

Abstract
NADPH oxidases (Nox enzymes) are critical mediators of both physiologic and pathophysiologic processes. Nox enzymes catalyze NADPH-dependent generation of reactive oxygen species (ROS), including superoxide and hydrogen peroxide. Until recently, Nox4 was proposed to be involved exclusively in normal physiologic functions. Compelling evidence, however, suggests that Nox4 plays a critical role in fibrosis, as well as a host of pathologies and diseases. These considerations led to a search for novel, small molecule inhibitors of this important enzyme. Ultimately, a series of novel tertiary sulfonylureas (23-25) was designed using pharmacophore modeling, synthesized, and evaluated for inhibition of Nox4-dependent signaling.
Keywords: NADPH oxidases, Nox4, reactive oxygen species (ROS), pharmacophore development, molecular modeling
Graphical abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
1. Introduction
The NADPH oxidase family of enzymes plays a role in a variety of physiological and pathophysiological responses. It consists of one single subunit (Nox5) and six multi-subunit enzymes (Nox1, Nox2, Nox3, Nox4, Duoxl, and Duox2). Of particular interest, Nox4 is widely distributed in a variety of tissues including kidney,1 lung,2 liver,3 as well as heart and vasculature.4 Nox4 influences multiple biological functions by constitutively generating H2O2.5
Recent work using cultured cells and genetically modified animals has shed new light on the biological functions of Nox4. Nox4 favors vasodilation and thus lowers blood pressure,6 enhances capillary angiogenesis in ischemic limbs,7 and inhibits angiotensin II-induced vascular inflammation and remodeling.8 On the other hand, a plethora of studies suggest that Nox4 also contributes to disease development, especially in situations involving ischemia or fibrosis. In fact, an increasing number of human studies indicate that biosynthesis of Nox4 is upregulated in various diseases including hypertension,9 cardiac hypertrophy,10 atherosclerosis,11 diabetic nephropathy,12 pulmonary hypertension,13 and pulmonary fibrosis.14 In streptozotocin-induced diabetic rats, expression of Nox4 is increased15 and deletion of Nox4 is reno-protective.16 Nox4 also contributes to the atherosclerotic phenotype in smooth muscle17 and potentially mediates cardiac hypertrophy in response to phenylephrine and pressure overload.18 Compared to wild-type mice, global Nox4 knockout animals showed attenuated liver injury, inflammation, and fibrosis after injury.19 Cardiac-specific Nox4 knockout mice have less apoptosis, hypertrophy, interstitial fibrosis, and better cardiac function.10b Nox4 has been shown to have important roles in pulmonary fibrosis as well.14 In addition, Nox4 in the hypothalamic paraventricular nucleus contributes to hypertension induced by aldosterone and salt in mice,20 and a small isoform of Nox4 mediates TLR4-induced apoptosis during renal ischemia/reperfusion injury.21 Other important pathophysiological roles of Nox4 include promoting the loss of bone mass in osteoporosis,22 contributing to lung vascular permeability induced by pseudomonas aeruginosa,23 promoting glomerular lesions in a mouse model of diabetic nephropathy,24 and mediating fibrosis formation in response to TGF.25 Moreover, Nox4 plays an important role in abnormal neuropharmacology by contributing to hypoxia-promoted tumor progression in glioblastoma multiforme,26 as well as increasing the severity of brain lesions in a model of ischemic stroke.27
Thus, small-molecule pharmacologic inhibitors of Nox4 have great potential for a wide spectrum of diseases. Developing Nox4 inhibitors has been an active area of research for decades.28 Only a few molecules, however, are currently known to target Nox4.29 GKT136901, which inhibits both Nox1 and Nox4, protects against bleomycin-induced pulmonary fibrosis.30 VAS2870 inhibits both Nox2 and Nox4 activity in vascular endothelial cells without scavenging superoxide.31 It has also been applied in an ischemia-reperfusion animal model leading to significantly reduced infarct size, improved neurological outcome and mortality by intrathecal injection.27 Similarly, fulvene-5 has been shown to inhibit Nox4 and Nox2 and to exert in vivo effects.32 Because Nox2 participates in host defense, systemic inhibition of Nox2 is not ideal. Another potential Nox4 inhibitor is grindelic acid, which belongs to the diarylheptanoid family. It is reported to be a Nox4 inhibitor with a low EC50 and no ROS scavenging ability. Although it does not appear to inhibit Nox2, its effect on other Nox enzymes and other flavoproteins remains to be validated.33 Other proposed Nox4 inhibitors include phenantridinones,34 but additional Nox4 inhibitors are desirable.
The design of novel Nox4 inhibitors with computational chemistry is appealing, and our approach has led to the discovery of three sulfonylureas that have shown promising biological activity. Our ultimate goal is to identify compounds with potential therapeutic benefit for ischemic and fibrotic diseases.
2. Results and discussion
2.1. Molecular modeling and chemistry
There are currently no reported X-ray structures of the NADPH oxidases. Without the advantage of 3D structural data of the macromolecular target, we pursued a pharmacophore-based approach35 that was coupled with energy-based calculations, molecular overlays, and chemical informatics. This combination in our hands over the years has yielded interesting compounds with promising biological results.36 The first iteration of pharmacophore modeling focused on lead identification. Multiple models were generated using the Phase module37 of Schrödinger Inc. software38 and scored in search of novel biologically active chemotypes. The subsequent iterations concentrated on lead optimization. The biological data obtained from the leads identified in the first iteration were used to improve the pharmacophore model which was then used to identify more potent compounds.
Strategy I
Multiple pharmacophore models were pursued using a series of compounds proposed by Borbély and coworkers as represented by 1 in Figure 1.34 Some models utilized diverse compounds, while others utilized representative compounds as proposed by Borbély et al. Better quality hits were obtained from the latter set of models. Some of the prioritized hits were known antioxidants like robinetin, 2, and catechin, 3 (Fig. 1).
Fig. 1.
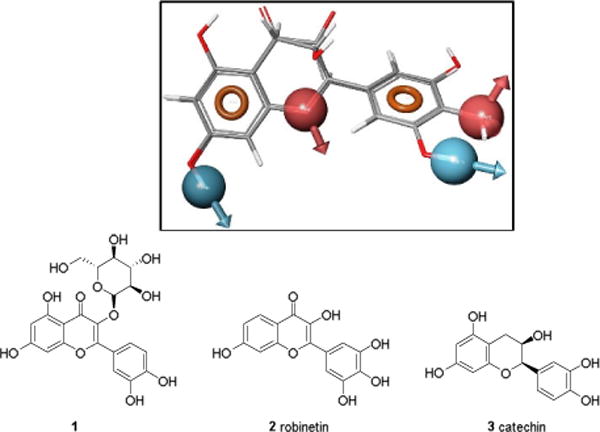
Two known antioxidants retrieved by the lead pharmacophore model (top) based on (1): robinetin (2) and catechin (3). The six-feature pharmacophore model included two hydrogen-bond acceptors (red), two hydrogen-bond donors (blue) and two aromatic rings (orange).
Following the biological testing of the selected lead compounds from these models, we could identify compounds that reduce ROS concentration, but apparently through scavenging and not through Nox4 enzyme inhibition. Due to a lack of evidence of enzyme inhibition, we abandoned further development of the models based on these compounds.
Strategy II
The second set of models was developed using existing patent data.39 Three representative compounds are shown in Fig. 2.
Fig. 2.
Three representative compounds.39
Based on the Nox4 inhibition structure activity relationships (SAR) reported in the patent literature for the compounds with a pyrazolo pyridine scaffold,39 the lead pharmacophore model was developed through the alignment of selected Nox4 inhibitors and subsequently used to retrieve compounds from our in-house 3D-structural databases. The three representative compounds are shown overlaid with the lead pharmacophore model in Fig. 3.
Fig. 3.
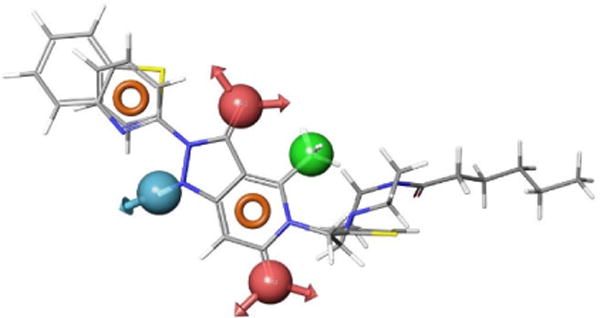
Compounds 4–6 overlaid with the pharmacophore model, AADHRR.9.
The six-feature pharmacophore model, AADHRR.9, is the ninth in a series of models that contain two H-bond acceptors (red), one H-bond donor (blue), one hydrophobic group (green), and two aromatic groups (orange). Whereas the model retrieved many compounds that contain a pyrazolo-pyridine scaffold (Fig. 4, left), we were interested in compounds possessing alternative scaffolds, as a means to identify active compounds with a lower molecular weight (Fig. 4, right).
