

We develop a novel synthetic biology platform for rapid, scalable expression of fungal biosynthetic genes and encoded metabolites.
Abstract
For decades, fungi have been a source of U.S. Food and Drug Administration–approved natural products such as penicillin, cyclosporine, and the statins. Recent breakthroughs in DNA sequencing suggest that millions of fungal species exist on Earth, with each genome encoding pathways capable of generating as many as dozens of natural products. However, the majority of encoded molecules are difficult or impossible to access because the organisms are uncultivable or the genes are transcriptionally silent. To overcome this bottleneck in natural product discovery, we developed the HEx (Heterologous EXpression) synthetic biology platform for rapid, scalable expression of fungal biosynthetic genes and their encoded metabolites in Saccharomyces cerevisiae. We applied this platform to 41 fungal biosynthetic gene clusters from diverse fungal species from around the world, 22 of which produced detectable compounds. These included novel compounds with unexpected biosynthetic origins, particularly from poorly studied species. This result establishes the HEx platform for rapid discovery of natural products from any fungal species, even those that are uncultivable, and opens the door to discovery of the next generation of natural products.
INTRODUCTION
Natural products are indispensable to modern medicine, with 73% of antibiotics, 49% of anticancer compounds, and 32% of all new drugs approved by the U.S. Food and Drug Administration between 1980 and 2012 being natural products or derivatives thereof (1). Fungi are prolific producers of therapeutically relevant natural products (2, 3), having yielded penicillin, the first widely used antibiotic; cyclosporine, the immunosuppressant that enabled widespread organ transplantation; and lovastatin, the progenitor of the statin class of cholesterol-lowering drugs. In all of these examples, compounds were isolated from laboratory cultures of single fungal isolates. Recent advances in genome sequencing have revealed that more than 5 million fungal species likely exist on Earth (4), with each species encoding as many as 80 natural product biosynthetic pathways (5, 6). However, despite the increased ease of DNA sequencing, fungal cultivation remains a bottleneck: only a fraction of the fungi in any given environmental sample have been cultured under laboratory conditions (7). Even within cultured species, the majority of biosynthetic gene clusters (BGCs) present in the genome are either transcriptionally silent or expressed at very low levels (8). The identification and expression of these BGCs thus present a major opportunity for the discovery of novel natural products.
Previous approaches for surveying transcriptionally silent, or cryptic, fungal BGCs for the production of novel compounds have included BGC activation within the native host through promoter or transcription factor manipulation (9–11), CRISPR-based genome editing (12), and epigenetic activation (13–15). These approaches, however, are limited to those BGCs whose native hosts are both culturable and genetically tractable. For cryptic BGCs within the genomes of several aspergilli, heterologous expression by cloning of large intact contigs into Aspergillus nidulans has yielded several new natural products (16).
Heterologous expression by complete BGC refactoring is an approach that is agnostic to the native host of a BGC, permitting access to cryptic BGCs from potentially any organism (17–19). We present HEx, an improved, scalable approach to heterologous expression of cryptic fungal BGCs (Fig. 1). HEx includes a set of bioinformatic tools to identify and prioritize BGCs in genome data; genetic tools to refactor BGCs for expression in Saccharomyces cerevisiae; S. cerevisiae background strains with improved growth and expression phenotypes; and synthetic biology tools to assemble and express synthetic DNA in the heterologous host (Figs. 2 to 4). Strains expressing BGCs were analyzed via untargeted metabolomics (20); if the compound appeared novel, select full structures were solved using liquid chromatography–mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR).
Fig. 1. Standard workflow for heterologous expression.
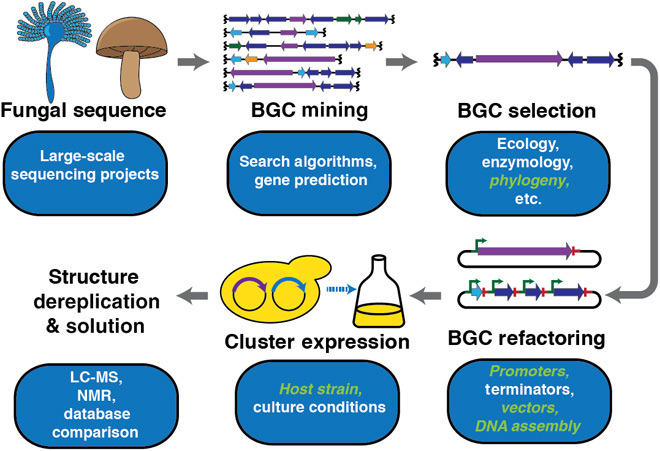
Aspects in green italics are addressed in this study.
Fig. 2. Tools developed for the HEx platform.

(A) eGFP expression from a series of PADH2-like promoters in cultures grown under both fermentative (YPD) and respiratory (YPE) conditions. All fluorescence intensities are reported as the mean of three biological replicates. Error bars represent 1 SD (n = 3). (B) Four fungal BGCs, two controls and two previously uncharacterized systems, each produce improved titers when heterologously expressed using PADH2-like promoters as compared to strong constitutive promoters. ND, not detected. Error bars represent 1 SD (n = 3). Quantitation for TC1 and TC3 was based on the sum of the integrations of extracted ion counts corresponding to the oxidized sesquiterpenoids outlined in table S4.
Fig. 4. DNA assembly by yeast homologous recombination.

(A) DNA assembly from commercially synthesized fragments and genetic parts using yeast homologous recombination. (B) Modified yeast plasmid preparation (exo+) leads to increased number of sequencing reads mapping to plasmid DNA. Dotted line marks the efficiency threshold to allow sequencing of 192 samples on a single MiSeq run. (C) Efficient assembly of up to 14 unique DNA parts can be achieved using the protocol outlined here. Data based on 78 unique assemblies.
Previous reports of complete pathway refactoring for expression in fungal hosts have demonstrated the utility of this approach but have been limited to the study of a single cluster (21–25). Here, we applied HEx to the expression of 41 cryptic fungal BGCs, 22 (54%) of which resulted in compounds not natively present in yeast (Figs. 5 and 6, Table 1, and Supplementary Text). The 41 BGCs were derived from diverse fungal species and include genes encoding either a membrane-bound, UbiA-like terpene cyclase (UTCs) (26) or a polyketide synthase (PKS) enzyme at their core. Two interesting biosynthetic insights were revealed from this study. First, UTCs represent a general class of biosynthetic enzymes present in a variety of both ascomycete and basidiomycete genomes. Second, a divergent form of PKSs identified in some basidiomycetes has the unusual property of incorporating amino acids in the absence of any nonribosomal peptide synthetase (NRPS) enzymes.
Fig. 5. PKS BGCs examined for this study.

All putative gene function abbreviations are listed in table S9. Cladogram was constructed as described in Materials and Methods. All plots are the chromatograms of the specified extracted ion in three biological replicates each of both the strain expressing the BGC and an empty vector control strain. Chromatograms are data collected with electrospray ionization in either positive (ESI+), negative (ESI−), or rapid polarity switching (RPS) mode or with multimode ionization in positive mode (MMI). Expression strains are outlined in table S10, and EICs of novel products are shown in the figs. S7 to S23.
Fig. 6. UbiA-type cyclases represent a general class of biosynthetic enzymes.
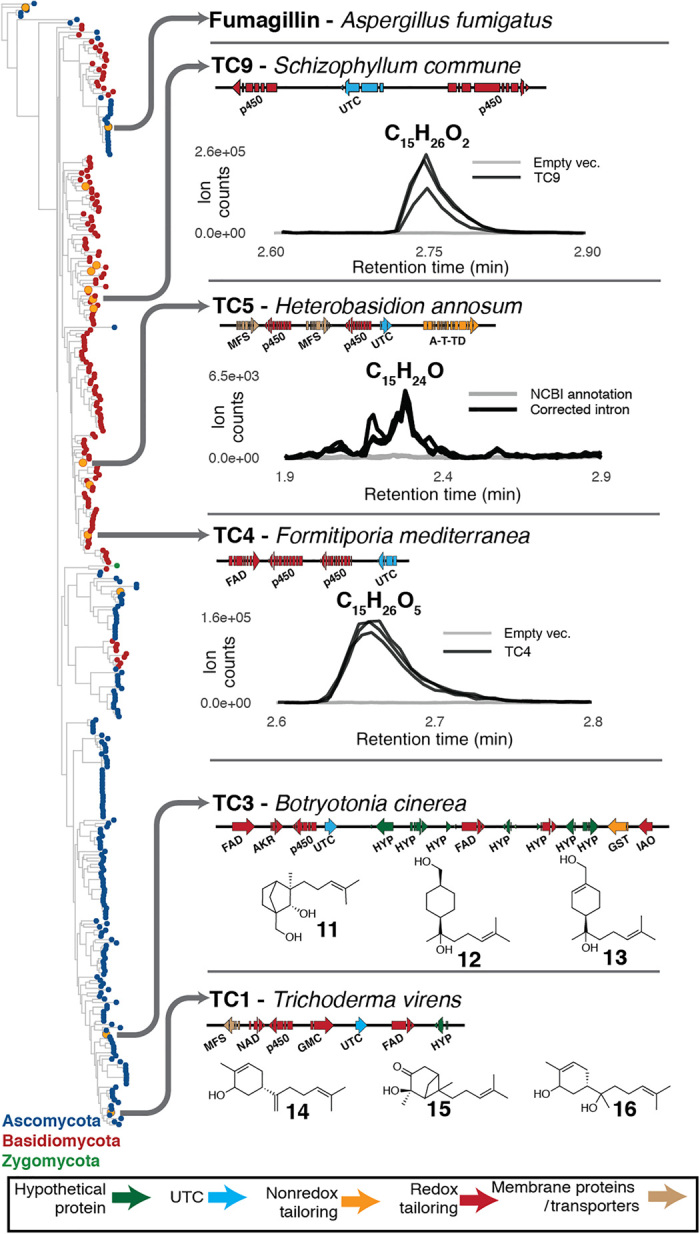
Putative enzyme activity abbreviations are listed in table S9. Cladogram generated using UTC cyclase sequence. The cyclases associated with all clusters examined in this study are denoted by orange tips in the cladogram.
