

Abstract
Glycosaminoglycans (GAGs) are extracellular matrix and/or cell surface sulfated glycans crucial to the regulation of various signaling proteins whose functions are essential in many pathophysiologycal systems. Because structural heterogeneity is high in GAG chains and purification is difficult, the use of structurally defined GAG oligosaccharides from natural sources as molecular models in both biophysical and pharmacological assays is limited. To overcome this obstacle, GAG-like oligosaccharides of well-defined structures are currently being synthezised by chemical and/or enzymatic means in many laboratories around the world. These synthetic GAG oligosaccharides serve as useful molecular tools in studies of GAG-protein interactions. Here, besides discussing the commonest routes utilized for the synthesis of GAG oligosaccharides, we also survey some libraries of these synthetic models currently available for research and discuss their activities in interaction studies with functional proteins, especially through the microarray approach.
Keywords: de novo synthesis, enzymatic synthesis, glycosaminoglycans, glycosaminoglycan-protein interactions, interactome, microarray, synthetic oligosaccharide libraries
1. Introduction
Glycosaminoglycans (GAGs) are sulfated polysaccharides found in the extracellular matrix and on surfaces of all human cells. GAGs are structurally composed of alternating hexosamine and uronic acid (UroA) or galactose (Gal). The hexosamine can be either glucosamine (GlcN), N-acetylgalactosamine (GalNAc) or their sulfated derivatives such as N-sulfoglucosamine (GlcNS). The UroA can be either glucuronic acid (GlcA) or iduronic acid (IdoA). GAGs are classified according to the type of monosaccharides in the chain and the glycosidic bond connecting them. The best known mammalian-derived GAG classes are heparin (Hp), heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), hyaluronan or hyaluronic acid (HA), and keratan sulfate (KS). The structure of these common GAG types are depicted in Figure 1.
Figure 1.
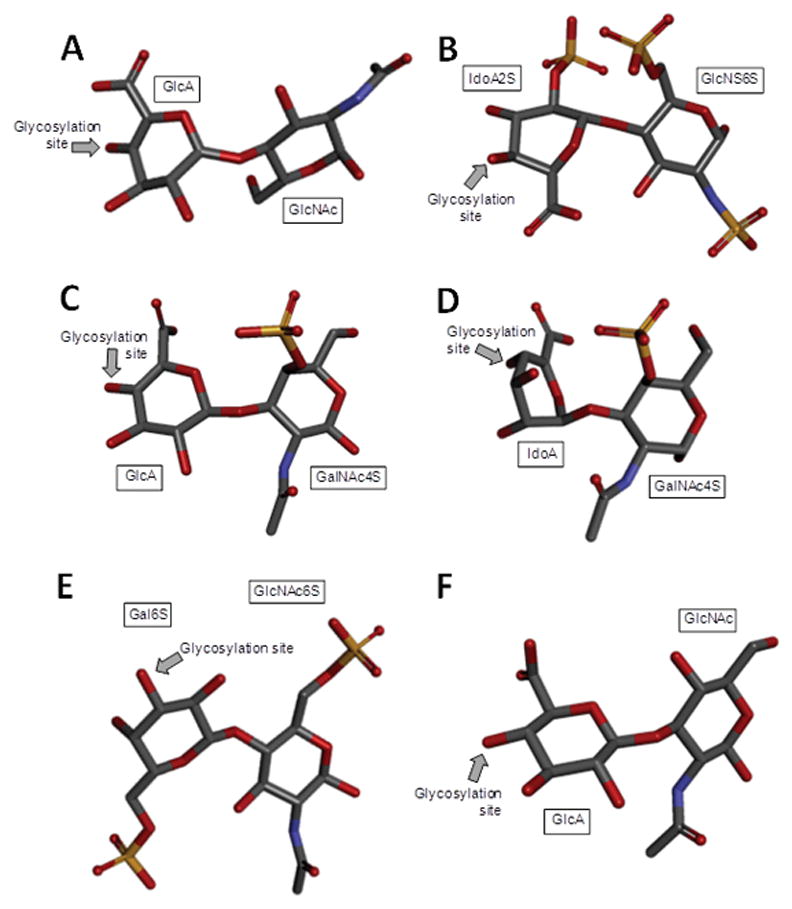
Stick model 3D representations of the commonest structures found in (A) heparan sulfate [D-GlcA-(β1→4)-D-GlcNAc] whose D-GlcNAc is β(1→4)-linked to the D-GlcA of the next disaccharide unit (extracted from PDB ID 3E7J);[2] (B) heparin: [L-IdoA2S-(α1→4)-D-GlcNS6S] whose D-GlcNS6S is α(1→4)-linked to the L-IdoA2S of the next disaccharide unit (extracted from PDB ID 1HPN);[4] (C) chondroitin 4-sulfate [D-GlcA-(β1→3)-D-GalNAc4S] whose GalNAc is β(1→4)-linked to the GlcA of next disaccharide unit (extracted from PDB ID 1OFM);[5] (D) dermatan sulfate [L-IdoA-(α1→3)-D-GalNAc4S] whose D-GalNAc4S is β(1→4)-linked to the L-IdoA of the next disaccharide unit (extracted from PDB ID 1OFL);[5] (E) keratan sulfate [D-Gal6S-(β1→4)-D-GlcNAc6S] whose D-GlcNAc6S is (β1→3)-linked to the D-Gal6S of the next disaccharide unit (extracted from PDB ID 1KES);[7] and (F) hyaluronan [D-GlcA-(β1→3)-D-GlcNAc] whose D-GlcNAc is (β1→4)-linked to the D-GlcA of the next disaccharide unit (extracted from PDB ID 2BVK).[10] The unsaturated uronic acids (Δ4,5UroA) of the original structures 3E7J, 1OFM and 1OFL were converted respectively to GlcA, GlcA and IdoA. The monosaccharide nomenclatures are GlcA for glucuronic acid; GlcNAc for N-acetylglucosamine; IdoA2S for 2-sulfated iduronic acid; GlcNS6S for N,6-disulfated glucosamine; GalNAc4S for 4-sulfated N-acetylgalactosamine; IdoA for iduronic acid; Gal6S for 6-sulfated galactose; GlcNAc6S for 6-sulfated N-acetylglucosamine. The arrows labeled as “Glycosylation site” indicate the positions of the glycosydic bonds of the disaccharide units. The atoms used in the representations are carbon (grey), nitrogen (blue), oxygen (red) and sulfur (yellow). The hydrogen atoms were omitted for visual simplification. 3D structure representations were created using Discovery Studio Visualizer v.4.5 software (BIOVIA, Dassault Systèmes). The monosaccharides are displayed in their commonest chair configurations: 4C1 for GlcA, GalNAc, GlcNAc and 1C4 for IdoA.
Hp and HS share the same disaccharide repeating unit, but differ in degrees of chain modification.[1] The unmodified Hp/HS backbone is named heparosan and is composed of [→4)-β-D-GlcA-(1→4)-α-D-GlcNAc-(1→] (Figure 1A), where GlcNAc stands for N-acetylglucosamine. HS undergoes less N-deacetylation/N-sulfation, a process that converts GlcNAc into GlcNS, when compared to Hp. Because N-deacetylation/N-sulfation is an activating factor for subsequent modifications, HS also undergoes less modification by way of epimerization (GlcA into IdoA) and O-sulfations at the C2 position of IdoA and the C3 and C6 positions of GlcNS (Figure 2). On the other hand, Hp chains undergo extensive modifications by the modifying enzymes, which in turn give rise to higher hetergeneity in its chains.[3] While Hp is mostly composed of [→4)-α-L-IdoA2S-(1→4)-α-D-GlcNS6S-(1→], where IdoA2S is the 2-sulfated IdoA (Figure 1B), HS is mostly composed of the unmodified [→4)-β-D-GlcA-(1→4)-α-D-GlcNAc-(1→] unit (Figure 1A).
Figure 2.
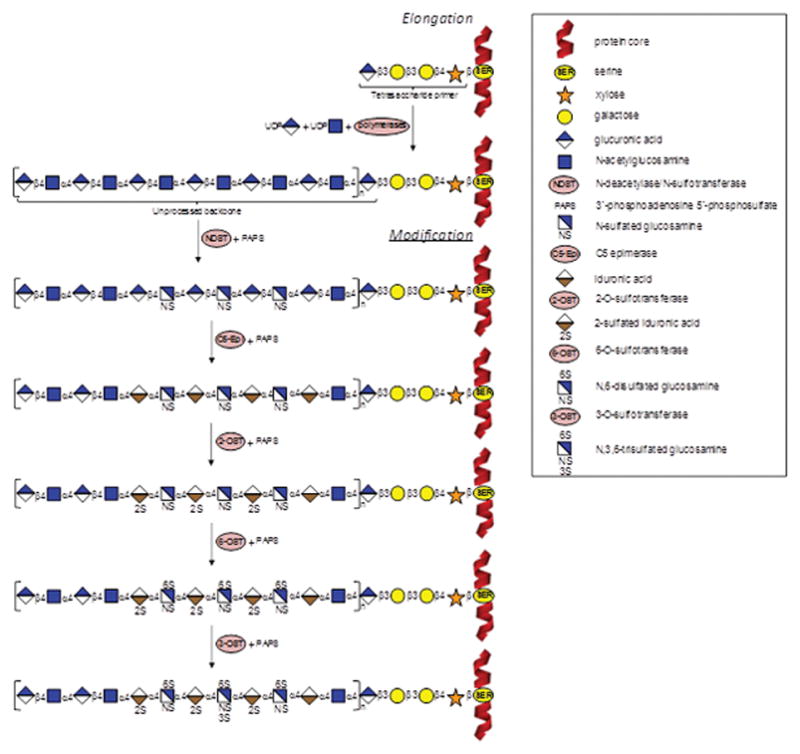
Schematic representation of heparin/heparan sulfate biosynthetic pathway. In the elongation stage, polymerization of the heparin/heparan sulfate chains is made by activities of polymerases (α-UDP-GlcNAc-glycosyltransferase and α-UDP-GlcA-glycosyltransferase) and thereafter by N-deacetylase/N-sulfotransferase (NDST) combined with the sulfate donor PAPS (3′-phosphoadenosine 5′-phosphosulfate). This process starts on a tetrasacchaide primer attached to a serine residue in the protein core. In the modification stage, C5-Epi (epimerase), 2-OST, 6-OST, and 3-OST (2-, 6- and 3-O-sulfotransferases) catalyze respectively the conversion of GlcA into IdoA, 2-sulfation at the IdoA, 6-sulfation at the GlcNS, and 3-sulfation at the GlcNS6S units. Heparin/heparan sulfate bears the same machinery of biosynthesis, varying only at the extensions of the reactions: heparin is much more processed than heparan sulfate.
