

Abstract
Neurons requires an extraordinarily high level of membrane trafficking activities because of enriched axonal terminals and dendritic branches. For that reason, defects in the membrane trafficking pathway are a hallmark of most, and may be all, neurodegenerative disorders. A major cellular membrane trafficking pathway is the Golgi apparatus (Golgi hereafter) – late endosome - lysosome axis for supplying lysosomal enzymes. This pathway is regulated by N-ethylmaleimide sensitive factor (NSF) ATPase. This review article is to discuss a novel hypothesis that brain ischemia inactivates NSF ATPase, resulting in a cascade of events of disruption of the Golgi – endosome - lysosome pathway, release of cathepsin B (CTSB), and induction of mitochondrial outer membrane permeabilization (MOMP) during the postischemic phase. This hypothesis is supported by recent studies demonstrating that NSF is trapped into inactive protein aggregates in neurons destined to die after brain ischemia. Consequently, Golgi, transport vesicles (TVs), and late endosomes (LEs) are accumulated and damaged, which is followed by CTSB release from these damaged structures. Moderate release of CTSB cleaves Bax-like BH3 protein (Bid) to become active truncated Bid (tBid). Active tBid is then translocated to the mitochondrial outer membrane, resulting in oligomerization of BCL2 Associated X Protein (Bax) forming the mitochondrial outer membrane pores, and releasing mitochondrial intramembranous proteins. Extensive CTSB release, however, can digest cellular proteins indiscriminately to induce cell death. Based on these new observations, we propose a novel hypothesis, i.e., brain ischemia leads to NSF inactivation, resulting in a massive buildup of damaged Golgi, TVs and LEs, fatal release of CTSB, induction of MOMP, and eventually brain ischemia-reperfusion injury.
Keywords: Brain ischemia-reperfusion injury, Membrane trafficking, NSF, Cathepsin B, Golgi, Transport vesicle, Late endosome, Lysosome, mitochondrial outer membrane permeabilization
Introduction
Transient cerebral brain ischemia or cardiac arrest leads to delayed neuronal death that occurs after 3 days of reperfusion. During the delayed period, all neurons appear perfectly normal under the light microscope [1–6]. Under electron microscopy (EM), however, massive buildup of damaged Golgi, transport vesicles (TVs), and late endosome (LE) takes place over time in neurons destined to die after transient cerebral or global ischemia [7–10].
Membrane trafficking
In a typical cell, the entire surface membrane turns over every hour via endocytosis and exocytosis [11]. The membrane trafficking activities of endocytic, exocytic, organelle delivery of newly synthesized proteins, and autophagy are mediated either via transport vesicles (TVs) or membranous organelles. In eukaryotic cells, membranous or secretory proteins are mostly synthesized in the endoplasmic reticulum (ER)-associated polyribosomes. After synthesis, they are translocated to the ER lumen and then transported into Golgi apparatus before being delivered to the destined or targeted organelles or cell membrane (Fig. 1A). At the destination, the delivery transport vesicle or organelle membrane fuses with the target organelle or cell membrane to assemble or deliver the cargo (Fig. 1A). Neurons have enriched axonal terminals and dendritic branches with extraordinarily large surface area, and thus require an extremely high level of endocytic, exocytic, the protein delivery, and autophagic membrane trafficking activities (Fig. 1A). This may be why defects in the membrane trafficking pathway preferentially affect neurons and are a hallmark of most, and maybe all, neurodegenerative disorders [12].
Fig. 1.

A: Cellular membrane trafficking encompasses 4 major types: (1) secretory or exocytic route, (2) endocytic pathway; (3) newly synthesized biomolecules delivery; and (4) autophagy pathway (Fig. 1A). ER = endoplasmic reticulum. V = transport vesicle. LE = late endosome. EE = early endosome. EL = endolysosome. AP = autophagosome. L = lysosome. B: Example of LE and lysosomal membrane fusion in the inset of A. Membrane fusion is mediated by active SNAREs tethering to bring two membranes together. After membrane fusion, SNAREs form inactive complexes and must be regenerated by NSF ATPase for the next round of fusion. During this process, NSF ATPase binds to inactive SNAREs via a SNAP adaptor to hydrolyze adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate for providing the energy. Brain ischemia leads to deposition NSF into inactive protein aggregates, thus stopping all membrane trafficking activities in postischemic neurons.
Membrane trafficking requires membrane-to-membrane fusion either between transport vesicles and organelles, or between two organelles, encompassing the following 4 major types: (1) to deliver organelle proteins; (2) exocytic or secretory pathway, (3) the endocytic pathway; and (4) the autophagy pathway (Fig. 1A). Among membrane trafficking structures, the late endosome (LE), representing the stomach of the cell [13,14], acts as a central hub receiving: (i) newly synthesized lysosomal proteins (e.g., cathepsins) from Golgi-derived transport vesicles (TVs), and (ii) degradative cargo from both the endocytic and autophagic pathways (Fig. 1A). LE (luminal pH=5.5) then fuses with lysosome to become a more acidic (pH<5) hybrid endolysosome or EL (=secondary lysosome) for the execution of degradation [13,14]. After digesting cargo, EL becomes a lysosome (L) that contains evenly distributed content. The lysosome is then recycled to fuse with LE again for the next round of digestive process (Fig. 1, A and B) [15].