Table 1. Summary of control and cryptic fungal BGCs examined in this study.
| ID | Type | Species of origin | Division | Productive? |
| IDT | Ctl | Aspergillus tubingensis | Ascomycota | Y |
| DHZ | Ctl | Hypomyces subiculosus | Ascomycota | Y |
| PKS1 | PKS | Coniothyrium sporulosum | Ascomycota | Y |
| PKS2 | PKS | Coniothyrium sporulosum | Ascomycota | Y |
| PKS3 | PKS | Acremonium Sp. KY4917 | Ascomycota | N |
| PKS4 | PKS | Aspergillus niger | Ascomycota | Y |
| PKS5 | PKS | Thielavia terrestris | Ascomycota | N |
| PKS6 | PKS | Trichoderma virens | Ascomycota | Y |
| PKS7 | PKS | Pseudogymnoascus pannorum | Ascomycota | N |
| PKS8 | PKS | Scedosporium apiospermum | Ascomycota | Y |
| PKS9 | PKS | Metarhizium anisopliae | Ascomycota | N |
| PKS10 | PKS | Cochliobolus heterostropus | Ascomycota | Y |
| PKS11 | PKS | Pseudogymnoascus pannorum | Ascomycota | N |
| PKS12 | PKS | Pseudogymnoascus pannorum | Ascomycota | N |
| PKS13 | PKS | Pseudogymnoascus pannorum | Ascomycota | Y |
| PKS14 | PKS | Verruconis gallopava | Ascomycota | Y |
| PKS15 | PKS | Moniliophthora roreri | Basidiomycota | Y |
| PKS16 | PKS | Punctularia strigosozonata | Basidiomycota | Y |
| PKS17 | PKS | Hydnomerulius pinastri | Basidiomycota | Y |
| PKS18 | PKS | Arthroderma gypseum | Ascomycota | Y |
| PKS19 | PKS | Setosphaeria turcica | Ascomycota | N |
| PKS20 | PKS | Pyrenophora teres | Ascomycota | Y |
| PKS21 | PKS | Cladophialophora yegresii | Ascomycota | N |
| PKS22 | PKS | Talaromyces cellulolyticus | Ascomycota | Y |
| PKS23 | PKS | Endocarpon pusillum | Ascomycota | Y |
| PKS24 | PKS | Talaromyces cellulolyticus | Ascomycota | Y |
| PKS25 | PKS | Moniliophthora roreri | Basidiomycota | N |
| PKS26 | PKS | Hypholoma sublateritium | Basidiomycota | N |
| PKS27 | PKS | Ceriporiopsis subvermispora | Basidiomycota | N |
| PKS28 | PKS | Moniliophthora roreri | Basidiomycota | Y |
| TC1 | UTC | Trichderma Virens | Ascomycota | Y |
| TC2 | UTC | Trichderma Virens | Ascomycota | N |
| TC3 | UTC | Botryotonia cinerea | Ascomycota | Y |
| TC4 | UTC | Formitiporia mediterranea | Basidiomycota | Y |
| TC5 | UTC | Heterobasidion annosum | Basidiomycota | Y |
| TC6 | UTC | Gelatoporia subvermispora | Basidiomycota | N |
| TC7 | UTC | Dichomitus squalens | Basidiomycota | N |
| TC8 | UTC | Pleurotus ostreatus | Basidiomycota | N |
| TC9 | UTC | Schizophyllum commune | Basidiomycota | Y |
| TC10 | UTC | Stereum hirsutum | Basidiomycota | N |
| TC11 | UTC | Stereum hirsutum | Basidiomycota | N |
| TC12 | UTC | Dichomitus squalens | Basidiomycota | N |
| TC13 | UTC | Dacryopinax primogenitus | Basidiomycota | N |
| Total 43 Productive 24 | ||||
Such a large-scale study of cryptic fungal natural products has not been possible until now and was enabled through recent breakthroughs in DNA sequencing and DNA synthesis, combined with the breakthroughs in the heterologous host developed here in the HEx platform. These results reveal that the unstudied fungal sequences accumulating in genome databases can be functionally characterized in a scalable way with HEx, enabling the production of novel molecules that have never been observed in nature and paving the way for discovery of the next generation of natural products throughout the fungal kingdom.
Results and Discussion
Characterization of genetic parts
HEx is enabled by a new panel of yeast promoters that we selected and characterized to satisfy two distinct design criteria. First, the expression of BGC genes should be regulatable and coordinated. Second, each promoter sequence should be sufficiently unique so as to be compatible with homologous recombination–based DNA assembly techniques. Small panels of promoters with fewer than four members that meet these criteria have been previously reported (27–30) but were insufficient for the expression of our chosen BGCs, which range from 3 to 14 genes in size.
Previous studies have demonstrated the utility of the yeast ADH2 promoter (PADH2) for the heterologous expression of a variety of biosynthetic genes (31–33). PADH2, which is repressed by glucose, is inactive during fermentative growth, with activation occurring only after diauxic shift. Thus, PADH2 is auto-inducible (34) in media containing glucose and other fermentable carbon sources that are converted to nonfermentable carbon sources.
To allow for the assembly and coordinated, auto-inducible expression of entire BGCs on a small number of plasmids, we identified and characterized a panel of sequence-divergent promoters functionally similar to PADH2. First, we identified 48 genes within a published S. cerevisiae transcriptome data set (35) that appeared coregulated with ADH2 in that their transcripts were weakly expressed in mid-log phase fermentative growth, but were highly abundant during respiration (table S2). We then constructed single-copy chromosomal integrations of the corresponding promoters driving expression of a green fluorescent protein (GFP) gene and measured fluorescence. Four promoters (PADH2, PMLS1, PPCK1, and PICL2) demonstrated the desired delayed induction phenotype with expression levels after 24 hours being similar to or greater than PTDH3 or PFBA1, two commonly used strong constitutive promoters (Fig. 2A) (36, 37). We also identified 23 promoters that were coregulated with PADH2, but whose degree of induction was slightly or significantly lower, thus providing opportunities for lower levels of coordinated gene expression (Fig. 2A, fig. S1, A to C, and table S2). Among these 20 promoters, no two have greater than 29% identity, demonstrating that these sequences are sufficiently unique to allow assembly using homology-based approaches. In addition, we characterized the promoters of the closest ADH2 homolog in each of five close relatives of S. cerevisiae, the sensu stricto Saccharomyces species (fig. S1, D to F). Three of these promoters (from Saccharomyces paradoxus, Saccharomyces bayanus, and Saccharomyces kudriavzevii) were functionally equivalent to the S. cerevisiae homolog (Fig. 2A), bringing the number of PADH2-like promoters to 30, with 7 having strengths at least equal to strong constitutive promoters. We refer to these PADH2-like promoters as the HEx promoters.
To study the utility of these promoters for BGC engineering, we chose to engineer versions of four BGCs on 2-μm plasmids, with expression of each gene driven by either a strong HEx promoter or a strong constitutive promoter. Two of these clusters, DHZ and IDT, were controls, selected as they are known to function in yeast and are the producers of the polyketide 7′,8′-dehydrozearalenol (DHZ; 1) and an indole diterpene (IDT; 2), respectively. In addition, we selected two uncharacterized BGCs containing a UbiA-type sesquiterpene cyclase (UTC; Fig. 2B, TC1 and TC3). Analysis of the control clusters demonstrated that production of compound 1 was detectable only when expression was driven by the HEx promoters and undetectable with constitutive promoters. Titers of 2 were 4.5-fold higher with HEx promoters than with constitutive expression. For the uncharacterized UTC-containing clusters, combined titers for the oxygenated sesquiterpenoids (table S5) produced by both clusters were significantly greater (20-fold for TC1, 100-fold for TC3) when refactored with PADH2-like sequences versus strong constitutive promoters. These results establish the broad utility of PADH2-like, delayed-induction HEx promoters as tools for the coordinated expression of multiple heterologous proteins in yeast.
Improved host strains
HEx promoters are active only under aerobic respiration, which necessitates their use in yeast host strains with functional mitochondria. We chose to use the well-characterized S288c-derived strains BY4741 and BY4742 (table S4) (38) as our starting point for host optimization. These and related strains have been used in previous heterologous expression studies in yeast with great success (17, 39) despite known mitochondrial genome stability defects present in all strains in the S288c lineage, as indicated by increased petite frequencies (40, 41). Because the alleles associated with mitochondrial genome instability have been characterized (40), we facilitated heterologous expression of fungal BGCs in yeast by generating strains in which these deficiencies, as well as vestigial defects in sporulation efficiency present in S288c-derived strains, were repaired.
These defects were repaired in an improved strain background named DHY (Fig. 3A). Alleles absent from all S288c-derived strains that lead to increased mitochondrial stability [SAL1 CAT5(91M) MIP1(661T) HAP1] (40, 41) and high sporulation [MKT1(30G) RME1(INS-308A) TAO3(1493Q)] were introduced (42). These alterations increased sporulation from 2 to 62% after 2 days while decreasing petite frequency from 52 to 2.5%. In addition, we deleted the PEP4 and PRB1 vacuolar protease encoding genes as in BJ5464, a strain with demonstrated improvements in heterologous protein production (43). We generated a panel of strains, both prototrophic and auxotrophic, of both mating types (MATa and MATα) with these nine beneficial changes. For BGC expression, we also integrated several genes for essential posttranslational modification enzymes. npgA, a holo–acyl carrier protein (ACP) synthase from A. nidulans has demonstrated flexibility for generation of a variety of holo-carrier proteins for both PKS-containing (39) and NRPS-containing (44) systems expressed in S. cerevisiae. In addition, because cytochrome P450s are ubiquitous in fungal BGCs, ATEG_05064, a cytochrome P450 reductase Aspergillus terreus, has been engineered into our strain background. The full panel of strains used in this study is listed in table S4.
Fig. 3. Description and characterization of DHY strains.
