differentially expressed during human lung development. Respir Res 2016; 17:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68▪▪.Jiang Z, Lao T, Qiu W, et al. A chronic obstructive pulmonary disease susceptibility gene, FAM13A, regulates protein stability of β-catenin. Am J Respir Crit Care Med 2016; 194:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that FAM13A may be essential for development of emphysema. Fam13a-deficient mice were shown to be resistant to development of emphysema, even after 6 months of exposure to cigarette smoke.
- 69.Eisenhut F, Heim L, Trump S, et al. FAM13A is associated with nonsmall cell lung cancer (NSCLC) progression and controls tumor cell proliferation and survival. Oncoimmunology 2017; 6:e1256526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corvol H, Rousselet N, Thompson KE, et al. FAM13A is a modifier gene of cystic fibrosis lung phenotype regulating rhoa activity, actin cytoskeleton dynamics and epithelial-mesenchymal transition. J Cyst Fibros 2017; doi: 10.1016/j.jcf.2017.11.003 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 71.Jiang Z, Knudsen NH, Wang G, et al. Genetic control of fatty acid β-oxidation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2017; 56:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ziółkowska-Suchanek I, Mosor M, Podralska M, et al. FAM13A as a novel hypoxia-induced gene in non-small cell lung cancer. J Cancer 2017; 8:3933–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med 2017; 5:869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huber O, Petersen I. 150th anniversary series: desmosomes and the hallmarks of cancer. Cell Commun Adhes 2015; 22:15–28. [DOI] [PubMed] [Google Scholar]
- 75.Gallicano GI, Kouklis P, Bauer C, et al. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 1998; 143:2009–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006; 116:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stokes DL. Desmosomes from a structural perspective. Curr Opin Cell Biol 2007; 19:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dubash AD, Kam CY, Aguado BA, et al. Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J Cell Biol 2016; 212:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leslie KO. Idiopathic pulmonary fibrosis may be a disease of recurrent, tractional injury to the periphery of the aging lung: a unifying hypothesis regarding etiology and pathogenesis. Arch Pathol Lab Med 2012; 136:591–600. [DOI] [PubMed] [Google Scholar]
- 80.van Moorsel CHM, Hoffman TW, van Batenburg AA, et al. Understanding idiopathic interstitial pneumonia: a gene-based review of stressed lungs. Biomed Res Int 2015; 2015:304186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang L, Chen Y, Cui T, et al. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/β-catenin signaling pathway in human lung cancer. Carcinogenesis 2012; 33:1863–1870. [DOI] [PubMed] [Google Scholar]
- 82.Zhu X, Han W, Xue W, et al. The association between telomere length and cancer risk in population studies. Sci Rep 2016; 6:22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsang AR, Wyatt HDM, Ting NSY, Beattie TL. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell 2012; 11:482–490. [DOI] [PubMed] [Google Scholar]
- 84.Victorelli S, Passos JF. Telomeres and cell senescence - size matters not. EBioMedicine 2017; 21:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Povedano JM, Martinez P, Flores JM, et al. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep 2015; 12:286–299. [DOI] [PubMed] [Google Scholar]
- 86.Birch J, Anderson RK, Correia-Melo C, et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol Cell Mol Physiol 2015; 309:L1124–L1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87▪▪.Snetselaar R, van Batenburg AA, van Oosterhout MFM, et al. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One 2017; 12:e0189467. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study with IPF lung biopsies, the positional correlation between short telomeres, fibrosis and alveolar type II cells is made.
- 88▪▪.Naikawadi RP, Disayabutr S, Mallavia B, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016; 1:e86704. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using mouse models, it is shown that telomere dysfunction in alveolar epithelial type II is sufficient for the development of pulmonary fibrosis.
- 89.Tomás-Loba A, Flores I, Fernández-Marcos PJ, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008; 135:609–622. [DOI] [PubMed] [Google Scholar]
- 90.Yuan P, Cao J, Abuduwufuer A, et al. Clinical characteristics and prognostic significance of TERT promoter mutations in cancer: a cohort study and a meta-analysis. PLoS One 2016; 11:e0146803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomasetti C, Marchionni L, Nowak MA, et al. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A 2015; 112:118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu AT, Xue L, Salfati EL, et al. GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat Commun 2018; 9:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest 2013; 123:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Córdoba-Lanús E, Cazorla-Rivero S, Espinoza-Jiménez A, et al. Telomere shortening and accelerated aging in COPD: findings from the BODE cohort. Respir Res 2017; 18:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stuart BD, Lee JS, Kozlitina J, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med 2014; 2:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Snetselaar R, van Moorsel CHM, Kazemier KM, et al. Telomere length in interstitial lung diseases. Chest 2015; 148:1011–1018. [DOI] [PubMed] [Google Scholar]
- 97.Gluckman PD, Low FM, Buklijas T, et al. How evolutionary principles improve the understanding of human health and disease. Evol Appl 2011; 4:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bihlmeyer NA, Brody JA, Smith AV, et al. Genetic diversity is a predictor of mortality in humans. BMC Genet 2014; 15:159. [DOI] [PMC free article] [PubMed] [Google Scholar]