As shown in Fig. 1B, the core elements of the membrane trafficking machinery are: (i) NSF (N-ethyl-maleimide sensitive factor ATPase); (ii) SNAP (soluble NSF attachment protein), and (iii) SNAREs (soluble NSF attachment protein receptors). NSF is the sole ATPase for regenerating active SNAREs after membrane-to-membrane fusion. SNAP is an adaptor for connecting NSF to SNAREs and also for stimulating the NSF ATPase activity. SNAREs are fusion proteins and divided into vesicle SNAREs (v-SNAREs) and target membrane SNAREs (t-SNAREs). Interaction between v- and t-SNAREs brings two cellular membranous organelles together, also known as SNAREs tethering, before membrane-to-membrane fusion (Fig. 1B). After membrane-to-membrane fusion, SNAREs form inactive complexes and must be regenerated by NSF ATPase for the next round of fusion (Fig. 1B). During this process, cytosolic free NSF ATPase interacts with SNAREs via an adaptor protein SNAP and hydrolyzes adenosine triphosphate (ATP) to dissociate the inactive SNARE complex into active SNAREs (Fig. 1B) [16]. To date, there is only a single form of NSF ATPase in most organisms except in Drosophila that expresses dNSF-1 and dNSF-2 [17]. For that reason, NSF deficiency will bring all neuronal membrane trafficking activities to a halt (Fig. 1B). In comparison, SNAREs are both redundant and abundant [11,16]. Three sets of SNAREs are relatively organelle-specific, including: (i) presynaptic SNAREs (e.g., syntaxin-1 and synaptobrevin); (ii) trans-Golgi network SNAREs [e.g., STX16 and Vti1a]; and (iii) late endosome or LE SNAREs (e.g., Vti1b) [15,16]. The SNARE organelle specificity may be utilized to investigate specific Golgi-to-LE, and LE-to-lysosome fusion events and organelle damage after brain ischemia [18].
Depletion of active NSF ATPase owing to its inactive deposition in neurons after brain ischemia
Only cytoplasmic free NSF hexamer ATPase is the active form for dissociating the inactive SNARE complex, whereas deposition of NSF into a Triton-insoluble fraction results in NSF inactivation [17]. This is consistent with the confocal microscopic results showing that NSF is depleted from the cytoplasm during the period of 0.5 – 72 h of reperfusion only from CA1 neurons destined to die after transient cerebral ischemia [18,19]. Depletion of the cytosolic ATPase can be viewed by confocal microscopy. Fig. 2 shows confocal microscopic images of CA1 neurons labeled with NSF antibody. Brain sections were obtained from a sham-operated control rat and rat subjected to 15 min of ischemia followed by 24 h of reperfusion. NSF is located in the peri-nuclear and dendritic truck (Sham, arrowheads), as well as in the neuropil (Sham, arrows) of sham-operated control CA1 neurons. NSF is mostly depleted from the CA1 neuronal perinuclear region and dendritic trunk (24h, arrowheads), but remains relatively at the sham control level in the CA1 neuropil (24h, arrows) at 24 h of reperfusion after transient cerebral ischemia. In contrast, NSF is less affected in surviving neurons such as those in the DG after the same period of transient cerebral ischemia [18,19]. Depletion of cytoplasmic NSF is owing to deposition of NSF into inactive protein aggregates, as demonstrated by the sucrose gradient centrifugation study [18]. The NSF-containing aggregates form very dense structures in vivo and cannot be accessed by NSF antibody during immunostaining of brain sections [18–20]. As mentioned, NSF is the sole ATPase to regenerate active SNAREs for controlling membrane trafficking [15–17]. Intracellular NSF deficiency can result in severe neuronal damage and eventually neuronal death [15–19].
Fig. 2.

Confocal microscopic images of CA1 neurons labeled with NSF antibody. Brain sections were obtained from a sham-operated control rat and rat subjected to 15 min of ischemia followed by 24 h of reperfusion. NSF is located in the peri-nuclear and dendritic truck (Sham, arrowheads), as well as in the neuropil (Sham, arrows) of sham-operated control CA1 neurons. NSF is mostly depleted from the CA1 neuronal perinuclear region and dendritic trunk (24h, arrowheads), but remains relatively at the sham control level in the CA1 neuropil (24h, arrows) at 24 h of reperfusion after transient cerebral ischemia.
Ultrastructural characterization of buildup of Golgi fragments, and transport vesicles (TVs), and late endosomes (LEs) after brain ischemia
Golgi and endosomal membranes are among the most dynamic cellular structures with the highest concentration of SNAREs, thus requiring the highest amount of NSF to regenerate active SNAREs [21]. In contrast, other membranes, such as the ER membranes, may have a relatively lower concentration of SNAREs, thus requiring a lower level of NSF for regenerating active SNAREs [21]. For that reason, inactivation of NSF affects mainly the Golgi – LE – lysosome axis. In cultured cells or cell free systems, inactivation of either NSF or the key SNAREs leads to massive buildup of Golgi fragments, TVs and enlarged LEs [21,22]. Expression of an activity-deficient mutant of syntaxin 11 (an endosomal SNARE protein) inhibits the late endosome to lysosome fusion in macrophages, resulting in an accumulation of enlarged LEs [22].