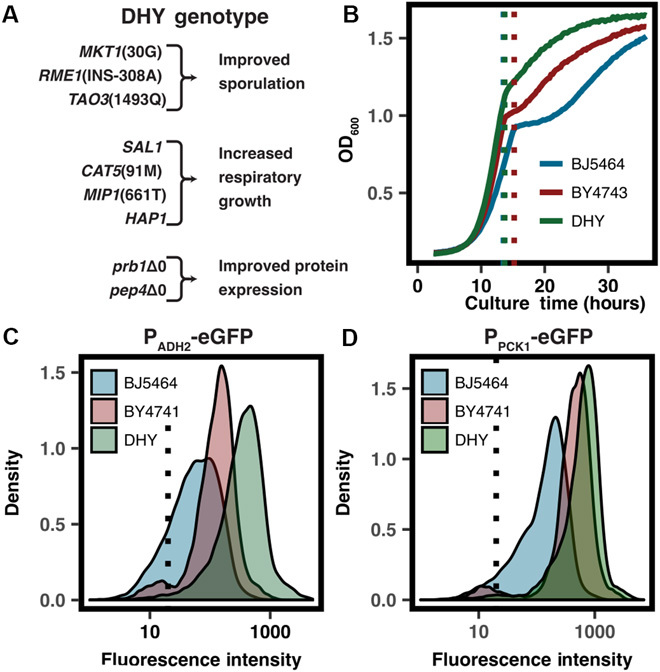
(A) Annotated genotype of the DHY yeast background. (B) DHY-derived yeast strain JHY702 shows improved growth, particularly after diauxic shift. Growth curves are representative of six biological replicates. Density plots for fluorescence intensity in multiple backgrounds show significantly improved eGFP expression when driven by both (C) PADH2 and (D) PPCK1. Density plots represent the fluorescence intensity of 104 individual cells.
The engineered DHY background exhibits improved respiratory growth as compared to both BY4741 and BJ5464 (Fig. 3B). GFP expression driven by PADH2 (Fig. 3C) and PPCK1 (Fig. 3D) also showed marked improvement in DHY. Not only was the mean expression significantly increased, but also a population of nonfluorescent cells prevalent in the BY4741 culture was undetectable in the DHY-derived strain, likely a result of the improved mitochondrial function during expression-inducing respiratory growth conditions.
Compared to commonly used laboratory strains, the improved genetic tractability, growth, and expression characteristics of the DHY background make it an ideal host strain for the HEx platform and for heterologous protein expression more generally.
High-throughput DNA assembly
Heterologous expression of microbial BGCs necessitates a high-throughput, low-cost means of assembling large, multigene constructs expressing cryptic BGCs. Yeast homologous recombination represents such an approach and has been previously applied to the refactoring of large BGCs for expression in model bacterial hosts using intact DNA from native-producing strains or environmental samples (45–49). In the absence of this native DNA to be used as a polymerase chain reaction (PCR) template, BGC sequence must be sourced from commercial vendors. At present, commercial providers of synthetic DNA supply gene-sized fragments at a relatively low cost, but do not offer cost-effective solutions for the larger DNAs required for studying BGCs, which are often >20 kb. In addition, synthesis of AT-rich yeast regulatory sequences, especially promoters, has proven challenging for commercial providers. We therefore purchased synthetic DNA encoding the protein-coding regions of BGC genes and developed an improved means to assemble these genes into large multigene cassettes including promoters, terminators, and expression vectors.
The strategy utilized here to design parts for cluster refactoring and assembly by yeast homologous recombination (YHR) is illustrated in Fig. 4A. DNAs for adjacent fragments were designed with 50 base pairs (bp) of overlapping sequence. In cases where a gene was small enough to be ordered as a single DNA fragment, overlapping sequence to both the flanking promoter and terminator was added, whereas with genes that were split into multiple DNA fragments, overlapping sequences were added to adjacent sequences. All clusters were refactored by building plasmids of seven or fewer genes each. Each gene within a plasmid was flanked by promoters and terminators used in the order defined in table S6. Placing overlapping sequences exclusively on the coding sequence fragments allowed for the same standard parts (promoters, terminators, and linearized vectors) to be generated in bulk and used in all assemblies (table S7). For assemblies involving three or more genes, an auxotrophic marker was placed between the second terminator and the third promoter with no marker present on the vector. By applying the constraint of the auxotrophic marker and origin of replication being on separate fragments, assembly of incorrect plasmids was significantly reduced.
Most previous YHR-based assembly techniques relied on passage of vectors through Escherichia coli to generate large amounts of DNA (50). We developed a protocol for the purification of plasmid DNA directly from yeast clones, wherein the majority of contaminating yeast genomic DNA was removed by exonuclease treatment. This procedure enabled high-throughput DNA sequencing libraries to be prepared directly from yeast colonies, simplifying the process of verifying that target plasmids were correctly assembled. Unmapped sequencing reads in exonuclease-treated samples mapped primarily to the native 2-μm plasmid present in the majority of laboratory yeast strains (cir+ strains). This hypothesis was confirmed by demonstrating that sequencing plasmid DNA out of Y800 (51), a strain lacking this plasmid (cir0), led to greater than 75% of reads mapping to the desired plasmid (Fig. 4B).
The HEx process is a simplified workflow with increased throughput and decreased cost relative to other YHR techniques. We found that assemblies of up to 14 unique DNA fragments can routinely be achieved with high efficiency (Fig. 4C). Overall, we have applied HEx to assemble 41 gene clusters and sequence >1000 yeast clones.
Expression of cryptic BGCs
To apply the HEx platform on a large scale, we chose to examine two classes of fungal BGCs: those encoding a PKS and those encoding a UTC as their core enzyme. Phylogenetic analysis has suggested that much chemical diversity remains to be discovered in fungal PKSs, with the possible existence of entirely unstudied classes (52). UTCs represent a newly discovered class of membrane-bound terpene cyclases, homologous to UbiA prenyltransferases, discovered during the recent elucidation of the biosynthesis of fumagillin (26).
We developed a computational pipeline to prioritize PKS- and UTC-containing BGCs for expression with HEx. We studied all 581 sequenced fungal genomes publicly available in the GenBank database of the National Center for Biotechnology Information (NCBI; as of July 2015). We analyzed each genome for BGCs using antiSMASH2 (53), identifying 3512 BGCs harboring an iterative type 1 PKS and 326 BGCs harboring a UTC homolog. We generated phylogenetic trees of each of these enzyme types, identified characterized homologs from the MIBiG (Minimum Information about a Biosynthetic Gene cluster) database (54), and selected BGCs from clades having few characterized members (Figs. 5 and 6). These BGCs were found in the genomes of both ascomycetes and basidiomycetes. Basidiomycetes have historically been more difficult than ascomycetes to culture with fewer tools for genetic manipulation available (55). As a result, BGCs from basidiomycetes are understudied, with only two PKS-containing clusters deposited in MIBiG as of writing, suggesting that these organisms represent a reservoir of understudied BGCs. All coding sequences were ordered as a series of fused exons with no codon optimization unless required for DNA synthesis (on average, approximately one change per 5000 bp). The start codons, stop codons, and intron/exon boundaries were exactly as deposited in GenBank (table S8).
To explore novel fungal PKSs, we began with the hypothesis that novel PKS sequence would lead to novel compounds. To select unusual PKS BGCs, we performed phylogenetic analysis of the ketosynthase sequences of all 3512 PKS sequences found in the 581 sequenced fungal genomes (Fig. 5 and fig. S7). We first identified sequences that existed in clades where few or no characterized BGCs were found. To further narrow the list to BGCs likely to produce a compound, we selected those whose genetic structure was conserved across three or more species and contained an in-cis or proximal in trans protein capable of releasing the polyketide from the carrier protein of the PKS (fig. S4). From the BGCs that met these criteria, we selected 28, containing between 3 and 11 genes, for characterization with HEx. Seven of these were derived from basidiomycete-specific clades (Fig. 5), whereas the remaining 21 were found in the genomes of ascomycetes.
Of the seven basidiomycete BGCs chosen, three (PKS16, PKS17, and PKS28) produced natural products. The production of 7 and 8 from PKS16, both of which are novel N-, S-bis-acylated amino acids, is unprecedented, because they incorporate an amino acid, but the cluster contains no NRPS gene (Fig. 5 and fig. S4). Similarly, PKS17 produces compound 6, a leucine O-methyl ester with an additional polyketide chain amidated to the amino ester. PKS28 produced a pair of compounds that were not structurally characterized but, on the basis of high-resolution mass spectral data, are likely to contain at least one nitrogen atom. To our knowledge, these are the first examples of fungal BGCs producing polyketide–amino acid hybrid compounds in the absence of NRPS-encoding genes.
Of the 21 ascomycete-derived PKS clusters, 13 produced compounds. The most notable was the PKS1 cluster, which only contained a PKS, a hydrolase, and the genes for three tailoring enzymes: a cytochrome P450 monooxygenase, a flavin-dependent monooxygenase, and a short-chain reductase (table S9). This cluster produced 9 and 10 as major products (Fig. 5) along with a variety of oligoesters. Compound 9, an asymmetric macrotriolide, results from the condensation of two triketides with a single diketide and closely resembles the macrosphelide family of fungal natural products, compounds with antimicrobial activity (56) whose BGCs have yet to be elucidated.
In addition to these novel compounds, two clusters produced known compounds and novel derivatives thereof. PKS15 produced orsellinic acid (3) as the major product, along with several other higher–molecular weight compounds. PKS23 produced 4 and 5 as major products, along with several additional putative products of higher mass. Compounds 4 and 5 are both precursors of phenalenone, a compound whose BGC was elucidated after the selection of PKS23 for the expression in this study (57). Together, these results demonstrate the power of the HEx platform to produce both novel and previously known compounds using unstudied BGCs derived from uncultivated fungi.
To study fungal UTCs, we constructed a phylogenetic tree based on the UbiA-type sesquiterpene cyclase Fma-TC from the fumagillin biosynthetic pathway (Fig. 6) (52). The cytochrome P450 monooxygenase Fma-P450 from the fumagillin pathway is a powerful enzyme catalyzing the 8e oxidation of bergamotene to generate a highly oxygenated product (9). We selected 13 UTC-containing BGCs spanning the entirety of the cladogram in Fig. 6, where a cytochrome P450 monooxygenase gene was proximal to the UTC gene (fig. S5A). Screening of strains expressing these clusters by LC/high-resolution mass spectrometry revealed novel spectral features consistent with oxidized sesquiterpenoids produced by five clusters (Fig. 4). The structures of the major compounds produced by TC1 (compounds 14, 15, and 16) and TC3 (compounds 11, 12, and 13) were elucidated by NMR (tables S16 to S21). Unique among these clusters is TC9 from the basidiomycete Schizophyllum commune, where the UTC alone produces a series of sesquiterpenols that, when placed in the context of the full cluster, are further oxidized by the two adjacent cytochrome P450 monooxygenases. These results demonstrate not only a series of structurally novel sesquiterpenoids but also that the membrane-bound UTCs represent a general class of terpene cyclase encoded in genomes of diverse fungi.