CS are composed of alternating 3-linked β-D-GalNAc and 4-linked β-D-GlcA units (Figure 1C). It shares much in common with DS, which has 4-linked α-L-IdoA instead of β-D-GlcA (Figure 1D), the result of an active C5 epimerization process during the biosynthesis of DS.[6] Both CS and DS are also sulfated. While CS can bear 4-O- and/or 6-O-sulfations at the GalNAc units, DS has 4-O-sulfation at GalNAc (Figure 1D) and occasional 2-O-sulfation at the IdoA unit.[6]
HA and KS are the least processed GAG types. KS is composed of alternating 4-linked β-D-GlcNAc and 3-linked β-D-Gal units (Figure 1E). Although O-sulfations can occur at the C6 positions of both monosaccharides, GlcNAc units are usually more sulfated than Gal units[8] in KS (Figure 1E). HA is composed of alternating 3-linked β-D-GlcNAc and 4-linked β-D-GlcA units and no further modifications occur after chain polymerization, making HA the only nonsulfated GAG type[9] (Figure 1F).
Because of GAGs’ extracellular location, high anionic character and rich structural diversity, GAGs are capable of interacting with and regulating many proteins whose functions are essential to pathophysiology. In particular, GAGs are known to modulcate wound repair;[11] coagulation;[12] thrombosis;[13] cancer growth and metastasis;[14] inflammation;[15] neovascularization;[16] tissue development,[17] regeneration[18] and repair;[19] cellular growth,[20] differentiation[21] and migration.[22] Although the high structural heterogeneity of GAGs is considered necessary in order to exert their regulatory role over such a vast collection of functional proteins, it poses a challenge for researchers interested in structure-function relationships of GAG. GAG-derived oligosaccharides of defined structures, especially those containg motifs capable of with binding and regulating proteins, are needed to understand the correlation between GAG structure and its biological activity. However, isolation of these biologically active GAG-derived oligosaccharides of well-defined structures through regular depolymerization and purification processes is still laborious. This greatly limits the use of GAG oligosaccharides derived from natural sources. To overcome this obstacle, GAG-like oligosaccharides of well-defined structures are currently being synthezised through chemical and/or enzymatic routes in several laboratories around the world. These synthetic GAG oligosaccharides serve as useful molecular tools for studies of GAG-protein interactions. In this report, after reviewing the current successful routes utilized for synthesis of GAG-like oligosaccharides and presenting some representative libraries available for protein interaction investigations, we also discuss the microarray technology currently utilized in interactomic studies of these synthetic GAG oligosaccharides.
2. Common synthetic routes and GAG oligosaccharide libraries
2.1 Synthesis of GAGs
Research on carbohydrate synthesis has increased markedly in the last decade.[23] Automated systems for carbohydrate synthesis are also being proposed.[23a,23b] In glycosaminoglycanomics, for example, GAG-like molecules (mostly oligosaccharides of well-defined structures and sizes) have been sucessfully synthesized.[24] GAG synthesis can be accomplished either by chemoenzymatic routes[24a–24d,24p,24r,24am,24ai,24as,24az] or based on modular assembly of pre-synthesized building blocks.[23,24e–24j,24l–24o,24ak,24al,24an–24ap,24r,24u,24v,24y] Module-based synthesis is sometimes employed in conjunction with additional chemical and/or enzymatic steps to increase yield and diversity of sulfation patterns.[24e,24f,25] Below, some advances in GAG synthesis are briefly presented. Discussion is separated by GAG species. Note the lack of discussion concerning KS. Protocols for synthesis of KS-related oligosaccharides have not been proposed so far. This comprises however a fertile research program in the field.
2.1.1. Hp and HS
Since Hp is the most investigated therapeutic GAG, easily available and frequently exploited as model compound to any other GAG type (especially HS) in both experimental and theoretical studies, more research has been carried out on Hp synthesis than any other GAG type.[24c–24aj] The second most studied GAG in terms of synthetic methodology is HS.[24q,24ak–24at] As seen in Figures 1A and 1B and stated above, HS and Hp are both composed of GlcN units as the hexosamine and a UroA. The biosynthetic system described in Figure 2 can be exploited for laboratorial synthesis of both Hp and HS molecules in vivo and some of these reactions are also exploited in the chemoenzymatic-based synthesis of Hp and HS oligosaccharides, as detailed below.
2.1.1.1 Modular synthesis of Hp
As reported by Haller and Boons, Hp fragments can be synthesized by modular strategy whereby GlcA moieties are introduced at later stages of the synthetic sequence through selective oxidation of the primary hydroxyl groups with 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) combined with sodium hypochlorite (NAClO) as co-oxidant.This strategy avoids many problems such as C5 epimerization, low glycosyl-donating properties of UroA derivatives and other issues caused by protecting group during the manipulation given the fact protection is sometimes not that selective.[24e] Group protection must be stereospecific in order to avoid heterogeneity in the products. Hence while Levulinoyl esters (Lev) were employed to protect the hydroxyl groups intended to be sulfated in the final products, acetyl (Ac) esters and benzyl (Bn) ethers can be used as permanent protecting groups.[24f]
Figure 3 schematically illustrates this synthetic strategy of selective oxidation and removal of protecting groups developed by Prof. Boons’ team.[24e] After modular assembly of the initial sulfated oligosaccharide, all protecting groups are removed, except the ones on C6 of GlcN. Then, the C6 hydroxyl groups of the glucosides and idosides are selectively oxidized to carboxylic acids by TEMPO and NaOCL and the remaining C6 protecting group is removed to give the target Hp-like oligosaccharide. The selection for the permanent protecting group (“P” in Figure 3) on the C6 positions of the GlcN residues is an important feature to this synthetic strategy and must be compatible with all subsequent steps and be removed without affecting the sulfates. Synthesis of Hp-like disaccharides and trisaccharides have been reported following this strategy[24e]
Figure 3.
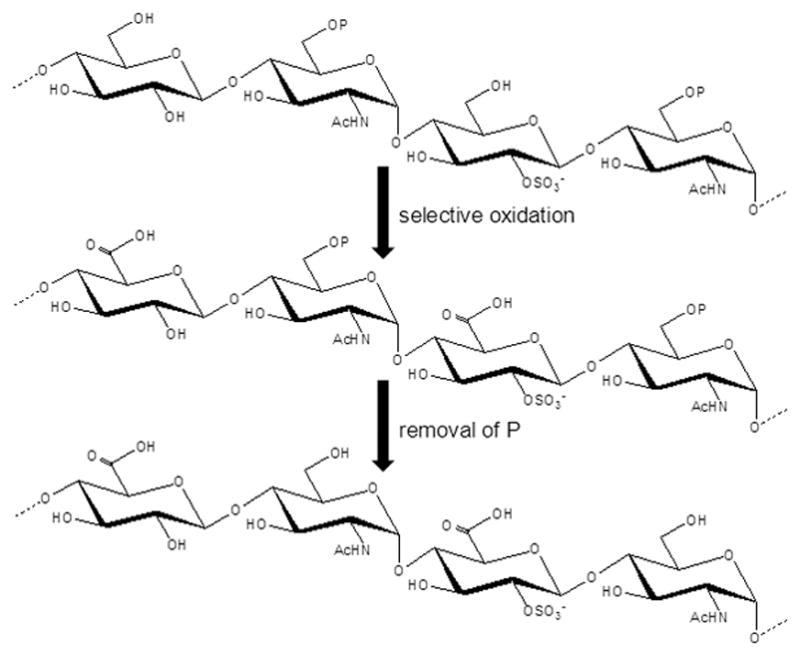
Selective oxidation and removal of protecting (P) groups during modular synthesis of Hp-like oligosaccharides.[24e] Ac stands for acetyl group.
The group of Prof. Peter Seeberger has shown that IdoA-containing building blocks can also be successfully synthesized by modular assemblies for GAG synthesis.[24g] This was achieved starting from the synthesis of methyl-3-O-benzyl-1,2-O-isopropylidene-α-L-idopyranosiduronate out of diacetone glucose. The product of the reaction was modified by nine additional reactive steps, resulting in a final yield of 36%. This idopyranosiduronate is quite useful as both glycosyl acceptor and intermediate molecule that can be further converted into IdoA trichloroacetimidate glycosyl donor for assembling IdoA-composed GAG types like Hp and HS.[24g] Following this protocol, disaccharides and trisaccharides have been obtained from six monosaccharide building blocks (four differentially protected GlcN, one IdoA and one GlcA).[24h] Production of specific protein-binding oligosaccharides, such as a hexasaccharide resembling the antithrombin (AT)-binding sequence, is possible by controlled stereoselective reactions. For instance, installation of α-GlcN linkage was well controlled by placing a conformational constraint on the UroA glycosyl acceptors thereby establishing a new method for stereochemical control. Combination of disaccharide mdules to form trans-UroA linkages was completely selective by virtue of C2 participating groups. Selectiveness in reactions can also achieved by adding either GAG monosaccharide type (GlcN or UroA) into the previously formed disaccharide or trisaccharide units.[24h]
A protocol for synthesis of a fully N-differentiated Hp-like oligosaccharide (containing amine, acetamide and N-sulfonate groups simultaneously) was successfully developed through a strategy of specific group protection. In particular, amine was protected as N-Cbz (carboxybenzyl), N-acetate as N-diacetate, N-sulfonate as azido groups. Free hydroxyls were masked as Bn ethers and O-sulfonate sites were protected by acetate esters. From this selective protection approach, Hp-like disaccharide and tetrasaccharide modules were synthesized.[24i] However, an undesirable β-linked glycosidic bond was unexpectedly generated at the late stages of the synthesis. Hence, an entirely α-composed Hp oligosaccharide chain can not be formed through this method.[24i]
Efficient de novo synthesis of UroA building blocks can also be achieved through stereoselective elongation of thiolacetal protected dialdehydes 1 (Figure 4A) and 2 (Figure 4B).[24o] The aldehydes 1 and 2 were prepared from D-xylose. In order to generate D-GlcA building block (Figure 4C), a highly stereoselective magnesium bromide diethyl etherate (MgBr2OEt2)-mediated Mukaiyama aldol addition to C4-aldehyde 1 can be carried out, whereas the L-IdoA-based building block (Figure 4D) can be prepared from MgBr2OEt2-mediated cyanation of the C5-aldehyde 2 (Figure 4B).[24o] Although the entire synthesis of UroA building blocks involve multiple subproducts and reactive steps, the yield of the final products are very high, about 30% of the starting material. [24o] Besides synthesis of UroA building blocks, orthogonally protected D-GlcN building blocks can also be synthesized for assembly of heparin/heparan sulfate oligosaccharides.[26] In this work D-GlcN building blocks was conveniently prepared from β-D-thiomannoside via a four-step reactive sequence involving regioselective O3 acylation, 4,6-O-benzylidenation, and C2 inversion via azide displacement of the C2-triflate.
