

Abstract
Premise of the Study
Targeted sequence capture can be used to efficiently gather sequence data for large numbers of loci, such as single‐copy nuclear loci. Most published studies in plants have used taxon‐specific locus sets developed individually for a clade using multiple genomic and transcriptomic resources. General locus sets can also be developed from loci that have been identified as single‐copy and have orthologs in large clades of plants.
Methods
We identify and compare a taxon‐specific locus set and three general locus sets (conserved ortholog set [COSII], shared single‐copy nuclear [APVO SSC] genes, and pentatricopeptide repeat [PPR] genes) for targeted sequence capture in Buddleja (Scrophulariaceae) and outgroups. We evaluate their performance in terms of assembly success, sequence variability, and resolution and support of inferred phylogenetic trees.
Results
The taxon‐specific locus set had the most target loci. Assembly success was high for all locus sets in Buddleja samples. For outgroups, general locus sets had greater assembly success. Taxon‐specific and PPR loci had the highest average variability. The taxon‐specific data set produced the best‐supported tree, but all data sets showed improved resolution over previous non‐sequence capture data sets.
Discussion
General locus sets can be a useful source of sequence capture targets, especially if multiple genomic resources are not available for a taxon.
Keywords: Buddleja, hybrid enrichment, Lamiales, PPR genes, Scrophulariaceae, single‐copy nuclear genes
Recent and rapid diversifications are common in the tree of life (Schluter, 2000; Madriñan et al., 2013), and large amounts of data are often required to resolve phylogenetic relationships (Rokas et al., 2003). Multiple independent loci are needed to accurately reconstruct species trees (Small et al., 2004; Leaché and Rannala, 2011) because gene trees from single loci may not reflect species relationships due to incomplete lineage sorting, unrecognized paralogy, and lateral gene transfer or hybridization between species (Maddison, 1997).
Until recently, most phylogenetic studies in plants have relied on a few loci, particularly from the easily amplified and variable plastid and nuclear ribosomal RNA regions. However, each of these regions represents only a single gene history (Small et al., 2004). Additional nuclear loci have been targeted for development in phylogenetic studies because many are relatively fast‐evolving and each nuclear locus potentially represents an independent gene history (Yuan et al., 2009). However, their traditional application using PCR and Sanger sequencing is often difficult because of the need to design primers and test loci for phylogenetic utility in each taxonomic group under study (Hughes et al., 2006; Zimmer and Wen, 2013).
The development of next‐generation sequencing technologies allows for the efficient sequencing of large portions of the genome, including in non‐model taxa (Egan et al., 2012; Twyford and Ennos, 2012; Soltis et al., 2013). Further increases in efficiency in time and cost can be achieved by combining multiplexing techniques with target enrichment, which reduces the proportion of the genome sequenced to subsets that are more likely to be useful (Grover et al., 2012). Various techniques have been developed for target enrichment, including methods based on restriction enzymes, transcriptomes, PCR, and sequence capture (Cronn et al., 2012; McCormack et al., 2013; Lemmon and Lemmon, 2013).
Targeted sequence capture, or hybrid enrichment, isolates target loci from a pool of fragmented genomic DNA using oligonucleotide probes, which can then be sequenced through next‐generation sequencing (Mamanova et al., 2010). Among the strengths of targeted sequence capture are the ability to target known loci, lower stringency in the matching of probes and targets compared to primers for PCR, and the ability to capture sequences even from degraded DNA (Cronn et al., 2012; Lemmon et al., 2012).
Choosing a set of loci to target is an early and crucial part of the sequence capture approach. A broadly used target locus set with universal probes, like ultraconserved elements in amniotes (Faircloth et al., 2012), has not been developed for plants because highly conserved sequences in plants are rare (Reneker et al., 2012; Zheng and Zhang, 2012; but see Buddenhagen et al., 2016). Instead, target locus sets of single‐copy nuclear loci with orthologs across a taxonomic group have been developed individually for groups, which requires multiple genomic and/or transcriptomic resources for taxa in the group and bioinformatic expertise (e.g., Mandel et al., 2014; Nicholls et al., 2015; Stephens et al., 2015; Heyduk et al., 2016). We refer to target locus sets identified with these methods as “taxon‐specific” because they comprise loci that are shown to be single‐copy and have orthologs specifically in the taxa whose genomic information is used. More recently, several pipelines for identifying taxon‐specific target loci that require less bioinformatic skills have been developed (Weitemier et al., 2014; Chamala et al., 2015; Schmickl et al., 2016), but multiple genomic resources are still needed.
Because multiple genomic resources are currently available for only a small number of taxa, and new resources can be expensive and time consuming to generate, the development of general target locus sets applicable in many taxa would facilitate the wider use of targeted sequence capture for plant phylogenomics. Several studies have identified loci that are putatively single‐copy and have orthologs across large clades of plants by examining genome and transcriptome data from distantly related species. These include the conserved ortholog set (COSII) in euasterids (Wu et al., 2006), shared single‐copy nuclear genes (APVO SSC genes) in angiosperms (Duarte et al., 2010), the pentatricopeptide repeat (PPR) gene family in angiosperms (Yuan et al., 2009), other low‐copy nuclear genes conserved across angiosperms (Zhang et al., 2012), and universal markers developed for individual families (e.g., Chapman et al., 2007; Curto et al., 2012). The utility of these general locus sets in comparison with taxon‐specific locus sets in targeted sequence capture and phylogenomics has not been evaluated (but see Granados Mendoza et al., 2015; Buddenhagen et al., 2016; Léveillé‐Bourret et al., 2018).
Once a target locus set is chosen, probes can be designed to capture the target loci. Probes can also be taxon specific or universal. However, because hybridization efficiency depends on sequence similarity between the probe and target (Cronn et al., 2012; Buddenhagen et al., 2016), and highly conserved sequences are rare in plants (Reneker et al., 2012), taxon‐specific probes designed using genomic data from one or more exemplar species in the clade under study usually perform better.
In this study, we identified four locus sets for targeted sequence capture in the genus Buddleja L. (Scrophulariaceae), a clade of 108 species with a crown age of ~20 Ma (Chau, 2017). We used one taxon‐specific set that was identified using genomic and transcriptomic data for two species of Buddleja, and three general sets consisting of COSII, APVO SSC, and PPR loci. We compared the four locus sets selected for probe design and evaluated the performance of the locus sets in Buddleja and outgroups in terms of the assembly of target sequences and sequence variation in loci. We also inferred phylogenetic relationships for Buddleja using concatenation and species tree approaches and compared support for relationships from different locus sets and from previous analyses using a non‐sequence capture data set.
METHODS
Whole‐genome shotgun sequencing of Buddleja globosa
One specimen of Buddleja globosa Hope (Washington Park Arboretum accession number: 179‐99‐A, voucher: R.G. Olmstead 2010‐46 [WTU]), a diploid species with a haploid (1C) genome size of 840 Mbp (Hanson et al., 2001), was selected for whole‐genome shotgun sequencing. DNA was extracted from fresh young leaves using a modified cetyltrimethylammonium bromide (CTAB) protocol (Doyle and Doyle, 1987) and purified through isopropanol precipitation. DNA was diluted to a concentration of 10 ng/μL and sheared by sonication in a Bioruptor (Diagenode Inc., Denville, New Jersey, USA) with a target size of 300 bp. The sequencing library was prepared with an Illumina TruSeq v2 DNA sample preparation kit (Illumina, San Diego, California, USA), and quality was checked with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, USA). The library was sequenced with 100‐bp paired‐end reads on one lane of an Illumina HiSeq 2000 (Illumina) at the QB3 Genomics Sequencing Laboratory at the University of California, Berkeley, USA. Reads were filtered and de novo assembled using CLC Genomics Server 5.0.2 (QIAGEN Bioinformatics, Redwood City, California, USA) with default parameters.
Selection of loci for targeted sequence capture and probe design
We took two approaches to selecting loci that are nuclear, single‐copy, and have orthologs across Buddleja for sequence capture. For the taxon‐specific approach, we used our genomic data for B. globosa and two transcriptomes for B. davidii Franch. (samples GRFT and XRLM) from the 1000 Plants (1KP) initiative (https://sites.google.com/a/ualberta.ca/onekp). Buddleja globosa and B. davidii are in different sections of Buddleja (Chau et al., 2017), so loci found in both species are likely to have orthologs throughout the genus. To select loci, we utilized a modified version of the marker development pipeline Sondovac (Schmickl et al., 2016). Briefly, the pipeline takes genome read data and removes any reads matching a plastome or mitochondriome reference. We used a plastome from Scrophularia takesimensis Nakai (GenBank accession no.: NC_026202), in the same family as Buddleja (Scrophulariaceae), and a mitochondriome from Salvia miltiorrhiza Bunge (GenBank accession no.: NC_023209), in the same order as Buddleja (Lamiales). Sondovac then removes duplicated transcripts from the transcriptome to identify single‐copy loci and finds genome reads matching the remaining unique transcripts, which are de novo assembled. Assembled contigs from genome reads are filtered for length (contig >180 bp, total length of all contigs for a transcript >600 bp) and uniqueness. Remaining contigs are compiled as target sequences. We used the pipeline separately with each of the two transcriptomes of B. davidii and then combined target sequences. Sequences with >90% sequence similarity were identified using cd‐hit‐est (Li and Godzik, 2006), and the longest sequence in each cluster was retained.