Including both PKS and UTC BGCs, we found that 19 of the 41 clusters studied did not produce detectable compounds. We hypothesized that gene annotation errors introduced by incorrect intron prediction was likely to be a common failure mode in the expression of cryptic fungal BGCs and therefore sought to rescue the production by improved intron annotation. Manual inspection of one UTC (TC5) that had yielded no products suggested an incorrect intron prediction at the 5′ terminus of the gene (fig. S5B). Correction of this intron led to a C-terminal protein sequence that aligned well with known functional UTCs. When tested in the HEx pipeline, the version with the corrected intron produced oxidized sesquiterpenoids (Fig. 6), confirming that incorrect intron prediction can be a failure mode in approaches that rely on publicly available gene annotations. These results illustrate the importance of careful gene curation and the need for improved eukaryotic gene prediction, particularly with sequences from taxa with few studied members. We anticipate this being particularly important for BGCs derived from basidiomycetes because introns are more common in this phylum than in filamentous ascomycetes (58).
CONCLUSION
Using the HEx platform developed here, we built strains expressing 41 cryptic fungal BGCs. Twenty-two (54%) of these clusters, derived from diverse ascomycete and basidiomycete fungal species, produced detectable levels of compounds not native to S. cerevisiae (Table 1). Ongoing and future studies will work to improve this success rate through a detailed analysis of those BGCs that failed. Testing multiple splice variants for ambiguous genes, quantifying transcript and protein expression levels for each gene, and ensuring phosphopantetheinylation of all ACP domains are among the approaches that may provide insight into the common failure modes of fungal BGCs refactored for expression in yeast. In addition, varying protein stoichiometry through building multiple versions of each refactored cluster with varying promoter strengths may also resurrect nonfunctional clusters or increase conversion of biosynthetic intermediates in those that produce multiple products.
A recent analysis of the diversity of natural products discovered over time has highlighted the need for innovative new approaches for molecule discovery (59). Here, by performing a large-scale survey of diverse BGCs from across the fungal kingdom, we have demonstrated such an approach. Using our platform, we identified a panel of novel natural products produced by enzymes with novel activities. Moreover, the genetic parts, improved host strains, and DNA assembly pipeline that comprise the HEx platform provide an improved means for accessing the vast biosynthetic potential encoding natural products with novel structures and bioactivities that exist within the multitude of cryptic BGCs present in fungal genomes.
MATERIALS AND METHODS
General materials and methods
Restriction enzymes were purchased from New England Biolabs (NEB). Cloning was performed in chemically competent E. coli DH10β (NEB, C3019l). Unless otherwise specified, PCR steps were performed using Q5 high-fidelity polymerase (NEB, M0491L) with programs set according to the manufacturer’s specifications. PCR primers were purchased from Integrated DNA Technologies. All yeast cultures grown under selective conditions were cultured in SD (synthetic dropout) media prepared with ingredients purchased from MP Biomedicals. Yeast dropout media were made using a dropout base (DOB) of 27 g/liter (4025-032) and the appropriate supplementary nutrients at the manufacturer-specified concentrations. Yeast-rich media were prepared using a YP (yeast extract and peptone) base consisting of yeast extract (10 g/liter; 212750, BD) and peptone (20 g/liter; 211677, BD). Carbon sources were dextrose (20 g/liter) in YPD and ethanol (30 g/liter) + glycerol (20 g/liter) in YPEG.
Generation of strains for HEx promoter characterization
All promoters were defined as the shorter of 500 bp upstream of the start codon of a gene or the entire 5′ intergenic region. All promoter sequences are listed in table S3. All promoters from S. cerevisiae were amplified from genomic DNA, whereas ADH2 promoters from all sensu stricto Saccharomyces were ordered as gBlocks from Integrated DNA Technologies. Minimal alterations were made to promoters from S. kudriavzevii and Saccharomyces mikitae to meet synthesis specifications. In all constructs, eGFP was cloned directly upstream of the terminator from the CYC1 gene (TCYC1). pRS415 was digested with Sac I and Sal I, and a Not I–eGFP–TCYC1 cassette was inserted by Gibson assembly generating pCH600. Digestion of pCH600 with Acc I and Pml I removed the CEN/ARS origin, which was replaced by 500-bp sequences flanking the ho locus using Gibson assembly to yield plasmid pCH600-HOint. Each of the promoters to be analyzed was amplified with appropriate assembly overhangs and inserted into pCH600-HOint digested with Not I to generate the pCH601 plasmid series. Digestion of the pCH601 plasmid series with Asc I generated linear integration cassettes, which were transformed into S. cerevisiae BY4741 by the LiAc/polyethylene glycol (PEG) method (60). Correct integration was confirmed by PCR amplification of promoters and Sanger sequencing.
Characterization of HEx promoters
For characterization, all strains were initially grown to saturation overnight in 100 μl of YPD media. These cells were then reinoculated at optical density (OD600) = 0.1 into 1 ml of fresh YPD and allowed to grow to OD600 = 0.4 to reach mid-log phase growth (approximately 6 hours). Each culture (500 μl) was pelleted by centrifugation and resuspended in YPE broth for YPE data, whereas YPD was used for YPD data. The 0-hour time point was collected immediately after resuspension. For each time point, 10 μl of culture was diluted in 2 ml of deionized water and sonicated for three short pulses at 35% output on a Branson Sonifier.
Expression data were collected on 10,000 cells for each replicate using a FACSCalibur flow cytometer (BD Bioscience) with the FL1 detector. Data were analyzed in R using the flowCore package.
Construction of improved HEx yeast strains
HEx strains are based on the BY4741/BY4742 background, which in turn is based on S288c (38). The strains were made in two stages: (i) creation of a core DHY set with restored sporulation and mitochondrial genome stability and (ii) creation of JHY derivatives modified for the HEx platform. All changes introduced in this study were confirmed by diagnostic PCR and sequencing.
A sporulation-restored strain set was built by crossing BY4710 (38) to a haploid derivative of YAD373 (41), a BY-based diploid that contains three quantitative trait loci (QTLs) that restore sporulation: MKT1(30G), RME1(INS-308A), and TAO3(1493Q). A spore clone from the resulting diploid was repaired for HAP1, which encodes a zinc-finger transcription factor localized to mitochondria and the nucleus. HAP1 is important for mitochondrial genome stability (61), and we inferred that it was also important for sporulation. S288c and derivatives contain a Ty1 insertion in the 3′ end of HAP1 that inactivates function. We excised the transposon using the delitto perfetto method (62) and confirmed repaired HAP1 function based on transcription of a CYC1p-lacZ reporter (63). The sporulation-restored HAP1-repaired strain and its auxotrophic and prototrophic derivatives were then used to create the DHY set of strains that were additionally restored for mitochondrial genome stability.
The above sporulation-restored strains were used to repair the poor mitochondrial genome stability known to be a problem with S288c and BY derivatives. Mitochondrial genome stability is essential for robust growth and ADH2p-like gene expression under conditions of respiration and for reducing the frequency of petite cells (slow-growing, respiration-defective cells that cannot grow on nonfermentable carbon sources). For a detailed description of the “mito-repair” method, see the construction of JHY650 (42). Briefly, we used the 50:50 genome editing method (64) to introduce the wild-type alleles of three genes shown to be important for mitochondrial genome stability by QTL analysis (40). The repaired QTLs are SAL1+ (repair of a frameshift), CAT5(91M), and MIP1(661T). Crosses with prototrophic and auxotrophic strains completed the DHY core set of about a dozen sporulation- and mitochondrial genome stability–restored strains that can be further modified as needed. DHY213 (table S4) is one such strain: It contains the seven desired changes described above, is otherwise congenic with BY4741, and was used in this study to create derivatives for the HEx platform (below and table S4).
Marker-free, seamless deletion of the complete PRB1 and PEP4 ORFs was performed using the 50:50 method (64). Integration of a 1609-bp ADH2p-npgA-ACS1t expression cassette on the chromosome was performed using a similar method used to integrate DNA segments with the recombinase directed indexing (REDI) method (42), except that URA3, not FCY1, was used as the counter-selectable marker. For an integration site, we replaced a 1166-bp cluster of three transposon long terminal repeats located centromere-distal to YBR209W on chromosome II (deletion of chrII 643438 to 644603). Two DNA segments were simultaneously inserted via homologous recombination at the integration site that had been cut with Sce I to create double strand breaks. One inserted segment was ADH2p-npgA (1448 bp), which was PCR-amplified from a BJ5464/npgA expression strain (npgA from A. nidulans) (65). We repaired the npgA 3′ end to wild type using a reverse PCR primer that replaced the npgA intron included previously (65) with the wild-type npgA 3′ sequence. To preclude recombination of the expression cassette with the native ADH2 locus, we used as the second DNA segment the 161-bp ACS1 terminator (not ADH2t), which was PCR-amplified from BY4741. The resulting strain (JHY692) was used in a similar fashion to replace only npgA with the CPR ORF (cytochrome P450 reductase, ATEG_05064 from A. terreus). Finally, a strain with both npgA and CPR expression cassettes (JHY702) was created by mating JHY692 and JHY705.
Preparation of fragments for plasmid assembly
Upon selection of a BGC for expression using the HEx platform, the coding sequences were downloaded as annotated in NCBI Genbank database. For each gene, overlapping sequences corresponding to the last 50 bp of the desired promoter and the first 50 bp of the desired terminator were added to the 5′ and 3′ termini, respectively. The regulatory sequences appended to each gene were dependent on position as defined in table S6. After the addition of assembly overhangs, all coding sequences longer than 4000 bases were split evenly into multiple fragments, each shorter than 4000 bp with 50-bp overhangs between adjacent parts. All sequences were then ordered as Genebits or GeneBytes from Gen9 (Cambridge, MA). Upon delivery, all fragments were amplified via PCR. Amplicons were purified using the QIAquick 96 PCR BioRobot kit (963141, Qiagen) prior to their use in plasmid assembly.
Similarly, regulatory cassettes containing fused terminators and promoters were amplified from the regulatory cassette plasmids described in table S6 and purified using the QIAquick PCR purification kit (28106, Qiagen).
