

Abstract
Astrocytes perform a wide array of physiological functions, including structural support, ion exchange, and neurotransmitter uptake. Despite this diversity, molecular markers that label subpopulations of astrocytes are limited, and mechanisms that generate distinct astrocyte subtypes remain unclear. Here we identified serine protease high temperature requirement A 1 (HtrA1), a bone morphogenetic protein 4 signaling regulated protein, as a novel marker of forebrain astrocytes, but not of neural stem cells, in adult mice of both sexes. Genetic deletion of HtrA1 during gliogenesis accelerates astrocyte differentiation. In addition, ablation of HtrA1 in cultured astrocytes leads to altered chondroitin sulfate proteoglycan expression and inhibition of neurite extension, along with elevated levels of transforming growth factor-β family proteins. Brain injury induces HtrA1 expression in reactive astrocytes, and loss of HtrA1 leads to an impairment in wound closure accompanied by increased proliferation of endothelial and immune cells. Our findings demonstrate that HtrA1 is differentially expressed in adult mouse forebrain astrocytes, and that HtrA1 plays important roles in astrocytic development and injury response.
SIGNIFICANCE STATEMENT Astrocytes, an abundant cell type in the brain, perform a wide array of physiological functions. Although characterized as morphologically and functionally diverse, molecular markers that label astrocyte subtypes or signaling pathways that lead to their diversity remain limited. Here, after examining the expression profile of astrocytes generated in response to bone morphogenetic protein signaling, we identify high temperature requirement A 1 (HtrA1) as an astrocyte-specific marker that is differentially expressed in distinct adult mouse brain regions. HtrA1 is a serine protease that has been linked to cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, a small blood vessel disease in humans. Understanding the role of HtrA1 during development and after injury will provide insights into how distinct astrocyte populations are generated and their unique roles in injury and disease.
Keywords: astrocyte heterogeneity, bone morphogenetic protein, brain injury, cerebrovascular disease, gliogenesis, HtrA1
Introduction
Although there are many molecular labels that identify morphologically, biochemically, and functionally diverse populations of neurons, molecular markers that label subpopulations of astrocytes are limited, and mechanisms that generate distinct astrocyte subtypes remain unclear. Astrocytes, stereotypically defined by their stellate morphology, are classically categorized as “fibrous” or “protoplasmic” based on morphological differences and anatomical locations (Golgi, 1885; Kimelberg, 2009). Molecular and electrophysiological analyses of developing and mature astrocytes have demonstrated heterogeneity among the astroglial population (Batter and Kessler, 1991; Chvatal et al., 1995; Zhou et al., 2006; Sosunov et al., 2014), which was made more evident by genomic scale expression profiling of astrocytes from different developmental time points and brain regions (Bachoo et al., 2004; Cahoy et al., 2008; Yeh et al., 2009; Zhang et al., 2014). However, researchers are just beginning to discover molecular markers that identify functionally unique or disease-associated astroglial subtypes (Ben Haim and Rowitch, 2017; John Lin et al., 2017; Liddelow et al., 2017). Additional functionally relevant molecular markers is essential for the understanding of astrocyte subtype development and function.
Molecular mechanisms of astrocyte specification and diversity have been studied extensively using cultured neural stem cells (NSCs) and glial restricted progenitors (GRPs; Raff et al., 1984; Rao et al., 1998; Herrera et al., 2001). Cells expressing the astrocytic intermediate filament glial fibrillary acidic protein (GFAP) can be generated from NSCs and GRPs by two different classes of cytokines, the leukemia inhibitory factor (LIF)/ciliary neurotrophic factor (CNTF) family and the bone morphogenetic proteins (BMPs; Lillien et al., 1988; Gross et al., 1996; Johe et al., 1996; Richards et al., 1996; Takizawa et al., 2001). Upon ligand binding, LIF/CNTF activates Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathways, whereas BMPs signal primarily through Smad pathways (Stahl et al., 1994; Liu et al., 1996). A transcriptional complex that contains both STAT and Smad proteins regulates GFAP expression, leading to the conclusion that both pathways contribute to astrogliogenesis through convergent mechanisms (Nakashima et al., 1999; Sun et al., 2001). However, while BMP signaling promotes the generation of stellate, postmitotic astrocytes from NSCs, LIF signaling promotes a bipolar, radial glia-like cell that maintains a stem cell phenotype (Bonaguidi et al., 2005). In addition to cellular and molecular differences, BMP and CNTF signaling generate functionally and molecularly distinct astrocytes that respond differently to injury when transplanted (Davies et al., 2008). These findings suggest that differential cytokine responsiveness plays an important role in astrocyte specification as well as in astrocyte diversity.
