

Abstract
Presynaptic voltage-gated K+ (Kv) channels in dorsal root ganglion (DRG) neurons are thought to regulate nociceptive synaptic transmission in the spinal dorsal horn. However, the Kv channel subtypes responsible for this critical role have not been identified. The Kv3.4 channel is particularly important because it is robustly expressed in DRG nociceptors, where it regulates action potential (AP) duration. Furthermore, Kv3.4 dysfunction is implicated in the pathophysiology of neuropathic pain in multiple pain models. We hypothesized that, through their ability to modulate AP repolarization, Kv3.4 channels in DRG nociceptors help to regulate nociceptive synaptic transmission. To test this hypothesis, we investigated Kv3.4 immunoreactivity (IR) in the rat cervical superficial dorsal horn (sDH) in both sexes and implemented an intact spinal cord preparation to investigate glutamatergic synaptic currents from second order neurons in the sDH under conditions that selectively inhibit the Kv3.4 current. We found presynaptic Kv3.4 IR in peptidergic and nonpeptidergic nociceptive fibers of the sDH. The Kv3.4 channel is hypersensitive to 4-aminopyridine and tetraethylammonium (TEA). Accordingly, 50 μm 4-aminopyridine and 500 μm TEA significantly prolong the AP, slow the maximum rate of repolarization in small-diameter DRG neurons, and potentiate monosynaptic excitatory postsynaptic currents (EPSCs) in dorsal horn laminae I and II through a presynaptic mechanism. In contrast, highly specific inhibitors of BK, Kv7, and Kv1 channels are less effective modulators of the AP and have little to no effect on EPSCs. The results strongly suggest that presynaptic Kv3.4 channels are major regulators of nociceptive synaptic transmission in the spinal cord.
SIGNIFICANCE STATEMENT Intractable neuropathic pain can result from disease or traumatic injury and many studies have been conducted to determine the underlying pathophysiological changes. Voltage-gated ion channels, including the K+ channel Kv3.4, are dysregulated in multiple pain models. Kv3.4 channels are ubiquitously expressed in the dorsal root ganglion (DRG), where they are major regulators of DRG excitability. However, little is known about the ionic mechanisms that regulate nociceptive synaptic transmission at the level of the first synapse in the spinal cord, which is critical to pain transmission in both intact and pathological states. Here, we show that Kv3.4 channels have a significant impact on glutamatergic synaptic transmission in the dorsal horn, further illuminating its potential as a molecular pain therapeutic target.
Keywords: Kv channel, pain transduction, spinal cord, synaptic transmission
Introduction
Glutamatergic synaptic transmission between primary nociceptors and secondary neurons in superficial layers of the dorsal horn is a critical step in the pain signaling pathway (Tao et al., 2005). However, our understanding of the presynaptic ion channels that regulate this process is limited (Tsantoulas and McMahon, 2014). Presynaptic voltage-gated K+ (Kv) channels are major regulators of synaptic transmission because they have a universal ability to regulate excitability in neural tissues (Dodson and Forsythe, 2004; Bean, 2007). In particular, high-voltage-activating Kv channels shape the repolarization of the action potential (AP) and therefore determine the activation of voltage-gated Ca2+ channels that are directly involved in vesicular neurotransmitter release at the nerve terminal. In the CNS, Kv3 channels are the best candidates for this role (Rudy and McBain, 2001; Ishikawa et al., 2003; Dodson and Forsythe, 2004; Goldberg et al., 2005; Kaczmarek and Zhang, 2017; Liu et al., 2017). Recent work demonstrated conclusively that Kv3.1/3.4 heteromultimers regulate AP duration in boutons of cerebellar stellate inhibitory interneurons and thereby help to determine evoked neurotransmitter release (Rowan et al., 2014, 2016; Rowan and Christie, 2017). However, whether a similar complex regulates nociceptive glutamatergic transmission in the spinal cord dorsal horn is not known. It is also important to know how cell signaling pathways associated with nociception might modulate key presynaptic Kv channels (Trimmer, 2014).
Previous work reported expression of multiple Kv channels, including Kv3.4, in adult DRG neurons (Gold et al., 1996; Rasband et al., 2001; Brooke et al., 2004a; Chien et al., 2007; Ritter et al., 2012, 2015a; Trimmer, 2014; Tsantoulas and McMahon, 2014; Liu et al., 2017). We have also determined that homomultimeric Kv3.4 channels underlie the majority of the high-voltage-activating K+ current in small-diameter dorsal root ganglion (DRG) neurons (Ritter et al., 2012, 2015b). Supporting this assessment, we found robust expression of Kv3.4 mRNA in these neurons, which dominates the small to negligible expression of the Kv3.1, Kv3.2, and Kv3.3 mRNAs (Ritter et al., 2012). In addition, siRNA knock-down nearly abolishes the Kv3.4 current in small-diameter DRG neurons and prolongs the duration of the AP, helping to demonstrate that Kv3.4 channels are major regulators of AP repolarization in the DRG (Ritter et al., 2012, 2015b). Moreover, Kv3.4 channels enhance their activity by undergoing switching from fast-inactivating A-type to slow-inactivating delayed rectifier-type upon phosphorylation of several serines within the channel's N-terminal inactivation domain (Covarrubias et al., 1994; Beck et al., 1998; Antz et al., 1999; Ritter et al., 2012; Zemel et al., 2017). In small-diameter DRG neurons, this mechanism shortens the AP, strongly suggesting that Kv3.4 channel activity drives repolarization of APs carrying nociceptive signaling (Ritter et al., 2012; Liu et al., 2017). Therefore, we hypothesized that Kv3.4 channels might ultimately determine nociceptive signaling at the level of the superficial dorsal horn (sDH) by regulating AP shape and duration, which would govern Ca2+-dependent glutamatergic vesicular release and the resulting excitatory postsynaptic current (EPSC).
To test this hypothesis, we investigated Kv3.4 immunoreactivity (IR) in the sDH, which receives Aδ- and C-fiber (nociceptive fiber) projections. Then, to probe the electrophysiological impact of the Kv3.4 channel, we implemented an ex vivo preparation of an intact cervical spinal cord, a method suitable for patch-clamp recordings from superficial second order dorsal horn neurons that receive nociceptive inputs. Under conditions that stimulate Aδ- and C-fibers, we tested the effects of relatively specific K+ channel inhibitors on the magnitude of EPSCs. Along with robust presynaptic Kv3.4 IR in the sDH, the electrophysiological results demonstrate that preferential inhibition of presynaptic Kv3.4 channels potentiates EPSCs in the sDH. Consistent with the hypothesis, inhibition of somatic Kv3.4 channels in the DRG also prolongs the AP by slowing the maximum rate of repolarization. The identification of the Kv3.4 channel as a significant player in the pain signaling pathway has implications in the pathophysiology of neuropathic pain induced by spinal cord injury and other nervous system diseases (Ritter et al., 2015a,b; Zemel et al., 2017).
Materials and Methods
Spinal cord preparation.
All animals were treated as approved by the institutional animal care and use committee of Thomas Jefferson University. Timed pregnant female Sprague Dawley rats (Taconic Farms) were maintained in the Thomas Jefferson University Animal Facility for 1 week before the birth of pups. For all experiments, rat pups were killed by overdose of ketamine (380 mg/kg), xylazine (40 mg/kg), and acepromazine (0.3 mg/kg), followed by decapitation. Cervical spinal cords were harvested from postnatal day 9 (P9) to P30 rat pups of either sex in a similar manner as described in previous studies (Pinto et al., 2008, 2010; Szucs et al., 2009). The spinal column was rapidly removed and placed in dissecting ACSF consisting of the following (in mm): 220 sucrose, 25 NaHCO3, 11 glucose, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, and 1.25 NaH2PO4 at room temperature bubbled with a 95% O2/5% CO2 gas mixture to oxygenate and adjust pH to 7.3–7.4. The spinal column was pinned down with the ventral side facing up and the ventral bony laminae were removed to expose the underlying spinal cord. The dorsal roots in the cervical region are ∼1–3 mm, so DRG attached to the dorsal roots were dissected out of the bony cavity intact to preserve as much root as possible for stimulation. Generally, segments C5–C8 were used for all experiments. The spinal cord with attached dorsal roots and DRGs was carefully lifted out of the spinal column and the cervical spinal cord region was trimmed from the rest of the cord. The dura mater was removed and ventral roots cut from the cord to reflect the dorsal roots medially, thereby exposing a strip of gray matter on the dorsolateral side of the cord corresponding to the dorsal horn. The pia mater was gently peeled off from the region of interest to allow access for patch electrodes and the DRG was removed from the dorsal root. The cleaned and trimmed cervical spinal cord was then pinned onto a beveled piece of elastomer compound eraser at an angle of ∼15° (see Fig. 3A) and transferred to an incubation chamber with oxygenated ACSF consisting of the following (in mm): 115 NaCl, 25 NaHCO3, 11 Glucose, 3 KCl, 2 CaCl2, 1 MgCl2, and 1 NaH2PO4 at room temperature until ready to transfer to the patch-clamp recording chamber. Compared with previous studies using this preparation, the use of cyanoacrylate glue was exempted in favor of small pins to keep the cord at the desired angle for illumination. In addition, this configuration allowed us to straighten out the natural curvature of the cervical cord.