Fig. 4.
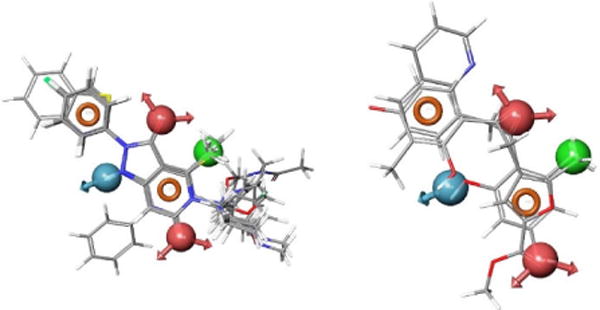
Sample of compounds retrieved by the lead pharmacophore model: those that contain the pyrazolo pyridine scaffold (left), and those do not contain the pyrazolo pyridine scaffold, but fit the pharmacophore model with a high score (right).
The first lead compound (7), retrieved from a commercially available chemical database, is displayed in Fig. 5. To validate the model, we synthesized and tested a series of related acetophenone derivatives (Fig. 6).
Fig. 5.
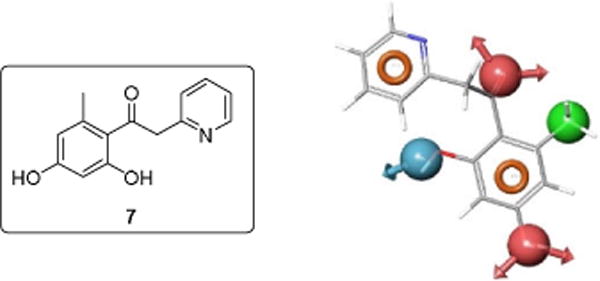
The commercially available lead compound (7) retrieved by the initial pharmacophore model is displayed along with the pharmacophore model.
Fig. 6.
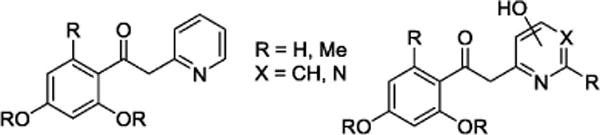
Acetophenone analogues derived from the lead compound (7) are shown.
We initially envisioned gaining access to this series of compounds through the Weinreb amide of suitably substituted benzoic acids. Unfortunately, steric crowding around the amide functionality precluded nucleophilic addition of the lithiated 2- and 4-methylheterocycles. Therefore, we took a less conventional approach via the acid chloride, as all other means failed. Thus, commercially available acid 8 was converted to the corresponding acid chloride 9 via treatment with thionyl chloride at 110°C for 4 hours (Scheme 1). The acid chloride was subsequently treated with lithiated 2-methylpyridine, 4-methylpyrimidine, or 3-(methoxymethoxy)-2,6-dimethylpyridine at −78°C to give the corresponding derivatives 10, 11 and 12, respectively.
Scheme 1.
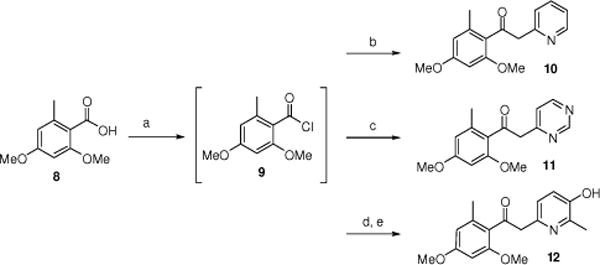
Synthesis of compounds 10–12. Reagents and conditions: (a) SOCl2, toluene, 110°C, 4 h; (b) 2-methylpyridine, TEA, THF, LDA, −78 °C, 2 h then rt-overnight (65%, 2 steps); (c) 4-methylpyrimidine, TEA, THF, LDA, −78 °C, 2 h then rt-overnight (40%, 2 steps); (d) 3-(methoxymethoxy)-2,6-dimethylpyridine, TEA, THF, LDA, −78 °C, 2 h then rt-overnight (45%, 2 steps); (e) 3N HCl (2 equiv.), EtOH, 80 °C, 2.5 h (80%).
Further acetophenone analogues were synthesized by addition of lithiated 2-methylpyridine or lithiated 4-methylpyrimidine to the MOM-protected ester 13 at −78 °C followed by removal of the protecting group to give resorcinols 14 and 15 (Scheme 2).
Scheme 2.
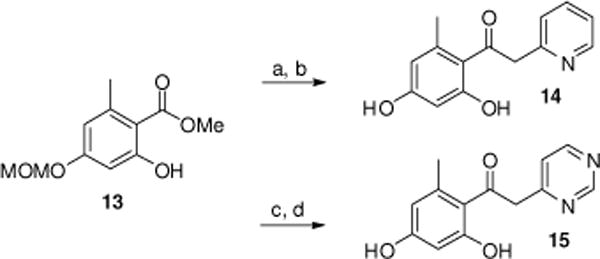
Synthesis of compounds 14–15. Reagents and conditions: (a) 2-methylpyridine, LDA, THF, −78 °C, 2 h (52%); (b) 3N HCl (2 equiv.), EtOH, 80 °C, 3 h (81%); (c) 4-methylpyrimidine, LDA, THF, −78 °C, 2 h (42%); (d) 3N HCl (2 equiv.), EtOH, 45 °C, 3 h (52%).
The final two acetophenone analogues were synthesized by addition of lithiated 2-methylpyridine to the Weinreb amide 16 or ester 18 to give ketone 17 or resorcinol 19, following hydrogenolysis under Pd/C conditions (Scheme 3). Biological evaluation of the acetophenone derivatives (10–12, 14, 15, 17, and 19) indicated peroxide scavenging activity, which prohibited further analysis as specific Nox4 inhibitors. Thus, we determined to further refine our lead pharmacophore model in order to eliminate peroxide scavenging capability.
Scheme 3.
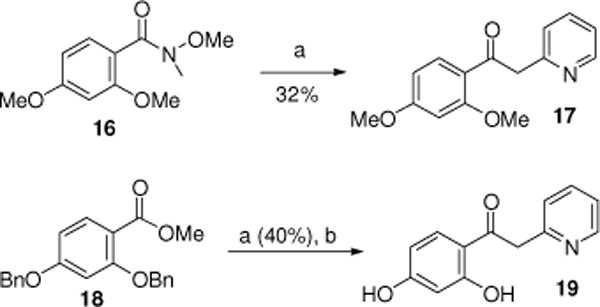
Synthesis of compounds 17 and 19. Reagents and conditions: (a) 2-methylpyridine, n-BuLi, THF, −20 °C, 2 h; (b) 10% Pd/C, EtOH, H2 (1 atm), overnight (48%).
Strategy III
Utilizing the biological assay results obtained from the leads from Strategy II, we then focused on the refinement of the lead pharmacophore model (Figs. 4 and 5). Our general goal was to eliminate the inherent scavenging behavior through structural modifications of the central region by reducing conjugation and removal of the phenolic functionality. By maintaining the aromatic pharmacophore features in the periphery, we systematically made changes by eliminating the central features one at a time. The features in the periphery were maintained because they presumably bind to the Nox4 enzyme. We envisioned utilizing sulfonylureas, which eliminated the requirement for the central H-bond acceptor. This resulted in the final model (Fig. 7). The modified version has the two aromatic regions connected by a sulfonylurea moiety. Based on this model, a subset of candidate sulfonylureas (Figs. 7 and 8) were evaluated in biological assays (in vitro), in an effort to validate the revised model and to find a novel lead compound for Nox4 inhibition. This model resulted in a series of sulfonylureas (Fig. 8) described below.
Fig. 7.
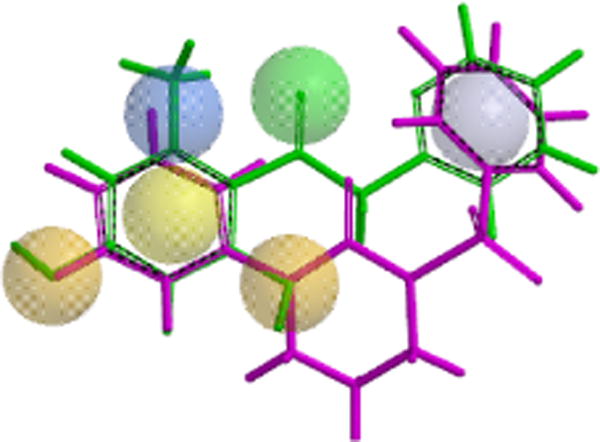
The molecular scaffold of the new sulfonylureas leads (purple) from the third, modified pharmacophore modeling strategy is superimposed on the original lead compound (green) and the pharmacophore in Spartan.
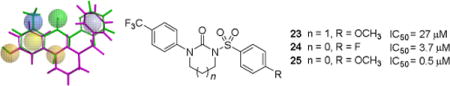
Fig. 8.