Our goals in this study were to find novel molecular markers expressed specifically in differentiated astrocytes, and to reveal genes that participate in cytokine-specific generation of astrocyte diversity during development. We identified the serine protease high temperature requirement A 1 (HtrA1) as an BMP signaling responsive molecular marker of astrocytes in the adult mouse forebrain. Here we show that HtrA1 is important for postnatal astrogliogenesis as well as for astrocytic morphology and extracellular matrix (ECM) protein regulation. In addition, deletion of HtrA1 increases microglia and endothelial cell proliferation after brain injury. Mutation in HtrA1 has been linked to the human small blood vessel disease cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Our demonstration of astrocyte specificity of HtrA1 also suggests CARASIL may be astrocytic in origin, and that HtrA1 functions in astrocytes may play important roles in cerebrovascular health.
Materials and Methods
Animals.
For the microarray analysis and subsequent expression characterization, CD1 mice (Harlan Laboratories) of either sex at the appropriate ages were used for NSC isolation and mRNA in situ hybridization (ISH). HtrA1-null mice (Supanji et al., 2013) were maintained as heterozygotes in a C57BL/6J background and genotyped by PCR with tail DNA. Generation of GFAP-Cre; BMPR1fx/fx mice has been previously described (Garcia et al., 2004; Sahni et al., 2010). All mice were housed in groups and maintained on a 12 h light/dark cycle with ad libitum access food and water. All experiments were performed with both male and female mice in each condition and analyzed without sex separation. Handling and treatment of animals were in accordance with Northwestern University Institutional Animal Care and Use Committee regulations and policies.
Transcriptome profiling by microarray.
Passage 1 dissociated NSCs were infected with retrovirus expressing green fluorescent protein (GFP) under the control of either CMV or GFAP promoters as previously described (Bonaguidi et al., 2005). Three days after infection, GFP-expressing cells in CMV-GFP retrovirus-transduced neurospheres were isolated by fluorescent-activated cell sorting (FACS) and processed for total RNA collection. GFAP-GFP retrovirus-transduced cells were plated for differentiation in EGF (1 ng/ml) with BMP4 (20 ng/ml) or LIF (20 ng/ml) plus Noggin (250 ng/ml; to block endogenous BMPs) for 7 d before GFP-expressing cells were isolated by FACS. Total RNA was immediately extracted after FACS from GFP-expressing cells using an RNAqueous Micro RNA isolation kit (Thermo Fisher Scientific). RNA from four replicates of all experimental conditions was examined by Nanodrop-1000 (Nanodrop Technologies) and Bioanalyzer (Agilent Technologies) before proceeding to cDNA amplification and labeling using the Ovation kit (NuGEN) according to the manufacturer's protocols. Gene expression profiles among the treatment groups were compared using the mouse WG-6 v1.1 microarray beadchip (Illumina). Hybridization and scanning of the beadchip array was performed by the Northwestern University genomic core facility with an Illumina BeadArray reader and preprocessed by the Illmina BeadStudio software. The microarray data has been deposited in the NCBI Gene Expression Omnibus (GEO; http://ncbi.nlm.nih.gov/geo/) and is accessible through GEO accession number GSE67942.
Quantitative real-time PCR.
Total RNA was isolated from mouse brain tissue or cultured cells using RNAqueous-4PCR RNA isolation kit (Thermo Fisher Scientific). cDNA was generated with 1 μg of total RNA using thermoscript reverse transcriptase (Thermo Fisher Scientific), and used in quantitative real-time PCRs (qRT-PCRs) with primers specific to desired candidate genes and SYBR Green master mix (Applied Biosystems). PCRs were performed in a Realplex2 thermal cycler (Eppendorf). Gene expression levels were standardized with GAPDH as the internal control, compared among experimental groups, and represented as fold changes relative to expression in the control group. At least three independent experiments were performed for all qRT-PCR studies.
