

Summary
Replication fork reversal is a rapidly emerging and remarkably frequent mechanism of fork stabilization in response to genotoxic insults. Here, we summarize recent findings that uncover key molecular determinants for reversed fork formation and describe how the homologous recombination factors BRCA1, BRCA2 and RAD51 protect these structures from extended nucleolytic degradation.
DNA replication forks are constantly challenged by DNA lesions and intrinsic replication obstacles. Replication fork reversal is a key protective mechanism that allows forks to reverse their course when they encounter DNA lesions and resume DNA synthesis without chromosomal breakage (Neelsen and Lopes, 2015). The fork-reversal model can be conceptually divided into two steps: (a) formation of reversed forks by the coordinated annealing of the two newly synthesized strands, leading to four-way junction structures resembling Holliday junctions, and (b) restart of the reversed forks. This gymnastic enables the original lesion either to be removed before reversed fork restart or bypassed through a template-switching mechanism (Neelsen and Lopes, 2015). Some of the key factors required for reversed fork restart have been identified, i.e., the human RECQ1 helicase or the DNA2 nuclease assisted by the WRN helicase (Berti et al., 2013; Thangavel et al., 2015). Conversely, our current understanding of the actual mechanism of reversed fork formation is limited.
The regressed arm of a reversed replication fork resembles a one-ended double-stranded break (DSB) and must be properly protected to prevent nuclease cleavage. However, the molecular determinants required to protect the integrity of regressed arms until forks are restarted are unknown. Here, we review recent articles that provide a fresh perspective on these important issues by defining a key function of two translocases of the SWI/SNF protein family, i.e., ZRANB3 and SMARCAL1, in reversed fork formation (Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017) and a key function of the breast cancer susceptibility proteins BRCA1 and BRCA2 in reversed fork protection (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017).
Using an elegant combination of single-molecule DNA fiber and electron microscopy approaches, Kolinjivadi et al. (2017), Taglialatela et al. (2017) and Vujanovic et al. (2017) demonstrate that the translocase activities of SMARCAL1 and ZRANB3 are required for replication fork reversal. In particular, Kolinjivadi et al. (2017) propose that SMARCAL1 remodels forks with persistent ssDNA gaps at the fork junction into reversed fork structures and Taglialatela et al. (2017) suggest that the RPA-binding activity of SMARCAL1 is required for its replication function, in agreement with previous biochemical studies (Betous et al., 2013). At the same time, Vujanovic et al. (2017) show that ZRANB3 interacts with polyubiquitinated PCNA to promote fork remodeling (Figure 1A). PCNA post-translational modifications are triggered by the formation of RPA-coated ssDNA at stalled forks and are key regulators of pathway choice between error-prone translesion DNA synthesis and error-free template switching mechanisms (Mailand et al., 2013). Indeed, the yeast Rad5 homologs HLTF and SHPRH are DNA translocases that promote PCNA polyubiquitination, and HLTF has been directly implicated in fork reversal via its HIRAN domain (Berti and Vindigni, 2016; Blastyak et al., 2010; Kile et al., 2015), further supporting the notion that PCNA polyubiquitination promotes replication fork remodeling.
Figure 1. Mechanisms of reversed replication fork formation.

(A) Accumulation of ssDNA at uncoupled replication forks coated by RPA promotes the recruitment of the E2–E3 ubiquitin conjugating enzymes, which mediate PCNA monoubiquitination at lysine 164 (K164), as well as UBC13-dependent K63-linked polyubiquitination of the same residue. Polyubiquitinylated PCNA interacts with the ZRANB3 translocase, which promotes replication fork reversal. (B) BRCA2 promotes RAD51 binding to replicating DNA. RAD51 directly interacts with Pol α to facilitate DNA synthesis and avoid gap formation. Persistent ssDNA gaps are remodeled into a reversed replication fork by the translocase activity of SMARCAL1. (C) Unstable RAD51 nucleofilaments and/or inefficient loading of RAD51 on ssDNA, such as in BRCA2-deficient or RAD51-T131P cells, does not impair fork reversal, either because fork reversal does not require extensive homology search at a distance or because the limited loading of RAD51 is sufficient to recruit specific translocases on the uncoupled fork.