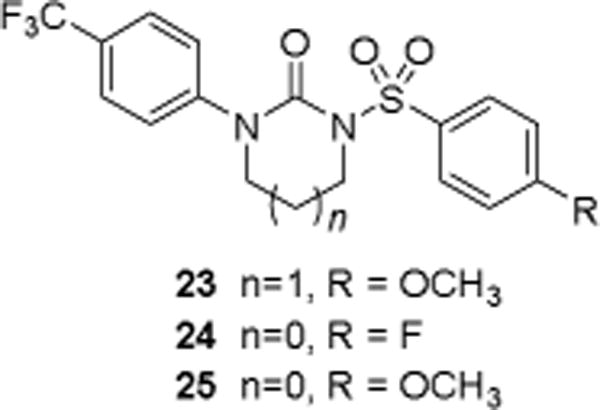
Three sulfonylureas (23-25) were prepared and tested to determine the validity of the pharmacophore model.
Synthesis of sulfonylurea compounds 23-25 is outlined in Scheme 4. Treatment of 4-trifluoromethyl aniline 20 with 2-chloroethyl isocyanate or with 3-chloropropyl isocyanate resulted in the formation of chlorosubstituted acyclic ureas 21 and 22. Sodium hydride (NaH)-mediated cyclization yielded cyclic urea derivatives, which were subsequently converted into the desired sulfonylurea products 23–25 upon reaction with 4-substituted benzenesulfonyl chlorides.
Scheme 4.
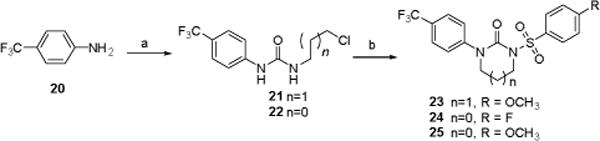
Synthesis of sulfonylureas. Reagents and conditions: (a) 2-chloroethyl isocyanate or 3-chloropropyl isocyanate, CH2Cl2, rt (87-89%); (b) i. NaH, ii. 4-methoxy benzenesulfonyl chloride or 4-fluoro benzenesulfonyl chloride (86-88%, 2 steps).
Figure 9 summarizes the progression of structures from the initial lead 7 to the sulfonylureas (23–25) possessing inhibitory activity against Nox4 but lacking ROS scavenging properties.
Fig. 9.
The three generations of potential Nox4 inhibitors beginning with compound 7 and resulting in the active sulfonylureas (23–25) are displayed. Each stage of the structural modifications corresponds to the iterative refinement of the pharmacophore models coupled with elimination of any molecular features responsible for hydrogen peroxide scavenging.
2.2. Biology
The primary products of the Nox enzymes are ROS. Nox enzymes directly produce superoxide, which is quickly dismutated to hydrogen peroxide. Many organic compounds are able to scavenge superoxide and hydrogen peroxide without affecting enzyme activity; thus, we first developed in vitro assays to measure these undesirable non-specific effects. Previous strategies for ROS detection using non-specific luminescent or fluorescent probes in cells or in cell-free systems40 tend to result in false positives. Recently, high-throughput screening approaches30a, 41 for monitoring H2O2 and superoxide have been developed that may represent an effective strategy to test inhibitors of various Nox isoforms.42 Nevertheless, it remains critical to rule out ROS scavenging and focus on the ability to suppress Nox enzyme activity.28a
To assess ROS scavenging, we measured the consumption of superoxide, generated by xanthine/xanthine oxidase, or exogenous hydrogen peroxide, by increasing concentrations of the compounds of interest. As shown in Fig. 10, compounds 23, 24, and 25 had no effect on superoxide and hydrogen peroxide in vitro at concentrations up to 100 μM.
Fig. 10.
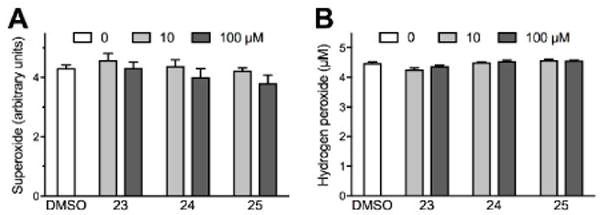
Sulfonylurea compounds minimally scavenge ROS in vitro. ROS scavenging by sulfonylurea compound 23, 24, or 25 was assessed in vitro. In panel A, superoxide was generated with xanthine/xanthine oxidase and measured using the cytochrome C assay. In panel B, 5 μM hydrogen peroxide was added exogenously and measured using the Amplex Red assay. ROS were measured in the presence of indicated concentrations of each compound or DMSO solvent. Bars represent means ± SEM of 4 (superoxide) or 3 (hydrogen peroxide) independent experiments. Differences between DMSO and sulfonylurea compounds are not significant.
To test the biological effectiveness of compounds that did not scavenge ROS, we measured upregulation of vascular smooth muscle alpha actin (α-SMA) by TGF-β treatment of human aortic smooth muscle cells (HASMCs) in culture. This effect of TGF-β has previously been shown to be Nox4-dependent.43 Cells were pretreated with a candidate inhibitor 30 minutes before exposure to TGF-β and harvested after 24 hours. Actin expression was measured by western blotting with a specific antibody. As shown in Fig. 11, compounds 23, 24 and 25 were capable of profoundly inhibiting Nox4-dependent signaling. These results suggest that sulfonylurea compounds may be direct Nox4 inhibitors.
Fig. 11.
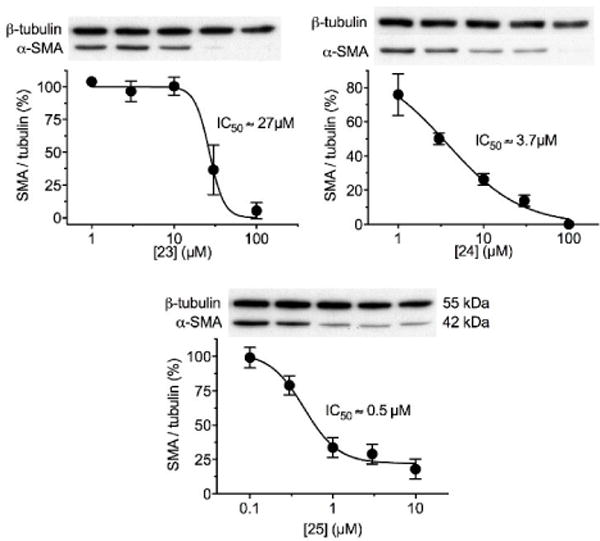
Sulfonylurea compounds inhibit Nox4-dependent signaling in cells. Human aortic smooth muscle cells in culture were incubated with indicated concentrations of compound 23, 24, or 25 and stimulated with 2 ng/ml TGF-β for 24 hours to induce a Nox4-dependent upregulation of smooth muscle alpha-actin (α-SMA). Protein expression was measured by Western blotting and normalized to β-tubulin. In the absence of inhibitor, α-SMA induction by TGF-β was maximal (100%). All three sulfonylurea compounds reduced α-SMA expression, allowing determination of each IC50 by non-linear regression, as indicated. Data points represent means ±SEM from 3-4 independent experiments.
TGF-β is known to significantly upregulate Nox4 expression in smooth muscle cells by 24 hours.43b To ensure that sulfonylurea compounds did not exert their effects by preventing Nox4 upregulation, we measured Nox4 mRNA in cells treated as in Fig. 11, using a qPCR assay described previously.44 In control cells stimulated with TGF-β alone, Nox4 mRNA was upregulated, as expected. However, preincubation with compounds 23, 24 and 25 in the same range of concentrations as in Fig. 11, did not affect Nox4 expression (results not shown).
Finally, to rule out a possible toxic effect of these compounds in cultured cells, we measured cell viability using a commercial mammalian cell live/dead assay. Stimulation with TGF-β alone had no effect on cell viability (99±0.61%, P=NS, n=4). However, cell viability was slightly reduced by preincubation with the highest concentration (100 μM) of compound 23 (92±3.09% viability, P<0.05, n=6), but not 24 (100 μM, 100±0.97% viability, P=NS, n=6) or 25 (10 μM, 97±2.50% viability, P=NS, n=6). It is thus extremely unlikely that the slight cytotoxicity of the highest concentration of compound 23 accounts for the complete inhibition of α-SMA induction shown in Fig. 11.
3. Conclusions
In this study, we report a novel strategy for identifying and synthesizing Nox4 inhibitors and demonstrate promising results on three test compounds with respect to their ability to inhibit Nox4 in a biological system. We took the novel approach of applying a pharmacophore model to the development of potential inhibitors. Our synthetic protocol is general, efficient, and applicable to a wide array of functional groups.