Figure 4.
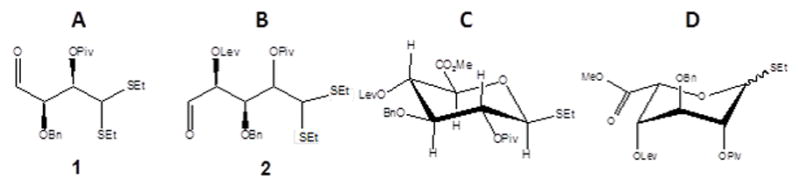
Structural representation of (A and B) dialdehydes 1 and 2 (compounds 12a and 17 of reference[24o]) utilized respectively for synthesis of (C) D-GlcA and (D) L-IdoA building blocks (compounds 16 and 22 of reference[24o]). The abbreviation of protecting groups are: Bn, benzyl; Lev, levulinoyl ether; Me, methyl, Piv, pivaloyl; Set, silyl ether.
Using similar strategies than Prof Seeberger which are based in introducing a mask carboxylate before the condensation process and exploring preformed disaccharides as building blocks, the group of Prof Martin-Lomas has prepared from 2001 to 2004, several Hp hexa-and octasacharides with alternative patterns of substituents.[24aab–24aaf] This strategy was also used by Guerrini and associates for preparing three Hp tetrasacharides in order to uncover the minimal binding sequence to fibroblast growth factor 1 (FGF-1).[24aag] Most of these systentic Hp oligosaccharides (tetra, hexa and ocatsaccharides) were used in binding studies with FGF.[24aab–aag] The substituent distribution of these Hp oligosaccharide libraries prepared by groups of Profs. Seeberger, Boons, Martin-Lomas and Guerrini, exemplifies the overall flexibility of the common synthetic approach of Hp oligosaccharides and their utility in GAG interactomics.
2.1.1.2 Chemoenzymatic synthesis of Hp
The precursor for both Hp and HS, heparosan, can be obtained as a capsular polysaccharide extracted from Escherichia coli K5 strain. Heparosan has been successfully employed as the starting material for the synthesis of these two GAG types.[24p,24s] This is because the bacterial heparosan is identical to unmodified precursors of mammalian HS and Hp and can be further modified through sequential chemoenzymatic reactions in vitro to produce the complex and highly processed GAG types of humans.[24p,24s] Alternatively, even the use of heparosan can be circumvented if enzymes for extending the GAG chain and their activated substrates are available. The chemoenzymatic approaches follows the rationale of GAG biosynthesis in the Golgi apparatus (Figure 2).[24p,24s] In fact all enzymes required for enzymatic synthesis of HS and Hp have been cloned and recombinantly expressed successfully. These enzymes include bacterial glycosyltransferases needed to elongate the heparosan backbone as well as modifying enzymes involved in processing of the GAG backbones such as N-deacetylase/N-sulfotransferase (NDST), C5-epimerase (C5-Epi) and a series of sulfotranferases (OST) responsible for catalyzing sulfation at different O-positions in IdoA and GlcN (2-OST, 6-OST, and 3-OST) using the sulfate donor 3′-phosphoadenosine-5′-phosphosulfate (PAPS). Recently, two Hp dodecasaccharides with promising activities in coagulation have been chemoenzymatically synthesized[24az] (Table 1). Due to the existence of some comprehensive reviews regarding the use of chemoenzymatic synthesis of GAGs,[24a,24c] including Hp and HS, here we opted to focus our attention more on the use of chemoenzymatically synthesized Hp-/HS-derived oligosaccharides utilized for protein interaction studies as further detailed in section 3. Table 1 displays some of the synthetic Hp oligosaccharide libraries.
Table 1.
Structure of some synthetic Hp oligosaccharides.
| Structures a |
|---|
| GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS3S6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS6S-GlcA-pNP[24az] |
| GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS6S-GlcA-pNP[24az] |
| IdoA2S-GlcNAc6S-IdoA2S-GlcNAc6S-IdoA2S-GlcNAc6S[24v] |
| IdoA-GlcNS6S-IdoA-GlcNS6S-IdoA-GlcNS6S[24v] |
| IdoA2S-GlcNS-IdoA2S-GlcNS-IdoA2S-GlcNS[24v] |
| GlcNS-IdoA2S-GlcNAc6S-IdoA-GlcNS-IdoA2S-OMe[24w] |
| GlcNS6S-IdoA2S-GlcNS6S-IdoA2S-GlcNS6S-IdoA2S-OPr[24aa,24ae] |
| GlcNS-IdoA2S-GlcNAc6S-IdoA-GlcNS-IdoA2S-OPr[24aa,24ae] |
| IdoA2S-GlcNS6S-IdoA-GlcNS-IdoA2S-GlcNAc6S-OPr[24aa,24ae] |
| GlcNS-IdoA2S-GlcNS-IdoA2S-GlcNS-IdoA2S-OPr[24aa,24ae] |
| GlcNS6S-IdoA-GlcNS6S-IdoA-GlcNS6S-IdoA-OPr[24aa,24ae] |
| GlcNAc6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-GlcA-AnMan[38] |
| GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-GlcA-AnMan[38] |
| GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-OMe (fondaparinux) |
GlcNS6S, N,6-disulfated glucosamine; GlcA, glucuronic acid; GlcNS3S6S, N,3,6-trisulfated glucosamine; IdoA2S, 2-sulfated iduronic acid; pNP, reducing-end anomeric O1-p-nitrophenyl; GlcNAc6S, 6-sulfated N-acetylglucosamine; IdoA, iduronic acid; GlcNS, N-sulfoglucosamine; OMe, reducing-end anomeric O1-methyl ether; OPr, reducing-end anomeric O1-propyl; AnMAn, anhydromannose.
2.1.1.3 Modular synthesis of HS
Six strategic monosaccharide building blocks can be employed for efficient synthesis of 20 different disaccharide moieties found in HS chains.[24ak] These monosaccharide building blocks are protected by a novel set of four orthogonal protecting groups: Lev, 9-fluorenylmethyl carbonate (Fmoc), tert-butyldiphenylsilyl ether (TBDPS) and allyl ether (All). [24ak] The duly protected disaccharide building blocks can be further converted into glycosyl donors and acceptors in modular synthesis of a wide range of well-defined HS oligosaccharides containing different sulfation patterns.[24s] Modular synthesis of HS oligosaccharides is further aided by the use of anomeric aminopentyl linker tethered to a perfluorodecyl tag, which makes possible the purification of highly polar intermediate derivatives through a fluorous solid-phase extraction (FSPE) process.[24ap] The fluorous-assisted methodology enable repeated glycosylation processes to be performed in order to drive reactions to completion of the aimed products, the HS oligosaccharides.[24ap] New GlcA donors were also specially developed for modular synthesis of HS oligosaccharides.[24ar] Unlike conventional GlcA donors, which usually perform poorly in glycosylation steps, the new type of GlcA donor can give high yields of coupling products via a selective protection reaction of the C2 hydroxyl group by permanent 4-acetoxy-2,2-dimethyl butanoyl (PivOAc) or temporarily by Lev at C4 hydroxyl modified with a selective removable 2-methyl-naphthyl (Nap) ether.[24ar] It has been shown that PivOAc esters can be removed without affecting sulfation esters, making the synthesis protocol very useful. The new GlcA donors do not demand further oxidation reactions and therefore allow easy modular preparation of HS-based disaccharide building blocks.[24ar]
Successful modular assembly of GAG molecules, especially HS oligosaccharides, chemically relies on a set of carefully chosen protecting groups.[24ah,24al] A key issue for selection of a set of protecting groups in modular synthesis of HS (and likely Hp) must meet some requirements as suggested: i) the C-2 hydroxyl-protecting groups of the GlcA and IdoA moieties should allow the stereoselective introduction of 1,2-trans-glycosidic linkages, whereas the C-2 amino group of the GlcN derivatives need to be derivatized in such a way that 1,2-cis-glycosidic linkages can be formed; ii) temporary protecting groups for the anomeric center and C-4 need to be selected for the preparation of glycosyl donors and acceptors; iii) a protecting group is required that can selectively be removed to reveal those hydroxyls that need sulfation; iv) the removal of the permanent protecting groups should be compatible with the presence of base and acid labile sulfate esters; v) the protecting group scheme should be applicable to synthesize each of disaccharide modules found in natural HS; and finally vi) a unified set of chemical conditions is required for the preparation of the various disaccharide modules, oligosaccharide assembly, sulfation and deprotection.[24al]
The protecting group strategy that have been developed for the HS disaccharide modules is summarized in Figure 5. Lev is employed for the hydroxyls that need sulfation. In HS, the C-3 and C-6 of GlcN and C-2 hydroxyls of GlcA/IdoA acid moiety can be sulfated and therefore depending on the sulfation pattern of a targeted disaccharide module, one or more of these positions will be protected as Lev esters. An important feature of the Lev ester is that when present at the C-2′ position, it will be able to direct the formation of 1,2-trans-glycosides by neighboring group participation. In case that the C-2′ position of a disaccharide module does not need sulfation, an Ac group is employed as a permanent protecting group. This ester can also perform neighboring group participation but is stable under the conditions used for the removal of Lev esters. An azido group will be used as an amino-masking functionality. This derivative does not perform neighboring group participation and therefore allows the introduction of α-glucosides. An azido-group can easily be reduced to an amine, which can either be acetylated or sulfated. The C-4′ hydroxyl, which is required for extension, will be protected as Fmoc. The Fmoc group can be removed with triethylamine (Et3N) in dichloromethane or dimethylformamide (DMF) without affecting the Lev ester whereas the Lev group can be cleaved with hydrazine buffered with acetic acid and these conditions do not affect the Fmoc carbonate. The anomeric center of the disaccharides will be protected as thexyldimethyl silyl (TDS) glycosides and this functionality can easily be removed by treatment with HF in pyridine without affecting the other protecting groups. The resulting lactol can then be converted into a trichloroacetimidate by employing K2CO3 and trichloroacetonitrile in DCM. Finally, Bn ethers are used as permanent protecting groups for the other hydroxyls.[24al]
Figure 5.