A key question for future studies would be to define whether there is a functional interaction between these different fork remodelers or whether different remodelers are required depending on the particular structure of the replication intermediate or type of replication challenge. Indeed, SMARCAL1, ZRANB3, and HLTF recognize different types of fork structures in vitro, suggesting that cells might use different factors depending on the particular type of replication intermediate (Betous et al., 2013; Hishiki et al., 2015; Kile et al., 2015). Along the same line, the studies of Kolinjivadi et al. (2017), Taglialatela et al. (2017), and Vujanovic et al. (2017) clearly show that SMARCAL1 or ZRANB3 depletion does not fully abrogate reversed fork formation, supporting the idea that fork reversal is not mediated by a single fork remodeler and that different structures might arise even when using the same type of replication challenge. Moreover, other DNA translocases, including RAD54 (Bugreev et al., 2011) and FANCM (Gari et al., 2008), can mediate fork reversal in vitro, suggesting that additional factors may contribute to this process.
The central recombinase factor RAD51 is also involved in reversed fork formation, pointing to a previously unappreciated role of this homologous recombination (HR) factor in replication fork remodeling (Zellweger et al., 2015). Interestingly, Kolinjivadi et al. (2017), Tagliatela et al. (2017), and Vujanovic et al. (2017) show that fork reversal is not impaired under conditions that lead to formation of unstable RAD51 nucleofilaments and/or inefficient loading of RAD51 on ssDNA (Figure 1C), such as in BRCA1/2-mutant cells or in cells expressing a dominant RAD51 mutant allele, RAD51-T131P, which was recently reported to destabilize RAD51 nucleofilaments (Wang et al., 2015). The molecular steps that allow replication forks to reverse under conditions of inefficient RAD51 loading remain to be defined. Possible scenarios are that the initial RAD51-mediated step of reversed fork formation does not require extensive homology search because of the close proximity of the two arms of the fork, or that RAD51 might work in a distinct fashion at forks compared to HR where its function is BRCA1/2-dependent and relies on the formation of stable nucleofilaments. Another important avenue of future investigation is to determine whether alternative HR mediators—e.g., MMS22L-TONSL (Piwko et al., 2016), the RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3) (Taylor et al., 2015), and RAD54 (Bugreev et al., 2011)—may assist RAD51 nucleofilament formation and replication fork reversal in the absence of BRCA2. Interestingly, Kolinjivadi et al. (2017) found that RAD51 interacts with the replicative polymerase Pol α and proposed a model whereby this interaction is important to stabilize Pol α at forks and avoid ssDNA gap formation at fork junctions that would otherwise be remodeled into reversed forks by SMARCAL1 under conditions of replication stress (Figure 1B). This model suggests an alternative scenario where destabilizing the RAD51 nucleofilament may indirectly promote fork reversal by favoring ssDNA gap accumulation at fork junctions and SMARCAL1 activity.
HR factors can also control MRE11 nuclease activity and ssDNA accumulation at stalled forks (Hashimoto et al., 2010), pointing to a tight link between fork remodeling and degradation. In particular, the breast cancer susceptibility proteins BRCA1 and BRCA2, as well as RAD51, are emerging as key regulators of replication fork degradation. In their absence, replication forks are extensively degraded by MRE11 (Schlacher et al., 2011; Ying et al., 2012). This degradation phenotype is emerging as one of the leading causes of BRCA-deficient tumor sensitivity to DNA-damaging chemotherapies (Ray Chaudhuri et al., 2016). However, the mechanisms leading to this extensive fork degradation phenotype, as well as the structure of the replication intermediates targeted by nucleases in the absence of BRCA1 or BRCA2, remained unclear. The studies reviewed here provide important clues on the molecular basis for this extended fork degradation phenotype (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). One of their major breakthroughs is the finding that unprotected reversed forks are the structures targeted by MRE11 in a BRCA-deficient background, suggesting that BRCA1 and BRCA2 play a crucial role in reversed fork protection. In particular, BRCA1 and BRCA2 are not required for reversed fork formation, but are instead required to stabilize the RAD51 filament on the regressed arms of already formed reversed replication forks, thereby protecting the open double-stranded end of the regressed arm from nucleolytic attack (Figure 2A). Altogether, these studies suggest that RAD51 has two distinct functions during replication stress: a BRCA-independent function in promoting the initial step of reversed fork formation, as discussed above, and a BRCA-dependent function whereby BRCA proteins protect the already formed reversed forks from nucleolytic degradation by stabilizing the RAD51 filament on the regressed arm. In BRCA2-deficient cells, this second function is lost, leading to the nascent strand degradation phenotype observed with BRCA2 mutants unable to stabilize RAD51 on ssDNA (Schlacher et al., 2011), with RAD51 mutants that destabilize the RAD51 nucleofilament (Kolinjivadi et al., 2017; Mijic et al., 2017; Zadorozhny et al., 2017), or with small molecules that inhibit RAD51 DNA binding activity (Taglialatela et al., 2017).