Figure 3.

Intact cervical spinal cord preparation for patch-clamp recordings of glutamatergic synaptic currents from the superficial dorsal horn. A, Schematic of the experimental setup representing its main components. Neurons in the superficial dorsal horn are visualized using oblique infrared LED illumination and a 40× immersion objective. The spinal cord (SC) was pinned at an angle of 10° to 15° on a piece of elastomer compound eraser. The spinal cord is represented by a cross-section of the cervical region with its axis perpendicular to the plane of the image. BSE, Bipolar stimulation electrode (suction electrode); DR, dorsal roots (one free and the other inside the suction electrode); PCE, patch-clamping electrode hooked up to a Multiclamp 700B amplifier. B, Images of lamina I neurons subjected to whole-cell patch clamping. Top, Infrared image. Bottom, Fluorescence image of neuron loaded with biocytin (conjugated with Alexa Fluor 488) through the PCE. C, Representative monosynaptic eEPSCs evoked consecutively by stimulating the DR (100 μA, 1 ms, 10 sweeps) while holding the neuron's VH at −70 mV (see Materials and Methods). The average trace is shown in black. D, Histogram of eEPSC peak amplitudes. The stimulus intensity ranged between 100 and 600 μA. E, Consecutive eEPSCs recorded before and after exposing the spinal cord to 1 μm CNQX (averages are displayed in black and red, respectively). F, Spontaneous glutamatergic synaptic currents at VH = −70 mV before (black) and after (red) exposure to 1 μm CNQX.
Dorsal horn neuron illumination and visualization.
Neurons in the dorsal horn were illuminated using oblique infrared LEDs and visualized using a RolerA-XR camera and Q-Capture Pro7 software (Pinto et al., 2008, 2010; Szucs et al., 2009; Hachisuka et al., 2016). The LED was mounted on a small micromanipulator (Narishige) placed on the microscope head stage and the x–y–z axes were adjusted until maximal contrast was achieved. Still images were taken using the Q-Capture Pro7 software. Neurons were selected for recording based on their location in laminae I and II of the superficial dorsal horn.
Preparation of acutely dissociated DRG neurons.
P7–P28 pups were killed as described above for spinal cord experiments and ganglia were harvested from all accessible levels and placed into Hanks' buffered saline solution (HBSS) with 10 mm HEPES. Ganglia were dissociated by treatment with 1.5 mg/ml collagenase in HBSS/HEPES solution for 30 min, followed by a 15–20 min treatment with 1 mg/ml trypsin in HBSS/HEPES solution. DRG neurons were then transferred to L-15 Leibovitz medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 24 mm NaHCO3, 38 mm glucose, and 2% penicillin–streptomycin and mechanically dissociated with a fire-polished Pasteur pipette. Neurons were plated onto poly-L-ornithine-coated coverslips and kept at 37°C for up to 48 h.
Electrophysiology.
Patch electrodes were made from Corning 7056 thin wall capillary glass (Warner Instruments) and pulled with a PIP5 micropipette puller (HEKA) or a P-97 micropipette puller (Sutter Instruments). Electrodes were fire polished to have tip resistances of 1–4 MΩ. Signals were amplified using a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2 kHz (4-pole Bessel), digitized at 10 kHz (Digidata 1440; Molecular Devices), and stored in a computer using Clampex version 10.2 software (Molecular Devices). Spinal cord recordings were obtained at room temperature in oxygenated ACSF and the internal pipette solution consisted of the following (in mm): 150 K-gluconate, 3 KCl, 1 MgCl2, 1 EGTA, and 10 HEPES, pH 7.3 with KOH. All spinal cord voltage-clamp recordings were conducted at holding potentials (VH) between −70 and −80 mV, the empirically determined reversal potential of Cl−, to minimize detection of any inhibitory postsynaptic currents (IPSCs). In some instances, recorded neurons were labeled using an Alexa Fluor-conjugated biocytin marker (Thermo Fisher Scientific) for visualization. A suction electrode was used to stimulate the dorsal root using an A-M Systems isolated pulse stimulator (model 2100). Typically, the dorsal roots were 1–3 mm in length and stimulated with pulses in the range of 100–600 μA, duration of 1 ms, and frequency of 0.1–1 Hz. Recordings of monosynaptic EPSCs were those that had no failures upon stimulation. Only monosynaptic EPSCs were chosen for further analysis.
AP experiments were performed on small-diameter DRG neurons (≤25 μm) as described previously (Ritter et al., 2012, 2015a; Zemel et al., 2017). In these experiments, the external solution consisted of the following (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES. The internal solution consisted of the following (in mm): 130 K-MES, 1 CaCl2, 1 EGTA, 10 HEPES, 2 Mg-ATP, and 0.3 Tris-GTP. Liquid junction potential (+15.2 mV for spinal cord recordings and +15.5 mV for DRG recordings) were calculated using Clampex version 10.5 software and were corrected offline.
Drugs and toxins.
All toxins and drugs were stored as concentrated stocks and added to the recording solution immediately before recording. Tetraethylammonium-Cl (TEA; Sigma-Aldrich), 4-aminopyridine (Sigma-Aldrich), α-dendrotoxin (α-DTX; Alomone Laboratories), iberiotoxin (IbTX; Smartox), and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX disodium salt; Alomone Laboratories) were dissolved in deionized water and XE991 (Alomone) was dissolved in DMSO. For DRG experiments, a 100 mm 4-aminopyridine stock solution was made and the pH was adjusted to ∼7.4 with HCl before use.
Immunohistochemistry.
Animals were killed as described previously and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde (Ritter et al., 2015a; Zemel et al., 2017). Cervical spinal cords were harvested and stored in 4% paraformaldehyde (1 d), followed by 0.1 m phosphate buffer (1 d), and finally in 30% sucrose-containing phosphate buffer (>3 d). The tissue was then embedded in tissue-freezing medium and 30 μm sections were cut. Sections were collected on glass slides and stored until further use.
Immunohistochemistry procedures were performed at room temperature. The slides with the sections were washed with PBS 3 times (5 min each). Sections were then blocked with 10% normal goat serum (NGS) in PBST (PBS containing 0.2% Triton X-100) for 1 h and then incubated with guinea pig anti-CGRP (1:1000; BMA Biomedicals), rabbit anti-Kv3.4 (1:100; Alomone Laboratories), or guinea pig anti-VGLUT2 (1:2000; Millipore) overnight at room temperature. Sections were then washed 3 times (5 min each) and incubated with goat anti-rabbit Alexa Fluor 488 (1:150; Abcam) or goat anti-guinea pig Alexa Fluor 568 (1:500; Thermo Fisher Scientific) in 5% NGS in PBST for 1 h at room temperature. The sections were then again washed with PBS 3 times (5 min each) and coverslips were added with FluorSave reagent (Calbiochem). Finally, the slides were allowed to dry at room temperature overnight and stored at 4°C. For double immunostaining of Kv3.4 and IB4, Alexa Fluor 594-conjugated isolectin GS-IB4 (2 μg/ml; Thermo Fisher Scientific) was incubated for 30 min after completion of overnight primary and secondary antibody treatment for Kv3.4 alone. The sections were imaged on a FluoView FV1000 confocal microscope (Olympus).
Data analysis and statistics.
Data processing and analysis were conducted in Clampfit version 10.5 (Molecular Devices) and Origin Pro version 9.1 (Origin Laboratory). Student's paired t test was used to evaluate differences in paired datasets. Details of the statistical analyses are provided in the corresponding figure legends and exact p-values are generally shown on the graphs. Phase plane plots for nociceptor action potentials were obtained by plotting the first derivative of the AP waveform versus the membrane potential of the AP waveform, which allows visualization of rate changes as a function of voltage. Values for means are presented as mean ± SEM throughout.