Could inactivation of NSF also lead to buildup of Golgi fragments, and TVs, and enlarged LEs after brain ischemia? EM studies demonstrate that this is the case. Massive accumulation of protein aggregate-associated Golgi fragments, TVs, and enlarged LEs is the most prominent ultrastructural change in neurons destined to die after brain ischemia [7–10]. As shown in Fig. 3, under the EM, most prominent changes in neocortical and CA1 neurons destined to undergo neuronal death after transient cerebral ischemia are massive accumulation of protein aggregate-associated Golgi fragments, TVs, and enlarged LEs. A sham-operated control CA1 neuron, stained with the osmium-uranium-lead, has normal rough endoplasmic reticulum (ER), polyribosome rosettes (small arrows), Golgi (G), mitochondria (M), endolysosomes (EL, also known as secondary lysosome), and LE (large arrows) (Fig. 3a). However, the intact Golgi structures are difficult to find in CA1 and some neocortical neurons after transient cerebral ischemia, because they are completely fragmented (Fig. 3b, Gf). Meanwhile, massive buildup of TVs (Fig. 3b, arrowheads) and enlarged LEs (Fig. 3b, large arrows), as well as monoribosomes (Fig. 3b, small arrows), were observed in postischemic neurons. The Golgi fragments (Gfs), TVs and LEs are often associated with protein aggregates (Fig. 3, Stars). The LE structures in the insets of Figs. 3a and 3b can be better viewed with a higher magnification of EM images shown in Figs. 3c and 3d. EL from the sham-operated control neuron have intact lipid membranes and contain partially digested cargo (Fig. 3c, EL) while LE contains multi-vesicular structures (Fig. 3c, arrow). In comparison, the LE structures from the postischemic neuron are enlarged, containing opaque vesicle structures, and have multiple membranous breaks or damage (Fig. 3d, arrowheads). As described above, accumulation of damaged Golgi fragments, TVs and LEs in neurons observed by EM may be a result of NSF deficiency after transient cerebral or global ischemia.
Fig. 3.

EM micrographs of hippocampal CA1 neurons stained with uranium-lead. CA1 tissue sections were obtained from rats subjected to non-ischemic sham surgery or 20 min of ischemia followed by 24 h of reperfusion. a: A sham neuron shows normal rough endoplasmic reticulum (ER), ribosomal rosettes (small arrows), mitochondria (M), Golgi apparatus (G), late endosome (large arrow), and endolysosome (EL, also known as secondary lysosome). b: A 24 h reperfused neuron shows accumulation of Golgi fragments (Gf), transport vesicles (arrowheads), enlarged late endosome (large arrows), monoribosomes (small arrows), and ribosomal aggregates (stars). c and d: Higher magnifications of the insets of “a and b” show that ELs and a LE (arrow) from the sham control neuron has intact lipid membranes, whereas an enlarged LE from the postischemic neuron has a number of membrane break damage (arrowheads). Scale bar = 0.5 μm.
Cathepsin B (CTSB)
In comparison with other cathepsins, cathepsin B (CTSB) shows the highest level of expression in neurons [23,24]. CTSB has three forms (Figs. 4 and 5): (i) 46kDa proCTSB located in Golgi/TVs, (ii) 33kDa CTSB in LE, and (iii) 24/25kDa CTSB in EL and lysosomes. After synthesis from the ER, the 46kDa proCTSB moves to Golgi where it is further modified, and then packed into TVs that move toward and fuse with LE via NSF-mediated machinery (Fig. 5). In the acidic environment of the LE’s lumen (~pH 5.5), the proregion of 46kDa proCTSB is cleaved to become a mature 33kDa LE CTSB (Fig. 5). The LE then fuses with lysosome (mediated also by the NSF machinery) to become endolysosome (EL=secondary lysosome) (Fig. 5). In the more acidic luminal environment of the EL’s (pH<5) and lysosomal (~pH 4.5) lumen, the 33kDa LE CTSB is further cleaved into a 24/25 kDa EL/lysosomal CTSB [24–27] (Figs. 4 and 5). 46kDa Golgi proCTSB, 33kDa LE CTSB and 24/25 kDa lysosomal CTSB all have proteolytic activities [24].
Fig. 4.

CTSB has three active forms: (i) 46 kDa Golgi CTSB located in Golgi/TVs, (ii) 33 kDa CTSB in late endosome (LE), and (iii) 24/25 kDa lysosome CTSB in endolysosome (EL) and lysosome (L).
Fig. 5.

Inactivation of NSF ATPase after brain ischemia leads to a cascade of events of massive buildup of Golgi fragments, TVs and enlarged LEs, fatal release of 33 kDa CTSB, and brain ischemia-reperfusion injury.