Our other approach for locus selection utilized three general locus sets, which comprise loci that are putatively single‐copy and have orthologs across large clades of plants. COSII were identified in the euasterid clade by comparing expressed sequence tag databases for several species of euasterids (in Solanum, Capsicum, and Coffea) and Arabidopsis thaliana (L.) Heynh. (Wu et al., 2006). Sequences for a subset of 369 COSII genes in Solanum lycopersicum L. were downloaded from the Sol Genomics Network (ftp://ftp.sgn.cornell.edu/COSII/Rasmus_s_cleantomatoseq.fasta). Duarte et al. (2010) identified a set of 959 APVO SSC genes shared broadly in angiosperms by comparing genome sequences of three eudicots (Arabidopsis, Populus, and Vitis) and one monocot (Oryza). Yuan et al. (2009) identified 127 loci in the PPR gene family that are single‐copy and intronless in both A. thaliana and Oryza sativa L. Coding sequences for APVO SSC and PPR loci in A. thaliana were downloaded from The Arabidopsis Information Resource (http://www.arabidopsis.org). Sequences in B. globosa for loci in each of the three locus sets were compiled by conducting BLASTN searches of sequences from Solanum or Arabidopsis against the assembled B. globosa contigs. Up to five of the top hits with a bit score greater than 70 were retained. For some loci, there were hits that overlapped from different contigs, and these were assembled using de novo assembly in Geneious v9.1.6 (Biomatters, Auckland, New Zealand). If hits in an assembly had pairwise identity <95%, all hits matching that locus were removed because this was inferred to be evidence of the presence of paralogs. Remaining hits and consensus sequences were filtered by length (individual sequence >120 bp, total length of all sequences for a locus >600 bp).
Filtered target sequences from all four locus sets were combined. For any sequences with >90% sequence similarity to another, only the longest sequence was retained as the target sequence using cd‐hit‐est (Li and Godzik, 2006). Some sequences from different locus sets had significant overlap and were assumed to be from the same locus. These were assembled using de novo assembly in Geneious v9.1.6 if pairwise identity >95%, and consensus sequences were used as the target sequences.
One probe set targeting all four locus sets was designed and manufactured by RAPiD Genomics (Gainesville, Florida, USA). Biotinylated RNA probes were 120 bp with 2× tiling density over target sequences. Additional checks were performed to eliminate probes targeting multi‐copy loci: probes with more than 10 hits to the assembled B. globosa genome or with more than 100 matching raw B. globosa reads were discarded.
Taxon sampling for targeted sequence capture
Fifty samples were chosen for targeted sequence capture and sequencing (Appendix 1). We sampled 46 species of Buddleja, including 21 of 24 species in section Alternifoliae, which had poor resolution in previous phylogenetic analyses with a seven‐locus data set (Chau et al., 2017), and at least one representative from each of the other sections of Buddleja. Additionally, we tested the performance of our probes in more distantly related outgroups. We included Teedia lucida Rudolphi (Scrophulariaceae), a member of the sister group to Buddleja; Scrophularia nodosa L. (Scrophulariaceae), in the same family as Buddleja; and Parmentiera aculeata (Kunth) Seem. (Bignoniaceae) and Lantana leonardiorum Moldenke (Verbenaceae), in the same order (Lamiales) as Buddleja. We also wanted to examine the effectiveness of this method for museum samples, so we included eight samples with DNA extracted from herbarium specimens.
DNA extraction, sequence capture, and sequencing
DNA was extracted from dried leaf tissue, either preserved in silica gel or from a herbarium specimen, using a modified CTAB protocol and purified by isopropanol precipitation. DNA was run on 1% agarose gels to assess DNA quality. DNA concentration was measured with a Qubit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Samples were diluted or concentrated to attain a concentration of 50 ng/μL, where possible, although some samples had concentrations as low as 2 ng/μL. Volumes of 35 to 50 μL were submitted for library preparation.
Library preparation, sequence capture, and sequencing (Capture‐Seq) were done by RAPiD Genomics. For each sample, 250 to 1000 ng of genomic DNA, where available, was fragmented to a target size of 400 bp. DNA from herbarium specimens was not additionally fragmented if gel images showed that the DNA was already degraded. DNA libraries were constructed by end‐repairing the sheared DNA, A‐tailing and adapter ligation, barcoding, and PCR amplification. Libraries were pooled in equimolar ratios by ploidy of species, and probes were hybridized to pools to enrich for targets. Enriched pools were combined in equimolar ratios for sequencing, and 100‐bp paired‐end reads were sequenced on ~16% of one lane of an Illumina HiSeq 3000 (Illumina).
Read processing and assembly
De‐multiplexed reads were obtained from RAPiD Genomics. Sequence quality was checked using FastQC v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Using modified scripts from the pipeline SqCL (https://github.com/singhal/SqCL), Trimmomatic 0.36 (Bolger et al., 2014) was used to remove adapters, barcodes, and poor quality bases using the setting LEADING:20 TRAILING:20 SLIDINGWINDOW:5:20 MINLEN:36.
Remaining paired and unpaired reads were assembled using the pipeline HybPiper (Johnson et al., 2016). Briefly, the reads_first.py script sorts reads by target sequence for each sample using the Burrows–Wheeler Alignment tool (BWA) (Li and Durbin, 2009), assembles mapped reads for each target sequence using the assembler SPAdes (Bankevich et al., 2012), and finally extracts the coding sequence from the assembled contig using Exonerate (Slater and Birney, 2005). For the target reference sequences used by BWA, we used our target sequences for probe design.
Assembled coding sequences from each sample were compiled and sorted by target sequence using the HybPiper script retrieve_sequences.py. If multiple long‐length contigs were assembled by SPAdes for a target sequence for a sample, one contig was chosen based on higher sequencing coverage depth or higher percent identity to the reference sequence. Data on lengths of assembled coding sequences for each target sequence and statistics on assembly efficiency were calculated using the scripts get_seq_lengths.py and hybpiper_stats.py. Multiple long‐length contigs for a target sequence for a sample, which may represent paralogs, were identified using the script paralog_investigator.py.
After removing two samples with failed sequencing, differences among locus sets for Buddleja samples in the average percentage of target sequences with an assembled sequence and in the average percentage of the total target length assembled were evaluated with one‐way analysis of variance tests blocked by sample and Tukey multiple comparison tests conducted in R (R Core Team, 2015).
Phylogenetic analyses
Any samples with assembled coding sequences for less than 50% of target sequences were excluded from further analyses. A custom script (Appendix S1) was used to filter out loci with missing data or with paralogs in any remaining sample.
For each sequence set, sequences were aligned with MAFFT (Katoh and Standley, 2013) using default parameters. Sites with more than 50% missing data were removed from alignments using the “clean” function in Phyutility (Smith and Dunn, 2008). Concatenated alignments were generated for each of the four locus sets using the “concat” function in Phyutility.
Because a target locus might comprise multiple target sequences, we created concatenated alignments for each locus using the “concat” function in Phyutility. The percentage of identical sites was calculated for each locus in Geneious v9.1.6 (Biomatters). Differences among locus sets in the average percentage of identical sites were evaluated with one‐way analysis of variance tests and Tukey multiple comparison tests in R.
Maximum likelihood (ML) trees were inferred for the concatenated alignments for each locus set using RAxML v8.0.7 (Stamatakis, 2014). We searched for the best‐scoring ML tree and conducted 100 rapid bootstraps. Data sets were unpartitioned, and we used the GTR + gamma model of rate heterogeneity.
Concatenated alignments were also used to infer species trees using singular value decomposition scores for species quartets (SVDquartets) (Chifman and Kubatko, 2014) in PAUP* v4.0a157 (Swofford, 2003). All possible quartets were evaluated. Trees were selected using Quartet FM (QFM) quartet assembly (Raez et al., 2014), and the multispecies coalescent tree model was used. Ambiguities were distributed. For each analysis, 100 bootstraps were performed.
RESULTS
Whole‐genome shotgun sequencing of Buddleja globosa
One lane of sequencing produced 292,788,924 100‐bp paired reads. After filtering, 257,538,046 reads (88%) remained and were used in the de novo assembly. Of these, 246,968,396 reads (84.4%) were assembled into 311,304 contigs that had a total length of 343,339,138 bp. Contigs ranged in length from 118 to 166,512 bp and had an N50 of 2390 bp. Raw reads are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject ID PRJNA419550, SRA SRP125846). Assembled contigs are available in the Dryad Digital Repository (available at https://doi.org/10.5061/dryad.v6q0p; Chau et al., 2018).