Stab wound injury.
Eight-to-10-week-old age-matched HtrA1 wild-type and mutant mice of both sexes were secured on a stereotaxic injection frame (Kopf, model 900) and anesthetized with isofluorane. The skull opening was made with a dental drill. Stab wound injury was performed with a scalpel in the right neocortex (bregma from −0.8 to −2.8 mm; laterolateral, 1.5 mm; 0.9 mm deep). Mice were killed at 3 and 7 d after injury for immunohistochemical (IHC) analyses. Postinjury proliferation was measured with the injection of a single dose of EdU (50 mg/kg body weight; Thermo Fisher Scientific) 4 h before death. To analyze the lesion area, coronal cryostat sections of 400 μm distance rostral and distal of the injury site were collected and examined to identify the lesion center, the location with the largest lesion area. Four sections 50 μm apart surrounding the lesion center were quantified for changes in lesion area, neurovascular morphology, and proliferation.
NSC and astrocyte cultures.
NSCs from the subventricular zone (SVZ) of postnatal day (P) 1 mice were isolated and cultured as neurospheres using standard protocols (Bonaguidi et al., 2005). Primary astrocytes were dissected from P2 mice and cultured as monolayers as previously described (McCarthy and de Vellis, 1980; Sahni et al., 2010). For coculture studies, astrocytes transduced with GFP-tagged scrambled shRNA or HtrA1 shRNA lentivirus were FACS purified then plated at 50,000 cells/well onto 12 mm poly-d-lysine (PDL)-coated glass coverslips in 10% fetal bovine serum (FBS) containing astrocyte growth media as described. At 7 d after FACS, astrocytes were either harvested for Western blot analyses or used for neuronal coculture studies. Before the start of cocultures, astrocytes were adapted for serum-free conditions by culturing in astrocyte growth media containing 5% FBS for 16 h. On the following day, 20,000 freshly dissected cortical neurons from P1 C57BL/6 mice were plated on top of the astrocytes, and cultured without serum in neurobasal media with N2 and B27 supplements along with penicillin–streptomycin–glutamine (Thermo Fisher Scientific) for 7 d before fixing with 4% paraformaldehyde (PFA) and proceeding with further analysis. For treatment of cultured astrocytes with recombinant HtrA1 (R&D Systems), serum-derived cultured astrocytes were adapted for serum-free conditions as described and incubated in serum-free astrocyte media with or without recombinant HtrA1 (250 ng/ml) for 3 h. Protein was then extracted and analyzed by Western blots. For generating human NSCs and astrocytes from embryonic stem cells or induced pluripotent stem cells, we followed the protocol described by Duan et al. (2015) with the following modifications: before Passage 6 (day 165), NSCs are treated with LIF (20 ng/ml) for 7 d to enrich for glial progenitors. Cells are then dissociated and plated at 100,000 cells/well on PDL-coated 12 mm glass coverslips for 3 d before switching to differentiation media with BMP4 (10 ng/ml) for 7 d followed by FGF1 (50 ng/ml) for 7 d. Cells are fixed (on day 185) with 4% PFA and used for immunocytochemical analyses.
Bioinformatics analysis.
Raw signal intensity data from Illumina BeadStudio version 3 for individual probes were analyzed by the Northwestern University bioinformatics core facility using the R/Bioconductor lumi package, which corrected for small sample size variance via an empirical Bayesian method and performed normalization with internal controls. Probes with all sample values near or at background levels were marked “absent” and removed, leaving 21,217 probes for further analysis. Probes marked as “Present” were examined for differential expression among experimental groups using stringent statistical criteria with Bonferroni FDR p values <0.05 and fold-change >2-fold.
ISH.