Neuronal release of 33 kDa LE CTSB after brain ischemia
Inhibition of NSF ATPase leads to a halt of the CTSB delivery from Golgi to LE, and then from LE to lysosome (Fig. 5), thus trapping CTSB inside Golgi, TV and LE structures, resulting in enlargement of these structures [28–33]. It has been shown that Golgi, LE, and lysosome structures have the highest level of NSF, SNAP and SNAREs to accommodate for the higher membrane trafficking activities [21,22]. Therefore, inactivation of NSF ATPase after transient cerebral ischemia may interfere mainly with the Golgi-to-LE and LE-to-lysosome membrane fusion, resulting in accumulation of 33kDa LE CTSB and concomitant reduction of 24/25 kDa lysosomal CTSB [18]. In addition, the 46kDa proCTSB can autocleave its own propeptide in Golgi and TVs to become a 33kDa CTSB [25,26], particularly in the postischemic acidosis condition [5,18], and thus may also contribute to the accumulation of 33 kDa CTSB and concomitant reduction of 46kDa Golgi CTSB.
This has been observed in a recent study showing that 33kDa CTSB is significantly increased, while 46 kDa Golgi and 24/25kDa lysosomal CTSB are correspondingly decreased in the cytosolic and intracellular microsomal membrane fraction prepared from the brain tissue samples after transient cerebral ischemia [18]. Therefore, 33kDa CTSB may be released not only from massively enlarged LEs, but also from Golgi and TVs (Fig. 5). However, CTSB may not be released from ELs or lysosomes, because EL/lysosomal CTSB is 24/25kDa. Release of 33kDa CTSB (that is an active protease) into the cytoplasm can further damage Golgi and LE, resulting in amplified release of 33kDa CTSB into the cytoplasm (Fig. 5).
In consistent with the biochemical analysis described above, confocal microscopic study further shows that CTSB antibody stains only small dots in non-ischemic sham-operated control CA1 neurons. In contrast, both the size and number of CTSB-immunostained structures are dramatically increased in CA1 neurons after transient cerebral ischemia [18]. Furthermore, CTSB-immunostained punctate pattern in sham-operated control CA1 neurons become diffused or evenly distributed throughout the cytoplasm of CA1 pyramidal neurons after transient cerebral ischemia [18]. These results suggest that CTSB may be released from the structures of Golgi fragments, TVs, and LEs (see Fig. 5).
Why CTSB is an important cathepsin after brain ischemia?
There are at least 15 cathepsins distributed in various organs. The cathepsins most commonly implicated in cell death are CTSB and cathepsin D (CTSD) [34,35]. Among cathepsins, CTSB is the most abundant cathepsin in neurons [22,23]. Our recent study shows that CTSB is dramatically increased in the cytoplasm of CA1 neurons after global brain ischemia while CTSD is unchanged (data not shown), suggesting that CTSB may be a major cathepsin released into the cytoplasm of neurons destined to die after transient cerebral ischemia.
Could CTSB inhibitors offer neuroprotection against brain ischemia-reperfusion injury?
Of relevance, at cytosolic neutral pH (pH≈7.4), CTSB exhibits proteolytic activity whereas most other cathepsins have minimal activities [13]. Furthermore, although not yet studied in mature brain ischemia models, CTSB knockout (KO) in mice protects neonatal hippocampal neurons from hypoxic-ischemic injury [36]. Therefore, CTSB KO in mice might also protect adult mature neurons from ischemia-reperfusion injury.
Previous studies show that the chemical agents CA-074me and E64d might offer neuroprotection by inhibiting CTSB after brain ischemia [37,38]. However, these inhibitors are not ideal therapeutic agents for the following reasons. First, treatment of cells with CA-074me or E-64d increases both lysosomal CTSB half-life and concentration by >3-fold, and thus may postpones CTSB release and result in more severe brain injury [39,40]. Second, E-64d is not a CTSB-specific inhibitor and recent studies indicate that CA-074me is not CTSB-specific either, despite previous claims [41–43]. Third, the inhibitory effects of CA-074me and E64d are pH-dependent; they are strong inhibitors at pH4.5 in the lysosomal luminal acidic environment, but ineffective inhibitors for released CTSB at the cytosolic neutral pH [44]. Because cytosolic CTSB is harmful while lysosomal CTSB is required for degradation of toxic aggregates and damaged subcellular organelles, the ideal inhibitors will be strong inhibitors for released CTSB at neutral pH, and weak inhibitors for lysosomal acidic luminal CTSB. Such inhibitors are not currently available.
CTSB Release induces mitochondrial outer membrane permeabilization (MOMP)
Previous studies show that moderate CTSB release induces MOMP by activation of the tBid – Bax or Bak pathway, resulting in cell death, as shown in Fig. 6 [45,46]. Bak is translationally repressed in mature neurons and thus its protein is not expressed under either normal or stress conditions [47], whereas both Bid and Bax are expressed in mature neurons [48]. The full-length Bid protein does not expose its mitochondrial anchoring segment and is inactive in the cytosol of healthy cells. Moderate cytoplasmic release of CTSB cleaves the Bid autorepression domain to produce active truncated Bid (tBid), resulting in tBid translocation to the mitochondrial outer membrane. tBid then induces oligomerization of Bax to form mitochondrial outer membrane pores (Fig. 6). As a result, MOMP is induced, leading to cell death (Fig. 6).