Locus sets for targeted sequence capture
In total, 2906 target sequences from 1049 loci with a total length of 1,010,028 bp were submitted for probe design (Table 1). Of these, 1880 target sequences from 708 loci with a total length of 580,437 bp were identified in Buddleja using the taxon‐specific approach. The remaining 1026 target sequences from 341 loci with a total length of 429,591 bp were in the three general locus sets. There were 67 COSII loci, 162 APVO SSC loci, and 112 PPR loci. The average target locus length was higher in the three general sets (COSII: 1119 bp, APVO SSC: 1079 bp, PPR: 1605 bp) than in the taxon‐specific set (820 bp). Target locus sequences are available in the Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p; Chau et al., 2018).
Table 1.
Characteristics of target locus sets for probe design
| Locus sets | Total target sequencesa | Total target locib | Total target length (bp) | Average target sequence length (bp) | Average target locus length (bp) |
|---|---|---|---|---|---|
| Taxon‐specific | 1880 | 708 | 580,437 | 309 | 820 |
| General | 1034 | 344 | 431,226 | 419 | 1260 |
| COSII | 280 | 67 | 74,988 | 268 | 1119 |
| APVO SSC | 572 | 162 | 174,848 | 306 | 1079 |
| PPR | 174 | 112 | 179,755 | 1033 | 1605 |
| Total | 2906 | 1049 | 1,010,028 | 348 | 963 |
A target sequence is a single consensus sequence from reads mapped to a target locus. There may be multiple target sequences for a target locus if sequences do not overlap.
A target locus is from a single transcript (taxon‐specific approach) or a single gene (general approach).
DNA quality, sequence capture, and sequencing
All but one of our samples (B. rinconensis (Mayfield) J. H. Chau) had at least 250 ng of DNA for library preparation (Appendix 2). All DNA from silica gel–preserved tissues were of high molecular weight. DNA from herbarium specimen tissue varied in quality, but most were degraded (Appendix S2).
Of our 50 samples, 48 were successfully sequenced (Appendix S2). One sample (B. rinconensis) had a very low amount of DNA available and produced no mapped reads. Another sample (B. macrostachya Benth.) had sufficient starting DNA, but sequencing failed for unknown reasons. For the 48 remaining samples, between 372,898 and 4,963,618 paired reads were produced (Appendix 2). There were no issues with read quality when checked in FastQC. On average, 96% of reads were retained after trimming for low‐quality bases and adapter and barcode sequences. Raw reads are available in the NCBI Sequence Read Archive (BioProject ID PRJNA419999, SRA SRP125765).
Read assembly
For each sample, between 44% and 49% of total reads were mapped to the target sequences (Appendix 2). For species of Buddleja, the HybPiper pipeline produced assembled coding sequences for 91% to 99% of all target sequences. For outgroups, the number of target sequences with assemblies decreased with increasing phylogenetic distance from Buddleja. For other Scrophulariaceae, 76% to 90% of target sequences had assemblies. For other families, 24% to 47% of target sequences had assemblies.
All locus sets had high assembly success in Buddleja samples, with an average of 94% to 98% of target sequences with assemblies (Table 2). Significant differences between locus sets were found in the average percentage of target sequences with assemblies (P < 0.01) and the average percentage of total target length assembled (P < 0.01). The PPR and taxon‐specific locus sets had the highest success, with an average of 98% of target sequences having an assembly for both data sets. The COSII locus set had the lowest success, with an average of 94% of sequences having an assembly.
Table 2.
Average performance of locus sets in assembly for 44 Buddleja samples (excludes two Buddleja samples with failed sequencing) and four outgroup samples.a
| Locus sets | Buddleja | Outgroup | ||
|---|---|---|---|---|
| No. of sequences | Total length | No. of sequences | Total length | |
| Taxon‐specific | 1845 (98%)A | 567,161 (98%)Y | 992 (53%) | 287,344 (50%) |
| General | 984 (95%) | 421,307 (98%) | 733 (71%) | 312,062 (72%) |
| COSII | 264 (94%)C | 72,207 (96%)Z | 184 (66%) | 48,390 (65%) |
| APVO SSC | 549 (96%)B | 170,224 (97%)Y | 425 (74%) | 130,148 (74%) |
| PPR | 171 (98%)A | 178,876 (100%)X | 124 (71%) | 133,524 (74%) |
| Total | 2829 (97%) | 988,468 (98%) | 1724 (59%) | 599,405 (59%) |
Shown are the average number of target sequences with assembled coding sequence and the average total length of assembled coding sequences. In parentheses are the percentages of total target sequences used for probe design. Superscript letters show significant differences in averages at the 0.05 level among locus sets for Buddleja samples from Tukey multiple comparison tests with blocking by sample.
For the four outgroup samples, there was a larger difference between assembly of taxon‐specific and general locus sets, with greater assembly success in general locus sets (Table 2). On average, 50% of taxon‐specific target sequences had assemblies, whereas 65% to 74% of target sequences in general locus sets had assemblies.
Samples from herbarium specimens performed similarly to samples from silica‐preserved tissue (Appendices 1 and 2). The average percentage of targets with assembled sequences was 96.1% in our seven samples from herbarium specimens with adequate DNA for library preparation versus 97.6% in samples from silica‐preserved tissue.
Sequence filtering for phylogenetic analyses
We used 44 samples in Buddleja and two outgroups in Scrophulariaceae with high assembly success in phylogenetic analyses. Of the 2906 target sequences, 1524 did not have an assembled coding sequence for at least one sample and 538 had paralogous sequences for at least one sample. These were removed from further analyses under our filtering criteria. Thus, 800 taxon‐specific target sequences and 400 general target sequences remained for phylogenetic analyses (Table 3).
Table 3.
Characteristics of assembled sequence data sets used for phylogenetic analyses.a
| Locus sets | Total sequencesb | Total loci | Average sequence length (bp) | Average locus length (bp) | Average total length: unaligned (bp) | Total length: aligned, trimmed (bp) | Average % variable sites |
|---|---|---|---|---|---|---|---|
| Taxon‐specific | 800 (43%) | 511 (72%) | 336 | 526 | 268,710 (46%) | 268,603 | 36.07%b |
| General | 400 (39%) | 261 (76%) | 605 | 928 | 242,161 (56%) | 242,359 | 30.55% |
| COSII | 82 (29%) | 50 (75%) | 346 | 567 | 28,332 (38%) | 28,380 | 27.96%B |
| APVO SSC | 217 (38%) | 128 (79%) | 429 | 728 | 93,194 (53%) | 93,253 | 28.56%B |
| PPR | 101 (58%) | 83 (74%) | 1194 | 1453 | 120,635 (67%) | 120,726 | 35.17%b |
| Total | 1200 (41%) | 772 (74%) | 425 | 661 | 510,579 (51%) | 510,962 | 34.20% |
Sequences with missing data or paralogous sequences in any sample out of 44 Buddleja samples and two outgroups used were removed from data sets. In parentheses are the percentages of total target sequences used for probe design. Superscript letters in the last column show significant differences in averages at the 0.05 level among locus sets from a Tukey multiple comparison test.
A sequence is assembled to a single target sequence. There may be multiple target sequences for a target locus if target sequences do not overlap.
The PPR locus set had the largest percentage of target sequences (58%) remaining after filtering, whereas the COSII locus set had the smallest percentage (29%). The taxon‐specific locus set had an intermediate percentage of target sequences (43%) retained, although the absolute number of taxon‐specific target loci (511) was higher than in any of the three general sets (50–128). The PPR locus set had the longest average length of filtered assembled loci (1453 bp), whereas the taxon‐specific locus set had the shortest average length (526 bp).
The PPR and taxon‐specific loci showed the greatest sequence variability (i.e., lowest percentage of identical sites). Variable sites comprised 35.17% and 36.07%, respectively, of the locus sequences on average, which was significantly higher than the average percentages for the other two general locus sets (P < 0.01). The COSII loci had the lowest percentage of variable sites (27.96%).
The trimmed concatenated alignments had a total length of 268,710 bp for the taxon‐specific locus set and 28,332 to 120,635 bp for the general locus sets. Concatenated alignments for each locus set are available in the Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p; Chau et al., 2018) and TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931).
Phylogenetic analyses
ML analyses with different locus sets produced trees with different levels of node support (Appendix 3). The tree from the taxon‐specific data set was substantially more well‐supported, with 93% of nodes with bootstrap support (BP) ≥ 90% (Fig. 1). Trees from general data sets had 61% to 75% of nodes with BP ≥ 90%, with the tree from COSII sequences being the least well‐supported. In section Alternifoliae, 100% of nodes had BP ≥ 90% with the taxon‐specific data set, and 78% to 89% of nodes had such high support with general data sets. Among ML trees from different locus sets, there were several well‐supported topological differences, including in the positions of species B. asiatica, B. alternifolia, B. crispa, and B. myriantha in section Alternifoliae.
Figure 1.