Brain tissue was collected from mice that were transcardially perfused with cold phosphate buffer saline (PBS) and 4% PFA followed by 2 h postfix with 4% PFA. Brains were cryoprotected in 30% sucrose overnight, and stored at −80°C in OCT compound (Tissue-Tek) until use. Cryosections measuring 20 μm (Leica CM3050) from desired brain regions were washed three times with PBS with 0.02% Triton X-100 (PBST) and treated with Proteinase K (1 μg/ml) for 5 min. Sections were then washed three times in PBST and fixed in 4% paraformaldehyde (PFA) and 0.1% glutaraldehyde for 30 min. Following three PBST washes, Digoxygenin (DIG)-labeled antisense mRNA probes (1:150 in hybridization buffer) were added onto the slides which were then covered with siliconized glass coverslips and placed in a humidified hybridization chamber at 68°C overnight. Slides were washed at 68°C on day 2 with high stringency wash buffer (5× SSC with 50% formamide and 0.1% SDS) twice and low stringency wash buffer (2× SSC with 50× formamide) three times, followed by three Tris-buffered saline with 0.1% Tween (TBST) washes before incubation overnight in anti-DIG alkaline phosphotase antibody (Roche; 1:2000) in 5% goat serum. On day 3, slides were washed in TBST+2 mm levamasole three times and transferred into NBT-BCIP (Roche) containing developing solution from 6 h to overnight. Upon signal detection, slides were dehydrated through an ethanol gradient, cleared in xylene, and mounted in Histomount (Zymed) for storage and analysis.
IHC.
Tissue sections measuring 10 μm were prepared and washed with PBST as described for the ISH protocol before incubation with primary antibodies diluted in PBST with 5% goat serum at 4°C overnight. Primary antibodies used were as follows: rabbit anti-GFAP (1:1000; Dako Cytomation), mouse anti-GFAP (1:1000; Sigma-Aldrich), chicken anti-GFAP (1:1000; Abcam), rabbit anti-GFP (1:1000; Thermo Fisher Scientific), chicken anti-GFP (1:1000; Abcam), mouse anti-CSPG (CS-56, 1:1000; Sigma-Aldrich), rabbit anti-collagen IV (1:500; Abcam), rat anti-CD31 (1:500; Millipore), mouse anti-PDGFRβ (1:1000; Abcam), rabbit anti-HtrA1 (1:200; Abcam). Following primary incubation, tissue sections were washed three times in PBST and then incubated in fluorescent secondary antibody (1:500; Thermo Fisher Scientific) in PBST with 5% normal goat serum and DAPI nuclear stain for 1 h at room temperature. After a second set of PBST washes, sections were mounted in ProLong Gold antifade reagent (Thermo Fisher Scientific). EdU staining followed secondary antibody washes and was performed according to the manufacturer's instructions (Thermo Fisher Scientific). Stained sections were imaged on a confocal microscope (Leica SP5).
Protein extraction and Western blotting.
Protein was extracted from astrocyte cultures using M-PER (Mammalian Protein Extraction Reagent; Pierce) with Halt protease and phosphatase inhibitor mixture (Thermo Fisher Scientific) and stored at −80°C until use. To correctly resolve chondroitin sulfate proteoglycans (CSPGs), including neurocan, samples were treated with Chondroitinase ABC (1:20 of 10 U/ml stock; Seikagaku Bioscience) at 37°C for 90 min before further processing. Ten micrograms of protein were boiled in strong denaturing conditions for 10 min and loaded onto 4–20% polyacrylamide gels (Biorad). Protein was transferred onto polyvinylidene difluoride membranes at 4°C for 1 h, followed by blocking in 5% nonfat dry milk in TBST for 1 h at room temperature. Membranes were then incubated with primary antibody in blocking solution overnight at 4°C. After washing, membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (1:2000 in blocking solution; Santa Cruz Biotechnology) and developed using Pierce Enhanced Chemiluminescent Western Blotting Substrate (Thermo Fisher Scientific). The following primary antibodies were used: sheep anti-neurocan (1:500; R&D Systems), mouse anti-CSPG (CS-56, 1:1000; Sigma-Aldrich), rabbit anti-HtrA1 (1:500; Abcam), mouse anti-BMP4 (1:1000; Millipore), rabbit anti-transforming growth factor-β (anti-TGFβ; 1:1000; Abcam), and mouse anti-GAPDH (1:5000; Millipore).
Lentiviral construct production and transduction.