Figure 2. Mechanisms of reversed replication fork protection and restart.
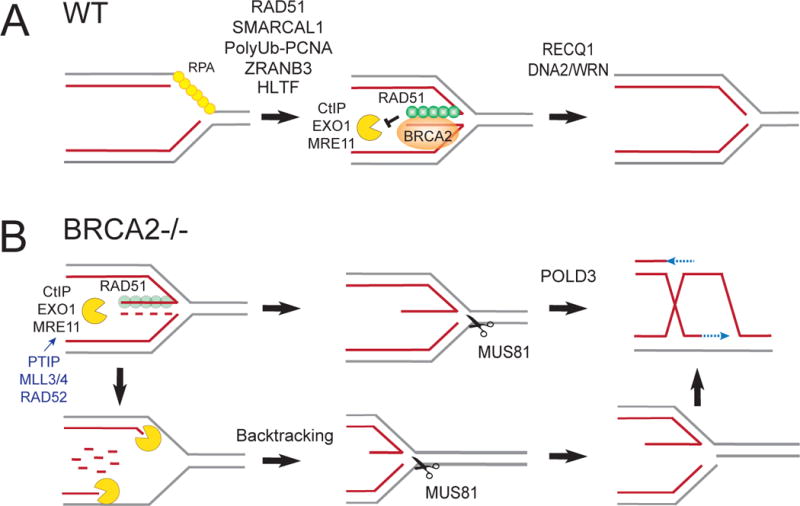
(A) BRCA2 stabilizes the RAD51 nucleofilament on the regressed arms, thereby preventing MRE11, CtIP and EXO1-dependent resection of the regressed arms. RECQ1 promotes restart of the reversed replication forks. DNA2/WRN also promote reversed fork restart through a mechanism that entails limited DNA2-dependent processing of the regressed arm. (B) Reversed forks are extensively degraded by MRE11, CtIP and EXO1 in BRCA2-deficient cells. PTIP, MLL3/4 and RAD52 promote MRE11 recruitment on the stalled forks. The initial limited degradation of regressed arm generates a reversed fork with a 3′ ssDNA flap that is a substrate for MUS81 cleavage. MUS81 cleavage produces a migrating bubble that promotes POLD3-dependent DNA synthesis. If MUS81 does not promptly cleave the partially resected forks, the nucleolytic degradation might quickly proceed to degrade nascent strands behind the junction, finally leading to extensively resected forks. After extended fork resection, reannealing of the parental strands might lead to “backtracking” of the fork. As a consequence of fork backtracking, a new reversal event may occur, possibly promoting a new MUS81 cleavage event.
Interestingly, Mijic et al. (2017) also provide new insight into the mechanism of MRE11 recruitment to stalled forks by showing that RAD52 is another factor, together with PTIP and MLL4 (Ray Chaudhuri et al., 2016), required to load MRE11 and prime MRE11-dependent fork resection in BRCA2-deficient cells. At the same time, Lemacon et al. (2017) further define the exact sequence of events leading to reversed fork resection in BRCA2-deficient cells by showing that the CtIP protein initiates MRE11-dependent degradation of the regressed arms, which is then extended by the EXO1 nuclease. Of note, the same study also elucidates the molecular steps that rescue the degraded forks upon drug withdrawal by showing that the initial resection of the regressed arm generates a reversed fork with a 3′-ssDNA tail that is then cleaved by MUS81 to permit POLD3-dependent rescue of the stalled forks in a BRCA2-deficient background (Figure 2B). Interestingly, a similar mechanism was recently proposed for completion of DNA replication at common fragile sites and telomeric loci (Dilley et al., 2016; Minocherhomji et al., 2015). This mechanism relies on a specialized form of DNA repair originally characterized in yeast and termed break-induced replication (BIR), whereby MUS81 cleavage of stalled replication forks produces a migrating bubble that drives POLD3-dependent DNA synthesis (Sakofsky and Malkova, 2017).