Results
Spinal cord Kv3.4 channels are present in presynaptic peptidergic and nonpeptidergic nociceptive fibers of the sDH
To probe the functional role of the Kv3.4 channel on nociceptive spinal synaptic signaling, it is important to demonstrate presynaptic Kv3.4 expression in the sDH. We conducted immunohistochemical analyses in rat pup spinal cord to assess the expression of Kv3.4, several markers of nociceptors (CGRP and IB4), and an established marker of the excitatory presynaptic compartment (VGLUT2) (see Materials and Methods). We observed Kv3.4 immunoreactivity in dorsal horn laminae I–III, where it colocalized with peptidergic calcitonin gene-related peptide (CGRP) and nonpeptidergic isolectin B4 (IB4) nociceptive fibers (Figs. 1A–C, 2A–C). In addition, Kv3.4 immunoreactivity colocalized with the presynaptic glutamatergic marker VGLUT2 (Fig. 2D). Therefore, Kv3.4 channels are expressed presynaptically in sDH laminae, where they colocalize with known markers of nociceptive primary afferents.
Figure 1.

Colocalization of the Kv3.4 channel with the peptidergic nociceptive marker CGRP. Immunohistochemical staining demonstrates colabeling of CGRP with Kv3.4 protein. B and C are magnified areas of A.
Figure 2.

Colocalization of the presynaptic Kv3.4 channel with the nonpeptidergic nociceptive marker isolectin B4 (IB4). A–C, Immunohistochemical staining demonstrating colabeling of IB4 with Kv3.4 protein. B and C are magnified areas of A. D, Colabeling of Kv3.4 protein with the glutamatergic presynaptic marker VGLUT2.
Characterization of cervical sDH neurons in an intact spinal cord preparation
To investigate spinal nociceptive synaptic transmission under conditions that preserve the integrity of neural circuitry in the cervical spinal cord, we implemented and characterized an optimized intact preparation developed previously to study the lumbar and thoracic regions (Pinto et al., 2008, 2010; Szucs et al., 2009). The cervical spinal cord presented a few challenges because of the short roots (1–3 mm) and cervical flexure (see Materials and Methods). A bipolar suction electrode applied to the selected dorsal root (C5–C8) was used for electrical stimulation and oblique LED illumination allowed visualization of sDH neurons (see Materials and Methods; Fig. 3A,B). We selected spinal cord neurons based on their location in laminae I or II of the sDH, and conducted whole-cell patch-clamp recordings using standard methods as described previously (Fig. 3C–F; Materials and Methods; Pinto et al., 2008, 2010; Szucs et al., 2009). Upon preferential electrical stimulation of C- and Aδ-fibers of the dorsal root (100–600 μA, 1 ms, 0.1 Hz), these neurons displayed robust monosynaptic evoked EPSCs (eEPSCs) sensitive to CNQX (1 μm), which indicates excitatory glutamatergic synaptic transmission most likely associated with nociceptive signaling (Fig. 3E, Table 1). In the absence of stimulation and at a holding potential approximately equal to the Cl− reversal potential (to nullify inhibitory synaptic currents), these neurons also exhibited spontaneous CNQX-sensitive EPSCs (sEPSCs) resulting from spontaneous quantal release of glutamate from C- and Aδ-fibers and interneurons synapsing on the nociceptive laminae of the sDH (Fig. 3F). Correspondingly, under current-clamp conditions and after stimulation of the dorsal roots, we observed subthreshold and suprathreshold EPSPs (Fig. 4A,B). Further supporting the healthy quality of the spinal cord preparation, the selected neurons also exhibited robust passive and active membrane parameters (Fig. 4C,D, Table 2).
Table 1.
EPSC properties
| n | ||
|---|---|---|
| Peak (pA) | 272.39 ± 31.57 | 79 |
| Rise time (ms) | 7.89 ± 0.65 | 79 |
| Latency (ms) | 21.19 ± 0.65 | 79 |
| Jitter (ms) | 1.63 ± 0.17 | 75 |
Peak was calculated as the amplitude of the EPSC waveform. Rise time was calculated as the time from 10% to 90% of the EPSC waveform. Latency was calculated as the time from the start of the stimulus to the peak amplitude. Jitter was calculated as the time variability in the start of the EPSC waveform across multiple traces.
Figure 4.

Spiking examples from neurons in the superficial dorsal horn. A, Subthreshold and suprathreshold responses evoked by a stimulus of 100 μA. B, Pair of APs exhibiting an afterdepolarization. This response was evoked by a brief 0.5 ms stimulus. C, Spontaneous spiking (resting membrane potential = −62 mV). D, Recording of passive and active responses evoked by sustained current injection (−20 to 30 pA). First active trace is shown in red.
Table 2.
Passive and active properties of second order dorsal horn neurons
| n | ||
|---|---|---|
| RMP (mV) | −74 ± 0.88 | 65 |
| Input resistance (GΩ) | 0.95 ± 0.07 | 53 |
| Capacitance (pF) | 37.56 ± 3.84 | 37 |
| Threshold (mV) | −55.65 ± 0.77 | 40 |
| AP amplitude (mV) | 96.6 ± 1.81 | 40 |
| ADP amplitude (mV) | 7.69 ± 0.7 | 24 |
| AHP (mV) | −76.79 ± 1.21 | 16 |
| APD50 (ms) | 1.86 ± 0.09 | 40 |
| APD90 (ms) | 0.68 ± 0.03 | 40 |
| Max depolarization rate (mV ms−1) | 142.09 ± 8.29 | 40 |
| Max repolarization rate (mV ms−1) | 60.24 ± 4.01 | 40 |
AP amplitude was calculated as the difference from the most negative membrane potential to the most positive membrane potential during an AP waveform. ADP amplitude was calculated from the most negative membrane potential to the peak of the ADP. Maximum depolarization and repolarization rates were determined from the derivative of the AP waveform.
RMP, Resting membrane potential; ADP, afterdepolarization.
Evoked EPSCs in the sDH are potentiated by TEA and 4-aminopyridine, but not by antagonists of BK, Kv7, and Kv1 channels
After the demonstration of Kv3.4 channel expression in presynaptic nerve terminals of the spinal cord dorsal horn and physiological validation of the intact spinal cord preparation, we set out to determine whether this Kv channel is a regulator of synaptic transmission in the sDH. Kv3 channels are hypersensitive to low, submillimolar concentrations of the well known K+ channel blockers 4-aminopyridine and TEA (Schröter et al., 1991; Vega-Saenz de Miera et al., 1992). To test the effects of these inhibitors on synaptic transmission, we held the membrane potential of the spinal cord neuron at −70 mV (∼ECl) and recorded eEPSCs upon strong stimulation of the dorsal root (100–600 μA) to excite high-threshold nociceptive fibers (C and Aδ). Under these conditions, the recorded eEPSC results from activation of spinal cord glutamatergic AMPA receptors (Fig. 3). Exposure to either 50 μm 4-aminopyridine or 500 μm TEA similarly potentiated the average peak of the eEPSCs by 47.14 ± 18.69% and 20.71 ± 8.19%, respectively (Fig. 5A,B). These results suggest that, through inhibition of presynaptic C-/Aδ-fiber K+ channels, the presynaptic AP is prolonged and, consequently, vesicular Ca2+-dependent glutamatergic release is enhanced.
Figure 5.

eEPSCs from superficial dorsal horn neurons are potentiated by submillimolar concentrations of TEA and 4-aminopyridine. A, B, Left and center, Consecutive monosynaptic eEPSCs recorded before and after (15–20 sweeps) exposing the spinal cord to 50 μm 4-aminopyridine (A) and 500 μm TEA (B). Averages are displayed in red. Right, Pooled paired measurements of peak EPSCs before (control) and after exposure to 4-aminopyridine (A) and TEA (B), with box plots showing the percentage change in peaks across paired experiments. Sample size and p-values of the paired Student's t test are shown on the graphs. Stimulation parameters are as indicated in the legend to Figure 2 and in the Materials and Methods. Each symbol in the graphs represents an independent response from a separate spinal cord (i.e., the sample size corresponds to number of animals examined). Percentage change box plots describe the datasets as follows: dashed and solid lines represent mean and median, respectively; lower and upper edges of the box represent the 25th and 75the percentiles, respectively; bottom and top whiskers correspond to 5th and 95the percentiles, respectively; and crosses represent minimum and maximum values.