Plasmid DNA (pGIPz)-containing short hairpin RNA (shRNA) sequences targeted to mouse HtrA1 were purchased (GE Dharmacon) and used to produce lentiviral particles as previously described (Bond et al., 2014). Viral transductions were performed on P1 NSCs dissociated into single cells at a density of 5 × 104/ml in 50 ml of neurosphere growth media for 12 h before lentivirus was added (at a titer of 1 × 108 colony formation units/ml) and incubated at 37°C for 48 h. The transduced cells were then isolated by FACS and plated for differentiation studies or protein isolation as previously described. Lentiviral transduction of P2 astrocytes was performed in 80% confluent monolayer cultures before Passage 2 for 48 h. Cells were then shaken and dissociated for FACS isolation, followed by plating at 5 × 104/ml in astrocyte growth media for subsequent studies.
Quantification and statistical analyses.
Images acquired on a confocal microscope (Leica SP5) were exported to National Institutes of Health ImageJ for cell counting and integrated pixel-density analysis. Measurement of astrocyte morphological properties was performed with Cell Profiler software (Carpenter et al., 2006) using the MeasureObjectSizeShape module. Quantification of endothelial cell density, branching, length, and uniformity in the neocortex was performed using the Angiotool software (Zudaire et al., 2011) with an intensity threshold of 11/255, vessel diameter of 15, and remove small particle setting of 40. Quantification results were presented as mean ± SEM, generated from ≥3 independent trials per experimental condition. Differences among experimental groups were evaluated with unpaired Student's t test for two group experiments and one-way ANOVA with Tukey's post hoc test for pairwise comparisons in three group experiments using Microsoft Excel with XL Toolbox plugin.
Results
BMP and LIF generate morphologically and molecularly distinct astrocytes
Previous work has demonstrated that BMP and LIF generate molecularly and morphologically distinct astrocytes (Bonaguidi et al., 2005). To examine the genome-wide molecular differences between astrocytes specified by BMP or LIF signaling, we cultured NSCs derived from P1 mouse brain during the period of active cortical astrogliogenesis. NSCs were transduced with a retrovirus containing GFP under the control of the mouse GFAP promoter, then differentiated in the presence of either BMP4 or LIF with Noggin, a BMP signaling inhibitor (Fig. 1A). After 7 d in culture, GFP-expressing cells in each experimental condition displayed clearly distinctive morphologies (Fig. 1B). Total mRNA from GFP-expressing cells was collected and used for gene expression profiling by a cDNA microarray that contained 45,281 probes for transcripts found in the mouse genome. To identify differentially expressed genes, probes with >2-fold change in intensity and a false discovery rate (FDR)-adjusted p value of <0.05 in pairwise comparisons were considered as positives and selected for further analysis. The number and the identities of differentially expressed genes in pairwise comparisons of each experimental group are summarized in Fig. 1-1 and detailed in Figs. 1-3, 1-4 and 1-5 Heatmap comparison of the 1108 differentially expressed genes among the three experimental groups revealed clearly opposing expression profiles between BMP4-induced astrocytes and the neurosphere-derived progenitors (Fig. 1C). Examination of known astrocytic genes also confirmed that many accepted markers of astrocytes are more highly expressed in the BMP4-treated group (Fig. 1-2), providing further support for candidates from BMP-induced genes as potential markers of differentiated astrocytes.
Figure 1.