Fig. 6.
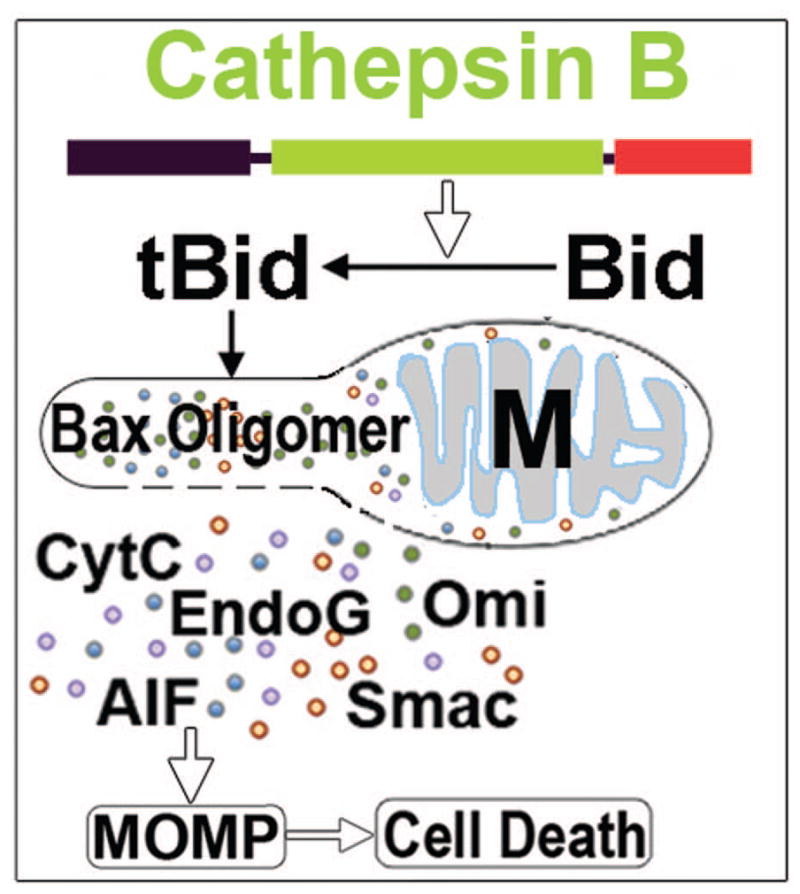
CTSB release induces mitochondrial outer membrane permeabilization (MOMP): Released CTSB converts Bid to active truncated Bid (tBid), and leads tBid and Bax mitochondrial (M) translocation and Bax oligomerization. Bax oligomerization forms mitochondrial outer membrane pore, resulting in MOMP. Consequently, mitochondrial intermembrane space proteins are released into the cytoplasm, eventually resulting in neuronal death after brain ischemia. CytC = cytochrome C. AIF = apoptosis inducing factor. EndoG = endonuclease G. HtrA2/Omi = a proapoptotic mitochondrial serine protease. SMAC/Diablo = Second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI.
MOMP has been detected in several paradigms of neuronal death and is commonly considered as the ‘point of no return’ [45]. A distinctive sign of MOMP is cytoplasmic release of proteins from the intermembrane space (between the inner and outer mitochondrial membranes) such as cytochrome C (CytC), apoptosis inducing factor (AIF), endonuclease G (EndoG), and Htra2 [49–52] (Fig. 7). CTSB-dependent Bid cleavage, as well as tBid and Bax translocation to mitochondrial outer membrane have been observed in in vivo and cell culture studies [53–56]. Bcl2 and Bcl-xL may also be involved in MOMP [53–56]. Extensive CTSB release, however, can digest proteins indiscriminately to directly induce cell death [46].
Fig. 7.

A: Ubiquitin immunogold EM micrographs of mitochondrion (M). EM micrographs of CA1 neurons from a sham-operated non-ischemic control rat and a rat subjected to 15 min ischemia followed by 24h of reperfusion. Ubiquitin immunogold labels mitochondrion and ubiquitinated protein aggregates at 24h of reperfusion (arrows). B: EM three-dimensional reconstruction of CA1 neuronal mitochondria in brain sections from sham-operated control and 24h reperfused rats. The red indicates the mitochondrial outer membrane. The yellow (arrows) represents the vacuole structures formed between the inner- and outer-mitochondrial membranes after brain ischemia. Scale bars in A and B = 0.5 μm. C: Western blotting of ubiquitin in the samples of detergent-salt insoluble mitochondrial pellets from sham-operated rats and rats subjected to 15 min of ischemia followed by 24 h of reperfusion. The molecular size is shown on the left.
Several recent studies indicate that a vacuole-like structure between mitochondrial inner and outer membranes is a distinctive morphological sign of MOMP [54,55]. This was observed by an immunogold EM study showing that mitochondrial outer membrane was associated with clusters of immunogold-labeled ubiquitinated proteins (Fig. 7, 24h, arrows), suggesting that neuronal mitochondrial outer membrane proteins were damaged and thus ubiquitinated at 24 h of reperfusion when CTSB was released after brain ischemia [24]. Many mitochondrial inner and outer membranes are separated to form vacuole-like structures, as observed in brain sections in vivo with the EM three-dimensional reconstruction technique (Fig. 7B, 24h, yellow color, arrows), suggesting damage to the mitochondrial outer membrane and induction of MOMP may occur in neurons displaying CTSB release after brain ischemia. Western blotting further shows the increased protein ubiquitination in the isolated mitochondrial fraction, indicating that mitochondrial proteins are indeed damaged after brain ischemia (Fig. 7C, 24h).