However, this result alone cannot rule out possible contributions of other K+ channels that are also significantly sensitive to TEA and/or 4-aminopyridine, such as Kv1, Kv7, and big-conductance Ca2+-activated K+ (BK) channels (Dodson and Forsythe, 2004). Expression of these K+ channels has also been reported in putative DRG nociceptors (Everill et al., 1998; Scholz et al., 1998; Rasband et al., 2001; Beekwilder et al., 2003; Zhang et al., 2003, 2010; Chi and Nicol, 2007; Rose et al., 2011; Zheng et al., 2013; Martinez-Espinosa et al., 2015; Liu et al., 2017). To rule out whether these K+ channels were contributing to the observed TEA and 4-aminopyridine effects, we tested α-DTX, XE991, and IbTX, which are highly selective antagonists of Kv1.1/1.2/1.6, Kv7, and BK channels, respectively. Upon exposing spinal cords to these antagonists, we observed no effect on the average peak of the eEPSCs, which is in contrast to the potentiating effects of TEA and 4-aminopyridine (Fig. 6A–C). In addition, α-DTX, XE991 and IbTX have little to no effect on jitter and rise time of the eEPSCs (Table 3). Given the differential effects of TEA and 4-aminopyridine against the other more specific K+ channel antagonists, we can conclude that inhibition of a Kv3-type channel is most likely responsible for the associated potentiation of the eEPSCs. Furthermore, Kv3.4 is the top candidate. In addition to its presynaptic expression in nociceptive afferents of the sDH (Figs. 1, 2), we have previously reported strong evidence demonstrating that Kv3.4 is the dominant Kv3 isoform in putative DRG nociceptors from rat pups (Ritter et al., 2012, 2015a,b).
Figure 6.

eEPSCs from superficial dorsal horn neurons are not affected by specific inhibitors of Kv7, BK, and Kv1 channels. A–C, Left and center, Consecutive monosynaptic eEPSCs recorded before and after (2–30 sweeps) exposing the spinal cord to the indicated K+ channel inhibitors (XE991, IbTX, and α-DTX). Averages are displayed in red. Right, Pooled paired measurements of peak EPSCs before (control) and after exposure to the indicated inhibitors. Sample size and p-values of the paired Student's t test are shown on the graphs. Stimulation parameters are as indicated in the legend to Figure 2 and in the Materials and Methods. Each symbol in the graphs represents an independent response from a separate spinal cord (i.e., the sample size corresponds to number of animals examined). Percentage change box plots are displayed to the right of summary data plots (legend to Fig. 5 describes box plot characteristics).
Table 3.
Effects of pharmacological compounds on EPSC properties
| 500 μm TEA |
50 μm 4-aminopyridine |
100 nm IbTX |
30 μm XE991 |
80 nm DTX |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| Peak (pA) | 323.91 ± 121.32 | 370.27 ± 123.30* | 193.91 ± 43.69 | 258.21 ± 49.75** | 473.19 ± 178.68 | 502.68 ± 191.33 | 367.66 ± 85.29 | 322.93 ± 123.56 | 470.52 ± 203.08 | 461.1 ± 179.54 |
| Rise Time (ms) | 8.59 ± 1.60 | 9.81 ± 1.69 | 8.45 ± 2.00 | 7.96 ± 1.44 | 9.23 ± 3.27 | 8.48 ± 2.85 | 13.75 ± 3.17 | 13.05 ± 3.25 | 7.34 ± 2.23 | 7.35 ± 2.24 |
| Latency (ms) | 20.27 ± 2.34 | 20.45 ± 2.41 | 22.44 ± 3.06 | 22.77 ± 3.31 | 22.07 ± 4.15 | 21.63 ± 3.98 | 25.15 ± 4.32 | 25.99 ± 4.80 | 21.31 ± 5.71 | 21.60 ± 5.61 |
| Jitter (ms) | 1.66 ± 0.36 | 1.71 ± 0.31 | 2.07 ± 0.73 | 1.56 ± 0.45 | 1.8 ± 0.38 | 1.6 ± 0.52 | 2.83 ± 1.03 | 3.58 ± 1.29 | 1.38 ± 0.58 | 2.05 ± 0.80 |
| n | 9 | 7 | 8 | 5 | 4 | |||||
Peak is the maximum amplitude of the EPSC waveform. Rise time was calculated as the time from 10% to 90% of the EPSC waveform. Latency was calculated as the time from the start of the stimulus to the peak amplitude. Jitter was calculated as the time variability in the start of the EPSC waveform across multiple traces.
*p ≤ 0.05;
**p ≤ 0.01.
TEA and 4-aminopyridine act presynaptically to potentiate the eEPSC
The results so far are consistent with a presynaptic role of the Kv3.4 channel. However, the average peak eEPSC resulting from consecutive stimulations might include both monosynaptic and polysynaptic responses. Confirming that low concentrations of TEA and 4-aminopyridine potentiate monosynaptic eEPSCs would add strong support to a presynaptic DRG mechanism involving regulation of the AP by Kv3.4. Therefore, from each stimulation run (a family of traces), we isolated consistent stable EPSC peaks (no failures) in each individual trace by-eye, segments that often coincided with the lowest variance around the peak (Fig. 7). This analysis consistently revealed similar monosynaptic peaks in individual traces, which were generally potentiated by low concentrations of TEA and 4-aminopyridine (Fig. 8A,B). In contrast, IbTX, α-DTX, and XE991 had little to no effect on monosynaptic eEPSC peaks (Fig. 8C–E).
Figure 7.

Determination of monosynaptic responses from individual eEPSC traces. The EPSCs depict the response to 4-aminopyridine (A) and to TEA (B). Representative eEPSC traces from examples displayed in Figure 5 demonstrate consistent monosynaptic peaks across multiple traces (dashed red lines). The by-eye identification of the peaks in individual traces was generally confirmed by determining the regions of the average trace with the lowest variance around the average peak. The magnitude of these peaks was used for the analysis of monosynaptic eEPSCs in Figure 8.
Figure 8.
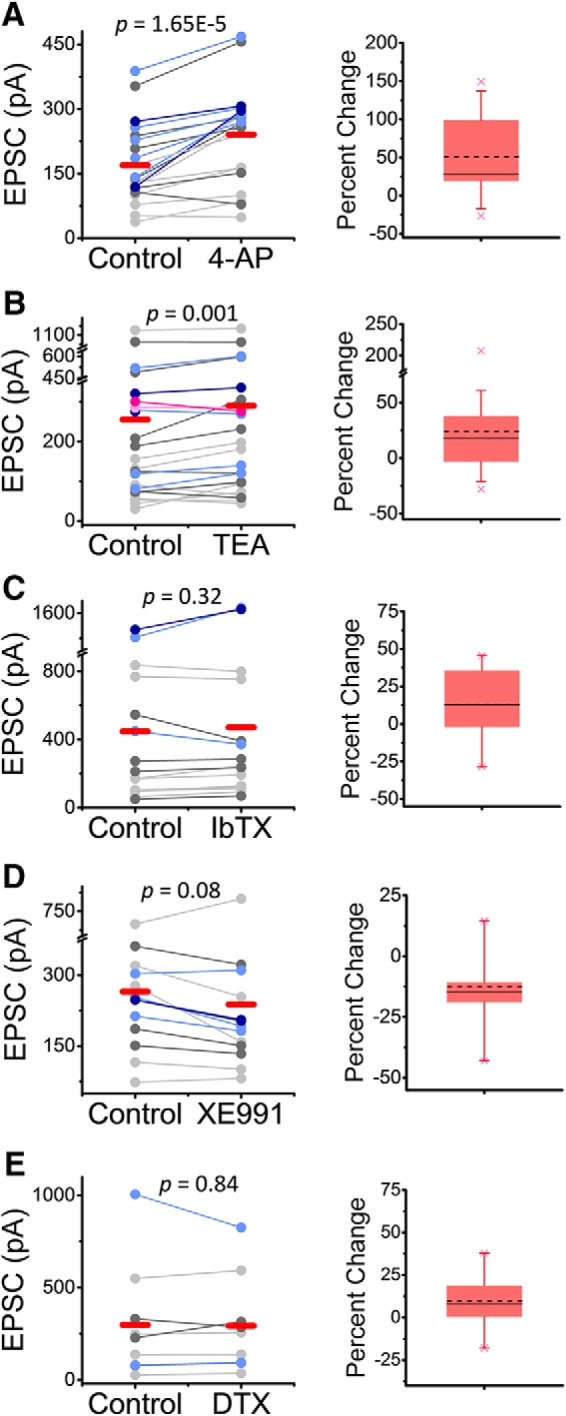
Submillimolar 4-aminopyridine and TEA consistently potentiate monosynaptic EPSCs. Pooled paired average peaks from the multipeak analysis (Fig. 7) before and after exposure to 50 μm 4-aminopyridine (A), 500 μm TEA (B), 100 nm IbTX (C), 30 μm XE991 (D), and 80 nm α-DTX (E). Color scheme displays the numerical order of peaks in a given recording (light gray = first peak, dark gray = second peak, light blue = third peak, dark blue = fourth peak, light pink = fifth peak, dark pink = sixth peak, averages shown in red). The p-values of the paired Student's t test are shown on the graphs. Percentage change box plots are displayed to the right of summary data plots (Fig. 5 legend describes box plot characteristics).