Genomic scale expression analysis by cDNA microarray revealed differential expression profiles of BMP-induced and LIF-induced astrocyte subtypes and identified novel astrocyte markers. A, Schematic diagram of gene expression microarray experimental design. B, Cellular morphology of each experimental group as visualized by GFP expression after viral transduction. LIF-induced cells acquire a radial morphology whereas BMP-induced cells acquire a stellate morphology after 7 d in culture. C, Heatmap of the top 1108 genes that are differentially expressed during astrogliogenesis with experimental groups and replicate numbers listed on the right of the heatmap. Opposing expression profiles are found between BMP-induced astrocytes and NSCs in neurospheres. LIF-induced GFAP+ cells are more similar to NSCs in overall profile than to BMP-induced astrocytes. Summary of differentially expressed genes, as well as expression of astrocytic genes, are listed in Figures 1-1, 1-2, 1-3, 1-4 and 1-5. D, The list of 14 genes expressed by BMP4-induced astrocytes that show ≥10-fold enrichment in astrocytes compared with neurons and oligodendrocytes. Blue highlights indicate genes known to be expressed by both astrocytes and NSCs, and gray highlights indicate genes that failed in vivo validation by ISH. Yellow highlights mark the final three candidates for further expression analyses. E, Expression analysis by microarray and qRT-PCR of Hopx, HtrA1, and F3. Expression levels are presented as fold change relative to the neurosphere group (FC; n = 4 per group). Comparison of qRT-PCR results between BMP and neurosphere groups revealed significant increases (*) of the three candidates in BMP astrocytes. F, Low-magnification ISH of Hopx, HtrA1, and F3 mRNA in P56 mouse neocortex and hippocampus (Allen Brain Atlas). G, High-magnification view of Hopx, HtrA1, and F3 mRNA in the dorsal cortex and the dentate gyrus of P60 mice as validated by in-house ISH. Hopx is found in both astrocytes (arrow) and NSCs in the SGZ (arrowhead) of the dentate gyrus (DG) but not in other cortical astrocytes. HtrA1 and F3 are found in astrocytes (arrows) but not in NSCs of the DG (arrowheads). All data presented as mean ± SEM. ABA, Allen Brain Atlas; Scale bars: B, 50 μm; F, 400 μm; G, 50 μm. *p < 0.01.
Summary of the number of differentially expressed genes identified by the microarray. 21217 probes that were identified as “present” were compared among experimental groups in pairwise fashion using listed criteria (N1, N2, N3). The number of differential expressed genes (N) was determined for each experimental group, which included genes that were induced (up) and suppressed (down). Download Figure 1-1, XLSX file (10.1KB, xlsx)
Expression of known astrocyte markers in Neurospheres, BMP induced, and LIF induced astrocytes. A summary of gene expression levels for fourteen commonly used astrocytes markers. While every marker is expressed in all three experimental groups, majority of the genes show strongest expression in BMP induced astrocytes. Download Figure 1-2, XLSX file (10.5KB, xlsx)
A list of differentially expressed genes identified by the comparison of BMP-induced astrocytes and neurosphere. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 830 genes are differentially expressed. 455 genes are expressed more highly in BMP-induced astrocytes compared to neurospheres. Conversely, 375 genes are expressed more highly in neurospheres compared to BMP-induced astrocytes. Download Figure 1-3, XLSX file (62.2KB, xlsx)
A list of differentially expressed genes identified by the comparison of BMP- and LIF-induced astrocytes. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 383 genes are differentially expressed. 257 genes are expressed more highly in BMP-induced astrocytes compared to LIF-induced astrocytes. Conversely, 124 genes are expressed more highly in LIF-induced astrocytes compared to BMP-induced astrocytes. Download Figure 1-4, XLSX file (34.8KB, xlsx)
A list of differentially expressed genes identified by the comparison of LIF-induced astrocytes and neurosphere. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 69 genes are differentially expressed. 57 genes are expressed more highly in LIF-induced astrocytes compared to neurospheres. Conversely, 12 genes are expressed more highly in neurospheres compared to LIF-induced astrocytes. Download Figure 1-5, XLSX file (16.5KB, xlsx)
BMP4-regulated microarray candidates revealed known and novel astrocyte-enriched genes
Our interest in the diversity among differentiated astrocytes in the cerebral cortex prompted us to focus first on genes upregulated in the BMP4-treated group. To identify astrocyte-enriched genes in this group, we compared 121 genes that were >5-fold higher in the BMP4 group relative to the neurosphere group and cross-referenced them with genes identified as 10-fold enriched in astrocytes compared with neurons and oligodendrocytes (Cahoy et al., 2008). Fourteen genes that fit these criteria were selected (Fig. 1D), and qRT-PCR was performed to independently verify gene-expression changes observed by the microarray (Fig. 1E; data not shown). mRNA expression in vivo was first inspected using the Allen Brain Atlas mouse brain expression database (Lein et al., 2007) and validated by in-house ISH (Fig. 1F,G). Three candidates, homeodomain-only protein (Hopx), high temperature requirement A1 (HtrA1), and F3, show expression patterns that resemble the distribution of astrocytes in the cerebral cortex or in the hippocampus (Fig. 1F,G). These findings suggest that select genes expressed by BMP4-induced astrocytes may identify subpopulations of astrocytes or distinguish astrocytes from NSCs in the adult mouse forebrain.