Concluding Remarks
It is well established that neuronal death after cardiac arrest or transient cerebral ischemia occurs in a delayed fashion selectively in some population of neurons such as hippocampal CA1 and neocortical pyramidal neurons [1–6]. The search for mechanisms underlying neuronal ischemia-reperfusion injury has been extensive, but the underlying mechanisms remain incompletely understood. Basic on the latest studies, this review article proposes a novel hypothesis that brain ischemia inactivates NSF ATPase, resulting in a cascade of events of disrupting the Golgi – endosome - lysosome pathway. Consequently, CTSB is released from damaged Golgi fragments, TVs and LEs into the cytoplasm, resulting in induction of MOMP. MOMP is commonly considered as cell damage that has reached a ‘point of no return’ [45]. Our latest studies also show that depletion of active NSF also occurs in neocortical neurons after 20 min of transient cerebral ischemia [18]. Therefore, the NSF inactivation-induced damaging events have emerged as a novel and common mechanism of brain ischemia-reperfusion injury. However, the details of the cascade of events resulting in fatal CTSB release and induction of MOMP after brain ischemia remain to be studied. Understanding the cascade of events of membrane trafficking damage, CTSB release, and induction of MOMP will provide novel insights into the mechanisms of brain ischemia-reperfusion injury and identify new therapeutic targets for its treatment.
Acknowledgments
Funding: This work was supported by National Institutes of Health (NIH) grants: NS36810, NS40407, and NS097875; by Veteran Affair Merit grant: I01BX001696; and by American Heart Association 0940042N-5 to B.R.H.
Common Abbreviations
- NSF
N-ethylmaleimide sensitive factor ATPase
- E329Q
a NSF deficient mutant
- SNAREs
soluble NSF attachment protein receptors
- SNAP
soluble NSF attachment protein
- CTSB
cathepsin B
- CTSD
cathepsin D
- TVs
transport vesicles
- LE
late endosome
- EL
endolysosome
- L
lysosome
- MOMP
mitochondrial outer membrane permeabilization
- Bax
BCL2 Associated X Protein
- Bid
Bax-like BH3 protein
- CytC
cytochrome C
- AIF
apoptosis inducing factor
- EndoG
endonuclease G
- Htra2
mitochondrial protease
- IRI
ischemia-reperfusion injury
- Tg
transgenic
- DG
dentate gyrus
- EM
electron microscopy
Footnotes
Compliance with Ethical Standards:
Conflict of Interest: Dong Yuan declares no conflict of interest. Chunli Liu and Bingren Hu are husband and wife
Ethical approval: This article does not contain any studies with human subjects. All the experimental procedures involving using animals were approved by the Animal Use and Care Committee in the University of Maryland School of Medicine.
Reference Cited
- 1.Smith ML, Auer RN, Siesjo BK. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol (Berl) 1984;64:319–332. doi: 10.1007/BF00690397. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi D, Baernstein A, Nichol G. Cardiac arrest: a public health perspective. Emerg Med Clin North Am. 2012 Feb;30(1):1–12. doi: 10.1016/j.emc.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984;64:139–147. doi: 10.1007/BF00695577. [DOI] [PubMed] [Google Scholar]
- 4.Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol. 1984;62:209–218. doi: 10.1007/BF00691854. [DOI] [PubMed] [Google Scholar]
- 5.Lin B, Ginsberg MD, Busto R. Hyperglycemic exacerbation of neuronal damage following forebrain ischemia: microglial, astrocytic and endothelial alterations. Acta Neuropathol. 1998;96:610–620. doi: 10.1007/s004010050942. [DOI] [PubMed] [Google Scholar]
- 6.Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol. 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- 7.Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. The Journal of Neuroscience. 2000;20:3191–1999. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjö BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134:1273–84. doi: 10.1016/j.neuroscience.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang F, Liu CL, Hu BR. Irreversible aggregation of protein synthesis machinery after focal brain ischemia. J Neurochem. 2006;98:102–12. doi: 10.1111/j.1471-4159.2006.03838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuvim MJ, Adachi R, Hoffenberg S, Dickey BF. Traffic control: Rab GTPases and the regulation of interorganellar transport. News Physiol Sci. 2001;16:56–61. doi: 10.1152/physiologyonline.2001.16.2.56. [DOI] [PubMed] [Google Scholar]
- 12.Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol Life Sci. 2013;70:2919–2934. doi: 10.1007/s00018-012-1201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Repnik U, Česen MH, Turk B. The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol. 2013;5:a008755. doi: 10.1101/cshperspect.a008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tjelle TE, Brech A, Juvet LK, Griffiths G, Berg T. Isolation and characterization of early endosomes, late endosomes and terminal lysosomes: their role in protein degradation. J Cell Sci. 1996;109(Pt 12):2905–2914. doi: 10.1242/jcs.109.12.2905. [DOI] [PubMed] [Google Scholar]