To test the presynaptic mechanism further, we also investigated the effects of the K+ channel antagonists on the paired-pulse ratio (PPR) and the amplitude of sEPSCs A change in the PPR demonstrates a presynaptic effect tied to vesicle depletion inducing synaptic depression (PPR = P2/P1 < 1) or presynaptic Ca2+ accumulation associated with synaptic facilitation (PPR > 1) (Fioravante and Regehr, 2011). In contrast, no PPR change would be more consistent with a postsynaptic effect that affects paired responses equally. Generally, we found that the PPR was <1 under control conditions, indicating synaptic depression. In the presence of 4-aminopyridine and TEA, the PPR was consistently decreased further (0.79 ± 0.23 to 0.58 ± 0.24 for 4-aminopyridine, 0.69 ± 0.11 to 0.48 ± 0.11 for TEA), which suggests exacerbated vesicle depletion resulting from a presynaptic effect of the inhibitors (Fig. 9A,B).
Figure 9.

Submillimolar 4-aminopyridine and TEA decrease the PPR. A, B, Paired pulse (interstimulus interval = 30–80 ms) EPSC recordings before and after (10 sweeps) exposing the spinal cord to 50 μm 4-aminopyridine (A) and 500 μm TEA (B). Right, Pooled paired measurements of the PPR (= P2/P1) before (control) and after exposure to 4-aminopyridine (A) and TEA (B). Sample size and p-values of the paired Student's t test are shown on the graphs. All recordings were conducted at VH = −70 mV. Stimulation parameters are as indicated in the legend to Figure 2 and in the Materials and Methods. Each symbol in the graphs represents an independent response from a separate spinal cord (i.e., the sample size corresponds to number of animals examined).
Desensitization of postsynaptic AMPA receptors could have contributed to the observed synaptic depression (Kirischuk et al., 2002; Chen et al., 2004; Christie et al., 2010). Therefore, to assess directly a possible postsynaptic phenomenon, we examined the effect of TEA on the amplitude of sEPSCs. The origin of the sEPSCs includes vesicle release from primary DRG nociceptive afferents and spinal interneurons. To isolate the sEPSCs mainly mediated by AMPA receptor channels, we recorded the spontaneous activity at ECl (−70 mV), as was done previously (Fig. 3). We observed significant spontaneous activity, which allowed robust measurements of sEPSC peak amplitudes before and after exposure to 500 μm TEA (>400 events; Fig. 10A). In three independent paired experiments, we found that the normalized amplitude histograms of the sEPSCs recorded before and after exposure to TEA were indistinguishable (Fig. 10B,C). These results ruled out a postsynaptic action of TEA, which could have been responsible for the TEA-dependent potentiation of the eEPSCs. In addition, there was no change in the frequency of events before and after exposure to 500 μm TEA (6.22 ± 2.59 events/s and 6.54 ± 3.37 events/s, before and after TEA, respectively; estimates derived from data presented in Fig. 10), demonstrating that TEA did not impact the level of spontaneous activity, a proxy measurement for presynaptic resting membrane potential. Based on the evidence provided by three independent experiments (monosynaptic potentiation, enhanced synaptic depression, and lack of effect on sEPSC amplitude and frequency), we conclude that submillimolar 4-aminopyridine and TEA potentiate glutamatergic synaptic transmission at a presynaptic level thorough the inhibition of Kv3.4 channels in DRG neurons.
Figure 10.

Submillimolar TEA does not affect sEPSCs. A, Representative sweeps of sEPSCs at −70 mV before and after exposing the spinal cord to 500 μm TEA (left and right, respectively). Magnified segments are also shown to demonstrate individual events. B, Relative frequency histograms of peak EPSC amplitudes from three independent recordings (three neurons each from three different spinal cords) before and after exposure to TEA. Relative frequency is the fraction of sEPSCs that falls into a given bin (bin size = 0.75 pA). C, Cumulative plots of sEPSC amplitudes corresponding to the data shown in B. In all three cases, the two-sample Kolmogorov–Smirnov test returned no difference between the control and TEA plots. The p-values are indicated on the plots.
DRG action potential is consistently modulated by the Kv channel inhibitors TEA and 4-aminopyridine
If presynaptic potentiation of the eEPSC results from prolonging the presynaptic AP upon inhibition of the Kv3.4 current in DRG neurons, then we would expect broadening of the somatic AP by submillimolar TEA and 4-aminopyridine and little and inconsistent effects of IbTX, α-DTX, and XE991 on somatic AP duration. This hypothesis, however, assumes that the somatic and presynaptic APs are shaped by a similar ensemble of ion channels and therefore are similarly regulated by Kv3.4. To test these ideas, we recorded somatic APs from acutely dissociated DRG neurons before and after the exposure to the selected K+ channel inhibitors at the same concentrations used in the spinal cord recordings (Fig. 11, Table 4). Whereas TEA and 4-aminopyridine consistently broadened the AP [AP duration at 50% of amplitude (APD50), AP duration at 90% of amplitude (APD90); p = 4.99E-4 to 0.042] and slowed the maximum rate of repolarization (p = 6.99E-4 to 0.037), the effects of IbTX on these properties were inconsistent but marginally prolonged the APD50 (p = 0.043). In contrast, XE991 and α-DTX did not affect the AP waveform (Fig. 11, Table 4). Overall, these results are consistent with a major direct role of Kv3.4 on the repolarization of the AP in DRG neurons, which secondarily regulates evoked Ca2+-dependent glutamatergic neurotransmission in the sDH of the spinal cord. The inconsistent results with IbTX suggest heterogeneity and quantitative differences in the relative contributions of Kv3.4 and BK channels to AP repolarization in the soma and nerve terminals of putative nociceptors.
Figure 11.

Analysis of primary nociceptor APs in the absence and presence of several K+ channel inhibitors. Left to right, Representative AP traces, phase plane plots, and changes in APD50, APD90, and maximum repolarization rate (derived from phase plane plots) before and after exposure to 50 μm 4-aminopyridine (A), 500 μm TEA (B), 100 nm IbTX (C), 30 μm XE991 (D), and 80 nm α-DTX (E). Averages are shown in black and p-values of the paired Student's t test are displayed on graphs. Additional properties are reported in Table 4.
Table 4.