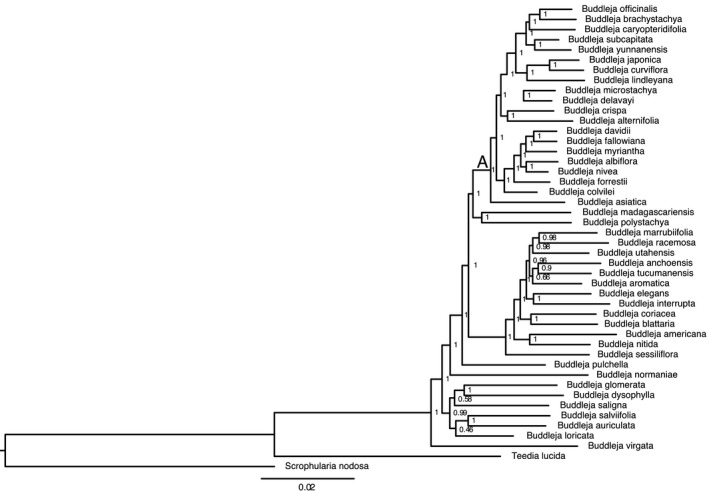
Maximum likelihood phylogram from RAxML analysis with concatenated sequences from taxon‐specific locus set. Values at nodes indicate bootstrap support. (A) indicates node subtending clade of section Alternifoliae.
The SVDquartets analyses produced trees with similar topologies to the ML analyses but with less support at the nodes (Appendix 4). Support varied from 65% of nodes with BP ≥ 90% in the tree from PPR sequences to 47% of nodes with BP ≥ 90% in the tree from COSII sequences. Topological incongruencies between trees from ML and SVDquartets analyses generally occurred at nodes weakly supported in the SVDquartets trees. All tree files are available in TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931).
DISCUSSION
Comparison of locus sets
We were able to develop a substantially greater number of taxon‐specific target loci than general target loci. This was not unexpected because closely related species, which were used to develop the taxon‐specific locus set, are expected to share more loci than distantly related species, which were used to develop the general locus sets.
Recovery of assembled coding sequences for our target sequences was high overall. In Buddleja, no sample had less than 80% of target sequences with an assembled sequence for any locus set. Although statistically significant differences were found among average assembly success in different locus sets, the difference was not large, ranging from 94% to 98%. Outgroups showed a different pattern, with general locus sets consistently outperforming the taxon‐specific locus set. This pattern is consistent with the fact that the taxon‐specific locus set was designed using genomic resources in Buddleja, thus it is unknown whether these loci are single‐copy or even present in taxa outside Buddleja. Conversely, the general locus sets include loci that have a high probability of being single‐copy and having orthologs across angiosperms or other large clades. Recovery efficiency of general loci should be affected primarily by the extent of sequence divergence between the probes designed using Buddleja genome data and the target sequences in other taxa. Although recovery of assembled sequences was lower overall for the outgroup taxa, even for one of the most distant outgroups, Parmentiera aculeata, a species in a different family that diverged from Buddleja approximately 53 Ma (Magallón et al., 2015), 56% to 72% of target sequences in the general locus sets were recovered. The other species in a different family, Lantana leonardiorum, had a lower assembly efficiency overall, which may be because the quantity of DNA available for library preparation was less than recommended for this sample. The general locus set that performed best varied among the different outgroup taxa. The APVO SSC locus set had the highest percentage of target sequences with an assembly in three outgroup taxa, whereas the PPR locus set had the highest percentage in one taxon.
Regarding phylogenetic informativeness, the loci in our taxon‐specific and PPR locus sets had significantly higher average percentages of variable sites than the COSII and APVO SSC locus sets. The PPR loci also had the greatest average length. In our phylogenetic trees from ML analyses with concatenated data, the taxon‐specific locus set produced the tree with the highest percentage of well‐supported nodes, probably because the taxon‐specific concatenated alignment is more than twice the length of the longest general locus data set. The three general locus sets still produced trees with at least 60% of nodes with high support in the ML analyses and at least 47% in the SVDquartets analyses. For section Alternifoliae, previous ML analyses using four nuclear loci and three plastid loci resolved only four nodes (22%) with BP ≥90% (Chau et al., 2017). With targeted sequence capture data sets, at least 78% of nodes had high support in ML analyses and at least 61% in SVDquartets analyses.
Incongruence was present among phylogenetic trees from different locus sets and different analysis methods, although many discordant relationships had low support. These may represent instances of rapid diversification or hybridization. Using phylogenetic methods that incorporate the multispecies coalescent will help account for incomplete lineage sorting and other processes that can mislead inference methods using concatenated data (Degnan and Rosenberg, 2009).
Recommendations for use of targeted sequence capture in plant phylogenomics
In groups where multiple genomic resources do not exist to design a taxon‐specific locus set for sequence capture, using general locus sets is a suitable alternative. In our ingroup, both general and taxon‐specific locus sets had high assembly success. In our outgroups, recovery of sequences was higher in general locus sets than in the taxon‐specific locus set. Information content of general locus sets was high, increasing resolution and support of phylogenetic relationships versus non‐sequence capture data sets. Although the total number of loci and total sequence length will likely be lower in general locus sets than in a taxon‐specific set, dozens to hundreds of loci can potentially still be targeted, which may be sufficient to resolve relationships (Leaché and Rannala, 2011).
Even in groups where taxon‐specific locus sets can be designed, researchers may consider adding general locus sets to their sequence capture targets. In addition to having greater or comparable assembly efficiency and information content per locus, utilizing general locus sets can facilitate the combination of data from different studies because known loci can be used in different taxa. In particular, the PPR loci have other characteristics that make them desirable for phylogenetic analysis, including a lack of introns that facilitates unambiguous alignment (Yuan et al., 2009). The PPR loci, with their greater average length and high proportion of variable sites, are also suited to the inference of well‐supported gene trees, which are necessary for a number of species tree methods (e.g., Accurate Species TRee ALgorithm [ASTRAL]; Mirarab and Warnow, 2015).
Designing a general target locus set for a group does not require multiple genomic resources, but some source of genomic sequence data is still necessary to design probes whose sequences will adequately complement targets in the group of interest. The most closely related taxon whose genome is available should be used, but in our study, probes designed for a different genus or different family from a sample were still able to recover 56% to 95% of sequences in general locus sets given adequate starting DNA. Many genomic resources for plants are now publicly available for probe design, including genomes (e.g., Phytozome; https://phytozome.jgi.doe.gov/pz/portal.html) and transcriptomes (e.g., 1KP initiative; https://sites.google.com/a/ualberta.ca/onekp/).
Targeted sequence capture is a suitable method even for samples from herbarium specimens or those that otherwise have degraded DNA. In our study, sequence recovery was nearly the same in our herbarium samples as in our silica‐preserved samples. For several of these herbarium samples, PCR amplification of low‐copy nuclear loci had not been successful (unpublished data), but the targeted sequence capture method generated large amounts of sequence data suitable for phylogenetic analyses.
Assembly of sequencing reads can be accomplished with a number of different programs and pipelines. Although HybPiper successfully generated assembled coding sequences for the vast majority of target sequences, it did not assemble separate contigs for paralogs of target sequences where they were expected to occur in polyploid species (data not shown). For groups where polyploidization or hybridization are important parts of the evolutionary history, testing of other assembly methods is suggested.
CONCLUSIONS
We show in this study that targeted sequence capture using general or taxon‐specific locus sets is an effective method for gathering sequence data for phylogenetic studies that can significantly increase resolution and support of relationships versus non‐sequence capture data sets consisting of only several loci. General target loci are simpler and require fewer resources to develop, but can be as effective as taxon‐specific loci in terms of assembly success and phylogenetic informativeness. Applying general locus sets widens the opportunity to use targeted sequence capture in more plant groups with few genomic resources.
DATA ACCESSIBILITY
Raw reads from whole genome shotgun sequencing of Buddleja globosa are available in the NCBI Sequence Read Archive, BioProject IDPRJNA419550, SRA SRP125846 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP125846). Assembled contigs from whole genome shotgun sequencing of Buddleja globosa are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p). Target locus sequences in Buddleja globosa used for probe design for targeted sequence capture are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p). Raw reads from sequencing after targeted sequence capture for 50 samples are available in the NCBI Sequence Read Archive, BioProject ID PRJNA419999, SRA SRP125765 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP125765). Concatenated alignments for each target locus set are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p) and TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931). Tree files from maximum likelihood and SVDquartets analyses are available on TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931).
Supporting information
ACKNOWLEDGMENTS
The authors thank A. Leaché, T. Gill, and J. Grummer for technical assistance in preparing genomic libraries, the use of laboratory and computing equipment, and advice on genomic laboratory techniques and phylogenomic analyses; P. Lu‐Irving for spearheading the initial foray into genomic work; M. Deyholos, D. Soltis, and the 1KP initiative for providing access to transcriptome data; and funding from the National Science Foundation (no. DEB‐1311111 [Doctoral Dissertation Improvement Grant] and no. DEB‐1020369).