Plasmid assembly by YHR
For each assembled plasmid, 500 ng of each coding sequence fragment was combined with 500 ng of each required regulatory cassette and 100 ng of the appropriate expression vector linearized with Sal1. This DNA mix was transformed into the assembly strain (primarily BY4743ΔDNL4) using a standard LiAc/PEG protocol (60). Briefly, a 5-ml culture of the assembly strain was grown to saturation overnight in YPD. Two milliliters of this culture was used to inoculate 50 ml of fresh YPD and grown to OD600 ≅ 1 (approximately 5 hours). Cell pellets were harvested by centrifugation for 10 min at 2800g and 4°C. Cells were then washed three times with 1 ml of 100 mM LiAc. The DNA fragment mix was brought up to a final volume of 74 μl with nuclease-free water and combined with 36 μl of 1 M LiAc and 10 μl of salmon sperm DNA solution (10 mg/ml; D1626; Sigma-Aldrich). The mixture was thoroughly mixed and combined with 240 μl of 50% (w/v) PEG solution (MW = 3350; P3640; Sigma-Aldrich) and mixed well. Five OD units of cells (cell pellet from 5 ml of OD600 = 1 culture) were resuspended in this transformation mix and incubated at 30°C for 30 min followed by 45 min at 42°C. Transformed cells were pelleted by centrifugation for 30 s at 10,000g. The supernatant was discarded, and cells were resuspended in 100 μl of nuclease-free water. This suspension and a 50× dilution thereof were spread on plates of the appropriate SD medium and incubated for 3 days at 30°C. Typical assembly transformations yield 5000 to 10,000 colonies.
Preparation of DNA for direct sequencing of yeast plasmid DNA
Yeast colonies were picked into 1.5 ml of the appropriate SD medium in a 2-ml deep-well block and incubated with shaking at 30°C and 1000 rpm until grown to saturation, typically 2 days. Cell pellets were harvested through centrifugation at 2800g for 20 min, and the supernatants were discarded. Plasmid DNA was isolated using a modified version of the QIAprep Turbo miniprep kit (27173/27191/27193, Qiagen). Zymolase (1000 U; Zymo Research E1004/5) and 80 mg of RNAse A (19101, Qiagen) were added to 400 ml of P1 buffer. Each pellet in the deep-well block was resuspended in 250 μl of this modified P1 and incubated with shaking for 2 to 3 hours. The remainder of the plasmid preparation was undertaken according to the manufacturer’s instructions with final elution in 100 μl of water.
Prepared plasmids were treated with exonuclease to remove any contaminating linear DNA. A volume of 22.5 μl of each prepared plasmid was combined with 1.5 μl of Exonuclease V (10 U/μl; NEB, M0345L), 3.0 μl of NEB Buffer 4 (NEB, B7004S), and 3.0 μl of ATP (10 mM; NEB, P0756S) followed by incubation at 37°C for 1 hour. Exonuclease reactions were quenched by the addition of 1 μl of EDTA (0.33 M) and incubation for 30 min at 70°C. Finally, reactions were purified using Sera-mag magnetic particles (1.5 mg/ml, bead/sample ratio = 1:1) with final elution in 25 μl of tris-Cl (10 mM).
Preparation of sequencing libraries from purified plasmid DNA
Sequencing libraries were prepared using a previously published variation on the Nextera system (FC-121-1031, Illumina) (66). Tagmentation was set up on ice as follows: 1 μl of purified, exonuclease-treated plasmid DNA (1 to 20 ng/μl) was combined with 1.25 μl of Nextera TD buffer and 0.25 μl of Nextera TDE enzyme before incubation at 55°C for 10 min in a prewarmed thermocycler. Tagmented samples were used directly for adapter and barcode addition by PCR. For each plate of samples, 12 column-wise master mixes were prepared as follows: 57.2 μl of Kapa HiFi Hotstart Ready Mix (2×; KM2602, Kapa Biosystems) and 45.6 μl of 5 μM Index primer. Similarly, eight row-wise primer master mixes were prepared by the combination of 85.8 μl of Kapa HiFi Hotstart Ready Mix with each of the row-wise index primers. Appropriate column-wise index and row-wise index master mixes (10 μl) were added directly to each tagmentation reaction, yielding a final reaction volume of 22.5 μl. These reactions were placed in a thermocycler, and the following program was run: 3 min at 72°C, 5 min at 98°C, 13 cycles of 10 s at 98°C, 30 s at 65°C, and 30 s at 72°C, followed by 2 min at 72°C before holding at 4°C. All 22.5 μl of each reaction in a plate were pooled and purified using Sera-mag magnetic particles (1.5 mg/ml, bead/sample ratio = 1:1) with final elution in 60 μl Tris-Cl (10 mM). Size selection for fragments from 200 to 600 bp was carried out on 30 μl of the pooled library using a Pippin prep (Sage Science Pippin Prep gel cassette 100 to 600 bp, CSD 2010) followed by quality assessment and quantitation by both Bioanaylzer (high-sensitivity chip, Agilent Technologies) and qPCR using the KAPA Illumina Platform Library quantification kit (KK4835, ABI Prism). Sequencing libraries were run on either an Illumina MiSeq (up to 192 samples) or an Illumina NextSeq (up to 384 samples) platform.
After sequence verification, 2 μl of the remaining yeast plasmid DNA was transformed into E. coli, and the resulting colonies were grown overnight in Terrific Broth + carbenicillin (100 mg/liter) before high-concentration plasmid isolation using either a QIAprep Spin Miniprep (27106, Qiagen) or a QIAprep 96 Turbo kit (120012, Qiagen).
Selection of cryptic fungal BGCs
We analyzed 581 public fungal genomes deposited in the Genbank database of the NCBI and applied the antiSMASH2 software (53) to search for type 1 PKS and UTC gene clusters. This analysis identified 3512 type 1 PKS gene clusters and 326 UbiA-like terpene gene clusters in 538 fungal genomes.
Phylogenetic analysis of both sequence sets was performed by building multiple sequence alignments of all protein sequences using MAFFT (67) and building phylogenetic trees as shown in Figs. 3 and 4 using FastTree 2 (68).
Twenty-eight of the 3512 sequenced type 1 PKS gene clusters and 13 of the 326 terpene gene clusters were selected for expression in yeast as described in the main text.
A full cladogram for the PKS proteins of all PKS-containing BGCs with all sequences deposited in MIBiG labeled is shown in fig. S7.
Construction and culture of production strains
Production strains were constructed by transforming plasmid DNA isolated out of E. coli (Qiagen miniprep 27106) into the appropriate expression host (JHY692 for PKS-containing plasmids, JHY705 for all others) using the Frozen-EZ Yeast Transformation II kit (Zymo Research T2001) followed by plating on the appropriate SD media (CSM-Leu for PKS-containing plasmids and CSM-Ura for all others). For BGCs encoded on at least two plasmids, three biological replicates for each haploid transformant were mated on YPD plates and incubated at 30°C for 4 to 16 hours before streaking for single colonies on CSM-Ura/-Leu and incubated at 30°C. All production strains used in this study are described in table S9.
Small-scale cultures for analysis were begun by picking three biological replicates of each production strain along with empty vector controls into 500 μl of the appropriate SD medium in a 1-ml deep-well block and grown for approximately 24 hours at 30°C. Overnight culture (50 μl) was used to inoculate 500 ul of each of the production media to be tested in the experiment (generally both YPD and YPE) in 1-ml deep-well blocks. All blocks were covered with gas-permeable plate seals (AB-0718, Thermo Fisher Scientific) and incubated at 30°C for 72 hours with shaking at 1000 rpm. Supernatants were clarified by centrifugation for 20 min at 2800g, and a minimum of 100 μl of clarified supernatant was stored for future analysis. The remainder of the supernatant was discarded, and the cell pellets were extracted by mixing with 400 μl of 1:1 ethyl acetate/acetone. Cell debris was precipitated by centrifugation for 20 min at 2800g, and 200 μl of the extraction solvent was pipetted to a fresh block and evaporated in a SpeedVac.
Before the analysis, all supernatants were passed through a 0.2-μm filter plate, whereas all cell pellet extracts were resuspended in 200 μl of high-performance liquid chromatography (HPLC)–grade methanol before filtering.
Analysis of small-scale cultures
LC-MS analysis was conducted on an Agilent 6545 quantitative time-of-flight mass spectrometer interfaced to an Agilent 1290 HPLC system. The ion source for most analyses was an electospray ionization source (dual-inlet Agilent Jet Stream or “dual AJS”). In some analyses, an Agilent Multimode Ion Source was also used for atmospheric pressure chemical ionization. The parameters used for both ionization sources are outlined in the Supplementary Materials.
The HPLC column for all analyses was a 50-mm × 2.1-mm Zorbax RRHD Eclipse C18 column with 1.8-μm beads (959757-902, Agilent). No guard column was used.
Gradient conditions were isocratic at 95% A from 0 to 0.2 min, with a gradient from 95% A to 5% A from 0.2 to 4.2 min, followed by isocratic conditions at 5% A from 4.2 to 5.2 min, followed by a gradient from 5% A to 95% A from 5.2 to 5.4 min, followed by isocratic reequilibration at 95% A from 5.4 to 6 min. For electrospray analyses, A was 0.1% (v/v) formic acid in water, and B was 0.1 % (v/v) formic acid in acetonitrile. For atmospheric-pressure chemical ionization analyses, B was substituted by 0.1% (v/v) formic acid in methanol.
Data analysis by untargeted metabolomics was performed with XCMS using optimal parameters determined by IPO (69).
For PKS-containing clusters, automated analyses were set to generate extracted ion chromatograms (EICs) for the top 100 spectral features as defined by both fold change and P value. These EICs were then manually inspected to identify the subset of automatically identified features that appear specific to the expressed BGC as defined by presence in each of three biological replicates of the production strain and absence from three biological replicates of a negative control strain (figs. S6 and S8 to S24).
Large-scale culture and compound isolation
For compound isolation, large-scale fermentation was carried out. The yeast strains were first struck out onto the appropriate SD agar plates and incubated for 48 hours at 30°C. A colony was then inoculated into 40 ml of SD medium and incubated at 28°C for 2 days with shaking at 250 rpm. This seed culture was used to inoculate 4 liters of YPD medium (1.5% glucose) and cultured for 3 days at 28°C and 250 rpm. Supernatants were then clarified by centrifugation and extracted with equal volume of ethyl acetate. Cell pellets were extracted with 1 liter of acetone. For compounds containing carboxylic acid groups, the pH value of the supernatant was adjusted to 3 by adding HCl before extraction. The organic phases were combined and evaporated to dryness. The residue was purified by ISCO-CombiFlash Rf 200 (Teledyne ISCO) with a gradient of hexane and acetone. After analysis by LC-MS, the fractions containing the target compounds were combined and further purified by semipreparative HPLC using C18 reverse-phase column. The purity of each compound was confirmed by LC-MS, and the structure was solved by NMR (tables S11 to S21 and figs. S25 to S79).