HtrA1 is expressed in mouse and human astrocytes
We chose to focus on the serine protease HtrA1 for further analysis due to its known roles in TGF-β family signaling and ECM protein organization (Oka et al., 2004; Hadfield et al., 2008; Chamberland et al., 2009). To better determine whether HtrA1 mRNA is localized to astrocytes, we investigated HtrA1 expression in vivo by mRNA ISH. HtrA1 mRNA in the adult mouse forebrain is strongly expressed in astrocytes located in the cerebral cortex and the hippocampus. HtrA1 mRNA is also present in the thalamus and the hypothalamus but its expression there is weaker (Fig. 2A,F). Colocalization with GFAP by IHC found that the majority of HtrA1+ cells in cortical layer 1 express GFAP (86.34 ± 5.42%; Fig. 2H), and that none of the HtrA1+ cells express the neuronal cell marker NeuN (0 of 1961; Fig. 2I). We also did not detect HtrA1 mRNA in oligodendrocytes or their precursors marked by Olig2 expression in the same cortical region during the period of cortical oligodendroglial differentiation (0 of 537; Fig. 2J). In deeper cortical layers, where most astrocytes lack GFAP expression but still express S100β and aldehyde dehydrogenase 1L1 (Aldh1L1), HtrA1 expression is limited to cells with stellate morphology (Fig. 2K, arrowheads) and does not overlap with NeuN or Olig2 (Fig. 2L,M, arrowheads). ISH also revealed a small difference in the level of HtrA1 mRNA expressed in the upper cortex (cortical layers 1–3) versus deeper cortex (cortical layers 4–6; Fig. 2C). This difference in HtrA1 transcript is also observed when comparing mRNA isolated from upper and deeper cortical tissue by qRT-PCR, which concurrently detected differential expression in GFAP but not in Tuj1 as expected (Fig. 2G). However, HtrA1 mRNA is rarely detected in GFAP-expressing white-matter astrocytes in the corpus callosum (Fig. 2C, dotted lines, N, arrowheads, N′), nor is it highly expressed in the medial thalamus or hypothalamus (Fig. 2E,F). These findings suggest that HtrA1 mRNA is differentially expressed by astrocytes in multiple forebrain regions.
Figure 2.

HtrA1 mRNA ISH reveals region and cell type-specific expression. A, Representative expression pattern of HtrA1 mRNA (blue) in P60 adult mouse forebrain (n = 8). Boxes indicate areas of interest in C–F. B, Schematic diagram of HtrA1-expressing cells in the adult mouse forebrain. Red dots represent areas where HtrA1 mRNA is detected. Boxes indicate areas of interest as shown in H–N′. C–F, Higher-magnification views of HtrA1 mRNA expression in dorsal neocortex (C), hippocampus (D), medial thalamus (E), and medial hypothalamus (F). Dotted lines demarcate boundaries between the cortex and corpus callosum in C and the hippocampus and thalamus in D. G, HtrA1 expression comparison between upper and deeper cortex by qRT-PCR shows a small (1.5-fold) but significant difference (p = 0.005, n = 6). GFAP and Tuj1 were used as positive and negative controls for differential expression respectively. H, I, HtrA1 mRNA with GFAP (H) or NeuN (I) immunostaining in cortical layers 1 and 2 of adult mouse cortex demonstrates that HtrA1 is specifically expressed in astrocytes but not neurons (arrowheads). J, HtrA1 mRNA (dark blue) with IF staining of Olig2 (green) in P14 mouse cerebral cortex. No HtrA1 expression was observed in Olig2-expressing oligodendrocyte progenitors and oligodendrocytes (white arrowheads) in the upper layers of the dorsal cortex where HtrA1 is strongly expressed. K, L, HtrA1 mRNA with GFAP (K) or NeuN (L) IHC in cortical layer 4 where HtrA1 mRNA is detected in GFAP-negative cells with stellate morphology (K, arrowheads) and does not colocalize with NeuN (L, arrowheads). M, Similarly, Olig2+ cells also lack HtrA1 expression in deeper cortical layers (white arrowheads). N, N′, HtrA1 mRNA is rarely detected in the corpus callosum (CC) where GFAP is strongly expressed (N, arrowheads). Dotted line demarcates the boundaries for CC in N′. O, O′, HtrA1 mRNA is not detected around the lateral ventricle (O′, white arrowhead), where GFAP-expressing adult NSCs reside in the SVZ (arrowheads). P, P′, HtrA1 mRNA is highly expressed in the dentate gyrus (DG) of the hippocampus. However, expression is sparse in the SGZ, where GFAP-expressing NSCs reside (P, arrowheads). Scale bars: A, 400 μm; C–F, 100 μm; H–N′, 50 μm; O–P′, 100 μm.