APPENDIX 1. Specimens used for targeted sequence capture, with voucher information, infrageneric or familial classification, and sample source (herbarium specimen or silica gel preserved).
| Species | Voucher (Herbarium)a | Section in Buddleja, or family if outside Buddleja | Herbarium sample? |
|---|---|---|---|
| Buddleja albiflora Hemsl. | J. Chau 260 (WTU, A) | Alternifoliae | No |
| Buddleja alternifolia Maxim. | J. Chau 262 (WTU, A) | Alternifoliae | No |
| Buddleja americana L. | L. Frost 148 (WTU) | Buddleja | No |
| Buddleja anchoensis Kuntze | J. Chau 224 (WTU, LPB) | Buddleja | No |
| Buddleja aromatica J. Rémy | J. Chau 206 (WTU, LPB) | Buddleja | No |
| Buddleja asiatica Lour. | J. Chau 157 (WTU) | Alternifoliae | No |
| Buddleja auriculata Benth. | J. Chau 246 (WTU) | Chilianthus | No |
| Buddleja blattaria J. F. Macbr. | J. Chau 101 (WTU) | Buddleja | No |
| Buddleja brachystachya Diels | (KUN 22547)b | Alternifoliae | Yes |
| Buddleja caryopteridifolia W. W. Sm. | J. Chau 171 (WTU) | Alternifoliae | No |
| Buddleja colvilei Hook. f. | J. Chau 42 (WTU) | Alternifoliae | No |
| Buddleja coriacea J. Rémy | J. Chau 194 (WTU, LPB) | Buddleja | No |
| Buddleja crispa Benth. | J. Chau 170 (WTU) | Alternifoliae | No |
| Buddleja curviflora Hook. & Arn. | R. Olmstead 2010‐49 (WTU) | Alternifoliae | No |
| Buddleja davidii Franch. | J. Chau 177 (WTU) | Alternifoliae | No |
| Buddleja delavayi L. F. Gagnep. | J. Chau 165 (WTU) | Alternifoliae | No |
| Buddleja dysophylla (Benth.) Radlk. | J. Chau 233 (WTU) | Chilianthus | No |
| Buddleja elegans Cham. & Schltdl. subsp. elegans | R. Olmstead 2010‐214 (ICN) | Buddleja | No |
| Buddleja fallowiana Balf. f. & W. W. Sm. | J. Chau 166 (WTU) | Alternifoliae | No |
| Buddleja forrestii Diels | J. Chau 161 (WTU) | Alternifoliae | No |
| Buddleja glomerata H. Wendl. | J. Chau 254 (WTU) | incertae sedis | No |
| Buddleja interrupta Kunth | J. Chau 123 (WTU) | Buddleja | No |
| Buddleja japonica Hemsl. | J. Wood 124‐2014 (A) | Alternifoliae | No |
| Buddleja lindleyana Fortune | J. Wood & K. Richardson 125‐2014 (A) | Alternifoliae | No |
| Buddleja loricata Leeuwenberg | J. Chau 253 (WTU) | Chilianthus | No |
| Buddleja macrostachya Benth. | J. Chau 159 (WTU) | Alternifoliae | No |
| Buddleja madagascariensis Lam. | J. Chau 256 (WTU) | Nicodemia | No |
| Buddleja marrubiifolia Benth. | M. Moore 1567 (WTU, MEXU) | Buddleja | No |
| Buddleja microstachya E. D. Liu & H. Peng | E. Liu 925 (KUN) | Alternifoliae | Yes |
| Buddleja myriantha Diels | J. Chau 158 (WTU) | Alternifoliae | No |
| Buddleja nitida Benth. | J. Chau 150 (WTU) | Buddleja | No |
| Buddleja nivea Duthie | R. Olmstead 2010‐47 (WTU) | Alternifoliae | No |
| Buddleja normaniae J. H. Chau | D. Riskind 23860 (TEX) | Buddleja | Yes |
| Buddleja officinalis Maxim. | J. Chau 179 (WTU) | Alternifoliae | No |
| Buddleja polystachya Fresen. | G. Simon 308 (MO) | Nicodemia | Yes |
| Buddleja pulchella N. E. Br. | I. Nanni 319 (NBG) | Pulchellae | Yes |
| Buddleja racemosa Torr. | J. Chau 324 (WTU) | Buddleja | No |
| Buddleja rinconensis (Mayfield) J. H. Chau | S. Aguilar Ruiz 164 (TEX) | Buddleja | Yes |
| Buddleja saligna Willd. | J. Chau 231 (WTU) | Chilianthus | No |
| Buddleja salviifolia (L.) Lam. | J. Chau 240 (WTU) | Salviifoliae | No |
| Buddleja sessiliflora Kunth | G. Webster 31455 (DAV) | Buddleja | Yes |
| Buddleja subcapitata E. D. Liu & H. Peng | H. Peng 5153 (KUN) | Alternifoliae | Yes |
| Buddleja tucumanensis Griseb. | J. Chau 212 (WTU, LPB) | Buddleja | No |
| Buddleja utahensis Coville | J. Chau 322 (WTU) | Buddleja | No |
| Buddleja virgata L. f. | J. Chau 180 (WTU) | Gomphostigma | No |
| Buddleja yunnanensis L. F. Gagnep. | J. Chau 178 (WTU) | Alternifoliae | No |
| Teedia lucida Rudolphi | J. Chau 318 (WTU) | Scrophulariaceae | No |
| Scrophularia nodosa L. | J. Chau 228 (WTU) | Scrophulariaceae | No |
| Parmentiera aculeata (Kunth) Seem. | S. Grose 93 (WTU) | Bignoniaceae | No |
| Lantana leonardiorum Moldenke | P. Lu‐Irving 2012‐105 (WTU) | Verbenaceae | No |
Herbaria acronyms are per Index Herbariorum (http://sweetgum.nybg.org/science/ih/).
Collector unknown. Herbarium accession number provided.
APPENDIX 2. Amount of DNA available for library preparation, sequencing output, and assembly efficiency for each sample used in targeted sequence capture.
| Sample | Total DNA (ng) | Total raw reads | Total trimmed reads (% of raw) | Total mapped reads (% of raw) | Total target sequences with assembled coding sequence (% of total target sequences) | Total length of assembled coding sequences for all target sequences (% of total target sequence length) | Taxon‐specific target sequences with assembled coding sequence (% of total taxon‐specific target sequences) | COSII target sequences with assembled coding sequence (% of total COSII target sequences) | APVO SSC target sequences with assembled coding sequence (% of total APVO SSC target sequences) | PPR target sequences with assembled coding sequence (% of total PPR target sequences) | Total length of assembled coding sequences for taxon‐specific target sequences (% of total taxon‐specific target sequence length) | Total length of assembled coding sequences for COSII target sequences (% of total COSII target sequence length) | Total length of assembled coding sequences for APVO SSC target sequences (% of total APVO SSC target sequence length) | Total length of assembled coding sequences for PPR target sequences (% of total PPR target sequence length) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buddleja albiflora | 2439 | 2,327,360 | 2,237,793 (96%) | 1,074,864 (46%) | 2867 (99%) | 1,005,867 (100%) | 1862 (99%) | 271 (97%) | 561 (98%) | 173 (99%) | 578,979 (100%) | 73,416 (98%) | 172,050 (98%) | 181,422 (101%) |
| Buddleja alternifolia | 2635 | 1,929,508 | 1,846,378 (96%) | 882,612 (46%) | 2814 (97%) | 985,218 (98%) | 1834 (98%) | 263 (94%) | 546 (95%) | 171 (98%) | 564,198 (97%) | 72,330 (96%) | 169,830 (97%) | 178,860 (100%) |