As with other enzyme inhibitors, the ideal Nox4 inhibitor would be selective, specific, efficacious, and lack toxicity. In this study, we ruled out ROS scavenging, demonstrated efficacy of inhibition of Nox4-dependent signaling using an in cellulo assay, and showed a lack of cytotoxicity. Based on these results, we identified three potential Nox4 inhibitors with IC50 values of 27 μM, 3.7 μM and 0.5 μM in cell-based assays. Additional work, however, will be necessary to more fully characterize these promising compounds. They will need to be tested against other Nox enzymes to determine selectivity and tested in a direct assay for Nox4 activity. Selectivity remains challenging due to limitations in specific assays and the presence of multiple Nox enzymes in most cell types.
4. Experimental Section
4.1. General
Glassware was dried in an oven (120 °C), heated under reduced pressure, and cooled under argon before use. Unless otherwise noted, materials obtained from commercial suppliers were used without further purification. Reactions were monitored by thin-layer chromatography on Analtech silica gel plates using UV-light and ceric sulfate or β-naphthol for visualization. Column chromatography was performed on silica gel (230–400 mesh) using hexanes and ethyl acetate as eluent. Evaporation of solvents was conducted under reduced pressure at 50 °C. FTIR spectra were recorded neat on a Perkin-Elmer Spectrum 65. NMR spectra were recorded on a Bruker Avance III 400 NMR spectrometer at 400 MHz (1H) and 100 MHz (13C), respectively. Deuterated chloroform was used as the solvent, unless otherwise noted, and spectra were calibrated against the residual solvent peak (7.24 ppm for 1H and 77.0 ppm for 13C). Chemical shifts (δ) and coupling constants (J) are given in ppm (parts per million) and Hz (Hertz), respectively. The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet. Low resolution ESI mass spectra were obtained on a Waters Acquity UPLC H-Class with PDA and SQ mass detectors using a Waters BEH C18 1.7 μm column (2.1×50mm). High resolution mass spectra were obtained on VG 70–70H or LC/MSD trapSL spectrometer operating at 70 eV using a direct inlet system.
4.2. Synthetic Procedures
4.2.1. 1-(2,4-Dimethoxy-6-methylphenyl)-2-(pyridin-2-yl)ethan-1-one (10) and 1-(2,4-dimethoxy-6-methylphenyl)-2-(pyrimidin-4-yl)ethan-1-one (11)
Thionyl chloride (79 mg, 0.663 mmol, 0.048 mL, 1.3 equiv.) was added dropwise to 2,4-dimethoxy-6-methylbenzoic acid (8, 100 mg, 0.510 mmol) dissolved in anhydrous toluene (5 mL) at rt in a two-neck, 10 mL RBF equipped with a condenser. The solution was heated to reflux for 4 h. The solvent was evaporated under vacuum, and the product (9) was used in the next step without purification.
The acyl chloride (9) was dissolved in dry THF (3 mL), cooled to −78 °C, and treated with triethylamine (0.142 mL, 1.02 mmol, 2 equiv.) with stirring. In a separate RBF, dry THF (3 mL) was added followed by diisopropylamine (0.16 mL, 1.121 mmol, 2.2 equiv.). This solution was cooled to −78 °C and treated with n-BuLi (0.67 mL, 1.070 mmol, 2.1 equiv.) and stirred for 10 mins. The 2-methylpyridine (104 mg, 0.111 mL, 1.12 mmol, 2.2 equiv.) was added dropwise. After 30 mins, the solution of the acid chloride was added via syringe to the lithiated 2-methylpyridine solution. The yellow solution was kept at −78 °C and allowed to warm slowly to rt overnight. The solvent was removed under vacuum. Dichloromethane (10 mL) was added and washed with water (5 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified on silica (1% MeOH:DCM) to give the product.
10: Yield 65%, 2 steps, 77:23 mixture of keto:enol tautomers; 1H NMR (400 MHz, CDCl3, major isomer) δ 8.55 (dd, J = 4.0, 0.8 Hz, 1H), 7.65 (dt, J = 8.0, 2.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.17 (dt, J = 6.0, 0.8 Hz, 1H), 6.30 (s, 2H), 4.33 (s, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 2.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 203.0, 161.4, 158.5, 155.4, 149.2, 144.1, 138.6, 136.4, 124.4, 121.7, 118.3, 107.3, 95.9, 55.6, 55.3, 53.8, 20.1; FTIR (neat): 2934, 2839, 1688, 1594, 1459, 1320, 1199, 1150, 1093, 808, 751, 620 cm−1; MS (ESI) m/z 272 [M+H]+.
11: Yield 40%, 2 steps, 55:45 mixture of keto:enol tautomers; 1H NMR (400 MHz, CDCl3, major isomer) δ 9.15 (bs, 1H), 8.67 (d, J = 4.8 Hz, 1H), 7.35 (d, J = 4.4 Hz, 1H), 6.31 (m, 1H), 4.29 (s, 2H), 3.82 (s, 6H), 2.24 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.9, 164.2, 161.8, 158.8, 156.6, 154.3, 139.3, 121.9, 119.7, 107.6, 95.9, 55.6, 55.4, 53.1, 20.3; FTIR (neat): 2997, 2921, 2842, 1629, 1577, 1466, 1316, 1200, 1000, 884, 744, 649 cm−1; MS (ESI) m/z 273 [M+H]+.
4.2.2. 1-(2,4-Dimethoxy-6-methylphenyl)-2-(5-hydroxy-6-methylpyridin-2-yl)ethan-1-one (12)
To a stirred solution of the 2,6-dimethylpyridin-3-ol (500 mg, 4.06 mmol) in DMF (15 mL) in a 50 mL flame-dried RBF, K2CO3 (0.617 g, 4.47 mmol) and MOM-Cl (0.37 mL, 4.87 mmol) were added successively. The solution was stirred at RT overnight. Water (30 mL) was added and the reaction mixture extracted with EtOAc (2 × 20 mL). The organic layer was washed with cold water (2 × 30 mL) and brine (30 mL) and dried over magnesium sulfate and concentrated under reduced pressure.
Thionyl chloride (79 mg, 0.663 mmol, 0.048 mL, 1.3 equiv.) was added dropwise to 2,4-dimethoxy-6-methylbenzoic acid (8, 100 mg, 0.510 mmol) dissolved in anhydrous toluene (5 mL) at rt in a two-neck, 10 mL RBF equipped with a condenser. The solution was heated to reflux for 4 h. The solvent was evaporated under vacuum, and the product (9) was used in the next step without purification.
The acyl chloride (9) was dissolved in dry THF (3 mL), cooled to −78 °C, and treated with triethylamine (0.142 mL, 1.09 mmol, 2 equiv.) with stirring. In a separate RBF, dry THF (3 mL) was added followed by DIA (0.16 mL, 1.12 mmol, 2.2 equiv.). This solution was cooled to −78 °C and treated with n-BuLi (0.73 mL, 1.121 mmol, 2.2 equiv.) and stirred for 10 mins. The MOM-protected pyridine (179 mg, 1.07 mmol, 2.1 equiv.) was added dropwise. After 30 mins, the acid chloride solution was added dropwise. The yellow solution was kept at −78 °C and allowed to warm slowly to rt overnight. The solvent was removed under vacuum, and dichloromethane (10 mL) was added and washed with water (10 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified on silica (2% MeOH: DCM) to give the MOM ether as a yellow solid.
A solution of the MOM ether (50 mg, 0.145 mmol), 3M HCl (0.097 mL, 0.290 mmol) in EtOH (2 mL) was stirred at 80 °C. After the reaction was completed (monitored by TLC), the organic solvent was directly removed under reduced pressure. The residue was taken up in water (1 mL) and the pH adjusted to 6 using sodium bicarbonate solution (1M). The product was extracted with dichloromethane (3 × 5 mL) and dried and concentrated. Further purification was achieved on silica with 40% (3:1 EtOH in ethyl acetate)/hexane.
12: Yield 80%; 63:27 mixture of keto-enol tautomers; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 9.54 (s, 1H), 7.00 (d, J = 8 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.44 (d, J = 2.0 Hz, 1H), 6.37 (d, J = 2.0 Hz, 1H), 4.10 (s, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 2.29 (s, 3H), 2.16 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 203.1, 161.1, 158.3, 149.7, 147.2, 143.3, 138.0, 123.8, 122.4, 122.3, 107.9, 96.3, 56.2, 55.6, 48.9, 23.3, 19.8; FTIR (neat): 2933, 2836, 1575, 1457, 1359, 1281, 1197, 1045, 949, 806, 618 cm−1; MS (ESI) m/z 302 [M+H]+.