Effects of pharmacological compounds on the DRG action potential
| 500 μm TEA |
50 μm 4-aminopyridine |
100 nm IbTX |
30 μm XE991 |
80 nm DTX |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| Capacitance (pF) | 12.88 ± 1.20 | 16.07 ± 4.52 | 13.83 ± 1.46 | 12.75 ± 1.47 | 12.73 ± 1.11 | |||||
| Diameter (μm) | 21.61 ± 0.93 | 20.78 ± 0.97 | 21.25 ± 1.02 | 21.43 ± 0.88 | 20.47 ± 0.97 | |||||
| RMP (mV) | −62.36 ± 4.42 | −65.50 ± 6.07 | −67.25 ± 2.30 | −71.50 ± 2.75* | −66.93 ± 2.90 | −70.79 ± 3.67** | −66.07 ± 2.45 | −64.93 ± 2.52 | −64.88 ± 3.05 | −69.88 ± 4.56 |
| IR (GΩ) | 0.55 ± 0.16 | 0.77 ± 0.19 | 1.06 ± 0.32 | 1.38 ± 0.33 | 0.76 ± 0.19 | 0.92 ± 0.23 | 0.93 ± 0.17 | 1.39 ± 0.29 | 0.89 ± 0.17 | 1.05 ± 0.15 |
| Threshold (mV) | −29.21 ± 3.04 | −32.64 ± 4.23 | −28.63 ± 2.08 | −31.88 ± 2.60* | −26.36 ± 3.82 | −29.21 ± 4.31* | −29.07 ± 1.81 | −30.36 ± 1.65 | −29.88 ± 2.60 | −35.00 ± 2.05* |
| Amplitude (mV) | 113.05 ± 2.93 | 111.91 ± 4.63 | 115.47 ± 2.41 | 116.98 ± 3.86 | 108.08 ± 4.96 | 110.23 ± 6.26 | 109.97 ± 3.98 | 107.89 ± 3.24 | 110.79 ± 4.55 | 107.27 ± 5.39 |
| AHP (mV) | −73.47 ± 2.35 | −74.49 ± 5.45 | −73.11 ± 1.54 | −75.68 ± 2.14 | −72.36 ± 2.24 | −76.85 ± 3.01* | −73.32 ± 2.48 | −73.35 ± 2.11 | −71.63 ± 1.91 | −76.34 ± 2.35 |
| APD50 (ms) | 4.33 ± 0.63 | 7.60 ± 1.27** | 4.43 ± 0.47 | 6.28 ± 0.83* | 4.15 ± 0.48 | 5.46 ± 0.90* | 4.73 ± 0.53 | 5.16 ± 0.62 | 3.80 ± 0.43 | 3.67 ± 0.50 |
| APD90 (ms) | 1.04 ± 0.05 | 1.30 ± 0.06*** | 1.09 ± 0.07 | 1.40 ± 0.11* | 1.05 ± 0.05 | 1.17 ± 0.10 | 1.13 ± 0.06 | 1.15 ± 0.07 | 1.05 ± 0.07 | 1.05 ± 0.09 |
| Max depolarization rate (mV/ms) | 79.84 ± 17.67 | 79.35 ± 21.17 | 75.42 ± 8.83 | 69.75 ± 8.76 | 68.26 ± 10.93 | 67.01 ± 11.73 | 61.28 ± 8.94 | 55.55 ± 6.12 | 77.82 ± 12.72 | 71.50 ± 11.87 |
| Max repolarization rate (mV/ms) | 38.37 ± 3.68 | 25.04 ± 2.30*** | 33.90 ± 2.64 | 25.94 ± 2.66* | 37.99 ± 3.81 | 35.13 ± 4.95 | 35.38 ± 3.75 | 32.76 ± 2.91 | 38.04 ± 3.15 | 40.65 ± 4.17 |
| n | 7 | 8 | 7 | 7 | 8 | |||||
RMP, Resting membrane potential; AHP, afterhyperpolarization.
*p < 0.05;
**p ≤ 0.01;
***p ≤ 0.001.
Discussion
The Kv3.4 channel is a major regulator of AP repolarization in small-diameter nociceptors in the DRG. Here, we investigated whether this regulation actually affects nociceptive signaling in the spinal cord. Consistent with a presynaptic localization, we found that Kv3.4 is expressed in excitatory presynaptic terminals in nociceptive afferents of the sDH, where it colocalizes with key molecular markers of the pain pathway (CGRP, IB4, and VGLUT2). Using an ex vivo preparation of the cervical spinal cord and several K+ channel inhibitors, we demonstrate that submillimolar concentrations of 4-aminopyridine and TEA potentiate monosynaptic glutamatergic eEPSCs, suggesting that inhibition of the presynaptic 4-aminopyridine/TEA-hypersensitive Kv3.4 is responsible for this potentiation. Strengthening this conclusion, these inhibitors also decreased the PPR but did not affect the amplitude and frequency of sEPSCs. In contrast, specific inhibition of other DRG K+ channels that also exhibit hypersensitivities to 4-aminopyridine and/or TEA did not affect the eEPSC. Further supporting a direct relationship between Kv3.4-dependent regulation of AP duration in the DRG and the eEPSC peak in the sDH, submillimolar TEA and 4-aminopyridine prolonged the presynaptic AP, whereas other specific K+ channel inhibitors induced little and inconsistent effects on the AP waveform.
Optimization of the ex vivo cervical spinal cord patch-clamping technique
Over the last decade, several studies have used intact organ spinal cord preparations to study electrophysiological and morphological parameters as well as local circuitry (Pinto et al., 2008, 2010; Szucs et al., 2009; Hachisuka et al., 2016). Compared with traditional slices, this technique has many advantages, including less damage to the spinal cord, which is especially important toward understanding the complex circuitry of the dorsal horn (Peirs and Seal, 2016). However, this technique has thus far been mainly applied to the lumbar and thoracic regions of the spinal cord. The cervical spinal cord is important, not only from a physiological perspective, but also from a relevant pathological viewpoint because the cervical region is the most common location of spinal cord injuries in humans. Therefore, we focused on optimizing this preparation to expand the application and relevance of the ex vivo spinal cord technique. By minimizing the pronounced flexure of the cervical region and ensuring the viability of short dorsal roots, we obtained a robust and reliable new preparation suitable for intact spinal cord patch-clamping experiments.
Presynaptic Kv3.4 channel regulates glutamatergic signaling in the superficial dorsal horn
Consistent with previous reports (Brooke et al., 2004a; Chien et al., 2007), we observed Kv3.4 expression in the neuropil of the sDH, where it colocalizes with markers of nociceptive fibers, CGRP, and IB4. In addition, our new results show that Kv3.4 found in the sDH is expressed presynaptically in glutamatergic axonal terminals, as determined by its colocalization with VGLUT2. Therefore, Kv3.4 is ideally present in the terminal axonal compartment to regulate nociceptive synaptic transmission through its ability to shape AP repolarization.
Generally, presynaptic Kv channels help to tune synaptic transmission by regulating the spiking properties of neurons (Dodson and Forsythe, 2004; Kaczmarek and Zhang, 2017). This information, coupled with previous work demonstrating that Kv3 channels are the primary regulators of AP repolarization in the CNS and that Kv3.4 is the dominant Kv3 channel in DRG neurons, suggests that this ion channel might play a significant role as a regulator of nociceptive synaptic activity at the level of the first synapse in the pain pathway (Goldberg et al., 2005; Ritter et al., 2012; Rowan et al., 2014, 2016; Liu et al., 2017; Rowan and Christie, 2017). New data described here strongly support this hypothesis by demonstrating that a Kv channel hypersensitive to 4-aminopyridine and TEA, such as Kv3.4, regulates the AP repolarization rate and duration in nociceptors and, consequently, the amplitude of the eEPSC in the sDH.
Because the Kv3.4 channel's main role is to help repolarize the AP in small-diameter DRG neurons, its inhibition would prolong the AP that ultimately reaches the nerve terminal of putative nociceptors. Therefore, if the AP evoked by electrical stimulation of the dorsal root is prolonged after inhibition of the Kv3.4 channel by either TEA or 4-aminopyridine, then activation of voltage-gated Ca2+ channels and the resulting Ca2+ entry into the nerve terminal are increased. Therefore, the probability of Ca2+-dependent vesicular glutamate release increases and the ensuing eEPSC is potentiated, which is consistent with accepted theories of quantal neurotransmission (Katz and Miledi, 1967; Mulkey and Zucker, 1991; Llinás et al., 1992; Borst et al., 1995; Bollmann and Sakmann, 2005). The results of this work are reminiscent of the role that presynaptic Kv3.4 might play at the mouse neuromuscular junction, where inhibition of this ion channel potentiates the end plate potential (Brooke et al., 2004b). In addition, the interpretation of our results gains additional support from the pattern of differential effects that K+ channel inhibitors have on the AP waveform in the DRG, which generally mirrors the effects on the eEPSC in the sDH. In particular, however, there are interesting differences possibly reflecting quantitatively different contributions of distinct K+ channels to AP repolarization in the soma and the nerve terminal. For instance, it appears that IbTX is capable of prolonging the AP by mainly increasing the APD50, but has no consistent effect on the eEPSC. In contrast, submillimolar 4-aminopyridine and TEA consistently lengthen the APD50 and APD90 and accordingly potentiate the monosynaptic eEPSC. The DRG AP results are consistent with previous studies (Li et al., 2007; Zhang et al., 2010; Liu et al., 2017).
To establish that the mechanism discussed above most likely involves presynaptic regulation by Kv3.4, submillimolar TEA and 4-aminopyridine also enhanced synaptic depression by decreasing the PPR. This is likely the result of increased vesicle depletion. Moreover, we found no evidence of a postsynaptic contribution because the amplitude of spontaneous EPSCs was not affected by submillimolar TEA. In addition, TEA had no effect on the spontaneous EPSC frequency, which, under the conditions of our experiments, might originate from spontaneous release and evoked release resulting from spontaneous depolarizations originating in the DRG and spinal interneurons. Therefore, TEA-hypersensitive K+ channels do not regulate resting membrane potential (Table 4) and spontaneous spiking, which is consistent with the interpretation of our results. Kv3.4 is a high-voltage-activating A-type Kv channel that is best suited to shaping AP repolarization, as established by recent work from us and others (Ritter et al., 2012; Rowan et al., 2014, 2016; Liu et al., 2017; Rowan and Christie, 2017).