All NMR spectra including 1H, 13C, COSY, HSQC, HMBC, and NOESY were obtained on a Bruker AV500 spectrometer with a 5-mm dual cryoprobe at the University of California, Los Angeles, Molecular Instrumentation Center. The NMR solvents used for these experiments were purchased from Cambridge Isotope Laboratories Inc.
Acknowledgments
Funding: This work was funded by NIH 1U01GM110706 and by a Burroughs Wellcome Fund Career Award at the Scientific Interface. Author contributions: M.E.H., C.J.B.H., M.T., Y.T., J.H., U.S., C.K., R.P.S., R.W.D., L.M.S., and R.S.A. conceived the project and designed all the experiments. C.J.B.H., M.T., U.S., J.H., A.M.C., C.R.F., H.-C.L., E.S., M.M., J.L., J.C., and M.E.H. performed all the experiments and analyzed the data. C.J.B.H., M.T., Y.F.L., B.N., D.I., Y.T., and M.E.H. selected BGCs for expression. J.R.H., G.A.V., M.M., C.J.B.H., and M.E.H. organized and curated the data. C.J.B.H., M.T., and M.E.H. wrote the manuscript. Competing interests: C.J.B.H., U.S., B.N., Y.T., M.M., and M.E.H. all own shares in Hexagon Bio Inc. C.R.F. owns shares in Ginkgo Bioworks Inc. B.N., C.J.B.H., U.S., M.E.H., and J.H. are inventors on a patent application related to this work filed by Stanford University (application no. PCT/US2017/062100; priority date 16 November 2016). C.J.B.H., U.S., and M.E.H. are also inventors on an additional patent application related to this work filed by Stanford University (application no. US20170275635A1; priority date 24 March 2016). All the other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaar5459/DC1
Supplementary Text
fig. S1. Characterization of S. cerevisiae PADH2-like promoters.
fig. S2. Cloning vectors used in this study.
fig. S3. Improving DNA assembly.
fig. S4. Schematics of all PKS-containing BGCs examined here.
fig. S5. UTC-containing BGCs examined here.
fig. S6. Volcano plot of all spectral features identified in the automated analysis of strains expressing PKS-containing BGCs.
fig. S8. All features produced by PKS1 in strain 132.
fig. S9. All features produced by PKS2 in strain 133.
fig. S10. All features produced by PKS4 in strain 255.
fig. S11. All features produced by PKS6 in strain 178.
fig. S12. All features produced by PKS8 in strain 164.
fig. S13. All features produced by PKS10 in strain 246.
fig. S14. All features produced by PKS13 in strain 206.
fig. S15. All features produced by PKS14 in strain 257.
fig. S16. All features produced by PKS15 in strain 247.
fig. S17. All features produced by PKS16 in strain 177.
fig. S18. All features produced by PKS17 in strain 176.
fig. S19. All features produced by PKS18 in strain 207.
fig. S20. All features produced by PKS20 in strain 208.
fig. S21. All features produced by PKS22 in strain 209.
fig. S22. All features produced by PKS23 in strain 241.
fig. S23. All features produced by PKS24 in strain 210.
fig. S24. All features produced by PKS28 in strain 240.
fig. S25. 1H NMR spectrum of compound 6 in CDCl3.
fig. S26. 13C NMR spectrum of compound 6 in CDCl3.
fig. S27. 1H-1H COSY spectrum of compound 6 in CDCl3.
fig. S28. HSQC spectrum of compound 6 in CDCl3.
fig. S29. HMBC spectrum of compound 6 in CDCl3.
fig. S30. 1H NMR spectrum of compound 7 in acetone-d6.
fig. S31. 13C NMR spectrum of compound 7 in acetone-d6.
fig. S32. 1H-1H COSY spectrum of compound 7 in acetone-d6.
fig. S33. HSQC spectrum of compound 7 in acetone-d6.
fig. S34. HMBC spectrum of compound 7 in acetone-d6.
fig. S35. 1H NMR spectrum of compound 8 in acetone-d6.
fig. S36. 13C NMR spectrum of compound 8 in acetone-d6.
fig. S37. 1H-1H COSY spectrum of compound 8 in acetone-d6.
fig. S38. HSQC spectrum of compound 8 in acetone-d6.
fig. S39. HMBC spectrum of compound 8 in acetone-d6.
fig. S40. 1H NMR spectrum of compound 9 in CDCl3.
fig. S41. 13C NMR spectrum of compound 9 in CDCl3.
fig. S42. 1H-1H COSY spectrum of compound 9 in CDCl3.
fig. S43. HSQC spectrum of compound 9 in CDCl3.
fig. S44. HMBC spectrum of compound 9 in CDCl3.
fig. S45. 1H NMR spectrum of compound 10 in CDCl3.
fig. S46. 13C NMR spectrum of compound 10 in CDCl3.
fig. S47. 1H-1H COSY spectrum of compound 10 in CDCl3.
fig. S48. HSQC spectrum of compound 10 in CDCl3.
fig. S49. HMBC spectrum of compound 10 in CDCl3
fig. S50. 1H NMR spectrum of compound 11 in CDCl3.
fig. S51. 13C NMR spectrum of compound 11 in CDCl3.
fig. S52. 1H NMR spectrum of compound 12 in CDCl3.
fig. S53. 13C NMR spectrum of compound 12 in CDCl3.
fig. S54. 1H-1H COSY spectrum of compound 12 in CDCl3.
fig. S55. HSQC spectrum of compound 12 in CDCl3.
fig. S56. HMBC spectrum of compound 12 in CDCl3.
fig. S57. NOESY spectrum of compound 12 in CDCl3
fig. S58. 1H NMR spectrum of compound 13 in CDCl3.
fig. S59. 13C NMR spectrum of compound 13 in CDCl3.
fig. S60. 1H-1H COSY spectrum of compound 13 in CDCl3.
fig. S61. HSQC spectrum of compound 13 in CDCl3.
fig. S62. HMBC spectrum of compound 13 in CDCl3.
fig. S63. NOESY spectrum of compound 13 in CDCl3.
fig. S64. 1H NMR spectrum of compound 14 in CDCl3.
fig. S65. 13C NMR spectrum of compound 14 in CDCl3.
fig. S66. 1H-1H COSY spectrum of compound 14 in CDCl3.
fig. S67. HSQC spectrum of compound 14 in CDCl3.
fig. S68. HMBC spectrum of compound 14 in CDCl3.
fig. S69. 1H NMR spectrum of compound 15 in CDCl3.
fig. S70. 13C NMR spectrum of compound 15 in CDCl3.
fig. S71. 1H-1H COSY spectrum of compound 15 in CDCl3.
fig. S72. HSQC spectrum of compound 15 in CDCl3.
fig. S73. HMBC spectrum of compound 15 in CDCl3.
fig. S74. HMBC spectrum of compound 15 in CDCl3.
fig. S75. 1H NMR spectrum of compound 16 in CDCl3.
fig. S76. 13C NMR spectrum of compound 16 in CDCl3.
fig. S77. 1H-1H COSY spectrum of compound 16 in CDCl3.
fig. S78. HSQC spectrum of compound 16 in CDCl3.
fig. S79. HMBC spectrum of compound 16 in CDCl3.
table S1. Ion source parameters used in this study.
table S2. Expression data for selected promoters drawn from genome-wide expression studies.
table S3. Sequences of HEx promoters
table S4. Background strains used throughout this study.
table S5. Features integrated for the determination of sesquiterpenoid titer in Fig. 2B.
table S6. Order of promoters and terminators used in the expression of all cryptic fungal BGCs examined in this study.
table S7. Standard part plasmids and expression vectors used for the assembly of cryptic BGCS in this study.
table S8. Coordinates of native loci from which all clusters examined here were derived along with the IDs of plasmids expressing the engineered cluster versions.
table S9. Abbreviations for the functional gene annotations used in Figs. 2, 5, and 6.
table S10. Strains expressing cryptic fungal BGCs analyzed here.
table S11. NMR data of compound 6.
table S12. NMR data of compound 7.
table S13. NMR data of compound 8.
table S14. NMR data of compound 9.
table S15. NMR data of compound 10.
table S16. NMR data of compound 11.
table S17. NMR data of compound 12.
table S18. NMR data of compound 13.
table S19. NMR data of compound 14.
table S20. NMR data of compound 15.
table S21. NMR data of compound 16.