4.2.3. 1-(2,4-Dihydroxy-6-methylphenyl)-2-(pyridin-2-yl)ethan-1-one (14) and 1-(2,4-dihydroxy-6-methylphenyl)-2-(pyrimidin-4-yl)ethan-1-one (15)
To an ice-cooled solution of ethyl 2,4-dihydroxy-6-methylbenzoate (300 mg, 1.529 mmol) in dichloromethane (10 mL), DIPEA (0.401 mL, 296 mg, 2.29 mmol) and MOM-Cl (0.116 mL, 123 mg, 1.59 mmol) were added successively. The solution was stirred at rt for 4 h. Saturated aqueous NH4Cl (15 mL) was added, and the reaction mixture was extracted with dichloromethane (3 × 15 mL). The organic layer was washed with water and brine and dried over magnesium sulfate and concentrated under reduced pressure. The off-white, solid residue was purified by flash chromatography to afford the MOM-ether (13) as a viscous liquid (silica, 40% EtOAc: hexanes).
13: Yield 80%; 1H NMR (400 MHz, CDCl3) δ 11.76 (s, 1H), 6.49 (d, J = 2.4 Hz, 1H), 6.38 (dd, J = 2.4, 0.8 Hz, 1H), 5.17 (s, 2H), 4.41 (q, J = 7.2 Hz, 2H), 3.46 (s, 3H), 2.52 (s, 3H), 1.41 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6, 165.2, 161.3, 143.3, 111.8, 106.4, 101.5, 93.8, 61.32, 56.3, 24.5, 14.2; FTIR (neat): 2985, 2938, 2828, 1613, 1575, 1398, 1263, 1217, 1022, 926, 866 cm−1.
In a RBF, dry THF (3 mL) was added followed by DIA (0.158 mL, 0.397 mL, 2.78 mmol, 4.2 equiv.). This solution was cooled to −78 °C and treated with n-BuLi (1.669 mL, 2.72 mmol, 4.1 equiv., 1.6 molar in hexane) and stirred for 10 mins. To this solution, 2-methylpyridine (0.262 mL, 247 mg, 2.65 mmol, 4 equiv.) was added dropwise. After 30 mins, a solution of methyl 2-hydroxy-4-(methoxymethoxy)-6-methylbenzoate (13, 150 mg, 0.663 mmol) was added dropwise to the pyridine solution. The yellow solution was kept at −78 °C and allowed to warm to rt over 2 h. The solvent was removed under vacuum, and dichloromethane (10 mL) was added and washed with water (5 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified on silica (2% MeOH: DCM) to give the MOM-ether as a yellow solid.
A solution of the MOM-ether (60 mg, 0.209 mmol) and 3M HCl (0.139 mL, 0.418 mmol) in EtOH (4 mL) was stirred at 80 °C. After the reaction was completed (monitored by TLC), the solvent was removed under reduced pressure. The residue was taken up in water (1 mL) and the pH adjusted to 6 using sodium bicarbonate solution (1M), extracted with dichloromethane (3 × 5 mL), dried and concentrated. Further purification was achieved on a flash column with 10 to 30% (1:3 ethanol in ethyl acetate)/hexane.
14: Yield 81%; 80:20 mixture of keto-enol tautomers; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 10.16 (bs, 1H), 9.65 (s, 1H), 8.45 (d, J = 4.4 Hz, 1H), 7.71 (dt, J = 7.6, 2.0 Hz, 1H), 7.26-7.21 (m, 2H), 6.18 (d, J = 2.0 Hz, 1H), 6.06 (d, J = 2.0 Hz, 1H), 4.28 (s, 2H), 2.01 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 202.8, 159.8, 157.7, 156.5, 149.3, 138.9, 136.7, 124.7, 122.1, 120.2, 109.6, 100.6, 53.5, 20.5; FTIR (neat): 2923, 2763, 1592, 1570, 1442, 1375, 1214, 1189, 973, 844, 723, 622 cm−1; MS (ESI) m/z 244 [M+H]+.
15: Yield 52%; 66:24 mixture of keto-enol tautomers; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 10.2 (s, 1H), 9.74 (s, 1H), 9.07 (d, J = 1.6 Hz, 1H), 8.71 (d, J = 5.2 Hz, 1H), 7.43 (dd, J = 4.8, 1.2 Hz, 1H), 6.20 (d, J = 2.0 Hz, 1H), 6.10 (d, J = 2.0 Hz, 1H), 4.32 (s, 2H), 2.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 201.0, 165.1, 160.2, 158.6, 158.2, 157.1, 139.4, 122.7, 119.6, 109.8, 100.6, 52.9, 20.8; FTIR (neat): 3082, 2923, 1605, 1548, 1434, 1350, 1248, 1159, 1051, 982, 820, 690 cm−1; MS (ESI) m/z 245 [M+H]+.
4.2.4. 1-(2,4-Dimethoxyphenyl)-2-(pyridin-2-yl)ethan-1-one (17)
In an oven dried 100 mL round-bottom flask was placed 2,4-dimethoxybenzoic acid (1.0 g, 5.49 mmol) and anhydrous dichloromethane (45 mL). The solution was stirred at room temperature and treated with 1-methylpiperidine (3.35 mL, 27 mmol). After stirring for 10 minutes the solution was cooled to 0 °C and pivaloyl chloride (0.79 g, 0.81 mL, 6.59 mmol) was added via syringe dropwise. The solution was stirred for 2 hours. N,O-Dimethyl hydroxylamine hydrochloride (0.803 g, 8.23 mmol) was added, and the mixture was stirred at room temperature for 24 h. The yellow solution was poured into HCl (80 mL, 1M). The organic layer was sequentially washed with saturated NaHCO3 and brine (80 mL) and dried over MgSO4. After concentration, the yellow oil was dried under vacuum to afford the amide (16) as a white solid.
16: Yield 80%; 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 8.4 Hz, 1H), 6.47-6.43 (m, 2H), 3.78 (s, 6H), 3.55 (bs, 3H), 3.23 (bs, 3H); 13C NMR (100 MHz, CDCl3) δ 161.8, 157.3, 129.0, 117.7, 104.3, 98.6, 60.9, 55.7, 55.4; FTIR (neat): 2964, 2935, 1642, 1606, 1513, 1311, 1289, 1118, 988, 938 cm−1.
Dry THF (2 mL) and 2-methylpyridine (41 mg, 0.44 mmol) were added to a 10 mL round-bottom flask and cooled to −78 °C. To this, n-BuLi (277 μL, 0.444 mmol) was added dropwise. A two-neck, round-bottom flask was charged with dry THF (2 mL) and N-2,4-trimethoxy-N-methylbenzamide (16, 100 mg, 0.444 mmol) was added. The solution was cooled to −78 °C with stirring. After 30 min, the lithiated 2-methylpyridine solution was added dropwise over 10 min via syringe pump. The reaction was stirred at −78 °C for 2 h and warmed to 0 °C. Water was added to the reaction, and the product was extracted with EtOAc (2 × 5 mL). The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography by eluting with 10 to 20% EtOAc in hexanes to afford the ketone (17) in 32% yield.
17: Yield 32%; 88:12 mixture of keto: enol tautomers; 1H NMR (400 MHz, CDCl3, major isomer) δ 8.48 (dd, J = 4.8 Hz, 0.8 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.56 (dt, J = 7.6 Hz, 2.0 Hz, 1H), 7.17 (d, J = 8.0 Hz, 1H), 7.08 (ddd, J = 7.6, 4.8 Hz, 0.8 Hz, 1H), 6.46 (dd, J = 8.8, 2.4 Hz, 1H), 6.36 (d, J = 2.4 Hz, 1H), 4.43 (s, 2H), 3.83 (s, 3H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 164.7, 161.0, 156.5, 149.3, 136.2, 133.2, 124.3, 121.5, 120.7, 105.3, 98.3, 55.6, 55.5, 52.8; FTIR (neat): 3006, 2940, 2838, 1661, 1592, 1502, 1435, 1258, 1110, 1024, 824, 750, 639 cm−1; MS (ESI) m/z 258 [M+H]+.
4.2.5. 1-(2,4-Dihydroxyphenyl)-2-(pyridin-2-yl)ethan-1-one (19)
Methyl 2,4-dihydroxybenzoate (3 g, 17.83 mmol), benzylbromide (6.71 g, 4.67 mL, 39.3 mmol), and potassium carbonate (5.4 g, 39.3 mmol) were added to acetonitrile (120 mL) and were stirred under argon at 60 °C for 48 hours. After 24 hours an additional 0.5 equiv. BnBr and 1 equiv. K2CO3 were added. The reaction mixture was filtered through Celite, and the solvent removed under vacuum. The resulting oil was purified by flash chromatography (silica: 10% EtOAc: hexanes) to give the product (18) as a white solid (4.8 g, 77%).
18: Yield 77%; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.4 Hz, 1H), 7.43-7.41 (m, 2H), 7.33-7.23 (m, 8H), 6.53-6.49 (m, 2H), 5.06 (s, 2H), 4.99 (s, 2H), 3.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 136.6, 136.1, 133.9, 128.7, 128.5, 128.2, 127.7, 127.5, 126.7, 113.2, 113.1, 106.1, 106.0, 101.5, 101.4, 70.5, 70.2, 51.7; FTIR (neat): 3066, 3035, 2835, 1724, 1604, 1505, 1378, 1188, 1129, 1008, 811 cm−1.