| Buddleja americana | 2097 | 1,903,722 | 1,836,865 (96%) | 885,319 (47%) | 2817 (97%) | 985,491 (98%) | 1844 (98%) | 259 (93%) | 543 (95%) | 171 (98%) | 566,616 (98%) | 71,232 (95%) | 168,741 (97%) | 178,902 (100%) |
| Buddleja anchoensis | 1332 | 1,541,712 | 1,485,689 (96%) | 714,972 (46%) | 2799 (96%) | 981,540 (97%) | 1842 (98%) | 253 (90%) | 534 (93%) | 170 (98%) | 564,861 (97%) | 70,035 (93%) | 168,069 (96%) | 178,575 (99%) |
| Buddleja aromatica | 2158 | 2,039,000 | 1,974,719 (97%) | 954,485 (47%) | 2845 (98%) | 999,882 (99%) | 1858 (99%) | 264 (94%) | 550 (96%) | 173 (99%) | 577,983 (100%) | 72,150 (96%) | 170,835 (98%) | 178,914 (100%) |
| Buddleja asiatica | 2629 | 1,783,174 | 1,711,379 (96%) | 820,709 (46%) | 2823 (97%) | 987,711 (98%) | 1840 (98%) | 265 (95%) | 547 (96%) | 171 (98%) | 566,256 (98%) | 72,336 (96%) | 170,622 (98%) | 178,497 (99%) |
| Buddleja auriculata | 1964 | 2,157,144 | 2,065,826 (96%) | 988,506 (46%) | 2856 (98%) | 992,868 (98%) | 1856 (99%) | 268 (96%) | 559 (98%) | 173 (99%) | 568,677 (98%) | 72,948 (97%) | 172,215 (98%) | 179,028 (100%) |
| Buddleja blattaria | 1656 | 1,447,556 | 1,386,802 (96%) | 663,387 (46%) | 2775 (95%) | 979,239 (97%) | 1812 (96%) | 259 (93%) | 534 (93%) | 170 (98%) | 558,126 (96%) | 71,358 (95%) | 168,093 (96%) | 181,662 (101%) |
| Buddleja brachystachya | 279 | 3,683,050 | 3,638,140 (99%) | 1,795,017 (49%) | 2820 (97%) | 987,081 (98%) | 1851 (98%) | 261 (93%) | 537 (94%) | 171 (98%) | 569,202 (98%) | 71,568 (95%) | 167,901 (96%) | 178,410 (99%) |
| Buddleja caryopteridifolia | 1446 | 2,052,498 | 1,979,901 (96%) | 954,372 (46%) | 2834 (98%) | 988,725 (98%) | 1851 (98%) | 260 (93%) | 551 (96%) | 172 (99%) | 567,498 (98%) | 71,904 (96%) | 170,613 (98%) | 178,710 (99%) |
| Buddleja colvilei | 1944 | 1,853,250 | 1,740,264 (94%) | 823,349 (44%) | 2817 (97%) | 983,865 (97%) | 1840 (98%) | 264 (94%) | 545 (95%) | 168 (97%) | 563,940 (97%) | 72,054 (96%) | 169,746 (97%) | 178,125 (99%) |
| Buddleja coriacea | 1690 | 1,498,552 | 1,432,719 (96%) | 683,814 (46%) | 2789 (96%) | 978,015 (97%) | 1827 (97%) | 257 (92%) | 536 (94%) | 169 (97%) | 560,007 (96%) | 70,983 (95%) | 168,309 (96%) | 178,716 (99%) |
| Buddleja crispa | 2714 | 2,530,742 | 2,444,030 (97%) | 1,179,451 (47%) | 2866 (99%) | 999,414 (99%) | 1862 (99%) | 269 (96%) | 564 (99%) | 171 (98%) | 574,776 (99%) | 73,164 (98%) | 172,704 (99%) | 178,770 (99%) |
| Buddleja curviflora | 1163 | 1,733,384 | 1,664,926 (96%) | 798,999 (46%) | 2815 (97%) | 987,141 (98%) | 1837 (98%) | 258 (92%) | 550 (96%) | 170 (98%) | 567,492 (98%) | 70,995 (95%) | 169,938 (97%) | 178,716 (99%) |
| Buddleja davidii | 1454 | 2,063,608 | 1,966,719 (95%) | 935,958 (45%) | 2855 (98%) | 992,937 (98%) | 1858 (99%) | 271 (97%) | 553 (97%) | 173 (99%) | 570,000 (98%) | 73,335 (98%) | 170,886 (98%) | 178,716 (99%) |
| Buddleja delavayi | 2422 | 2,207,958 | 2,111,416 (96%) | 1,008,612 (46%) | 2856 (98%) | 996,837 (99%) | 1861 (99%) | 267 (95%) | 555 (97%) | 173 (99%) | 573,270 (99%) | 72,663 (97%) | 171,246 (98%) | 179,658 (100%) |
| Buddleja dysophylla | 1201 | 2,937,822 | 2,830,220 (96%) | 1,362,500 (46%) | 2860 (98%) | 991,176 (98%) | 1855 (99%) | 270 (96%) | 561 (98%) | 174 (100%) | 566,532 (98%) | 73,176 (98%) | 172,419 (99%) | 179,049 (100%) |
| Buddleja elegans | 2856 | 1,197,350 | 1,160,891 (97%) | 561,792 (47%) | 2634 (91%) | 947,574 (94%) | 1760 (94%) | 229 (82%) | 480 (84%) | 165 (95%) | 545,775 (94%) | 66,249 (88%) | 158,847 (91%) | 176,703 (98%) |
| Buddleja fallowiana | 1916 | 2,138,936 | 2,051,696 (96%) | 983,118 (46%) | 2847 (98%) | 992,382 (98%) | 1860 (99%) | 267 (95%) | 549 (96%) | 171 (98%) | 570,099 (98%) | 72,942 (97%) | 170,547 (98%) | 178,794 (99%) |
| Buddleja forrestii | 1292 | 2,462,226 | 2,361,212 (96%) | 1,131,268 (46%) | 2857 (98%) | 998,178 (99%) | 1857 (99%) | 269 (96%) | 558 (98%) | 173 (99%) | 574,725 (99%) | 72,873 (97%) | 171,513 (98%) | 179,067 (100%) |
| Buddleja glomerata | 528 | 2,369,896 | 2,271,643 (96%) | 1,088,307 (46%) | 2854 (98%) | 988,509 (98%) | 1851 (98%) | 271 (97%) | 561 (98%) | 171 (98%) | 565,074 (97%) | 72,657 (97%) | 172,119 (98%) | 178,659 (99%) |
| Buddleja interrupta | 1813 | 1,267,806 | 1,206,474 (95%) | 572,778 (45%) | 2791 (96%) | 977,706 (97%) | 1836 (98%) | 254 (91%) | 529 (92%) | 172 (99%) | 560,952 (97%) | 70,728 (94%) | 167,193 (96%) | 178,833 (99%) |
| Buddleja japonica | 2759 | 2,113,824 | 2,018,901 (96%) | 964,282 (46%) | 2813 (97%) | 987,528 (98%) | 1838 (98%) | 259 (93%) | 545 (95%) | 171 (98%) | 566,673 (98%) | 71,724 (96%) | 169,590 (97%) | 179,541 (100%) |
| Buddleja lindleyana | 2914 | 2,451,874 | 2,376,319 (97%) | 1,151,239 (47%) | 2836 (98%) | 993,366 (98%) | 1845 (98%) | 264 (94%) | 557 (97%) | 170 (98%) | 570,303 (98%) | 71,826 (96%) | 172,080 (98%) | 179,157 (100%) |
| Buddleja loricata | 1140 | 2,786,804 | 2,670,518 (96%) | 1,279,501 (46%) | 2877 (99%) | 995,751 (99%) | 1860 (99%) | 279 (100%) | 564 (99%) | 174 (100%) | 568,935 (98%) | 74,520 (99%) | 172,779 (99%) | 179,517 (100%) |
| Buddleja macrostachya | 2729 | 7148 | 2191 (31%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Buddleja madagascariensis | 1851 | 2,601,922 | 2,463,259 (95%) | 1,165,370 (45%) | 2873 (99%) | 996,018 (99%) | 1868 (99%) | 271 (97%) | 564 (99%) | 170 (98%) | 571,761 (99%) | 72,933 (97%) | 172,755 (99%) | 178,569 (99%) |
| Buddleja marrubiifolia | 1338 | 1,551,386 | 1,477,662 (95%) | 703,133 (45%) | 2835 (98%) | 989,523 (98%) | 1853 (99%) | 268 (96%) | 544 (95%) | 170 (98%) | 568,347 (98%) | 72,963 (97%) | 169,788 (97%) | 178,425 (99%) |
| Buddleja microstachya | 1854 | 4,963,618 | 4,864,924 (98%) | 2,381,190 (48%) | 2891 (99%) | 1,007,031 (100%) | 1874 (100%) | 275 (98%) | 568 (99%) | 174 (100%) | 580,857 (100%) | 73,947 (99%) | 173,415 (99%) | 178,812 (99%) |
| Buddleja myriantha | 2604 | 2,099,840 | 2,007,289 (96%) | 958,589 (46%) | 2850 (98%) | 991,965 (98%) | 1862 (99%) | 266 (95%) | 551 (96%) | 171 (98%) | 570,192 (98%) | 72,762 (97%) | 170,481 (98%) | 178,530 (99%) |
| Buddleja nitida | 2435 | 2,274,192 | 2,189,504 (96%) | 1,052,472 (46%) | 2852 (98%) | 992,940 (98%) | 1854 (99%) | 268 (96%) | 559 (98%) | 171 (98%) | 569,679 (98%) | 72,636 (97%) | 171,603 (98%) | 179,022 (100%) |