REFERENCES AND NOTES
- 1.Newman D. J., Cragg G. M., Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Schueffler A., Anke T., Fungal natural products in research and development. Nat. Prod. Rep. 31, 1425–1448 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Wiemann P., Keller N. P., Strategies for mining fungal natural products. J. Ind. Microbiol. Biotechnol. 41, 301–313 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Blackwell M., The fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 98, 426–438 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Inglis D. O., Binkley J., Skrzypek M. S., Arnaud M. B., Cerqueira G. C., Shah P., Wymore F., Wortman J. R., Sherlock G., Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 13, 91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khaldi N., Seifuddin F. T., Turner G., Haft D., Nierman W. C., Wolfe K. H., Fedorova N. D., SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 47, 736–741 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller M., Zengler K., Tapping into microbial diversity. Nat. Rev. Microbiol. 2, 141–150 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Brakhage A. A., Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Lin H.-C., Tsunematsu Y., Dhingra S., Xu W., Fukutomi M., Chooi Y. H., Cane D. E., Calvo A. M., Watanabe K., Tang Y., Generation of complexity in fungal terpene biosynthesis: Discovery of a multifunctional cytochrome P450 in the fumagillin pathway. J. Am. Chem. Soc. 136, 4426–4436 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergmann S., Funk A. N., Scherlach K., Schroeckh V., Shelest E., Horn U., Hertweck C., Brakhage A. A., Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster. Appl. Environ. Microbiol. 76, 8143–8149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeh H.-H., Ahuja M., Chiang Y. M., Oakley C. E., Moore S., Yoon O., Hajovsky H., Bok J. W., Keller N. P., Wang C. C., Oakley B. R., Resistance gene-guided genome mining: Serial promoter exchanges in Aspergillus nidulans reveal the biosynthetic pathway for Fellutamide B, a proteasome inhibitor. ACS Chem. Biol. 11, 2275–2284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber J., Valiante V., Nødvig C. S., Mattern D. J., Slotkowski R. A., Mortensen U. H., Brakhage A. A., Functional reconstitution of a fungal natural product gene cluster by advanced genome editing. ACS Synth. Biol. 6, 62–68 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Adpressa D. A., Stalheim K. J., Proteau P. J., Loesgen S., Unexpected biotransformation of the HDAC inhibitor vorinostat yields aniline-containing fungal metabolites. ACS Chem. Biol. 12, 1842–1847 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Henke M. T., Soukup A. A., Goering A. W., McClure R. A., Thomson R. J., Keller N. P., Kelleher N. L., New aspercryptins, lipopeptide natural products, revealed by HDAC inhibition in Aspergillus nidulans. ACS Chem. Biol. 11, 2117–2123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao X.-M., Xu W., Li D., Yin W. B., Chooi Y. H., Li Y. Q., Tang Y., Hu Y., Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. Engl. 54, 7592–7596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clevenger K. D., Bok J. W., Ye R., Miley G. P., Verdan M. H., Velk T., Chen C., Yang K., Robey M. T., Gao P., Lamprecht M., Thomas P. M., Islam M. N., Palmer J. M., Wu C. C., Keller N. P., Kelleher N. L., A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat. Chem. Biol. 13, 895–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ro D.-K., Paradise E. M., Ouellet M., Fisher K. J., Newman K. L., Ndungu J. M., Ho K. A., Eachus R. A., Ham T. S., Kirby J., Chang M. C., Withers S. T., Shiba Y., Sarpong R., Keasling J. D., Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Galanie S., Thodey K., Trenchard I. J., Filsinger Interrante M., Smolke C. D., Complete biosynthesis of opioids in yeast. Science 349, 1095–1100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown S., Clastre M., Courdavault V., O’Connor S. E., De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci. U.S.A. 112, 3205–3210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith C. A., Want E. J., O’Maille G., Abagyan R., Siuzdak G., XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Chiang Y.-M., Oakley C. E., Ahuja M., Entwistle R., Schultz A., Chang S. L., Sung C. T., Wang C. C., Oakley B. R., An efficient system for heterologous expression of secondary metabolite genes in Aspergillus nidulans. J. Am. Chem. Soc. 135, 7720–7731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heneghan M. N., Yakasai A. A., Halo L. M., Song Z., Bailey A. M., Simpson T. J., Cox R. J., Lazarus C. M., First heterologous reconstruction of a complete functional fungal biosynthetic multigene cluster. Chembiochem 11, 1508–1512 (2010). [DOI] [PubMed] [Google Scholar]
- 23.T. Itoh, T. Kushiro, I. Fujii, Fungal Secondary Metabolism (2012), pp. 175–182. [Google Scholar]
- 24.Nielsen M. T., Nielsen J. B., Anyaogu D. C., Holm D. K., Nielsen K. F., Larsen T. O., Mortensen U. H., Heterologous reconstitution of the intact geodin gene cluster in Aspergillus nidulans through a simple and versatile PCR based approach. PLOS ONE 8, e72871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith D. J., Burnham M. K. R., Edwards J., Earl A. J., Turner G., Cloning and heterologous expression of the penicillin biosynthetic gene cluster from Penicillium chrysogenum. Nat. Biotechnol. 8, 39–41 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Lin H.-C., Chooi Y. H., Dhingra S., Xu W., Calvo A. M., Tang Y., The fumagillin biosynthetic gene cluster in Aspergillus fumigatus encodes a cryptic terpene cyclase involved in the formation of β-trans-bergamotene. J. Am. Chem. Soc. 135, 4616–4619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labbé S., Thiele D. J., Copper ion inducible and repressible promoter systems in yeast. Methods Enzymol. 306, 145–153 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Rönicke V., Graulich W., Mumberg D., Müller R., Funk M., Use of conditional promoters for expression of heterologous proteins in Saccharomyces cerevisiae. Methods Enzymol. 283, 313–322 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Johnston M., Davis R. W., Sequences that regulate the divergent GAL1-GAL10 promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 4, 1440–1448 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinhandl K., Winkler M., Glieder A., Camattari A., Carbon source dependent promoters in yeasts. Microb. Cell Fact. 13, 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee K. M., DaSilva N. A., Evaluation of the Saccharomyces cerevisiae ADH2 promoter for protein synthesis. Yeast 22, 431–440 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Reeves C. D., Hu Z., Reid R., Kealey J. T., Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol. 74, 5121–5129 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chooi Y.-H., Hong Y. J., Cacho R. A., Tantillo D. J., Tang Y., A cytochrome P450 serves as an unexpected terpene cyclase during fungal meroterpenoid biosynthesis. J. Am. Chem. Soc. 135, 16805–16808 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studier F. W., Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L. M., Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J., Shao Z., Zhao H., Nair N., Wen F., Xu J.-H., Zhao H., Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Biotechnol. Bioeng. 109, 2082–2092 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Lee M. E., DeLoache W. C., Cervantes B., Dueber J. E., A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 4, 975–986 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D., Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Bond C., Tang Y., Li L., Saccharomyces cerevisiae as a tool for mining, studying and engineering fungal polyketide synthases. Fungal Genet. Biol. 89, 52–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimitrov L. N., Brem R. B., Kruglyak L., Gottschling D. E., Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics 183, 365–383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deutschbauer A. M., Davis R. W., Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat. Genet. 37, 1333–1340 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Smith J. D., Schlecht U., Xu W., Suresh S., Horecka J., Proctor M. J., Aiyar R. S., Bennett R. A., Chu A., Li Y. F., Roy K., Davis R. W., Steinmetz L. M., Hyman R. W., Levy S. F., St. Onge R. P., A method for high-throughput production of sequence-verified DNA libraries and strain collections. Mol. Syst. Biol. 13, 913 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones E. W., Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol. 194, 428–453 (1991). [DOI] [PubMed] [Google Scholar]
- 44.Cacho R. A., Tang Y., Reconstitution of fungal nonribosomal peptide synthetases in yeast and in vitro. Methods Mol. Biol. 1401, 103–119 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Luo Y., Huang H., Liang J., Wang M., Lu L., Shao Z., Cobb R. E., Zhao H., Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat. Commun. 4, 2894 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamanaka K., Reynolds K. A., Kersten R. D., Ryan K. S., Gonzalez D. J., Nizet V., Dorrestein P. C., Moore B. S., Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. U.S.A. 111, 1957–1962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang H.-S., Charlop-Powers Z., Brady S. F., Multiplexed CRISPR/Cas9- and TAR-mediated promoter engineering of natural product biosynthetic gene clusters in yeast. ACS Synth. Biol. 5, 1002–1010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bilyk O., Sekurova O. N., Zotchev S. B., Luzhetskyy A., Cloning and heterologous expression of the grecocycline biosynthetic gene cluster. PLOS ONE 11, e0158682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng Z., Kim J. H., Brady S. F., Fluostatins produced by the heterologous expression of a TAR reassembled environmental DNA derived type II PKS gene cluster. J. Am. Chem. Soc. 132, 11902–11903 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shao Z., Zhao H., Zhao H., DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 37, e16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burns N., Grimwade B., Ross-Macdonald P. B., Choi E.-Y., Finberg K., Roeder G. S., Snyder M., Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev. 8, 1087–1105 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Li Y. F., Tsai K. J. S., Harvey C. J. B., Li J. J., Ary B. E., Berlew E. E., Boehman B. L., Findley D. M., Friant A. G., Gardner C. A., Gould M. P., Ha J. H., Lilley B. K., McKinstry E. L., Nawal S., Parry R. C., Rothchild K. W., Silbert S. D., Tentilucci M. D., Thurston A. M., Wai R. B., Yoon Y., Aiyar R. S., Medema M. H., Hillenmeyer M. E., Charkoudian L. K., Comprehensive curation and analysis of fungal biosynthetic gene clusters of published natural products. Fungal Genet. Biol. 89, 18–28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blin K., Medema M. H., Kazempour D., Fischbach M. A., Breitling R., Takano E., Weber T., antiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medema M. H., Kottmann R., Yilmaz P., Cummings M., Biggins J. B., Blin K., de Bruijn I., Chooi Y. H., Claesen J., Coates R. C., Cruz-Morales P., Duddela S., Düsterhus S., Edwards D. J., Fewer D. P., Garg N., Geiger C., Gomez-Escribano J. P., Greule A., Hadjithomas M., Haines A. S., Helfrich E. J., Hillwig M. L., Ishida K., Jones A. C., Jones C. S., Jungmann K., Kegler C., Kim H. U., Kötter P., Krug D., Masschelein J., Melnik A. V., Mantovani S. M., Monroe E. A., Moore M., Moss N., Nützmann H.