Dry THF (9 mL) and 2-methylpyridine (167 mg, 0.178 mL, 1.79 mmol) were added to a 25 mL round-bottom flask and cooled to −78 °C. To this, n-BuLi (1.12 mL, 1.79 mmol) was added dropwise. A 50 mL, two-neck round-bottom flask was charged with dry THF (9 mL) and methyl 2,4-bis(benzyloxy)benzoate (18, 250 mg, 0.78 mmol) was added and cooled to −78 °C with stirring. After 30 mins, the lithiated 2-methylpyridine solution was added dropwise over 2 min. The reaction was stirred at −78 °C for 10 min and water was added. After warming to rt, the product was extracted with EtOAc and the aqueous layer was back-extracted with EtOAc (2 × 10 mL). The combined organic layers were dried and concentrated. The bisbenzyl product was purified by flash chromatography (40%).
The bisbenzyl ketone (120 mg, 0.293 mmol) was dissolved in EtOH and 10% Pd/C (15.5 mg,) was added. The mixture was stirred at rt under 1 atmosphere of H2 overnight. The reaction mixture was filtered and purified by column chromatography by eluting with 1-2% MeOH in dichloromethane to give the product as a yellow solid (48%).
19: Yield 48%; 1H NMR (400 MHz, DMSO-d6,) δ 12.4 (s, 1H), 10.14 (bs, 1H), 8.46 (m, 1H), 7.71 (dd, J = 9.2, 2.4 Hz, 1H), 7.64-7.59 (m, 1H), 7.25 (d, J = 7.6 Hz, 1H), 7.15-7.12 (m, 1H), 6.32-6.30 (m, 1H), 6.24-6.23 (m, 1H), 4.33 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 200.8, 165.5, 165.4, 155.32, 149.5, 136.7, 133.2, 124.2, 122.0, 112.6, 108.6, 103.1, 47.7; FTIR (neat): 2924, 2870, 2755, 1613, 1587, 1415, 1344, 1227, 1124, 945, 842, 761 cm−1; MS (ESI) m/z 230 [M+H]+.
4.2.6. 1-(2-Chloroethyl)-3-(4-(trifluoromethyl)phenyl)urea (21) and 1-(3-Chloropropyl)-3-(4-(trifluoromethyl)phenyl)urea (22)
A 50 mL round-bottom flask was charged with a solution of 4-trifluoromethyl aniline (20, 1.61 g, 10 mmol, 1.0 equiv.) in CH2Cl2 (20 mL). 2-Chloroethyl isocyanate or 3-chloropropyl isocyanate (10 mmol, 1.0 equiv.) was added, and the reaction was stirred at rt for 72 h. The white precipitate was filtered, washed with CH2Cl2 (10 mL), and dried under high vacuum. The compound was used in subsequent steps without any further purification.
21: Compound 21 was synthesized as described in the general procedure using 3-chloropropyl isocyanate (1.2 g, 10 mmol, 1.0 equiv). The desired product was isolated as a white solid (2.51 g, 89 % yield). The crude solid was used without further purification. 1H NMR (400 MHz, DMSO-d6): δ 8.92 (s, 1H), 7.60-7.53 (m, 4H), 6.44-6.42 (t, J = 4.0 Hz, 1H), 3.68-3.65 (t, J1 = 8.0, 4.0 Hz, 2H), 3.25-3.20 (m, 2H), 1.93-1.86 (m, 2 H). 13C NMR (100 MHz, DMSO-d6): 155.4, 144.7, 126.5, 126.3, 123.8, 121.6, 121.3, 117.7, 43.5, 37.1, 33.0. FTIR (neat): 3321, 2969, 1695, 1637, 1596, 1557, 1522, 1409, 1310, 1230, 1180, 1156, 1062, 1013 cm−1.
22: Compound 22 was synthesized as described in the general procedure using 2-chloroethyl isocyanate (1.05 g, 10 mmol, 1.0 equiv). The desired product was isolated as a white solid (2.32 g, 87 % yield). The crude solid was used without further purification. 1H NMR (400 MHz, DMSO-d6): δ 9.11 (s, 1H), 7.61-7.55 (m, 4H), 6.56 (t, 1H), 3.68-3.65 (t, J = 8.0 Hz, 2H), 3.46-3.41 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 155.2, 144.5, 126.4, 126.4, 123.7, 121.8, 121.5, 117.8, 44.7, 41.7. FTIR (neat): 3379, 2970, 2929, 1737, 1650, 1598, 1562, 1408, 1323, 1243, 1157, 1064, 1013 cm−1.
4.2.7. 1-((4-Methoxyphenyl)sulfonyl)-3-(4-(trifluoromethyl) phenyl)-tetrahydropyrimidin-2(1H)-one (23), 1-((4-Fluorophenyl)sulfonyl)-3-(4-(trifluoromethyl)phenyl)-tetrahydropyrimidin-2(1H)-one (24), and 1-((4-Methoxyphenyl)sulfonyl)-3-(4-(trifluoromethyl)phenyl)-imidazolidin-2-one (25)
A round bottom flask (10 mL) equipped with a magnetic stir bar was charged with a solution of the acyclic urea (0.25 mmol, 1.0 equiv.) in THF (2 mL). The solution was cooled to 0 °C for 15 mins. NaH (30 mg, 0.75 mmol, 3.0 equiv., 60% suspension in oil) was slowly added. The reaction was stirred for 15 mins at 0 °C and allowed to warm to rt with stirring for 12 h. The appropriate arylsulfonyl chloride (0.28 mmol, 1.1 equiv.) was added, and the reaction was stirred at rt for an additional 4 h. The reaction mixture was poured into a separatory funnel containing ethyl acetate and 1 M HCl (10 mL each). The layers were separated. The organic layer was washed with distilled H2O (2 × 10 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuum. The crude product was purified using silica gel flash column chromatography. Gradient elution from 20-40% ethyl acetate in hexanes furnished the purified products as white crystalline solids.
23: Compound 23 was synthesized as described in the general procedure using acyclic urea 21 (70 mg, 0.25 mmol, 1.0 equiv.) and 4-methoxybenzenesulfonyl chloride (58 mg, 0.28 mmol, 1.1 equiv). The desired cyclic sulfonylurea 23 was isolated as a white crystalline solid (88 mg, 85% overall yield). 1H NMR (400 MHz, CDCl3) δ 8.02–7.93 (m, 2H), 7.56 (t, J = 12.6 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 6.99–6.93 (m, 2H), 4.14–4.04 (m, 2H), 3.86 (d, J = 3.4 Hz, 3H), 3.70 (dd, J = 13.0, 7.0 Hz, 2H), 2.29–2.21 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 163.6, 151.0, 145.4, 145.3, 131.2, 130.9, 126.2, 125.1, 122.3, 113.9, 55.7, 49.1, 45.4, 22.9. FTIR (neat): 2924, 2855, 1739, 1667, 1595, 1578, 1519, 1497, 1476, 1422, 1324, 1283, 1265, 1175, 1107, 1092, 1065, 1019 cm−1; HRESIMS m/z 415.0936 (M+H)+ (calcd for C18H18F3N2O4S, 415.0939).
24: Compound 24 was synthesized as described in the general procedure using acyclic urea 22 (70 mg, 0.25 mmole, 1.0 equiv.) and 4-fluorobenzenesulfonyl chloride (54 mg, 0.28 mmol, 1.1 equiv.). The desired cyclic sulfonylurea 24 was isolated as a white crystalline solid (91 mg, 90 % overall yield). 1H NMR (400 MHz, CDCl3) δ 8.04 (ddd, J = 8.1, 5.1, 2.5 Hz, 2H), 7.58 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 7.20–7.12 (m, 2H), 4.15–4.06 (m, 2H), 3.72 (dd, J = 11.6, 5.8 Hz, 2H), 2.32–2.20 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 166.8, 164.3, 150.8, 145.0, 135.6, 131.6, 128.8, 126.2, 125.1, 122.4, 116.1, 49.0, 45.5, 22.8. FTIR (neat): 2922, 2853, 1668, 1614, 1591, 1518, 1491, 1476, 1426, 1412, 1328, 1287, 1239, 1206, 1225, 1123, 1175, 1155, 1086, 1035, 1011 cm−1; HRESIMS m/z 403.0735 (M+H)+ (calcd for C17H15F4N2O3S, 403.0740).