Role of other K+ channels expressed in the DRG
The DRG expresses multiple K+ channels exhibiting differential cellular and subcellular distributions in heterogeneous populations of primary sensory neurons and, moreover, they share major differences in terms of their gating properties and mechanisms of modulation (Gold et al., 1996; Safronov et al., 1996; Rasband et al., 2001; Zhang et al., 2003, 2010; Chi and Nicol, 2007; Chien et al., 2007; Phuket and Covarrubias, 2009; Duan et al., 2012; Zheng et al., 2013). Therefore, they play a myriad of roles along the peripheral sensory pathway, regulating resting membrane potential and AP properties (shape, repolarization rate, propagation, latency to first spike, interspike interval, afterhyperpolarization, etc.). By using a battery of specific K+ channel antagonists (α-DTX, XE991, IbTX) against DRG K+ channels sharing sensitivities to submillimolar concentrations of TEA and/or 4-aminopyridine, we ruled out major possible contributions of several DRG K+ channels (Kv1.1, Kv1.2, Kv1.6, Kv7, and BK channels) to nociceptive synaptic transmission in the sDH. Although BDS-I is thought to be a specific Kv3.4 peptide inhibitor, we did not use it in these experiments because it also potentiates Nav1.7, a critical voltage-gated Na+ channel expressed in primary sensory neurons, with high potency (Diochot et al., 1998; Liu et al., 2012). It is also unlikely that DRG Kv1.4 and Kv4 channels contribute presynaptically to synaptic transmission in the sDH because they are low-voltage activating, highly resistant to TEA, and only modestly sensitive to 4-aminopyridine. Furthermore, the expression of Kv4.1 and Kv4.3 is limited to the soma of rat DRG neurons (Gold et al., 1996; Chien et al., 2007; Phuket and Covarrubias, 2009; Yunoki et al., 2014). In addition, other channels, such as Kv2 and Slack channels, are also expressed in the DRG but are unlikely to be candidates because they are relatively insensitive to TEA and 4-aminopyridine (Patel et al., 1997; Bocksteins et al., 2009; Lu et al., 2015). Overall, these results strongly suggest that Kv3.4, the most likely target of submillimolar TEA and 4-aminopyridine, is a major presynaptic regulator of excitatory neurotransmission from glutamatergic C- and Aδ-fibers in the sDH.
Implications and perspective
Previous work and the new results presented here collectively constitute compelling evidence for the presynaptic role of the Kv3.4 channel as a significant regulator of glutamatergic synaptic signaling in the spinal cord nociceptive pathway. These findings help to explain how spinal cord injury (SCI)-induced dysfunction of the Kv3.4 channel in primary nociceptors can lead to intractable neuropathic pain. Therefore, the Kv3.4 channel is an attractive target that might help in the development of more effective interventions to alleviate persistent pain induced by SCI and other nervous system diseases associated with pathological pain. Any manipulations that increase Kv3.4 activity in the DRG might have beneficial analgesic effects.
Footnotes
This work was supported by the Vickie and Jack Farber Foundation (M.C.), the Dean's Transformational Science Award (M.C.), the National Institutes of Health (Grant NS079855 to M.C. and Grant NS079702 to A.C.L.), the Dubbs Fellowship Fund (T.M.), Sigma Xi (GIAR Grant G20141015648241 to T.M.), Autifony Therapeutics, Ltd. (M.C.), and the Luso-American Development Foundation (V.P.). We thank Drs. Matthew Dalva, Melanie Elliott, Ethan Goldberg, David Ritter, and Benjamin Zemel, and members of the Covarrubias laboratory for helpful comments and feedback on previous versions of this manuscript; members of the Dalva laboratory for sharing reagents; and Dr. Bruce Bean for providing helpful tips regarding the effects of Kv channel inhibitors on the AP in the DRG.
The authors declare no competing financial interests.
References
- Antz C, Bauer T, Kalbacher H, Frank R, Covarrubias M, Kalbitzer HR, Ruppersberg JP, Baukrowitz T, Fakler B (1999) Control of K+ channel gating by protein phosphorylation: structural switches of the inactivation gate. Nat Struct Biol 6:146–150. 10.1038/5833 [DOI] [PubMed] [Google Scholar]
- Bean BP. (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8:451–465. 10.1038/nrn2148 [DOI] [PubMed] [Google Scholar]
- Beck EJ, Sorensen RG, Slater SJ, Covarrubias M (1998) Interactions between multiple phosphorylation sites in the inactivation particle of a K+ channel: insights into the molecular mechanism of protein kinase C action. J Gen Physiol 112:71–84. 10.1085/jgp.112.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekwilder JP, O'Leary ME, van den Broek LP, van Kempen GT, Ypey DL, van den Berg RJ (2003) Kv1.1 channels of dorsal root ganglion neurons are inhibited by n-butyl-p-aminobenzoate, a promising anesthetic for the treatment of chronic pain. J Pharmacol Exp Ther 304:531–538. 10.1124/jpet.102.042135 [DOI] [PubMed] [Google Scholar]
- Bocksteins E, Raes AL, Van de Vijver G, Bruyns T, Van Bogaert PP, Snyders DJ (2009) Kv2.1 and silent kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am J Physiol Cell Physiol 296:C1271–C1278. 10.1152/ajpcell.00088.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B (2005) Control of synaptic strength and timing by the release-site Ca2+ signal. Nat Neurosci 8:426–434. 10.1038/nn1417 [DOI] [PubMed] [Google Scholar]
- Borst JG, Helmchen F, Sakmann B (1995) Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol 489:825–840. 10.1113/jphysiol.1995.sp021095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke RE, Atkinson L, Batten TF, Deuchars SA, Deuchars J (2004a) Association of potassium channel Kv3.4 subunits with pre- and post-synaptic structures in brainstem and spinal cord. Neuroscience 126:1001–1010. 10.1016/j.neuroscience.2004.03.051 [DOI] [PubMed] [Google Scholar]
- Brooke RE, Moores TS, Morris NP, Parson SH, Deuchars J (2004b) Kv3 voltage-gated potassium channels regulate neurotransmitter release from mouse motor nerve terminals. Eur J Neurosci 20:3313–3321. 10.1111/j.1460-9568.2004.03730.x [DOI] [PubMed] [Google Scholar]
- Chen G, Harata NC, Tsien RW (2004) Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci U S A 101:1063–1068. 10.1073/pnas.0307149101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi XX, Nicol GD (2007) Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J Neurophysiol 98:2683–2692. 10.1152/jn.00437.2007 [DOI] [PubMed] [Google Scholar]
- Chien LY, Cheng JK, Chu D, Cheng CF, Tsaur ML (2007) Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci 27:9855–9865. 10.1523/JNEUROSCI.0604-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie LA, Russell TA, Xu J, Wood L, Shepherd GM, Contractor A (2010) AMPA receptor desensitization mutation results in severe developmental phenotypes and early postnatal lethality. Proc Natl Acad Sci U S A 107:9412–9417. 10.1073/pnas.0908206107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Wei A, Salkoff L, Vyas TB (1994) Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron 13:1403–1412. 10.1016/0896-6273(94)90425-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Schweitz H, Béress L, Lazdunski M (1998) Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J Biol Chem 273:6744–6749. 10.1074/jbc.273.12.6744 [DOI] [PubMed] [Google Scholar]
- Dodson PD, Forsythe ID (2004) Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci 27:210–217. 10.1016/j.tins.2004.02.012 [DOI] [PubMed] [Google Scholar]
- Duan KZ, Xu Q, Zhang XM, Zhao ZQ, Mei YA, Zhang YQ (2012) Targeting A-type K+ channels in primary sensory neurons for bone cancer pain in a rat model. Pain 153:562–574. 10.1016/j.pain.2011.11.020 [DOI] [PubMed] [Google Scholar]