| Buddleja nivea | 3077 | 2,043,774 | 1,961,831 (96%) | 941,137 (46%) | 2845 (98%) | 993,144 (98%) | 1857 (99%) | 267 (95%) | 550 (96%) | 171 (98%) | 570,342 (98%) | 72,723 (97%) | 170,832 (98%) | 179,247 (100%) |
| Buddleja normaniae | 555 | 1,761,248 | 1,723,674 (98%) | 843,232 (48%) | 2715 (93%) | 953,595 (94%) | 1767 (94%) | 250 (89%) | 533 (93%) | 165 (95%) | 538,533 (93%) | 70,212 (94%) | 167,562 (96%) | 177,288 (99%) |
| Buddleja officinalis | 2421 | 2,847,288 | 2,723,927 (96%) | 1,302,699 (46%) | 2874 (99%) | 998,727 (99%) | 1868 (99%) | 272 (97%) | 561 (98%) | 173 (99%) | 573,645 (99%) | 73,590 (98%) | 172,356 (99%) | 179,136 (100%) |
| Buddleja polystachya | 1762 | 2,852,964 | 2,753,780 (97%) | 1,328,296 (47%) | 2855 (98%) | 993,105 (98%) | 1856 (99%) | 266 (95%) | 560 (98%) | 173 (99%) | 569,733 (98%) | 72,453 (97%) | 171,951 (98%) | 178,968 (100%) |
| Buddleja pulchella | 813 | 4,035,056 | 3,975,394 (99%) | 1,955,353 (48%) | 2855 (98%) | 996,246 (99%) | 1863 (99%) | 267 (95%) | 552 (97%) | 173 (99%) | 573,279 (99%) | 72,552 (97%) | 171,012 (98%) | 179,403 (100%) |
| Buddleja racemosa | 1934 | 1,686,714 | 1,606,473 (95%) | 764,316 (45%) | 2852 (98%) | 991,569 (98%) | 1856 (99%) | 268 (96%) | 555 (97%) | 173 (99%) | 568,218 (98%) | 72,747 (97%) | 171,498 (98%) | 179,106 (100%) |
| Buddleja rinconensis | 53 | 16,406 | 7280 (44%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Buddleja saligna | 1691 | 3,294,204 | 3,184,952 (97%) | 1,539,951 (47%) | 2874 (99%) | 995,121 (99%) | 1862 (99%) | 275 (98%) | 565 (99%) | 172 (99%) | 569,283 (98%) | 73,962 (99%) | 172,983 (99%) | 178,893 (100%) |
| Buddleja salviifolia | 1310 | 2,476,222 | 2,361,059 (95%) | 1,124,310 (45%) | 2855 (98%) | 992,979 (98%) | 1855 (99%) | 269 (96%) | 560 (98%) | 171 (98%) | 568,737 (98%) | 73,218 (98%) | 172,296 (99%) | 178,728 (99%) |
| Buddleja sessiliflora | 458 | 865,438 | 836,550 (97%) | 404,077 (47%) | 2568 (88%) | 930,696 (92%) | 1719 (91%) | 227 (81%) | 458 (80%) | 164 (94%) | 532,899 (92%) | 66,048 (88%) | 154,200 (88%) | 177,549 (99%) |
| Buddleja subcapitata | 489 | 2,879,700 | 2,796,956 (97%) | 1,357,879 (47%) | 2844 (98%) | 991,950 (98%) | 1860 (99%) | 265 (95%) | 549 (96%) | 170 (98%) | 571,227 (98%) | 72,093 (96%) | 169,941 (97%) | 178,689 (99%) |
| Buddleja tucumanensis | 1704 | 2,650,974 | 2,568,142 (97%) | 1,242,037 (47%) | 2862 (98%) | 1,002,648 (99%) | 1867 (99%) | 269 (96%) | 556 (97%) | 170 (98%) | 580,179 (100%) | 72,552 (97%) | 171,471 (98%) | 178,446 (99%) |
| Buddleja utahensis | 1306 | 1,811,458 | 1,744,501 (96%) | 838,026 (46%) | 2840 (98%) | 990,624 (98%) | 1848 (98%) | 266 (95%) | 553 (97%) | 173 (99%) | 567,846 (98%) | 72,684 (97%) | 171,354 (98%) | 178,740 (99%) |
| Buddleja virgata | 1369 | 3,910,310 | 3,770,843 (96%) | 1,817,916 (46%) | 2862 (98%) | 985,062 (98%) | 1843 (98%) | 278 (99%) | 567 (99%) | 174 (100%) | 558,453 (96%) | 74,376 (99%) | 173,133 (99%) | 179,100 (100%) |
| Buddleja yunnanensis | 2234 | 2,246,266 | 2,147,602 (96%) | 1,025,852 (46%) | 2861 (98%) | 995,664 (99%) | 1856 (99%) | 271 (97%) | 562 (98%) | 172 (99%) | 570,924 (98%) | 73,509 (98%) | 172,338 (99%) | 178,893 (100%) |
| Teedia lucida | 2529 | 1,669,018 | 1,606,112 (96%) | 773,791 (46%) | 2620 (90%) | 915,036 (91%) | 1652 (88%) | 263 (94%) | 543 (95%) | 162 (93%) | 498,321 (86%) | 71,223 (95%) | 168,546 (96%) | 176,946 (98%) |
| Scrophularia nodosa | 2099 | 1,921,010 | 1,837,964 (96%) | 880,448 (46%) | 2210 (76%) | 758,007 (75%) | 1309 (70%) | 229 (82%) | 524 (92%) | 148 (85%) | 378,360 (65%) | 58,569 (78%) | 157,818 (90%) | 163,260 (91%) |
| Parmentiera aculeata | 2227 | 1,271,530 | 1,232,236 (97%) | 598,303 (47%) | 1359 (47%) | 469,827 (47%) | 684 (36%) | 156 (56%) | 409 (72%) | 110 (63%) | 187,875 (32%) | 39,657 (53%) | 121,932 (70%) | 120,363 (67%) |
| Lantana leonardiorum | 391 | 372,898 | 350,065 (94%) | 164,457 (44%) | 707 (24%) | 254,751 (25%) | 322 (17%) | 86 (31%) | 225 (39%) | 74 (43%) | 84,819 (15%) | 24,111 (32%) | 72,294 (41%) | 73,527 (41%) |
APPENDIX 3. Maximum likelihood phylograms from RAxML analyses with concatenated sequences from individual locus sets: (A) taxon‐specific, (B) conserved ortholog set (COSII), (C) shared single‐copy nuclear (APVO SSC) genes, and (D) pentatricopeptide repeat (PPR) genes. Values at nodes indicate bootstrap support.

APPENDIX 4. Best trees from SVDquartets analyses with concatenated sequences from individual locus sets: (A) taxon‐specific, (B) conserved ortholog set (COSII), (C) shared single‐copy nuclear (APVO SSC) genes, and (D) pentatricopeptide repeat (PPR) genes. Values at nodes indicate bootstrap support.

Chau, J. H. , Rahfeldt W. A., and Olmstead R. G.. 2018. Comparison of taxon‐specific versus general locus sets for targeted sequence capture in plant phylogenomics. Applications in Plant Sciences 6(3): e1032.
LITERATURE CITED
- Bankevich, A. , Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., Lesin V. M., et al. 2012. SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddenhagen, C. , Lemmon A. R., Lemmon E. M., Bruhl J., Cappa J., Clement W. L., Donoghue M. J., et al. 2016. Anchored phylogenomics of angiosperms I: Assessing the robustness of phylogenetic estimates. bioRxiv https://doi.org/10.1101/086298. [Google Scholar]
- Chamala, S. , García N., Godden G. T., Krishnakumar V., Jordon‐Thaden I. E., De Smet R., Barbazuk W. B., et al. 2015. MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Applications in Plant Sciences 3: 1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M. A. , Chang J. C., Weisman D., Kesseli R. V., and Burke J. M.. 2007. Universal markers for comparative mapping and phylogenetic analysis in the Asteraceae (Compositae). Theoretical and Applied Genetics 115: 747–755. [DOI] [PubMed] [Google Scholar]
- Chau, J. H. 2017. Systematics of Buddleja (Scrophulariaceae): Phylogenetic relationships, historical biogeography, and phylogenomics. Ph.D. dissertation, University of Washington, Seattle, Washington, USA.