- 15.Luzio JP, Gray SR, Bright NA. Endosome-lysosome fusion. Biochem Soc Trans. 2010;38:1413–1416. doi: 10.1042/BST0381413. [DOI] [PubMed] [Google Scholar]
- 16.Hong W, Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014;24:35–43. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Mohtashami M, Stewart BA, Boulianne GL, Trimble WS. Analysis of the mutant Drosophila N-ethylmaleimide sensitive fusion-1 protein in comatose reveals molecular correlates of the behavioural paralysis. J Neurochem. 2001;77:1407–417. doi: 10.1046/j.1471-4159.2001.00363.x. [DOI] [PubMed] [Google Scholar]
- 18.Yuan D, Liu C, Hu B. Dysfucntion of membrane trafficking leads to CTSB release after brain ischemia. Transl Stroke Res. 2017 doi: 10.1007/s12975-017-0572-0. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu CL, Hu BR. Alterations of N-Ethylmaleimide-Sensitive ATPase Following Transient Cerebral Ischemia. Neuroscience. 2004;128:767–774. doi: 10.1016/j.neuroscience.2004.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang K, Liu C, Kuluz J, Hu B. Alterations of CaMKII after hypoxia-ischemia during brain development. J Neurochem. 2004;91:429–437. doi: 10.1111/j.1471-4159.2004.02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalal S, Rosser MFN, Cyr DM, Hanson PI. Distinct Roles for the AAA ATPases NSF and p97 in the Secretory Pathway. In: Glick B, editor. Molecular Biology of the Cell. Vol. 15. 2004. pp. 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Offenhauser C, Lei N, Roy S, Collins BM, Stow JL, Murray RZ. Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic. 2011;12:762–773. doi: 10.1111/j.1600-0854.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- 23.Petanceska S, Burke S, Watson SJ, Devi L. Differential distribution of messenger RNAs for cathepsins B, L and S in adult rat brain: an in situ hybridization study. Neuroscience. 1994;59:729–738. doi: 10.1016/0306-4522(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 24.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pungerčar JR, Caglič D, Sajid M, et al. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. The FEBS journal. 2009;276:660–668. doi: 10.1111/j.1742-4658.2008.06815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunwar AJ, Rickmann M, Backofen B, Browski SM, Rosenbusch J, Schöning S, Fleischmann T, Krieglstein K, Fischer von Mollard G. Lack of the endosomal SNAREs vti1a and vti1b led to significant impairments in neuronal development. PNAS U S A. 2011;108:2575–80. doi: 10.1073/pnas.1013891108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 1988;85:7852–7856. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz R, Mayorga LS, Weidman PJ, Rothman JE, Stahl PD. Vesicle fusion following receptor-mediated endocytosis requires a protein active in Golgi transport. Nature. 1989;339:398–400. doi: 10.1038/339398a0. [DOI] [PubMed] [Google Scholar]
- 30.Wattenberg BW, Raub TJ, Hiebsch RR, Weidman PJ. The activity of Golgi transport vesicles depends on the presence of the N-ethylmaleimide-sensitive factor (NSF) and a soluble NSF attachment protein (alpha SNAP) during vesicle formation. J Cell Biol. 1992;118:1321–32. doi: 10.1083/jcb.118.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acharya U, Jacobs R, Peters JM, Watson N, Farquhar MG, Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- 32.Naslavsky N, McKenzie J, Altan-Bonnet N, Sheff D, Caplan S. EHD3 regulates early-endosome-to-Golgi transport and preserves Golgi morphology. J Cell Sci. 2009;122:389–400. doi: 10.1242/jcs.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullock BM, Bright NA, Fearon CW, Gray SR, Luzio J. Fusion of Lysosomes with Late Endosomes Produces a Hybrid Organelle of Intermediate Density and Is NSF Dependent. The Journal of Cell Biology. 1998;140:591–601. doi: 10.1083/jcb.140.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gómez-Sintes R, Ledesma MD, Boya P. Lysosomal cell death mechanisms in aging. Ageing Res Rev. 2016;S1568–1637:30024–30031. doi: 10.1016/j.arr.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Serrano-Puebla A, Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann N Y Acad Sci. 2016;1371:30–44. doi: 10.1111/nyas.12966. [DOI] [PubMed] [Google Scholar]
- 36.Ni J, Wu Z, Peterts C, Yamamoto K, Qing H, Nakanishi H. The Critical Role of Proteolytic Relay through Cathepsins B and E in the Phenotypic Change of Microglia/Macrophage. J Neurosci. 2015;35:12488–12501. doi: 10.1523/JNEUROSCI.1599-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Wang J, Song X, Wei R, He F, Peng G, Luo B. Protective mechanisms of CA074-me (other than cathepsin-B inhibition) against programmed necrosis induced by global cerebral ischemia/reperfusion injury in rats. Brain Res Bull. 2016;120:97–105. doi: 10.1016/j.brainresbull.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, Kominami E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain-cathepsin hypothesis’. Eur J Neurosci. 1998;10:1723–1733. doi: 10.1046/j.1460-9568.1998.00184.x. [DOI] [PubMed] [Google Scholar]