25: Compound 25 was synthesized as described in the general procedure from acyclic urea 22 (67 mg, 0.25 mmol, 1.0 equiv.) and 4-methoxybenzenesulfonyl chloride (58.0 mg, 0.28 mmol, 1.1 equiv.). The desired cyclic sulfonylurea 25 was isolated as a white crystalline solid (80 mg, 80 % overall yield). 1H NMR (400 MHz, CDCl3): δ 8.072 (d, J = 2.00, 8.00 Hz, 2H), 7.60 (m, 4H), 7.05-7.02 (m, 2H), 4.08-4.04 (m, 2H), 3.82 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 182.8, 164.2, 151.7, 141.5, 130.7, 129.1, 126.2, 122.6, 117.9, 114.3, 55.7, 42.1, 41.0, 30.9. FTIR (neat): 2908, 2849, 1719, 1617, 1593, 1576, 1524, 1496, 1467, 1397, 1358, 1313, 1296, 1260, 1178, 1153, 1089, 1069, 1022 cm−1; HRESIMS m/z 423.0600 (M+Na)+ (calcd for C17H15F3N2O4SNa, 423.0602).
4.3 Biological Evaluation
4.3.1 Superoxide scavenging assay
We modified an assay based on the measurement of superoxide-dependent changes in cytochrome C absorbance at 550 nm.45 Briefly, superoxide was generated in vitro in phosphate buffered saline solution, using hypoxanthine (100 μM)/xanthine oxidase (6×103 U/mL) in the presence of catalase (200 U/mL) to eliminate H2O2. Compounds under study were added at various concentrations (1–100 μM) and remaining superoxide was measured with cytochrome C (46 μM). Superoxide dismutase (575 U/mL) was used as a positive control in separate samples measured at the same time. Following a 1 min stabilization period, absorbance was measured at 550 nm in a microplate reader (Biotek), using a kinetic program (1 read/min) for 15 min. The linear slope representing the rate of superoxide production was used to calculate the percentage of superoxide scavenging.
4.3.2 Hydrogen peroxide scavenging assay
Hydrogen peroxide was measured in vitro, using an Amplex Red assay kit (Invitrogen), according to the manufacturer’s instructions. This assay measures the oxidation of Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) to fluorescent resorufin in the presence of horse radish peroxidase. Briefly, H2O2 (5 mM) was added to various concentrations (1–100 μM) of the compounds under study in reaction buffer. After an incubation of 5 min, Amplex Red was added to measure remaining H2O2. Reactions were incubated for 30 min at room temperature, protected from light. Resorufin fluorescence was measured in a microplate reader (Biotek) using excitation at 530 nm and emission at 590 nm.
4.3.3 Cell culture
Human aortic smooth muscle cells (HASMs) were purchased from Invitrogen, grown as recommended by the supplier and used before passage 10 for experiments. Mouse aortic smooth muscle cells (MASMs) were isolated from C57BL/6J mice and grown as described previously46 for less than 10 passages.
4.3.4 Western blotting
Potential Nox4 inhibitory activity of compounds under study was measured with an indirect assay, based on the upregulation of vascular smooth muscle alpha actin by TGF-β in cultured HASMs. This effect of TGF-β was previously shown to be Nox4-dependent.43b Cells were grown to 80% confluence and incubated with low serum (0.1% fetal bovine serum) for 24h. Cells were incubated with various concentrations (1–100 μM) of candidate inhibitors for 30 min, before addition of TGF-β (2 ng/ml). Cells exposed to TGF-β alone were used as positive controls. After 24 hours, cells were lysed in 1% triton buffer with protease inhibitors. Protein lysates were separated using SDS-PAGE and transferred to PVDF membranes. Blots were blocked, incubated with mouse monoclonal primary antibodies against smooth muscle alpha actin (A2547, Sigma) and beta tubulin (T4026, Sigma), which served as a loading control. Detection was performed using a horse radish peroxidase conjugated secondary antibody (NA931, GE Healthcare Life Sciences) and enhanced chemiluminescence.
4.3.5 Reverse transcription and quantitative PCR
Total RNA samples (2 μg), purified from cultured MASMs, using the RNeasy Plus kit (Qiagen, Chatsworth, CA), were used to prepare cDNA. Reactions were carried out at 42 °C, using random 15-mer primers and ProtoScript II reverse transcriptase (New England Biolabs, Beverly, MA). Following enzyme inactivation at 65 °C, cDNA samples were purified using the QIAquick kit (Qiagen).
Quantitative PCR was performed with a LightCycler instrument in glass capillaries (Roche Applied Science, Indianapolis, IN), using Platinum Taq DNA polymerase and SYBR green (Invitrogen, Carlsbad, CA). Nox4 was measured using primers 5′- CTGGTCTGACGGGTGTC-TGCATGGTG-3′ and 5′-CTCCGCACAATAAAGGCACAAAGGTCCAG-3′ in the presence of 4 mM MgCl2, and annealing at 65 °C. The housekeeping 18S rRNA was used for normalization and measured using primers 5′-GAATTGACGGAAGGGCACCACCAG-3′ and 5′-GTGCAGC-CCCGGACATCTAAGG-3′ in the presence of 3 mM MgCl2 with annealing at 60 °C. Data analysis was performed using the mak3i module of the qpcR software in the R environment, as described previously.44
4.3.6 Cell viability assay
HASMs were incubated with the compounds under study and TGF-β for 24 hours, as described above, before measuring viability using the Live/Dead Viability/Cytotoxicity kit (Invitrogen), by fluorescence microscopy. This assay relies on the uptake of calcein/AM (2 μM), hydrolyzed to green fluorescent calcein in live cells, and labeling of nuclear DNA with red fluorescent ethidium homodimer-1 (4 μM), due to loss of plasma membrane integrity in dead cells. Cells were incubated with both vital dyes for 10 min at room temperature before measuring fluorescence at 485±10 nm and 530 ±12.5 nm for calcein and ethidium, respectively. Untreated cells were used as negative controls and cells incubated for 30 min with 70% ethanol were used as positive controls for cytotoxicity.
4.3.7 Statistics
Prism software, version 7 (GraphPad) was used to perform calculations. Reactive oxygen species scavenging data were analyzed using one-way ANOVA, followed by Dunnett’s multiple comparisons test. Inhibition of smooth muscle alpha actin expression was analyzed using non-linear regression.
4.4 Computational
4.4.1 Pharmacophore modeling and database searches
The details of pharmacophore model generation were provided in the main text. The initial pharmacophore modeling was carried using the Phase module in the Schrödinger software. Additional pharmacophore modeling was carried out with Spartan 10 (V.1.1.0).
The database searches were performed flexibly, with conformations generated on-the-fly while keeping the initial conformations stored in the database. The following settings were used for searching:
Generate conformations during search
Keep existing conformers
Number of conformers per rotatable bond = 10
Maximum number of conformers per structure = 100
Sampling = Thorough
Amide bonds = Vary conformation
Relative Energy window = 10.0 kcal/mol
Skip conformer generation for structures with > 15 rotatable bonds
For Matching options, Intersite Distance Matching Tolerance (IDMT) was used to tighten or relax the fitting requirements. If the search retrieved more than 1,000 hits, the search was set to stop. The search was then resubmitted with a smaller IDMT until a hitlist of less than 1,000 was achieved. Follow-up searches were pursued with reduced IDMT until only a handful of compounds were retrieved. All searches with 1,000 hits and 0 hits were removed from the Project Table. This systematic tightening of the IDMT is analogous to shrink-wrapping. The typical starting IDMT value was 2.0 Å. Hit Treatment options are left at the default values with the number of hits at 1,000 used as a termination point.
Each pharmacophore model was initially subjected to searches of databases with known drugs; first with the Drugs in the Market database (from Zinc), which contains 4,356 compounds that are either marketed drugs or natural products. Then the search was followed with the Binding Database of 660,806 compounds with measured binding affinities. Once the pharmacophore models were prioritized, based on the results obtained by searching these two databases, those high priority models were subjected to database searches with in-house built databases including:
LeadsNow database of 2,819,898 compounds was extracted from the Zinc database as “immediately available” compounds.
Aldrich Database of 279,000 commercially available compounds.