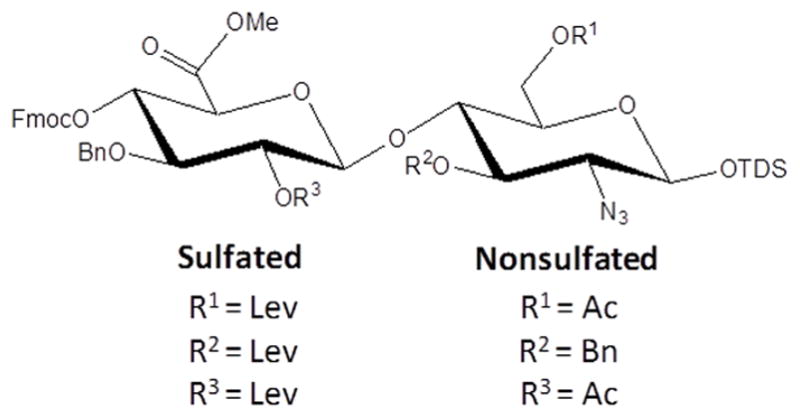
Orthogonal protecting groups for disaccharide building blocks.[24al] Lev, Ac, Bn, TDS and Fmoc represents respectively Levulinoyl, acetyl, benzyl, thexyldimethyl silyl and 9-fluorenylmethyl carbonate groups.
2.1.1.4 Chemoenzymatic synthesis of HS
Protocols to synthesize HS oligosaccharide on the bases of chemoenzymatic reactions have been established.[24a,24b,24s,24am,24ap,24as] Comprehensive reviews in the area have been published.[24a,24b,24s] Similar to Hp, the synthesis starts with extension of a primer, which is usually a reactive disaccharide, by bacterial polymerases using UDP-GlcNAc and UDP-GlcA as sugar donors. After this initial step, moderate incubation in the sequential order of the resultant polymer with (1) NDST+PAPS, (2) C5-epimerase, (3) 2-OST+PAPS, (4) 6-OST+PAPS, and finally (5) 3-OST+PAPS leads to production of HS oligosaccharides (Figure 2).[24am] These enzymatic reactions, which can be optimized to maximize the yield for any particular product, are often combined with chemical modifications to generate a large set of HS oligosaccharides (Table 2).[24a] Figure 4 illustrates some of these specific chemoenzymatic reactions involved in synthesis of HS oligosaccharides.[24am] Examples of HS-based oligosaccharides built from this method of synthesis are N-sulfated octasaccharides (Figure 6A), heptasaccharide carrying an internal Ido2S unit (Figure 6B), and deca-, undeca- and dodecasaccharides containing the trisulfated GlcNS3S6S units.[24am] Another example is the 2-O-sulfated GlcA-containing HS hexasaccharides recently produced by Hsieh and coworkers.[24aq] As seen for the modular-based synthesis of HS oligosaccharides,[24ap] fluorous tag can also be fruitfully used in chemoenzymatic-based systhesis of HS tetrasaccharide and hexasaccharide.[24as] Besides facilitating purification steps via FSPE, it has been reported that this tag does not interfere with any enzyme involved in the elongation and modification of HS chains.[24as] Table 2 displays some of the synthetic HS oligosaccharide libraries.
Table 2.
Structure of some synthetic HS oligosaccharides.
| Structures a |
|---|
| 48 HS-based disaccharides with the following structures: GlcNR3R16R2-GlcA2R3 and GlcNR3R16R2-IdoA2R3 where R = H or Acetyl or SO3−; R1, R2 and R3 = H or SO3− [39] |
| GlcA-GlcNAc6S-GlcA-GlcNAc6S-(CH2)5-NH2[24ao] |
| GlcA-GlcNAc6S-IdoA-GlcNAc6S-(CH2)5-NH2[24ao] |
| GlcA-GlcNAc6S-IdoA2S-GlcNAc6S-(CH2)5-NH2[24ao] |
| GlcA-GlcNS6S-GlcA-GlcNS6S-(CH2)5-NH2[24ao] |
| GlcA-GlcNS6S-IdoA-GlcNS6S-(CH2)5-NH2[24ao] |
| GlcA-(GlcNS6S-GlcA)n-GlcNAc-OMe where n = 1 or 2[24as] |
| IdoA-GlcNS6S-IdoA2S-GlcNS6S-(CH2)-NH2[40] |
GlcN, glucosamine; GlcA, glucuronic acid; IdoA, iduronic acid; GlcNAc6S, 6-sulfated N-acetylglucosamine; IdoA2S, 2-sulfated iduronic acid; GlcNS6S, N,6-disulfated glucosamine; GlcNAc, N-acetylglucosamine; OMe, reducing-end anomeric O1-methyl ether.
Figure 6.

Schematic representation for the synthesis of (A) N-sulfated octasaccharides and (B) a heptasaccharide carrying an internal IdoA2S unit. Reagents and recovery yield of each step are as follows: a, KfiA (α-UDP-GlcNAc-glycosyltranferase), UDP-GlcNAc or UDP-GlcNTFA (trifluoro acetylglucosamine), pmHS2 (α-UDP-GlcA-glycosyltransferase), and UDP-GlcA. The purification yield by fluorous column was 80%, whereas the purification yield without fluorous column (B) was about 40%. b, methanol/triethylamine/water (2:1:2), N-sulfotranferase, PAPS (3′-phosphoadenosine 5′-phosphosulfate). Recovery yield was 40–50%. c, KfiA and UDP-GlcNTFA. Recovery yield was 50%. d, C5-epi/2-OST, PAPS. Recovery yield was 40%. Modified with permission.[24am]
2.1.3 CS/DS
Modular automated synthesis of both CS[24au,24av] and DS[24ay] has been recently achieved. In this regard, two synthetic new CS hexasaccharides have been built through this method.[24au] These CS oligosaccharides were prepared on a solid support that is equipped with a photolabile linker. The linker cleavage from the resin is performed in a continuous-flow photoreactor under chemically mild conditions. [24au] Likewise, DS-like oligosaccharides can be produced in good yields based on the systematic combination of orthogonally protected D-GalNAc and L-IdoA building blocks together with the photo-cleavable linker used for CS oligosaccharide production.[24av]
Recently, a new enzyme-based method to synthesize a library of CS oligosaccharides consisting of 15 different structures has been reported.[24aw] This library covers 4-O-sulfated and 6-O-sulfated oligosaccharides with the size ranging from trisaccharide to nonasaccharide. This new route has allowed incorporation of unnatural monosaccharides such as 6-O-sulfo 2-azido galactosamine into CS oligosaccharides.[24aw] In another recent work, CS-related glycomimetics have been produced.[24ax] In this work the authors have described the synthesis of the disaccharide unit of the CS-E (GlcA-GalNAc(4,6-di-sulfated) in a multivalent presentation.[24ax] The work of Maza and associates has also produced some synthetic CS/DS-like oligosaccharides.[27] Figure 7 illustrates an example in which a a CS/DS-related tetrasaccharide has been created by using three major building blocks.[27] Both enzymatic[28] and chemical synthesis[27] have been proposed for production of CS/DS oligosaccharides including the use of automated system developed by the Seeberger group, by which two CS hexasaccharides have been produced.[29] Table 3 displays some of these synthetic CS/DS-like oligosaccharides.
Figure 7.
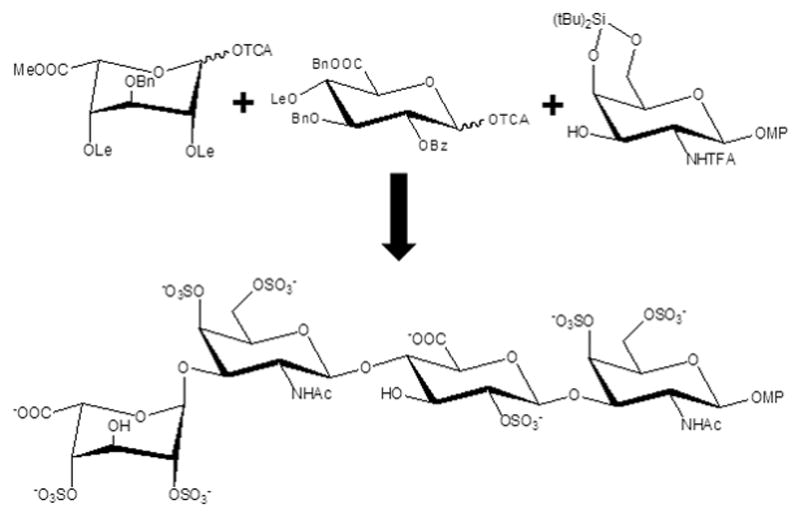
Building blocks used for the synthesis of a CS/DS tetrasaccharide.[27] The chemical groups are the following: OTCA, reducing-end anomeric O1-trichloroacetyl; Bn, benzyl; Me, methyl; Lev, Levulinoyl esters; Bz, benzyl; NHTFA, N-triflouracetyl; OMP, reducing-end anomeric O1-methoxyphenyl ether; Si(tBu)2, di-t-butylsilyl.
Table 3.
Structure of some synthetic CS/DS-like oligosaccharides.
| Structures a |
|---|
| GlcA2Bz3Bn4Ac6Me-(GalNTCA4R26R1-GlcA2Bz3Bn6Me)2-GalNTCA4R26R1-(CH2)-NHCbz where R1 = SO3− and R2 = H[24au] |
| GlcA2Bz3Bn4Ac6Me-(GalNTCA4R26R1-GlcA2Bz3Bn6Me)2-GalNTCA4R26R1-(CH2)-NHCbz where R1 = H and R2 = SO3− [24au] |
| IdoA2Bz4Ac6Me-GalNTCA4SO3−)n-O-(CH2)5-NHCbz where n = 1 or 2[24ay] |
| GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc6S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc-GlcA-GalNAc4S-GlcA-pNP[28] |
| GlcA-GalNTFA-GlcA-GalNAc-GlcA-pNP2[28] |
| GlcA-GalNTFA6S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNH2-GlcA-GalNAc-GlcA-pNP[28] |
| GlcA-GalNH26S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAz-GlcA-GalNAc-GlcA-pNP[28] |
| GlcA-GalNAz6S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc6S-GlcA-GalNAc6S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc-GlcA-GalNAc4S-GlcA-GalNAc4S-GlcA-pNP[28] |
| GlcA-GalNAc-GlcA-GalNAc-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc-GlcA-GalNAc6S-GlcA-GalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc6S-GlcA-GalNAc6S-GlcA-GalNAc6S-GlcAGalNAc6S-GlcA-pNP[28] |
| GlcA-GalNAc-GlcA-GalNAc4S-GlcA-GalNAc4S-GlcAGalNAc4S-GlcA-pNP[28] |
| IdoA2S4S-GalNAc4S6S-GlcA2S-GalNAc4S6S-OMP[27] |
| GalNAc4S6S-GlcA-(CH2)3-NH2[27] |
GlcA2Bz3Bn4Ac6Me, 2-benzoyl 3-Benzyl 4-Acetyl 6-Methyl glucuronic acid; GalNTCA, N-trichloroacetylgalactosamine; GlcA2Bz3Bn6Me, 2-benzoyl 4-acetyl 6-methyl glucuronate; IdoA2Bz4Ac6Me, 2-benzoyl 4-acetyl 6-methyl iduronate; GlcA, glucuronic acid; GalNAc6S, 6-sulfated galactosamine; pNP, reducing-end anomeric O1-p-nitrophenyl; GalNAc, N-acetylgalactosamine; GalNTFA, N-trifluoracetyl galactosamine; GalNTFA6S, 6-sulfated N-trifluoracetyl galactosamine; GalNH2, galactosamine; GalNH26S, 6-sulfated galactosamine; GalNAz, N-azide galactosamine; GalNAz6S, 6-sulfated N-azide galactosamine; GalNAc4S, 4-sulfated N-acetyl galactosamine; IdoA2S4S, 2,4-disulfated iduronate; GalNAc4S6S, 4,6-disulfated galactosamine; OMP, reducing-end anomeric O1-methylphenol.