- 39.Hook GR, Yu J, Sipes N, Pierschbacher MD, Hook V, Kindy MS. The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury. J Neurotrauma. 2014;31:515–529. doi: 10.1089/neu.2013.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katunuma N. Posttranslational Processing and Modification of Cathepsins and Cystatins. Journal of Signal Transduction. 2010 doi: 10.1155/2010/375345. Article ID 375345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montaser M, Lalmanach G, Mach L. CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol Chem. 2002;383:1305–1308. doi: 10.1515/BC.2002.147. [DOI] [PubMed] [Google Scholar]
- 42.Mihalik R, Imre G, Petak I, Szende B, Kopper L. Cathepsin B-independent abrogation of cell death by CA-074-OMe upstream of lysosomal breakdown. Cell Death Differ. 2004;11:1357–1360. doi: 10.1038/sj.cdd.4401493. [DOI] [PubMed] [Google Scholar]
- 43.Ryu JH, Na JH, Ko HK, You DG, Park S, Jun E, Yeom HJ, Seo DH, Park JH, Jeong SY, Kim IS, Kim BS, Kwon IC, Choi K, Kim K. Non-invasive optical imaging of cathepsin B with activatable fluorogenic nanoprobes in various metastatic models. Biomaterials. 2014;35:2302–2311. doi: 10.1016/j.biomaterials.2013.11.080. [DOI] [PubMed] [Google Scholar]
- 44.Cathers BE, Barrett C, Palmer JT, Rydzewski RM. pH Dependence of inhibitors targeting the occluding loop of cathepsin B. Bioorg Chem. 2002;30:264–275. doi: 10.1016/s0045-2068(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 45.Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;32:22–37. doi: 10.1016/j.arr.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 46.Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;824:22–33. doi: 10.1016/j.bbapap.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Jakobson M, Jakobson M, Llano O, Palgi J, Arumäe U. Multiple mechanisms repress N-Bak mRNA translation in the healthy and apoptotic neurons. Cell Death Dis. 2013;4:e777. doi: 10.1038/cddis.2013.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peña-Blanco A, García-Sáez AJ. Bax Bak and beyond: mitochondrial performance in apoptosis. FEBS J. 2017 Jul 29; doi: 10.1111/febs.14186. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 49.Groth-Pedersen L, Jäättelä M, Nylandsted J. A Method to Monitor Lysosomal Membrane Permeabilization by Immunocytochemistry. Cold Spring Harb Protoc. 2015;2015:904–907. doi: 10.1101/pdb.prot086181. [DOI] [PubMed] [Google Scholar]
- 50.Vanwalleghem G, Fontaine F, Lecordier L, Tebabi P, Klewe K, Nolan DP, Yamaryo-Botté Y, Botté C, Kremer A, Burkard GS, Rassow J, Roditi I, Pérez-Morga D, Pays E. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat Commun. 2015;6:8078. doi: 10.1038/ncomms9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamacher-Brady A, Brady NR. Bax/Bak-dependent, Drp1-independent Targeting of X-linked Inhibitor of Apoptosis Protein (XIAP) into Inner Mitochondrial Compartments Counteracts Smac/DIABLO-dependent Effector Caspase Activation. J Biol Chem. 2015;290:22005–22018. doi: 10.1074/jbc.M115.643064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dave Z, Byfield M, Bossy-Wetzel E. Assessing mitochondrial outer membrane permeabilization during apoptosis. Methods. 2008;46:319–323. doi: 10.1016/j.ymeth.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Goyal S, Amar SK, Dubey D, Pal MK, Singh J, Verma A, Kushwaha HN, Ray RS. Involvement of cathepsin B in mitochondrial apoptosis by p-phenylenediamine under ambient UV radiation. J Hazard Mater. 2015;300:415–425. doi: 10.1016/j.jhazmat.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 54.Amantini C, Morelli MB, Santoni M, Soriani A, Cardinali C, Farfariello V, Eleuteri AM, Bonfili L, Mozzicafreddo M, Nabissi M, Cascinu S, Santoni G. Sorafenib induces cathepsin B-mediated apoptosis of bladder cancer cells by regulating the Akt/PTEN pathway. The Akt inhibitor, perifosine, enhances the sorafenib-induced cytotoxicity against bladder cancer cells. Oncoscience. 2015;2:395–409. doi: 10.18632/oncoscience.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kilinc M, Gürsoy-Ozdemir Y, Gürer G, Erdener SE, Erdemli E, Can A, Dalkara T. Lysosomal rupture, necroapoptotic interactions and potential crosstalk between cysteine proteases in neurons shortly after focal ischemia. Neurobiol Dis. 2010;40:293–302. doi: 10.1016/j.nbd.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Hsu SF, Hsu CC, Cheng BC, Lin CH. Cathepsin B is involved in the heat shock induced cardiomyocytes apoptosis as well as the anti-apoptosis effect of HSP-70. Apoptosis. 2014;19:1571–1580. doi: 10.1007/s10495-014-1033-9. [DOI] [PubMed] [Google Scholar]