- Everill B, Rizzo MA, Kocsis JD (1998) Morphologically identified cutaneous afferent DRG neurons express three different potassium currents in varying proportions. J Neurophysiol 79:1814–1824. 10.1152/jn.1998.79.4.1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21:269–274. 10.1016/j.conb.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD (1996) Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol 75:2629–2646. 10.1152/jn.1996.75.6.2629 [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B (2005) Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci 25:5230–5235. 10.1523/JNEUROSCI.0722-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachisuka J, Baumbauer KM, Omori Y, Snyder LM, Koerber HR, Ross SE (2016) Semi-intact ex vivo approach to investigate spinal somatosensory circuits. Elife 5: pii: e22866. 10.7554/eLife.22866 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, Takahashi T (2003) Distinct roles of Kv1 and Kv3 potassium channels at the calyx of held presynaptic terminal. J Neurosci 23:10445–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek LK, Zhang Y (2017) Kv3 channels: enablers of rapid firing, neurotransmitter release, and neuronal endurance. Physiol Rev 97:1431–1468. 10.1152/physrev.00002.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1967) The timing of calcium action during neuromuscular transmission. J Physiol 189:535–544. 10.1113/jphysiol.1967.sp008183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Clements JD, Grantyn R (2002) Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons in rat collicular cultures. J Physiol 543:99–116. 10.1113/jphysiol.2002.021576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Gao SB, Lv CX, Wu Y, Guo ZH, Ding JP, Xu T (2007) Characterization of voltage-and Ca2+-activated K+ channels in rat dorsal root ganglion neurons. J Cell Physiol 212:348–357. 10.1002/jcp.21007 [DOI] [PubMed] [Google Scholar]
- Liu P, Jo S, Bean BP (2012) Modulation of neuronal sodium channels by the sea anemone peptide BDS-I. J Neurophysiol 107:3155–3167. 10.1152/jn.00785.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PW, Blair NT, Bean BP (2017) Action potential broadening in capsaicin-sensitive DRG neurons from frequency-dependent reduction of Kv3 current. J Neurosci 37:9705–9714. 10.1523/JNEUROSCI.1703-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Silver RB (1992) Microdomains of high calcium concentration in a presynaptic terminal. Science 256:677–679. 10.1126/science.1350109 [DOI] [PubMed] [Google Scholar]
- Lu R, Bausch AE, Kallenborn-Gerhardt W, Stoetzer C, Debruin N, Ruth P, Geisslinger G, Leffler A, Lukowski R, Schmidtko A (2015) Slack channels expressed in sensory neurons control neuropathic pain in mice. J Neurosci 35:1125–1135. 10.1523/JNEUROSCI.2423-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Wu J, Yang C, Gonzalez-Perez V, Zhou H, Liang H, Xia XM, Lingle CJ (2015) Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. Elife 4: pii: e10013. 10.7554/eLife.10013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Zucker RS (1991) Action potentials must admit calcium to evoke transmitter release. Nature 350:153–155. 10.1038/350153a0 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honoré E (1997) Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J 16:6615–6625. 10.1093/emboj/16.22.6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Seal RP (2016) Neural circuits for pain: recent advances and current views. Science 354:578–584. 10.1126/science.aaf8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuket TR, Covarrubias M (2009) Kv4 channels underlie the subthreshold-operating A-type K+-current in nociceptive dorsal root ganglion neurons. Front Mol Neurosci 2:3. 10.3389/neuro.02.003.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto V, Szûcs P, Derkach VA, Safronov BV (2008) Monosynaptic convergence of C- and Adelta-afferent fibres from different segmental dorsal roots on to single substantia gelatinosa neurones in the rat spinal cord. J Physiol 586:4165–4177. 10.1113/jphysiol.2008.154898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto V, Szucs P, Lima D, Safronov BV (2010) Multisegmental Adelta- and C-fiber input to neurons in lamina I and the lateral spinal nucleus. J Neurosci 30:2384–2395. 10.1523/JNEUROSCI.3445-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS (2001) Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A 98:13373–13378. 10.1073/pnas.231376298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DM, Ho C, O'Leary ME, Covarrubias M (2012) Modulation of Kv3.4 channel N-type inactivation by protein kinase C shapes the action potential in dorsal root ganglion neurons. J Physiol 590:145–161. 10.1113/jphysiol.2011.218560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DM, Zemel BM, Hala TJ, O'Leary ME, Lepore AC, Covarrubias M (2015a) Dysregulation of Kv3.4 channels in dorsal root ganglia following spinal cord injury. J Neurosci 35:1260–1273. 10.1523/JNEUROSCI.1594-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DM, Zemel BM, Lepore AC, Covarrubias M (2015b) Kv3.4 channel function and dysfunction in nociceptors. Channels 9:209–217. 10.1080/19336950.2015.1056949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K, Ooi L, Dalle C, Robertson B, Wood IC, Gamper N (2011) Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 152:742–754. 10.1016/j.pain.2010.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJM, Christie JM (2017) Rapid state-dependent alteration in Kv3 channel availability drives flexible synaptic signaling dependent on somatic subthreshold depolarization. Cell Rep 18:2018–2029. 10.1016/j.celrep.2017.01.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, DelCanto G, Yu JJ, Kamasawa N, Christie JM (2016) Synapse-level determination of action potential duration by K channel clustering in axons. Neuron 91:370–383. 10.1016/j.neuron.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Tranquil E, Christie JM (2014) Distinct Kv channel subtypes contribute to differences in spike signaling properties in the axon initial segment and presynaptic boutons of cerebellar interneurons. J Neurosci 34:6611–6623. 10.1523/JNEUROSCI.4208-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ (2001) Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24:517–526. 10.1016/S0166-2236(00)01892-0 [DOI] [PubMed] [Google Scholar]
- Safronov BV, Bischoff U, Vogel W (1996) Single voltage-gated K+ channels and their functions in small dorsal root ganglion neurones of rat. J Physiol 493:393–408. 10.1113/jphysiol.1996.sp021391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz A, Gruss M, Vogel W (1998) Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol 513:55–69. 10.1111/j.1469-7793.1998.055by.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröter KH, Ruppersberg JP, Wunder F, Rettig J, Stocker M, Pongs O (1991) Cloning and functional expression of a TEA-sensitive A-type potassium channel from rat brain. FEBS Lett 278:211–216. 10.1016/0014-5793(91)80119-N [DOI] [PubMed] [Google Scholar]
- Szucs P, Pinto V, Safronov BV (2009) Advanced technique of infrared LED imaging of unstained cells and intracellular structures in isolated spinal cord, brainstem, ganglia and cerebellum. J Neurosci Methods 177:369–380. 10.1016/j.jneumeth.2008.10.024 [DOI] [PubMed] [Google Scholar]
- Tao YX, Gu J, Stephens RL Jr (2005) Role of spinal cord glutamate transporter during normal sensory transmission and pathological pain states. Mol Pain 1:30. 10.1186/1744-8069-1-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. (2014) Ion channels and pain: important steps towards validating a new therapeutic target for neuropathic pain. Exp Neurol 254:190–194. 10.1016/j.expneurol.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulas C, McMahon SB (2014) Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci 37:146–158. 10.1016/j.tins.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Saenz de Miera E, Moreno H, Fruhling D, Kentros C, Rudy B (1992) Cloning of ShIII (Shaw-like) cDNAs encoding a novel high-voltage-activating, TEA-sensitive, type-A K+ channel. Proc Biol Sci 248:9–18. 10.1098/rspb.1992.0036 [DOI] [PubMed] [Google Scholar]
- Yunoki T, Takimoto K, Kita K, Funahashi Y, Takahashi R, Matsuyoshi H, Naito S, Yoshimura N (2014) Differential contribution of Kv4-containing channels to A-type, voltage-gated potassium currents in somatic and visceral dorsal root ganglion neurons. J Neurophysiol 112:2492–2504. 10.1152/jn.00054.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemel BM, Muqeem T, Brown EV, Goulão M, Urban MW, Tymanskyj SR, Lepore AC, Covarrubias M (2017) Calcineurin dysregulation underlies spinal cord injury-induced K+ channel dysfunction in DRG neurons. J Neurosci 37:8256–8272. 10.1523/JNEUROSCI.0434-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Gopalakrishnan M, Shieh CC (2003) Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience 122:1003–1011. 10.1016/j.neuroscience.2003.08.035 [DOI] [PubMed] [Google Scholar]
- Zhang XL, Mok LP, Katz EJ, Gold MS (2010) BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci 31:450–462. 10.1111/j.1460-9568.2009.07060.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Fang D, Liu M, Cai J, Wan Y, Han JS, Xing G (2013) Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 154:434–448. 10.1016/j.pain.2012.12.005 [DOI] [PubMed] [Google Scholar]