- Chau, J. H. , O'Leary N., Sun W.‐B., and Olmstead R. G.. 2017. Phylogenetic relationships in tribe Buddlejeae (Scrophulariaceae) based on multiple nuclear and plastid markers. Botanical Journal of the Linnean Society 184: 137–166. [Google Scholar]
- Chau, J. H. , Rahfeldt W. A., and Olmstead R. G.. 2018. Data from: Comparison of taxon‐specific versus general locus sets for targeted sequence capture in plant phylogenomics. Dryad Digital Repository https://doi.org/10.5061/dryad.v6q0p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman, J. , and Kubatko L.. 2014. Quartet inference from SNP data under the coalescent. Bioinformatics 30: 3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn, R. , Knaus B. J., Liston A., Maughan P. J., Parks M., Syring J. V., and Udall J.. 2012. Targeted enrichment strategies for next‐generation plant biology. American Journal of Botany 99: 291–311. [DOI] [PubMed] [Google Scholar]
- Curto, M. A. , Puppo P., Ferreria D., Nogueira M., and Meimberg H.. 2012. Development of phylogenetic markers from single‐copy nuclear genes for multi locus, species level analyses in the mint family (Lamiaceae). Molecular Phylogenetics and Evolution 63: 758–767. [DOI] [PubMed] [Google Scholar]
- Degnan, J. H. , and Rosenberg N. A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology and Evolution 24: 332–340. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , and Doyle J. L.. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Duarte, J. M. , Wall P. K., Edger P. P., Landherr L. L., Ma H., Pires J. C., Leebens‐Mack J., et al. 2010. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evolutionary Biology 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, A. N. , Schlueter J., and Spooner D. M.. 2012. Applications of next‐generation sequencing in plant biology. American Journal of Botany 99: 175–185. [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. , McCormack J. E., Crawford N. G., Harvey M. G., Brumfeld R. T., and Glenn R. C.. 2012. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Systematic Biology 61: 717–726. [DOI] [PubMed] [Google Scholar]
- Granados Mendoza, C. , Naumann J., Samain M.‐S., Goetghebeur P., De Smet Y., and Wanke S.. 2015. A genomic‐scale mining strategy for recovering novel rapidly‐evolving nuclear single‐copy genes for addressing shallow‐scale phylogenetics in Hydrangea . BMC Evolutionary Biology 15: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover, C. E. , Salmon A., and Wendel J. F.. 2012. Target sequence capture as a powerful tool for evolutionary analysis. American Journal of Botany 99: 312–319. [DOI] [PubMed] [Google Scholar]
- Hanson, L. , McMahon K. A., Johnson M. A. T., and Bennett M. D.. 2001. First nuclear DNA C‐values for 25 angiosperm families. Annals of Botany 87: 251–258. [DOI] [PubMed] [Google Scholar]
- Heyduk, K. , Trapnell D. W., Barrett C. F., and Leebens‐Mack J.. 2016. Phylogenomic analyses of species relationships in the genus Sabal (Arecaceae) using targeted sequence capture. Biological Journal of the Linnean Society 117: 106–120. [Google Scholar]
- Hughes, C. E. , Eastwood R. J., and Bailey C. D.. 2006. From famine to feast? Selecting nuclear DNA sequence loci for plant species‐level phylogeny reconstruction. Philosophical Transactions of the Royal Society B 361: 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Gardner E. M., Liu Y., Medina R., Goffinet B., Shaw A. J., Zerega N. J. C., et al. 2016. HybPiper: Extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Applications in Plant Sciences 4: 1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaché, A. D. , and Rannala B.. 2011. The accuracy of species tree estimation under simulation: A comparison of methods. Systematic Biology 60: 126–137. [DOI] [PubMed] [Google Scholar]
- Lemmon, A. R. , Emme S. A., and Lemmon E. M.. 2012. Anchored hybrid enrichment for massively high‐throughput phylogenomics. Systematic Biology 61: 727–744. [DOI] [PubMed] [Google Scholar]
- Lemmon, E. M. , and Lemmon A. R.. 2013. High‐throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics 44: 99–121. [Google Scholar]
- Léveillé‐Bourret, É. , Starr J. R., Ford B. A., Lemmon E. M., and Lemmon A. R.. 2018. Resolving rapid radiations within angiosperm families using anchored phylogenomics. Systematic Biology 67: 94–112. [DOI] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , and Godzik A.. 2006. Cd‐hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Maddison, W. P. 1997. Gene trees in species trees. Systematic Biology 46: 523–536. [Google Scholar]
- Madriñan, A. , Cortés A. J., and Richardson J. E.. 2013. Páramo is the world's fastest evolving and coolest biodiversity hotspot. Frontiers in Genetics 4: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallón, S. , Gómez‐Acevedo S., Sánchez‐Reyes L. L., and Hernández‐Hernández T.. 2015. A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist 207: 437–453. [DOI] [PubMed] [Google Scholar]
- Mamanova, L. , Coffey A. J., Scott C. E., Kozarewa I., Turner E. H., Kumar A., Howard E., et al. 2010. Target‐enrichment strategies for next‐generation sequencing. Nature Methods 7: 111–118. [DOI] [PubMed] [Google Scholar]
- Mandel, J. R. , Dikow R. B., Funk V. A., Masalia R. R., Staton S. E., Kozik A., Michelmore R. W., et al. 2014. A target enrichment method for gathering phylogenetic information from hundreds of loci: An example from the Compositae. Applications in Plant Sciences 2: 1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, J. E. , Hird S. M., Zellmer A. J., Carstens B. C., and Brumfield R. T.. 2013. Applications of next‐generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution 66: 526–538. [DOI] [PubMed] [Google Scholar]
- Mirarab, S. , and Warnow T.. 2015. ASTRAL‐II: Coalescent‐based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31: i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, J. A. , Pennington R. T., Koenen E. J. M., Hughes C. E., Hearn J., Bunnefeld L., Dexter K. G., et al. 2015. Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Frontiers in Plant Science 6: 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2015. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Available at http://www.R-project.org/ [accessed 26 May 2017]. [Google Scholar]
- Raez, R. , Bayzid M. S., and Rahman M. S.. 2014. Accurate phylogenetic tree reconstruction from quartets: A heuristic approach. PLoS One 9: e104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneker, J. , Lyons E., Conant G. C., Pires J. C., Freeling M., Shyu C.‐R., and Korkin D.. 2012. Long identical multispecies elements in plant and animal genomes. Proceedings of the National Academy of Sciences USA 109: E1183–E1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas, A. , Williams B. L., King N., and Carroll S. B.. 2003. Genome‐scale approaches to resolving incongruence in molecular phylogenetics. Nature 425: 798–804. [DOI] [PubMed] [Google Scholar]
- Schluter, D. 2000. The ecology of adaptive radiation. Oxford University Press, New York, New York, USA. [Google Scholar]
- Schmickl, R. , Liston A., Zeisek V., Oberlander K., Weitemier K., Straub S. C. K., Cronn R. C., et al. 2016. Phylogenetic marker development for target enrichment from transcriptomic and genome skim data: The pipeline and its application in southern African Oxalis (Oxalidaceae). Molecular Ecology Resources 16: 1124–1135. [DOI] [PubMed] [Google Scholar]
- Slater, G. , and Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, R. L. , Cronn R. C., and Wendel J. F.. 2004. Use of nuclear genes for phylogeny reconstruction in plants. Australian Systematic Botany 17: 145–170. [Google Scholar]
- Smith, S. A. , and Dunn C. W.. 2008. Phyutility: A phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24: 715–716. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E. , Gitzendanner M. A., Stull G., Chester M., Chanderbali A., Chamala S., Jordon‐Thaden I., et al. 2013. The potential of genomics in plant systematics. Taxon 62: 886–898. [Google Scholar]
- Stamatakis, A. 2014. RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, J. D. , Rogers W. L., Mason C. M., Donovan L. A., and Malmberg R. L.. 2015. Species tree estimation of diploid Helianthus (Asteraceae) using target enrichment. American Journal of Botany 102: 910–920. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L. 2003PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland, Massachusetts, USA. [Google Scholar]
- Twyford, A. D. , and Ennos R. A.. 2012. Next‐generation sequencing as a tool for plant ecology and evolution. Plant Ecology and Diversity 5: 411–413. [Google Scholar]
- Weitemier, K. , Straub S. C. K., Cronn R. C., Fishbein M., Schmickl R., McDonnell A., and Liston A.. 2014. Hyb‐Seq: Combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences 2: 1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Mueller L. A., Crouzillat D., Pétiard V., and Tanksley S. D.. 2006. Combining bioinformatics and phylogenetics to identify large sets of single‐copy orthologous genes (COSII) for comparative evolutionary and systematic studies: A test case in the euasterid plant clade. Genetics 174: 1407–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Y.‐W. , Liu C., Marx H. E., and Olmstead R. G.. 2009. The pentatricopeptide repeat (PPR) gene family, a tremendous resource for plant phylogenetic studies. New Phytologist 182: 272–283. [DOI] [PubMed] [Google Scholar]
- Zhang, N. , Zeng L., Shan H., and Ma H.. 2012. Highly conserved low‐copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytologist 195: 923–937. [DOI] [PubMed] [Google Scholar]
- Zheng, W.‐X. , and Zhang C.‐T.. 2012. Ultraconserved elements between the genomes of the plants Arabidopsis thaliana and rice. Journal of Biomolecular Structure and Dynamics 26: 1–8. [DOI] [PubMed] [Google Scholar]
- Zimmer, E. A. , and Wen J.. 2013. Using nuclear gene data for plant phylogenetics: Progress and prospects. Molecular Phylogenetics and Evolution 65: 774–785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads from whole genome shotgun sequencing of Buddleja globosa are available in the NCBI Sequence Read Archive, BioProject IDPRJNA419550, SRA SRP125846 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP125846). Assembled contigs from whole genome shotgun sequencing of Buddleja globosa are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p). Target locus sequences in Buddleja globosa used for probe design for targeted sequence capture are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p). Raw reads from sequencing after targeted sequence capture for 50 samples are available in the NCBI Sequence Read Archive, BioProject ID PRJNA419999, SRA SRP125765 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP125765). Concatenated alignments for each target locus set are available on Dryad Digital Repository (https://doi.org/10.5061/dryad.v6q0p) and TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931). Tree files from maximum likelihood and SVDquartets analyses are available on TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S21931).