2.1.4 HA
HA has the simplest structure among all GAG types (Figure 1F vs 1A-E). Therefore, synthesis of this GAG should be easier but because of the low yields of the end products and the high cost of the sugar nucleotides substrates (UDP-GlcNAc and UDP-GlcA) required, enzyme-based synthesis have been limited and been surpassed by the chemistry-based synthesis. Moreover due to the absence of sulfation in HA, HA’s affinity and specificity for GAG-binding proteins are low compared to other GAGs. Nonetheless, certain synthetic HA-based oligosaccharides have been generated and routes of synthesis have been proposed.[24a,30]
3. Microarray technology in GAG interactomics
3.1. Overview
Introduction of array-based high-throughput screening of molecular interactions in the early 1990s has made significant impacts on biochemistry and cell-biology. For the first time, rapid screening of molecular interactions can be carried out with relatively small amounts of material. Although originally developed for nucleic acids, microarray technology was quickly generalized to other biopolymers including peptides and polysaccharides with equal success. The technology is especially suited for polysaccharides since the highly diverse nature of glycan structures means study of protein-glycan interactions benefit more from high throughput techniques. In addition, glycan synthesis is usually more challenging than other biopolymers. As a result, the amount of glycan available for experimentation is usually low. The small material demand of microarrays means more experiments can be completed with only a modest amount of material. These reasons have made the microarray an ideal technology for high throughput screening of glycan-mediated interactions involving proteins and cells.
The first report of glycan microarray, or glycoarray, occurred in 2002.[31] Its popularity among glycobiology researchers has led to the creation of Consortium for Functional Glycomics (www.functionalglycomics.org), which continues to supply the community with access to glycoarrays. The sources of glycans on these microarrays vary. Many came from natural sources while others are produced by de novo synthesis. The expanded use of chemoenzymatic approaches in glycan synthesis has also greatly increased the number of glycans available in glycoarray analysis. Several strategies have been used to immobilize the glycans on microarrays. In particular, synthetic glycans can often be functionalized with an amine group at the reducing end, allowing them to be attached to glass slides with NHS activated carboxyl groups. For natural glycans, hydrazide activated surfaces can be used to attach glycans using their reducing end aldehyde.[32,33] The quantity of material required to prepare the microarrays is usually minute. Modern microarray printers require only femtomole quantity of material for each spot, allowing the manufacturing of large quantities of microarrays from a small amount of material. It is also common for each glycan to be printed at different concentrations, forming a concentration gradient that can be used to estimate dissociation constants under some circumstances. However, precise measurement of binding affinities remains challenging because of lack of reliable method to estimate the density of the ligand on the microarray surface.
Although the number of glycans available on glycoarrays has greatly increased, the number of GAG oligosaccharides on these microarrays is still small. This is partly because purification and synthesis of GAG continues to be nontrivial. The first report of dedicated GAG microarray created using synthetic GAG oligosaccharides appeared in 2006.[24l] Although the microarray contained only a very limited collection of GAG building blocks and Hp di-, tetra- and hexa-saccharides containing the disaccharide unit IdoA2S-GlcNS6S, it already produced interesting results by showing that the GAG-binding protein FGF-1 has specific interactions with GAGs. Around the same time, the group of Hsieh-Wilson also reported creation of GAG microarrays prepared from chemically modified natural GAG polymers.[34,35] Subsequent studies greatly expanded the repertoire of GAG oligosaccharides available in a glycoarray. For instance, groups of Xuefei Huang and Jian Liu collaborated to synthesize several GAG oligosaccharides by assembling the GAG carbohydrate backbone using de no synthesis and then sulfating them using sulfotransferases (Figure 2).[36] This method allowed them to create nine Hp hexasaccharides with differing sulfation patterns for microarray study. The Liu group went on to create a microarray of fourteen HS heptasaccharide using only chemoenzymatic synthesis in a separate study.[37] In that study, the authors also attempted to quantify the density of oligosaccharide on the surface by digesting the immobilized oligosaccharides with Hp lyases and quantifying the re-leased disaccahrides. Their conclusion was that immobilization efficiency ranged from 1 to 10 %. Recently, the most extensive GAG microarray created to date has just been published by the group of Geert-Jan Boons.[24ah] Through a combination of de novo synthesis and enzymatic sulfation methods, the authors prepared 47 tetrasaccharide with diverse sulfation density and distribution. This is the largest assembly of GAG tetrasaccharides to date and demonstrates the power of the hybrid synthetic approach, which overcomes the difficulty of working with sulfated building blocks encountered in conventional synthetic chemistry. It should be noted that although none of the microarrays is readily available to the glycobiology community currently, access to these microarrays will become more routine in the future.
3.2. Alternative Approaches to Microarray
Although microarray is the most facile method for screening GAG-protein interactions, the amount of oligosaccharide immobilized is usually difficult to quantify, thereby preventing accurate quantitation of dissociation constants. An alternative approach was adopted by the group of Pedro Nieto for high-throughput screening of protein-GAG interactions. In particular, Maza et al. used competitive binding between a GAG oligosaccharide and a fluorescently labeled GAG ligand with known protein affinity to estimate the affinity of any GAG oligosaccharide for the target protein.[27] Because the inhibition is estimated using the fluorescence polarization of the fluorescently labeled ligand, the throughput of the assay can be easily increased by using a fluorescent microplate reader. With such a method, Maza et al. studied the interaction of FGF-2 with several synthesized GAG oligosaccharides, including several tetrasaccharides containing CS/DS-like motifs. The results showed that highly sulfated CS-E like GAGs bind FGF-2 with equal affinity as Hp oligosaccharides.
Another alternative technology to microarrays was introduced by the Luminex corporation. Luminex pioneered the use of color coded polystyrene microspheres that can be coated with ligands such as GAG oligosaccharides through amide coupling. To detect protein binding, the coated beads are mixed with phycoerythrin labeled target protein. If the target protein binds the ligand-covered bead, the complex can be identified by a flow-cytometry instrument that can simultaneously detect both the color coding of the bead and the fluorescence of the target protein. Because the instrument can distinguish between up to 500 different types of color-coded beads, the technology allows the use of up to 500 different ligands simultaneously. While it may not be as high throughput as large microarrays, the technology does allow considerable flexibility in the number of ligands users can use.
3.3. Microarray in GAG interactomics
The scarcity of synthesized GAG microarrays means a relatively small number of GAG-protein interactions have been studied using them. However, these studies offer excellent insights into structure-activity relationships of GAG-protein interactions. Figure 8 illustrates a simplified scheme of the microarray technology employed in studies of GAG interactomics. The well-known GAG-binding growth factor FGF-1 was one of the first proteins to be studied using GAG microarray prepared from synthesized oligosaccharides.[24l] Although this initial study used a GAG microarray with only a limited set of IdoA2S-GlcNS6S containing oligosaccharides that differed only in their lengths, it did reveal that interaction of FGF-1 with Hp may not simply depend on size of GAGs since the monosaccharide 2-O-sulfated iduronate binds FGF-1 with higher affinity than fully sulfated disaccharide and tetrasaccharides. However, the study did not quantify the amount of GAG immobilized in each spot, therefore the results may be skewed by differences in concentrations of immobilized GAGs. In a later study, Shipp et al. studied GAG specificity of a set of proteins using a microarray of selectively desulfated Hp polymers.[35] Their results showed 2-O-sulfated IdoA along with GlcNS were important to the binding of FGF-2, but 6-O-sulfation on GlcN did not appear to be important. Interestingly, the same study showed that 6-O-sulfation is as important as 2-O-sulfation and N-sulfation for binding of FGF-1, revealing important differences in the binding specificity of FGF growth factors. The same study also showed that FGF-17 has GAG preferences similar to FGF-2 while FGF-16’s GAG specificity resembled that of FGF-1.
Figure 8.

Schematic illustration of preparation and usage of GAG microarrays. GAG microarrays are most often prepared by spotting synthetic GAG oligosaccharides functionalized with amine at the reducing end onto NHS-activated glass slides. Solutions containing fluorescently labeled GAG-binding proteins are then incubated with the microarray. After the incubation, excess GAG-binding proteins are washed away and affinity of the protein for each spot is determined by the fluorescent intensity.
FGF-2 was also the subject of study by Dulaney et al.[36] Using an array of nine Hp oligosaccharides, they showed that FGF-2 has the highest affinity for fully sulfated Hp hexasaccharides and tetrasaccharides, but removal of either 2-O-sulfation on IdoA, 6-O-sulfation on GlcN or N-sulfation on GlcN weakened FGF-2 binding greatly. This may seem to contradict the results of Shippe et al. However, because the Dulaney study used oligosaccharides while Shippe et al. used heterogeneous polymers, the results may not be directly comparable since length of GAG is also an important determinant of GAG-protein interactions. Most recently, Zong et al. probed the GAG specificity of FGF-2 using a microarray containing 47 synthesized GAG tetrasaccharides.[24ah] Their results showed presence of 2-O-sulfated IdoA at either the non-reducing end or in the middle of the polysaccharide will greatly enhance the binding, in agreement with the initial study by de Paz et al.[24l] Interestingly, the same study seems to indicate that, although the presence of 3-O-sulfation in GlcNS alone can significantly enhance binding to FGF-2, presence of both 3-O- and 6-O-sulfation in GlcNS weakens binding. The study also analyzed the GAG specificity of several chemokines. It showed the chemokine CCL2 have the same preference for 3-O-sulfated GlcNS as FGF-2, but the chemokine CCL7 appears to be a promiscuous GAG-binding protein with significant affinities for a large number of highly sulfated tetrasaccharides. The most interesting GAG specificity profile was produced by the chemokine CXCL10, which showed a high degree of selectivity towards the glycans on the microarray. In particular, it has the strongest affinity for a medium sulfation density tetrasaccharide GlcA-GlcNS6S-IdoA-GlcNS6S. The authors attributed this to the presence of both 6-O-sulfated GlcNS and the right combination of IdoA and GlcA in the backbone. Interestingly, CXCL10’s affinity for highly sulfated tetrasaccharides is weaker and a preference for 3-O-sulfated GlcNS was detected for these tetrasaccharides. Because of the large number of tetrasaccharides in the array the study by Zong et al. was able to produce considerable amount of insight into the complexity of GAG-protein interactions and demonstrated how small changes in protein sequence can lead to drastically different GAG specificities.