Supplementary Material
Acknowledgments
This research was partially funded by Union University. The authors are grateful for the HRESMS data provided by D. R. Phillips and C.-W. Chou [Proteomics and Mass Spectrometry (PAMS) Facility, NIH grant 1S10RR1028859] at the University of Georgia, Department of Chemistry, Athens, Georgia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Geiszt M, Kopp JB, Várnai P, Leto TL. Proc Natl Acad Sci USA. 2000;97:8010. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Vliet A. Free Radical Biol Med. 2008;44:938. doi: 10.1016/j.freeradbiomed.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Jiang JX, Török NJ. Advances in Hepatology. 2014;2014:742931. doi: 10.1155/2014/742931. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang S, Kisseleva T, Brenner DA. Frontiers in physiology. 2016;7:1. doi: 10.3389/fphys.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen F, Haigh S, Barman S, Fulton DJR. Frontiers in physiology. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nisimoto Y, Diebold BA, Cosentino-Gomes D, Lambeth JD, Lambeth JD. Biochemistry. 2014;53:5111. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Atertio Thromb Vasc Biol. 2011;31:1368. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 7.Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF. Circulation. 2011;124:731. doi: 10.1161/CIRCULATIONAHA.111.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Circul Res. 2012;110:1217. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 9.Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Stroke. 2004;35:584. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- 10.(a) Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel J-L, Hasenfuss G, Shah AM. J Am Coll Cardiol. 2003;41:2164. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]; (b) Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15565. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorescu D, Weiss D, Lassègue B, Clempus RE, Szöcs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Circulation. 2002;105:1429. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 12.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. The Journal of biological chemistry. 2001;276:1417. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 13.(a) Mittal M, Roth M, König P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MAR, Fink L, Hänze J, Seeger W, Grimminger F, Schmidt HHHW, Weissmann N. Circul Res. 2007;101:258. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]; (b) Sanders KA, Hoidal JR. Circul Res. 2007;101:224. doi: 10.1161/CIRCRESAHA.107.158246. [DOI] [PubMed] [Google Scholar]; (c) Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, Han W, Orfi L, Szantai-Kis C, Keri G, Szabadkai I, Barabutis N, Rafikova O, Rafikov R, Black SM, Jonigk D, Giannis A, Asmis R, Stepp DW, Ramesh G, Fulton DJR. Atertio Thromb Vasc Biol. 2014;34:1704. doi: 10.1161/ATVBAHA.114.303848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. Nat Med. 2009;15:1077. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Arozal W, Watanabe K, Veeraveedu PT, Ma M, Thandavarayan RA, Suzuki K, Tachikawa H, Kodama M, Aizawa Y. Biol Pharm Bull. 2009;32:1411. doi: 10.1248/bpb.32.1411. [DOI] [PubMed] [Google Scholar]; (b) Liu Y, Lei S, Gao X, Mao X, Wang T, Wong GT, Vanhoutte PM, Irwin MG, Xia Z. Clinical Science. 2012;122:161. doi: 10.1042/CS20110176. [DOI] [PubMed] [Google Scholar]
- 16.(a) Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. J Biol Chem. 2005;280:39616. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]; (b) Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX. Diabetes. 2010;59:1528. doi: 10.2337/db09-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HHHW, Coughlan MT, Cooper ME, Forbes JM, Jandeleit-Dahm KAM. Physiological reports. 2014;2:e12192. doi: 10.14814/phy2.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu S, Chamseddine AH, Carrell S, Miller FJ., Jr Redox biology. 2014;2:642. doi: 10.1016/j.redox.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie L-H, Tian B, Sadoshima J. Circul Res. 2013;112:651. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan T, Kisseleva T, Brenner DA. PLOS ONE. 2015;10:e0129743. doi: 10.1371/journal.pone.0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue B, Beltz TG, Johnson RF, Guo F, Hay M, Johnson AK. American journal of physiology Heart and circulatory physiology. 2012;302:H733. doi: 10.1152/ajpheart.00873.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mkaddem SB, Bens M, Vandewalle A. Oncotarget. 2010;1:741. doi: 10.18632/oncotarget.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Pietsch B, Hofbauer LC, Brandes RP, Schröder K. The Journal of clinical investigation. 2013;123:4731. doi: 10.1172/JCI67603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu P, Mohan V, Mansoor S, Tiruppathi C, Sadikot RT, Natarajan V. Am J Respir Cell Mol Biol. 2013;48:477. doi: 10.1165/rcmb.2012-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HHHW, Jandeleit-Dahm KA. Journal of the American Society of Nephrology : JASN. 2014;25:1237. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang F, Liu G-S, Dusting GJ, Chan EC. Redox biology. 2014;2:267. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh C-H, Shyu W-C, Chiang C-Y, Kuo J-W, Shen W-C, Liu R-S. PLoS ONE. 2011;6:e23945. doi: 10.1371/journal.pone.0023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HHHW. PLoS Biol. 2010;8:e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Jaquet V, Scapozza L, Clark RA, Krause K-H, Lambeth JD. Antioxidants & Redox Signaling. 2009;11:2535. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]; (b) Drummond GR, Selemidis S, Griendling KK, Sobey CG. Nature Reviews Drug Discovery. 2011;10:453. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim J-A, Neupane GP, Lee ES, Jeong B-S, Park BC, Thapa P. Expert Opinion on Therapeutic Patents. 2011;21:1147. doi: 10.1517/13543776.2011.584870. [DOI] [PubMed] [Google Scholar]; (d) Kleniewska P, Piechota A, Skibska B, Gorąca A. Archivum Immunologiae et Therapiae Experimentalis. 2012;60:277. doi: 10.1007/s00005-012-0176-z. [DOI] [PubMed] [Google Scholar]
- 29.Cifuentes-Pagano E, Csanyi G, Pagano PJ. Cell Mol Life Sci. 2012;69:2315. doi: 10.1007/s00018-012-1009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Laleu Bt, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon Ln, Houngninou-Molango S, Gradia A, Duboux G, Merlot Cd, Heitz F, Szyndralewiez Cd, Page P. J Med Chem. 2010;53:7715. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]; (b) Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CRJ, Burns KD, Touyz RM, Hebert RL. AJP: Renal Physiology. 2010;299:F1348. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 31.(a) Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HHHW, Morawietz H. Biochem Biophys Res Commun. 2006;344:200. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]; (b) Tenfreyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer A, Vantler M, Bekhite M, Wartenberg M, Sauer H, Rosenkranz S. Cardiovascular Research. 2006;71:331. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 32.Bhandarkar SS, Jaconi M, Fried LE, Bonner MY, Lefkove B, Govindarajan B, Perry BN, Parhar R, Mackelfresh J, Sohn A, Stouffs M, Knaus U, Yancopoulos G, Reiss Y, Benest AV, Augustin HG, Arbiser JL. The Journal of clinical investigation. 2009;119:2359. doi: 10.1172/JCI33877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kofler P, Pircher H, von Grafenstein S, Diener T, Höll M, Liedl K, Siems K, Jansen-Dürr P. Planta Med. 2013;79:244. doi: 10.1055/s-0032-1328129. [DOI] [PubMed] [Google Scholar]
- 34.Borbély Gb, Szabadkai In, Horváth Zn, Markó Pt, Varga Zn, Breza Nr, Baska F, Vántus T, Huszár Mn, Geiszt Ms, Hunyady Ls, Buday Ls, Őrfi Ls, Kéri Gr. J Med Chem. 2010;53:6758. doi: 10.1021/jm1004368. [DOI] [PubMed] [Google Scholar]
- 35.Güner OF. Pharmacophore Perception, Development, and Use in Drug Design. 1. International University Line; La Jolla, CA: 2000. [Google Scholar]
- 36.(a) Phillips RS, Anderson AD, Gentry HG, Güner OF, Bowen JP. Bioorganic and Medicinal Chemistry Letters. 2017;27:1705. doi: 10.1016/j.bmcl.2017.02.080. [DOI] [PubMed] [Google Scholar]; (b) Zhong H, Wees MA, Faure TD, Carrillo C, Arbiser J, Bowen JP. ACS Med Chem Lett. 2011;2:455. doi: 10.1021/ml200031m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. Journal of Computer-Aided Molecular Design. 2006;20:647. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 38.Schrödinger. Small-Molecule Drug Discovery Suite 2014-1. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 39.Page P, Orchard M, Fioraso-Cartier L, Mottironi B. Pyrazolo pyridine derivatives as NADPH oxidase inhibitors. 2013 [Google Scholar]
- 40.(a) Liochev SI, Fridovich I. Arch Biochem Biophys. 1997;337:115. doi: 10.1006/abbi.1997.9766. [DOI] [PubMed] [Google Scholar]; (b) Zielonka J, Lambeth JD, Kalyanaraman B. Free Radical Biol Med. 2013;65:1310. doi: 10.1016/j.freeradbiomed.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaggini F, Laleu B, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. Biorg Med Chem. 2011;19:6989. doi: 10.1016/j.bmc.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 42.Zielonka J, Cheng G, Zielonka M, Ganesh T, Sun A, Joseph J, Michalski R, O’Brien WJ, Lambeth JD, Kalyanaraman B. J Biol Chem. 2014;289:16176. doi: 10.1074/jbc.M114.548693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HHH, Lassègue B, Griendling KK. Atertio Thromb Vasc Biol. 2007;27:42. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin-Garrido A, Brown DI, Lyle AN, Dikalova A, Seidel-Rogol B, Lassègue B, Martín AS, Griendling KK. Free Radical Biol Med. 2011;50:354. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, Gleason RL, Jr, Taylor WR, Hart CM, Griendling KK, Lassegue B. Arterioscler Thromb Vasc Biol. 2013;33:2154. doi: 10.1161/ATVBAHA.113.301913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quick KL, Hardt JI, Dugan LL. J Neurosci Methods. 2000;97:139. doi: 10.1016/s0165-0270(00)00179-5. [DOI] [PubMed] [Google Scholar]
- 46.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, Griendling KK. Arterioscler Thromb Vasc Biol. 2009;29:480. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.