A common conclusion that emerges from all these studies is that protecting the reversed forks from nucleolytic degradation is an essential step to ensure genome stability. However, Mijic et al. (2017) showed that impairing the initial formation of reversed forks leads to increased chromosomal breakage, suggesting that fork reversal is an important transaction to preserve replication fork integrity, whereas Taglialatela et al. (2017) suggest that depletion of factors required fork reversal is accompanied by a marked reduction of genomic instability. This apparent discrepancy likely reflects the use of cancer cells in one study (Mijic et al., 2017) and mammalian epithelial cells in the other (Taglialatela et al., 2017), or possibly other differences in the experimental set-up, and should be the subject of further investigation in the future. In this regard, another recent study suggests that the role of BRCA2 in homologous recombination, but not in stalled replication fork protection, is primarily associated with supporting human mammary epithelial cell viability and preventing replication stress, a hallmark of precancerous cell lesions (Feng and Jasin, 2017). These studies suggest the tantalizing scenario that the HR function of BRCA2 is critical in primary cells to maintain genome stability and prevent cancer onset (Feng and Jasin, 2017), whereas the function of BRCA2 in replication fork stability becomes essential in cancer cells for modulating sensitivity/resistance to chemotherapeutics (Ray Chaudhuri et al., 2016).
The studies on the mechanisms of reversed fork formation and protection highlighted in this review shed new light on central processes for the maintenance of chromosome stability and also raise several questions to be addressed in the future: How do different fork remodelers functionally collaborate to mediate replication fork reversal? Is the choice DNA-damage specific? Which are the other factors required for reversed fork protection and stabilization, or for extended fork degradation in BRCA-deficient cells? How do cells cope with these extensively degraded forks? What is the fate of the replisome during fork reversal? Are there differences in these mechanisms between primary and cancer cells, which could be exploited clinically? Can we use this knowledge to define the precise mechanisms by which BRCA proteins suppress breast and ovarian cancer? We believe that these studies will spark the interest of many labs to address these pressing questions and provide a clear mechanistic link between fork reversal and genomic instability as a function of different genetic backgrounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nature structural & molecular biology. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Vindigni A. Replication stress: getting back on track. Nature structural & molecular biology. 2016;23:103–109. doi: 10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell reports. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyak A, Hajdu I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Molecular and cellular biology. 2010;30:684–693. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, Rossi MJ, Mazin AV. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic acids research. 2011;39:2153–2164. doi: 10.1093/nar/gkq1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539:54–58. doi: 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Jasin M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature communications. 2017;8:525. doi: 10.1038/s41467-017-00634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nature structural & molecular biology. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishiki A, Hara K, Ikegaya Y, Yokoyama H, Shimizu T, Sato M, Hashimoto H. Structure of a Novel DNA-binding Domain of Helicase-like Transcription Factor (HLTF) and Its Functional Implication in DNA Damage Tolerance. The Journal of biological chemistry. 2015;290:13215–13223. doi: 10.1074/jbc.M115.643643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Molecular cell. 2015;58:1090–1100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Molecular cell. 2017;67:867–881 e867. doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nature communications. 2017;8:860. doi: 10.1038/s41467-017-01180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat Rev Mol Cell Biol. 2013;14:269–282. doi: 10.1038/nrm3562. [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature communications. 2017;8:859. doi: 10.1038/s41467-017-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. doi: 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Piwko W, Mlejnkova LJ, Mutreja K, Ranjha L, Stafa D, Smirnov A, Brodersen MM, Zellweger R, Sturzenegger A, Janscak P, et al. The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. The EMBO journal. 2016;35:2584–2601. doi: 10.15252/embj.201593132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Malkova A. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Critical reviews in biochemistry and molecular biology. 2017;52:395–413. doi: 10.1080/10409238.2017.1314444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Molecular cell. 2017;68:414–430 e418. doi: 10.1016/j.molcel.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MRG, Spirek M, Chaurasiya KR, Ward JD, Carzaniga R, Yu X, Egelman EH, Collinson LM, Rueda D, Krejci L, et al. Rad51 Paralogs Remodel Pre-synaptic Rad51 Filaments to Stimulate Homologous Recombination. Cell. 2015;162:271–286. doi: 10.1016/j.cell.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. DNA2 drives processing and restart of reversed replication forks in human cells. The Journal of Cell Biology. 2015;208:545–562. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Molecular cell. 2017;67:882–890 e885. doi: 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Molecular cell. 2015;59:478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer research. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- Zadorozhny K, Sannino V, Belan O, Mlcouskova J, Spirek M, Costanzo V, Krejci L. Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell reports. 2017;21:333–340. doi: 10.1016/j.celrep.2017.09.062. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. The Journal of cell biology. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]