Besides FGF and chemokines, the well-known anticoagulation serpin antithrombin (AT) has also been studied using GAG microarray by Yang et al.[37] The array consists of fourteen heptasaccharides, many of which contained 3-O-sulfated GlcNS, which is known to be recognized by AT. The array results confirmed this preference since only heptasaccharides containing 3-O-sulfated GlcNS showed significant affinity for the protein. The array was also used to test the specificity of the sulfotransferase 3-OST (Figure 2). Interestingly, it has highest affinity for 3-O-sulfate containing heptasaccharides, which are also products of the enzyme. Its affinity for highly sulfated heptasaccharides without 3-O-sulfation are significantly lower even if sulfation density is maintained.
4. Conclusions
In this review article we have highlighted the importance of the combination between synthetic GAG oligosaccharide libraries and the microarray technology in the progress of the current GAG interactomics. Synthetic oligosaccharides have been created in many laboratories around the world and are becoming more and more available for biochemical and biophysical investigations of GAGs, especially those involving interactions with GAG-binding proteins. These oligosaccharides can be synthesized by chemoenzymatic methods or de novo synthesis through modular assembly. Some structures of these synthetic GAG oligosaccharide libraries are presented in Tables 1–4, respectively for Hp, HS, CS/DS and HA. These tables are far from surveying the entire synthetic GAG oligosaccharide libraries described in the literature. The primary goal of showing these tables is to illustrate the most common types of synthetic GAG oligosaccharide structures available for biophysical and pharmaceutical studies, especially those concerning structure-activity relationship of GAGs through investigations of GAG-protein interactions. Among all, the most studied oligosaccharide is the commercially available Hp pentasaccharide named fondaparinux (Table 1). The combination of synthetic GAG oligosaccharides and microarray technology has greatly enhanced the ability of the glycobiology community to study the GAG specificities in interactions with GAG-binding proteins. Although the microarrays available currently contained only small oligosaccharides with a limited number of sulfate configurations, the data already generated much insight into the complex and dynamic nature of protein-GAG interactions. With additional improvements in synthetic methodology, it is hoped that longer GAG oligosaccharides with even more diverse sulfate distributions can be made available, so that GAG microarray experiments can be as simple and common place as their nucleic acid counterparts.
Table 4.
Structure of some synthetic HA oligosaccharides.[24a]
| Structures a |
|---|
| GlcA-GlcNAc-GlcA-GlcNAc-OMP |
| GlcNAc-GlcA-GlcNAc-GlcA-(1)-OMP |
| GlcA-GlcNAc-OMP |
| GlcA-GlcNAc-GlcA-OMP |
| GlcNAc-GlcA-OMP |
| GlcNAc-GlcA-GlcNAc-GlcA-OMe |
| GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-OMe |
| GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-OMe |
| GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-OMP |
| GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-OMP |
| GlcA-GlcNAc-OMe |
| GlcNAc-GlcA-GlcNAc-OMe |
GlcA, glucuronic acid; GlcNAc, N-acetyl glucosamine; OMP, reducing-end anomeric O1-methoxyphenyl ether; OMe, reducing-end O1-methyl ether.
Acknowledgments
Profs. Vitor H. Pomin and Xu Wang acknowledge the managing editor David Peralta of ChemMedChem and the guest editors Nathan Brown and Andreas Bender of the Special Issue “Chemoinformatics in Drug Discovery” for the invitation to participate in the special issue with an article.
Biographies
 Vitor H. Pomin (M.S., Ph.D.) is assistant professor of Biological Chemistry, Biochemistry, Glycobiology, and NMR Spectroscopy at Institute of Medical Biochemistry Leopoldo de Meis, Federal University of Rio de Janeiro, Brazil, since May 2011. He pursued his undergraduate studies in Biological Sciences and graduate studies in Biological Chemistry at the same university. He received his Diploma of Licentiate, M.S., and Ph.D. in 2003, 2005 and 2008, respectively. He pursued a post-doctorate experience at the Complex Carbohydrate Research Center, University of Georgia, United States, until April 2011. Dr. Pomin has published around 60 articles in peer-reviewed journals and 13 book chapters, besides serving as main editor in 6 academic/scientific books. He is currently editorial member and frequent peer-reviewer in many internationally recognized journals like Biopolymers, Carbohydrate Polymers, Carbohydrate Research, Biochimica et Biophysica Acta-General Subjects, Marine Drugs, PlosOne, Journal of Biological Chemistry, Glycoconjugate Journal, Current Protein & Peptide Science, ChemPlusChem, Scientific Reports, Analytical Chemistry, Glycobiology, Biochemistry and many others. He conducts research mostly on glycobiology (especially sulfated glycans), structural (glyco)biology and NMR spectroscopy.
Vitor H. Pomin (M.S., Ph.D.) is assistant professor of Biological Chemistry, Biochemistry, Glycobiology, and NMR Spectroscopy at Institute of Medical Biochemistry Leopoldo de Meis, Federal University of Rio de Janeiro, Brazil, since May 2011. He pursued his undergraduate studies in Biological Sciences and graduate studies in Biological Chemistry at the same university. He received his Diploma of Licentiate, M.S., and Ph.D. in 2003, 2005 and 2008, respectively. He pursued a post-doctorate experience at the Complex Carbohydrate Research Center, University of Georgia, United States, until April 2011. Dr. Pomin has published around 60 articles in peer-reviewed journals and 13 book chapters, besides serving as main editor in 6 academic/scientific books. He is currently editorial member and frequent peer-reviewer in many internationally recognized journals like Biopolymers, Carbohydrate Polymers, Carbohydrate Research, Biochimica et Biophysica Acta-General Subjects, Marine Drugs, PlosOne, Journal of Biological Chemistry, Glycoconjugate Journal, Current Protein & Peptide Science, ChemPlusChem, Scientific Reports, Analytical Chemistry, Glycobiology, Biochemistry and many others. He conducts research mostly on glycobiology (especially sulfated glycans), structural (glyco)biology and NMR spectroscopy.
 Xu Wang (Ph.D.) has been an assistant professor in the School of Molecular Sciences at Arizona State University, Tempe, USA since 2011. After receiving his PhD in Biochemistry from University of Alberta in Edmonton, Canada in 2005, he joined the group of James H. Prestegard at the Complex Carbohydrate Research Center in Athens, Georgia USA as a post-doctoral associate before joining the faculty at Arizona State Univeristy. His research interest is in structural and mechanism studies of glycosaminoglycan-binding proteins, including chemokines, bacterial adhesins and cytokines. He has published more than 30 peer-reviewed articles and has reviewed manuscripts for many journals including Journal of Biological Chemistry, Glycobiology, Biochemistry and many others.
Xu Wang (Ph.D.) has been an assistant professor in the School of Molecular Sciences at Arizona State University, Tempe, USA since 2011. After receiving his PhD in Biochemistry from University of Alberta in Edmonton, Canada in 2005, he joined the group of James H. Prestegard at the Complex Carbohydrate Research Center in Athens, Georgia USA as a post-doctoral associate before joining the faculty at Arizona State Univeristy. His research interest is in structural and mechanism studies of glycosaminoglycan-binding proteins, including chemokines, bacterial adhesins and cytokines. He has published more than 30 peer-reviewed articles and has reviewed manuscripts for many journals including Journal of Biological Chemistry, Glycobiology, Biochemistry and many others.
Footnotes
Conflict of Interest
Vitor H. Pomin and Xu Wang declare no conflict of interest.