-W., Pan G., Pati A., Petras D., Reen F. J., Rosconi F., Rui Z., Tian Z., Tobias N. J., Tsunematsu Y., Wiemann P., Wyckoff E., Yan X., Yim G., Yu F., Xie Y., Aigle B., Apel A. K., Balibar C. J., Balskus E. P., Barona-Gómez F., Bechthold A., Bode H. B., Borriss R., Brady S. F., Brakhage A. A., Caffrey P., Cheng Y.-Q., Clardy J., Cox R. J., De Mot R., Donadio S., Donia M. S., van der Donk W. A., Dorrestein P. C., Doyle S., Driessen A. J., Ehling-Schulz M., Entian K.-D., Fischbach M. A., Gerwick L., Gerwick W. H., Gross H., Gust B., Hertweck C., Höfte M., Jensen S. E., Ju J., Katz L., Kaysser L., Klassen J. L., Keller N. P., Kormanec J., Kuipers O. P., Kuzuyama T., Kyrpides N. C., Kwon H.-J., Lautru S., Lavigne R., Lee C. Y., Linquan B., Liu X., Liu W., Luzhetskyy A., Mahmud T., Mast Y., Méndez C., Metsä-Ketelä M., Micklefield J., Mitchell D. A., Moore B. S., Moreira L. M., Müller R., Neilan B. A., Nett M., Nielsen J., O’Gara F., Oikawa H., Osbourn A., Osburne M. S., Ostash B., Payne S. M., Pernodet J.-L., Petricek M., Piel J., Ploux O., Raaijmakers J. M., Salas J. A., Schmitt E. K., Scott B., Seipke R. F., Shen B., Sherman D. H., Sivonen K., Smanski M. J., Sosio M., Stegmann E., Süssmuth R. D., Tahlan K., Thomas C. M., Tang Y., Truman A. W., Viaud M., Walton J. D., Walsh C. T., Weber T., van Wezel G. P., Wilkinson B., Willey J. M., Wohlleben W., Wright G. D., Ziemert N., Zhang C., Zotchev S. B., Breitling R., Takano E., Glöckner F. O., Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 11, 625–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadler M., Hoffmeister D., Fungal natural products—The mushroom perspective. Front. Microbiol. 6, 127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi M., Kim Y. P., Hiraoka H., Natori M., Takamatsu S., Kawakubo T., Masuma R., Komiyama K., Omura S., Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 48, 1435–1439 (1995). [DOI] [PubMed] [Google Scholar]
- 57.Gao S.-S., Duan A., Xu W., Yu P., Hang L., Houk K. N., Tang Y., Phenalenone polyketide cyclization catalyzed by fungal polyketide synthase and flavin-dependent monooxygenase. J. Am. Chem. Soc. 138, 4249–4259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kupfer D. M., Drabenstot S. D., Buchanan K. L., Lai H., Zhu H., Dyer D. W., Roe B. A., Murphy J. W., Introns and splicing elements of five diverse fungi. Eukaryot. Cell 3, 1088–1100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pye C. R., Bertin M. J., Lokey R. S., Gerwick W. H., Linington R. G., Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. U.S.A. 114, 5601–5606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gietz R. D., Schiestl R. H., High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Mattoon J. R., Caravajal E., Guthrie D., Effects of hap mutations on heme and cytochrome formation in yeast. Curr. Genet. 17, 179–183 (1990). [DOI] [PubMed] [Google Scholar]
- 62.Storici F., Resnick M. A., The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409, 329–345 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Gaisne M., Bécam A.-M., Verdière J., Herbert C. J., A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 36, 195–200 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Horecka J., Davis R. W., The 50:50 method for PCR-based seamless genome editing in yeast. Yeast 31, 103–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee K. K. M., Da Silva N. A., Kealey J. T., Determination of the extent of phosphopantetheinylation of polyketide synthases expressed in Escherichia coli and Saccharomyces cerevisiae. Anal. Biochem. 394, 75–80 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Baym M., Kryazhimskiy S., Lieberman T. D., Chung H., Desai M. M., Kishony R., Inexpensive multiplexed library preparation for megabase-sized genomes. PLOS ONE 10, e0128036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Price M. N., Dehal P. S., Arkin A. P., FastTree 2—Approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Libiseller G., Dvorzak M., Kleb U., Gander E., Eisenberg T., Madeo F., Neumann S., Trausinger G., Sinner F., Pieber T., Magnes C., IPO: A tool for automated optimization of XCMS parameters. BMC Bioinformatics. 16, 118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Redden H., Alper H. S., The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 6, 7810 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bellí G., Garí E., Piedrafita L., Aldea M., Herrero E., An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 26, 942–947 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christianson T. W., Sikorski R. S., Dante M., Shero J. H., Hieter P., Multifunctional yeast high-copy-number shuttle vectors. Gene 110, 119–122 (1992). [DOI] [PubMed] [Google Scholar]
- 73.Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., El Bakkoury M., Foury F., Friend S. H., Gentalen E., Giaever G., Hegemann J. H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D. J., Lucau-Danila A., Lussier M., M’Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J. L., Riles L., Roberts C. J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R. K., Véronneau S., Voet M., Volckaert G., Ward T. R., Wysocki R., Yen G. S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R. W., Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaar5459/DC1
Supplementary Text
fig. S1. Characterization of S. cerevisiae PADH2-like promoters.
fig. S2. Cloning vectors used in this study.
fig. S3. Improving DNA assembly.
fig. S4. Schematics of all PKS-containing BGCs examined here.
fig. S5. UTC-containing BGCs examined here.
fig. S6. Volcano plot of all spectral features identified in the automated analysis of strains expressing PKS-containing BGCs.
fig. S8. All features produced by PKS1 in strain 132.
fig. S9. All features produced by PKS2 in strain 133.
fig. S10. All features produced by PKS4 in strain 255.
fig. S11. All features produced by PKS6 in strain 178.
fig. S12. All features produced by PKS8 in strain 164.
fig. S13. All features produced by PKS10 in strain 246.
fig. S14. All features produced by PKS13 in strain 206.
fig. S15. All features produced by PKS14 in strain 257.
fig. S16. All features produced by PKS15 in strain 247.
fig. S17. All features produced by PKS16 in strain 177.
fig. S18. All features produced by PKS17 in strain 176.
fig. S19. All features produced by PKS18 in strain 207.
fig. S20. All features produced by PKS20 in strain 208.
fig. S21. All features produced by PKS22 in strain 209.
fig. S22. All features produced by PKS23 in strain 241.
fig. S23. All features produced by PKS24 in strain 210.
fig. S24. All features produced by PKS28 in strain 240.
fig. S25. 1H NMR spectrum of compound 6 in CDCl3.
fig. S26. 13C NMR spectrum of compound 6 in CDCl3.
fig. S27. 1H-1H COSY spectrum of compound 6 in CDCl3.
fig. S28. HSQC spectrum of compound 6 in CDCl3.
fig. S29. HMBC spectrum of compound 6 in CDCl3.
fig. S30. 1H NMR spectrum of compound 7 in acetone-d6.
fig. S31. 13C NMR spectrum of compound 7 in acetone-d6.
fig. S32. 1H-1H COSY spectrum of compound 7 in acetone-d6.
fig. S33. HSQC spectrum of compound 7 in acetone-d6.
fig. S34. HMBC spectrum of compound 7 in acetone-d6.
fig. S35. 1H NMR spectrum of compound 8 in acetone-d6.
fig. S36. 13C NMR spectrum of compound 8 in acetone-d6.
fig. S37. 1H-1H COSY spectrum of compound 8 in acetone-d6.
fig. S38. HSQC spectrum of compound 8 in acetone-d6.
fig. S39. HMBC spectrum of compound 8 in acetone-d6.
fig. S40. 1H NMR spectrum of compound 9 in CDCl3.
fig. S41. 13C NMR spectrum of compound 9 in CDCl3.
fig. S42. 1H-1H COSY spectrum of compound 9 in CDCl3.
fig. S43. HSQC spectrum of compound 9 in CDCl3.
fig. S44. HMBC spectrum of compound 9 in CDCl3.
fig. S45. 1H NMR spectrum of compound 10 in CDCl3.
fig. S46. 13C NMR spectrum of compound 10 in CDCl3.
fig. S47. 1H-1H COSY spectrum of compound 10 in CDCl3.
fig. S48. HSQC spectrum of compound 10 in CDCl3.
fig. S49. HMBC spectrum of compound 10 in CDCl3
fig. S50. 1H NMR spectrum of compound 11 in CDCl3.
fig. S51. 13C NMR spectrum of compound 11 in CDCl3.
fig. S52. 1H NMR spectrum of compound 12 in CDCl3.
fig. S53. 13C NMR spectrum of compound 12 in CDCl3.
fig. S54. 1H-1H COSY spectrum of compound 12 in CDCl3.
fig. S55. HSQC spectrum of compound 12 in CDCl3.
fig. S56. HMBC spectrum of compound 12 in CDCl3.
fig. S57. NOESY spectrum of compound 12 in CDCl3
fig. S58. 1H NMR spectrum of compound 13 in CDCl3.
fig. S59. 13C NMR spectrum of compound 13 in CDCl3.
fig. S60. 1H-1H COSY spectrum of compound 13 in CDCl3.
fig. S61. HSQC spectrum of compound 13 in CDCl3.
fig. S62. HMBC spectrum of compound 13 in CDCl3.
fig. S63. NOESY spectrum of compound 13 in CDCl3.
fig. S64. 1H NMR spectrum of compound 14 in CDCl3.
fig. S65. 13C NMR spectrum of compound 14 in CDCl3.
fig. S66. 1H-1H COSY spectrum of compound 14 in CDCl3.
fig. S67. HSQC spectrum of compound 14 in CDCl3.
fig. S68. HMBC spectrum of compound 14 in CDCl3.
fig. S69. 1H NMR spectrum of compound 15 in CDCl3.
fig. S70. 13C NMR spectrum of compound 15 in CDCl3.
fig. S71. 1H-1H COSY spectrum of compound 15 in CDCl3.
fig. S72. HSQC spectrum of compound 15 in CDCl3.
fig. S73. HMBC spectrum of compound 15 in CDCl3.
fig. S74. HMBC spectrum of compound 15 in CDCl3.
fig. S75. 1H NMR spectrum of compound 16 in CDCl3.
fig. S76. 13C NMR spectrum of compound 16 in CDCl3.
fig. S77. 1H-1H COSY spectrum of compound 16 in CDCl3.
fig. S78. HSQC spectrum of compound 16 in CDCl3.
fig. S79. HMBC spectrum of compound 16 in CDCl3.
table S1. Ion source parameters used in this study.
table S2. Expression data for selected promoters drawn from genome-wide expression studies.
table S3. Sequences of HEx promoters
table S4. Background strains used throughout this study.
table S5. Features integrated for the determination of sesquiterpenoid titer in Fig. 2B.
table S6. Order of promoters and terminators used in the expression of all cryptic fungal BGCs examined in this study.
table S7. Standard part plasmids and expression vectors used for the assembly of cryptic BGCS in this study.
table S8. Coordinates of native loci from which all clusters examined here were derived along with the IDs of plasmids expressing the engineered cluster versions.
table S9. Abbreviations for the functional gene annotations used in Figs. 2, 5, and 6.
table S10. Strains expressing cryptic fungal BGCs analyzed here.
table S11. NMR data of compound 6.
table S12. NMR data of compound 7.
table S13. NMR data of compound 8.
table S14. NMR data of compound 9.
table S15. NMR data of compound 10.
table S16. NMR data of compound 11.
table S17. NMR data of compound 12.
table S18. NMR data of compound 13.
table S19. NMR data of compound 14.
table S20. NMR data of compound 15.
table S21. NMR data of compound 16.