References
- 1.a) Sasisekharan R, Venkataraman G. Curr Opin Chem Biol. 2000;4:626–63. doi: 10.1016/s1367-5931(00)00145-9. [DOI] [PubMed] [Google Scholar]; b) Rabenstein DL. Nat Prod Rep. 2002;19:312–33. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 2.Shaya D, Zhao W, Garron ML, Xiao Z, Cui Q, Zhang Z, Sulea T, Linhardt RJ, Cygler M. J Biol Chem. 2010;285:20051–20061. doi: 10.1074/jbc.M110.101071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugahara K, Kitagawa H. IUBMB Life. 2002;54:163–175. doi: 10.1080/15216540214928. [DOI] [PubMed] [Google Scholar]
- 4.Mulloy B, Forster MJ, Jones C, Davies DB. Biochem J. 1993;1:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michel G, Pojasek K, Li Y, Sulea T, Linhardt RJ, Raman R, Prabhakar V, Sasisekharan R, Cygler M. J Biol Chem. 2004;279:32882–32896. doi: 10.1074/jbc.M403421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugahara K, Tadahisa Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Curr Opin Struct Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Arnott S, Guss JM, Hukins DWL. J Mol Biol. 1974;88:175–184. doi: 10.1016/0022-2836(74)90303-9. [DOI] [PubMed] [Google Scholar]
- 8.Pomin VH. Int J Biol Macromol. 2015;72:282–289. doi: 10.1016/j.ijbiomac.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 9.Almond A. Cell Mol Life Sci. 2007;64:1591–1596. doi: 10.1007/s00018-007-7032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almond A, DeAngelis PL, Blundell CD. J Mol Biol. 2006;358:1256–1269. doi: 10.1016/j.jmb.2006.02.077. [DOI] [PubMed] [Google Scholar]
- 11.a) Kosir MA, Quinn CC, Wang W, Tromp G. J Surg Res. 2000;92:45–52. doi: 10.1006/jsre.2000.5840. [DOI] [PubMed] [Google Scholar]; b) Ghatak S, Maytin EV, Mack JA, Hascall VC, Atanelishvili I, Rodriguez RM, Markwald RR, Misra S. Int J Cell Biol. 2015;2015:1–20. doi: 10.1155/2015/834893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bourin MC, Lindahl U. Biochem J. 1993;289:313–330. doi: 10.1042/bj2890313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang IK, Hursting MJ. Circulation. 2005;111:2671–2683. doi: 10.1161/CIRCULATIONAHA.104.518563. [DOI] [PubMed] [Google Scholar]
- 14.a) Afratis N, Gialeli C, Nikitovic D, Tsegenidis T, Karousou E, Theocharis AD, Pavão MSG, Tzanakakis GN, Karamanos NK. FEBS J. 2012;279:1177–1197. doi: 10.1111/j.1742-4658.2012.08529.x. [DOI] [PubMed] [Google Scholar]; b) Liu D, Shriver Z, Qi Y, Venkataraman G, Sasisekharan R. Semin Thromb Hemost. 2002;28:67–78. doi: 10.1055/s-2002-20565. [DOI] [PubMed] [Google Scholar]
- 15.Pomin VH. Eur J Med Chem. 2015;92:353–369. doi: 10.1016/j.ejmech.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Pardue EL, Ibrahim S, Ramamurthi A. Organogenesis. 2008;4:203–214. doi: 10.4161/org.4.4.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esko JD, Kimata K, Lindahl U. Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. Chapter 16. Cold Spring Harbor; NY: 2009. Available from http://www.ncbi.nlm.nih.gov/books/NBK1900/ [PubMed] [Google Scholar]
- 18.Salbach J, Rachner TD, Rauner M, Hempel U, Anderegg U, Franz S, Simon JC, Hofbauer LC. J Mol Med. 2012;90:625–635. doi: 10.1007/s00109-011-0843-2. [DOI] [PubMed] [Google Scholar]
- 19.Peplow PV. Thromb Haemost. 2005;94:4–16. doi: 10.1160/TH04-12-0812. [DOI] [PubMed] [Google Scholar]
- 20.Yamada Y, Sugahara K. Curr Drug Discov Technol. 2008;5:289–301. doi: 10.2174/157016308786733564. [DOI] [PubMed] [Google Scholar]
- 21.Smith RA, Meade K, Pickford CE, Holley RJ, Merry CL. Biochem Soc Trans. 2011;39:383–387. doi: 10.1042/BST0390383. [DOI] [PubMed] [Google Scholar]
- 22.Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AEI, Wells TNC, Parks WC, Wight TN, Martin TR, Frevert CW. J Immunol. 2010;184:2677–2685. doi: 10.4049/jimmunol.0903274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Werz DB, Seeberger PH. Chem Eur J. 2005;11:3194–3206. doi: 10.1002/chem.200500025. [DOI] [PubMed] [Google Scholar]; b) Seeberger PH, Werz DB. Nat Rev Drug Discov. 2005;4:751–763. doi: 10.1038/nrd1823. [DOI] [PubMed] [Google Scholar]; c) Boltje TJ, Buskas T, Boons GJ. Nat Chem. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yin J, Eller S, Collot M, Seeberger PH. Beilstein J Org Chem. 2012;8:2067–2071. doi: 10.3762/bjoc.8.232. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Prudden AR, Chinoy ZS, Wolfert MA, Boons GJ. Chem Commun. 2014;50:7132–7135. doi: 10.1039/c4cc02222j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Karst NA, Linhardt RJ. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891. [DOI] [PubMed] [Google Scholar]; b) Avci FY, DeAngelis PL, Liu J, Linhardt RJ. Chapter 15 in Front Mod Carb Chem. 2007;960:253–284. [Google Scholar]; c) DeAngelis PL, Liu J, Linhardt RJ. Glycobiology. 2013;23:764–777. doi: 10.1093/glycob/cwt016. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu J, Linhardt RJ. Nat Prod Rep. 2014;31:129–157. doi: 10.1039/c4np00076e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Haller M, Boons GJ. J Chem Soc Perkin Trans 1. 2001:814–822. [Google Scholar]; f) Haller MF, Boons GJ. Eur J Org Chem. 2002:2033–2038. [Google Scholar]; g) Lohman GJS, Hunt DK, Högermeier JA, Seeberger PH. J Org Chem. 2003;68:7559–7561. doi: 10.1021/jo0340760. [DOI] [PubMed] [Google Scholar]; h) Orgueira HA, Batolozzi A, Schell P, Litjens REJN, Palmacci ER, Seeberger PH. Chem Eur J. 2003;9:140–149. doi: 10.1002/chem.200390009. [DOI] [PubMed] [Google Scholar]; i) Lohman GJS, Seeberger PH. J Org Chem. 2004;69:4081–4093. doi: 10.1021/jo035732z. [DOI] [PubMed] [Google Scholar]; j) Noti C, Seeberger PH. Chem Biol. 2005;12:731–756. doi: 10.1016/j.chembiol.2005.05.013. [DOI] [PubMed] [Google Scholar]; k) de Kort M, Buijsman RC, van Boeckel CA. Drug Discov Today. 2005;10:769–779. doi: 10.1016/S1359-6446(05)03457-4. [DOI] [PubMed] [Google Scholar]; l) de Paz JL, Noti C, Seeberger PH. J Am Chem Soc. 2006;128:2766–2767. doi: 10.1021/ja057584v. [DOI] [PubMed] [Google Scholar]; m) Noti C, de Paz JL, Polito L, Seeberger PH. Chem Eur J. 2006;12:8664–8666. doi: 10.1002/chem.200601103. [DOI] [PubMed] [Google Scholar]; n) de Paz JL, Noti C, Böhm F, Werner S, Seeberger PH. Chem Biol. 2007;14:879–887. doi: 10.1016/j.chembiol.2007.07.007. [DOI] [PubMed] [Google Scholar]; o) Adibekian A, Bindschädler P, Timmer MSM, Noti C, Schützenmeister N, Seeberger PH. Chem Eur J. 2007;13:4510–4522. doi: 10.1002/chem.200700141. [DOI] [PubMed] [Google Scholar]; p) Zhang Z, McCallum SA, Xie J, Nieto L, Corzana F, Jiménez-Barbero J, Chen M, Liu J, Linhardt RJ. J Am Chem Soc. 2008;130:12998–13007. doi: 10.1021/ja8026345. [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Peterson S, Frick A, Liu J. Nat Prod Rep. 2009;26:610–627. doi: 10.1039/b803795g. [DOI] [PubMed] [Google Scholar]; r) Xu Y, Pempe EH, Liu J. J Biol Chem. 2012;287:29054–29061. doi: 10.1074/jbc.M112.358523. [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Li P, Sheng J, Liu Y, Li J, Liu J, Wang F. Med Res Rev. 2013;3:665–692. doi: 10.1002/med.21263. [DOI] [PubMed] [Google Scholar]; t) Wang Z, Li J, Cheong S, Bhaskar U, Akihiro O, Zhang F, Dordick JS, Linhardt RJ. J Biotechnol. 2011;156:188–96. doi: 10.1016/j.jbiotec.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; u) Nonaka M, Bao X, Matsumura F, Götze S, Kandasamy J, Kononov A, Broide DH, Nakayama J, Seeberger PH, Fukuda M. Proc Natl Acad Sci USA. 2014;111:8173–8178. doi: 10.1073/pnas.1319870111. [DOI] [PMC free article] [PubMed] [Google Scholar]; v) Yu F, Roy S, Arevalo E, Schaeck J, Wang J, Holte K, Duffner J, Gunay NS, Capila I, Kaundinya GV. Anal Bioanal Chem. 2014;406:3079–89. doi: 10.1007/s00216-014-7729-4. [DOI] [PubMed] [Google Scholar]; w) Canales A, Angulo J, Ojeda R, Bruix M, Fayos R, Lozano R, Giménez-Gallego G, Martín-Lomas M, Nieto PM, Jiménez-Barbero J. J Am Chem Soc. 2005;127:5778–9. doi: 10.1021/ja043363y. [DOI] [PubMed] [Google Scholar]; x) Canales-Mayordomo A, Fayos R, Angulo J, Ojeda R, Martín-Pastor M, Nieto PM, Martín-Lomas M, Lozano R, Giménez-Gallego G, Jiménez-Barbero J. J Biomol NMR. 2006;35:225–39. doi: 10.1007/s10858-006-9024-y. [DOI] [PubMed] [Google Scholar]; y) Canales A, Lozano R, López-Méndez B, Angulo J, Ojeda R, Nieto PM, Martín-Lomas M, Giménez-Gallego G, Jiménez-Barbero J. FEBS J. 2006;273:4716–27. doi: 10.1111/j.1742-4658.2006.05474.x. [DOI] [PubMed] [Google Scholar]; z) Nieto L, Canales Á, Fernández IS, Santillana E, González-Corrochano R, Redondo-Horcajo M, Cañada FJ, Nieto P, Martín-Lomas M, Giménez-Gallego G, Jiménez-Barbero J. Chembiochem. 2013;14:1732–44. doi: 10.1002/cbic.201300313. [DOI] [PubMed] [Google Scholar]; aa) Muñoz-García JC, García-Jiménez MJ, Carrero P, Canales A, Jiménez-Barbero J, Martín-Lomas M, Imberty A, de Paz JL, Angulo J, Lortat-Jacob H, Nieto PM. Glycobiology. 2014;24:1004–9. doi: 10.1093/glycob/cwu071. [DOI] [PubMed] [Google Scholar]; ab) García-Jiménez MJ, Gil-Caballero S, Canales Á, Jiménez-Barbero J, de Paz JL, Nieto PM. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18061293. pii: E1293. [DOI] [PMC free article] [PubMed] [Google Scholar]; ac) Chang CH, Lico LS, Huang TY, Lin SY, Chang CL, Arco SD, Hung SC. Angew Chem Int Ed Engl. 2014;53:9876–9879. doi: 10.1002/anie.201404154. [DOI] [PubMed] [Google Scholar]; ad) Laguri C, Sapay N, Simorre JP, Brutscher B, Imberty A, Gans P, Lortat-Jacob H. J Am Chem Soc. 2011;133:9642–9645. doi: 10.1021/ja201753e. [DOI] [PubMed] [Google Scholar]; ae) Muñoz-García JC, Solera C, Carrero P, de Paz JL, Angulo J, Nieto PM. Org Biomol Chem. 2013;11:8269–75. doi: 10.1039/c3ob41789a. [DOI] [PubMed] [Google Scholar]; af) Nieto L, Canales Á, Giménez-Gallego G, Nieto PM, Jiménez-Barbero J. Chemistry. 2011;17:11204–11209. doi: 10.1002/chem.201101000. [DOI] [PubMed] [Google Scholar]; ag) de Paula VS, Pomin VH, Valente AP. J Biol Chem. 2014;289:22969–79. doi: 10.1074/jbc.M114.572529. [DOI] [PMC free article] [PubMed] [Google Scholar]; ag) Xu Y, Pempe EH, Liu J. J Biol Chem. 2012;287:29054–29061. doi: 10.1074/jbc.M112.358523. [DOI] [PMC free article] [PubMed] [Google Scholar]; ah) Zong C, Venot A, Li X, Lu W, Xiao W, Wilkes JL, Salanga CL, Handel TM, Wang L, Wolfert MA, Boons GJ. J Am Chem Soc. 2017;139:9534–9543. doi: 10.1021/jacs.7b01399. [DOI] [PMC free article] [PubMed] [Google Scholar]; ai) Yang J, Hsieh PH, Liu X, Zhou W, Zhang X, Zhao J, Xu Y, Zhang F, Linhardt RJ, Liu J. Chem Commun (Camb) 2017;53:1743–1746. doi: 10.1039/c6cc08204a. [DOI] [PMC free article] [PubMed] [Google Scholar]; aj) Puvirajesinghe TM, Turnbull JE. Microarrays (Basel) 2016;5 doi: 10.3390/microarrays5010003. pii: E3. [DOI] [PMC free article] [PubMed] [Google Scholar]; ak) Prabhu A, Venot A, Boons GJ. Org Lett. 2003;5:4975–4978. doi: 10.1021/ol0359261. [DOI] [PubMed] [Google Scholar]; al) Arungundram S, Al-Mafraji K, Asong J, Leach FE, Amster IJ, Venot A, Turnbull JE, Boons GJ. J Am Chem Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]; am) Liu R, Xu Y, Chen M, Weïwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. J Biol Chem. 2010;285:34240–34249. doi: 10.1074/jbc.M110.159152. [DOI] [PMC free article] [PubMed] [Google Scholar]; an) Nguyen TKN, Arungundram S, Tran VM, Raman K, Al-Mafraji K, Venot A, Boons GJ, Kuberan B. Mol Biosyst. 2012;8:609–614. doi: 10.1039/c1mb05221g. [DOI] [PMC free article] [PubMed] [Google Scholar]; ao) Leach FE, III, Arungundram S, Al-Mafraji K, Venot A, Boons GJ, Amster IJ. Int J Mass Spectrom. 2012;330–332:152–159. doi: 10.1016/j.ijms.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; ap) Zong C, Venot A, Dhamale O, Boons GJ. Org Lett. 2013;15:342–345. doi: 10.1021/ol303270v. [DOI] [PMC free article] [PubMed] [Google Scholar]; aq) Hsieh PH, Xu Y, Keire DA, Liu J. Glycobiology. 2014;24:681–692. doi: 10.1093/glycob/cwu032. [DOI] [PMC free article] [PubMed] [Google Scholar]; ar) Dhamale OP, Zong C, Al-Mafraji K, Boons GJ. Org Biomol Chem. 2014;12:2087–2098. doi: 10.1039/c3ob42312c. [DOI] [PMC free article] [PubMed] [Google Scholar]; as) Cai C, Dickinson DM, Li L, Masuko S, Suflita M, Schultz V, Nelson SD, Bhaskar U, Liu J, Linhardt RJ. Org Lett. 2014;16:2240–2243. doi: 10.1021/ol500738g. [DOI] [PMC free article] [PubMed] [Google Scholar]; at) Gao Q, Chen CY, Zong C, Wang S, Ramiah A, Prabhakar P, Morris LC, Boons GJ, Moremen KW, Prestegard JH. ACS Chem Biol. 2016;11:3106–3113. doi: 10.1021/acschembio.6b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]; au) Eller S, Collot M, Yin J, Hahm HS, Seeberger PH. Angew Chem Int Ed. 2013;52:5858–5861. doi: 10.1002/anie.201210132. [DOI] [PubMed] [Google Scholar]; av) Liang CF, Hahm HS, Seeberger PH. Methods Mol Biol. 2015;1229:3–10. doi: 10.1007/978-1-4939-1714-3_1. [DOI] [PubMed] [Google Scholar]; aw) Liu J, Li J, Su G. Angew Chem Int Ed Engl. 2017 in press. [Google Scholar]; ax) Domínguez-Rodríguez P, Reina JJ, Gil-Caballero S, Nieto PM, de Paz JL, Rojo J. Chemistry. 2017 doi: 10.1002/chem.201701890. in press. [DOI] [PubMed] [Google Scholar]; ay) Kandasamy J, Schuhmacher F, Hahm HS, Klein JC, Seeberger PH. Chem Comm. 2014;50:1875–1877. doi: 10.1039/c3cc48860h. [DOI] [PubMed] [Google Scholar]; az) Xu Y, Chandarajoti K, Zhang KX, Pagadala V, Dou W, Hoppensteadt DM, Sparkenbaugh EM, Cooley B, Daily S, Key NS, Severynse-Stevens D, Fareed J, Linhardt RJ, Pawlinski R, Liu J. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aan5954. pii: eaan5954. [DOI] [PMC free article] [PubMed] [Google Scholar]; aaa) Mende M, Bednarek C, Wawryszyn M, Sauter P, Biskup MB, Schepers U, Bräse S. Chem Rev. 2016;116:8193–8255. doi: 10.1021/acs.chemrev.6b00010. [DOI] [PubMed] [Google Scholar]; aab) de Paz JL, Angulo J, Lassaletta JM, Nieto PM, Redondo—Horcajo M, Lozano RM, Gimenez-Gallego G, Martin-Lomas M. ChemBioChem. 2001;2:673–685. doi: 10.1002/1439-7633(20010903)2:9<673::AID-CBIC673>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; aac) Ojeda R, Ângulo J, Nieto PM, Martin-Lomas M. Can J Chem. 2002;80:917–936. [Google Scholar]; aad) Lucas R, Angulo J, Nieto PM, Martin-Lomas M. Org Biomol Chem. 2003;1:2253–2266. doi: 10.1039/b303115b. [DOI] [PubMed] [Google Scholar]; aae) Angulo J, Ojeda R, de Paz JL, Lucas R, Nieto PM, Lozano RM, Redondo-Horcajo M, Gimenez-Gallego G, Martin-Lomas M. ChemBioChem. 2004;5:55–61. doi: 10.1002/cbic.200300696. [DOI] [PubMed] [Google Scholar]; aaf) de Paz JL, Martin-Lomas M. J Org Chem. 2005:1849–1858. [Google Scholar]; aag) Guerrini M, Agulles T, Bisio A, Hricovini M, Lay L, Naggi A, Poletti L, Sturiale L, Torri G, Casu B. Biochem Biophys Res Commun. 2002;292:222–230. doi: 10.1006/bbrc.2002.6634. [DOI] [PubMed] [Google Scholar]
- 25.Farrugia BL, Lord MS, Melrose J, Whitelock JM. Molecules. 2015;20:4254–76. doi: 10.3390/molecules20034254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emmadi M, Kulkarni SS. Nat Protoc. 2013;8:1870–89. doi: 10.1038/nprot.2013.113. [DOI] [PubMed] [Google Scholar]
- 27.Maza S, Mar Kayser M, Macchione G, López-Prados J, Angulo J, de Paz JL, Nieto PM. Org Biomol Chem. 2013;11:3510–25. doi: 10.1039/c3ob40306h. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Su G, Liu J. Angew Chem Int Ed. 2017;56:11784–11787. doi: 10.1002/anie.201705638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C-F, Hahm HS, Seeberger PH. Chapter 1 – Automated Synthesis of Chondroitin Sulfate Oligosaccharides. In: Balagurunathan Kuberan, et al., editors. Glycosaminoglycans: Chemistry and Biology, Methods in Molecular Biology. Vol. 1229. 2015. pp. 3–10. [DOI] [PubMed] [Google Scholar]
- 30.a) Dinkelaar J, Codée JD, van den Bos LJ, Overkleeft HS, van der Marel GA. J Org Chem. 2007;72:5737–5742. doi: 10.1021/jo070704s. [DOI] [PubMed] [Google Scholar]; b) Dinkelaar J, Gold H, Overkleeft HS, Codée JD, van der Marel GA. J Org Chem. 2009;74:4208–4216. doi: 10.1021/jo9003713. [DOI] [PubMed] [Google Scholar]; c) Lu X, Kamat MN, Huang L, Huang X. J Org Chem. 2009;74:7608–7617. doi: 10.1021/jo9016925. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mukherjee C, Liu L, Pohl NL. Adv Synth Catal. 2014;356:2247–2256. doi: 10.1002/adsc.201400269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koizumi Y, Kelly JJ, Nakagawa T, Urakawa H, El-Fantroussi S, Al-Muzaini S, Fukui M, Urushigawa Y, Stahl DA. Appl Environ Microbiol. 2002;68:3215–25. doi: 10.1128/AEM.68.7.3215-3225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MR, Shin II. Org Lett. 2005;7:4269–72. doi: 10.1021/ol051753z. [DOI] [PubMed] [Google Scholar]
- 33.Zhi ZL, Powell AK, Turnbull JE. Anal Chem. 2006;78:4786–93. doi: 10.1021/ac060084f. [DOI] [PubMed] [Google Scholar]
- 34.Rogers CJ, Clark PM, Tully SE, Abrol R, Garcia KC, Goddard WA, 3rd, Hsieh-Wilson LC. Proc Natl Acad Sci USA. 2011;108:9747–52. doi: 10.1073/pnas.1102962108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shipp EL, Hsieh-Wilson LC. Chem Biol. 2007;14:195–208. doi: 10.1016/j.chembiol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 36.Dulaney SB, Xu Y, Wang P, Tiruchinapally G, Wang Z, Kathawa J, El-Dakdouki MH, Yang B, Liu J, Huang X. J Org Chem. 2015;80:12265–79. doi: 10.1021/acs.joc.5b02172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Hsieh PH, Liu X, Zhou W, Zhang X, Zhao J, Xu Y, Zhang F, Linhardt RJ. J Liu, Chem Commun (Camb) 2017;53:1743–1746. doi: 10.1039/c6cc08204a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chappell EP, Liu J. Bioorg Med Chem. 2013;21:4786–92. doi: 10.1016/j.bmc.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 39.Hu YP, Zhong YQ, Chen ZG, Chen CY, Shi Z, Zulueta MM, Ku CC, Lee PY, Wang CC, Hung SC. J Am Chem Soc. 2012;134:20722–7. doi: 10.1021/ja3090065. [DOI] [PubMed] [Google Scholar]
- 40.Gao Q, Chen CY, Zong C, Wang S, Ramiah A, Prabhakar P, Morris LC, Boons GJ, Moremen KW, Prestegard JH. ACS Chem Biol. 2016;11:3106–3113. doi: 10.1021/acschembio.6b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]