More interestingly, we found that HtrA1 mRNA is not expressed in adult mouse brain NSCs of the anterior SVZ (Fig. 2O,O′, arrowheads), making HtrA1 a rare astrocyte marker that distinguishes between postmitotic astrocytes and adult NSCs. It is sparsely distributed in the subgranular zone (SGZ) of the hippocampus (Fig. 2P,P′), unlike transcripts enriched in hippocampal NSCs, such as Hopx (Fig. 1G). Examination of HtrA1 mRNA with GFAP IHC revealed that the great majority of the HtrA1+ cells (92.3 ± 5.4%) in the SGZ do not display radial morphology, a hallmark of hippocampal NSCs. Together, mRNA expression analyses by ISH suggest HtrA1 may be a potential marker of specific populations of differentiated astrocytes in the adult mouse forebrain.
We also examined HtrA1 protein distribution by performing immunocytochemistry (ICC) in GFAP+ cells derived from NSCs differentiated for 7 d in BMP4 or LIF with Noggin. Corroborating the qRT-PCR data, we observed markedly higher levels of HtrA1 protein in BMP4-induced GFAP+ cells compared with LIF with Noggin-induced GFAP+ cells. (Fig. 3A). HtrA1 protein is detected in both cytosol and nuclei of cultured astrocytes, consistent with previous reports of HtrA1 subcellular localization in other cell types (Wang et al., 2012). This expression pattern is eliminated when the antibody was preabsorbed with a HtrA1 peptide (Fig. 3A). Similar protein expression is also detected in FBS-derived primary mouse astrocytes (Fig. 3B), where HtrA1 immunofluorescent (IF) signal diminishes when transitioned into a serum-free media and is upregulated with BMP4 treatment (Fig. 3B,C). Western blot analysis of HtrA1 protein in these conditions also reveals similar expression changes (Fig. 3D). These findings suggest that HtrA1 protein expression is regulated by BMP4 signaling in postnatal astrocytes.
Figure 3.

Characterization of HtrA1 expression in cultured mouse and human astrocytes. A, Representative IF images of P1 NSCs differentiated for 7 DIV in either LIF plus Noggin or BMP4. HtrA1 antibody (green) detected both cytosolic and nuclear signals in both LIF plus Noggin-differentiated and BMP4-differentiated GFAP-expressing cells (red). However, staining signal is significantly stronger in BMP4-induced astrocytes. Elevated HtrA1 immunoreactivity in BMP4-induced astrocytes is eliminated when a HtrA1 peptide is used for blocking the antibody before primary incubation. B, Representative IF images of HtrA1 protein (green) and GFAP (red) detected in primary astrocytes isolated from P2 mouse cortex using standard culturing protocol with 10% FBS (left). Serum withdrawal for 24 h (middle) followed by the addition of BMP4 for 24 h (right) increases the expression level of HtrA1. C, Quantification of HtrA1 IF revealed a significant difference in HtrA1 protein levels between serum-free and BMP4-treated primary astrocytes (n = 3, p = 0.008), A.U., Arbitrary Unit. D, Representative micrograph of Western blots showing HtrA1 protein expression in astrocytes cultured in FBS containing media, serum-free media (SF), or serum-free media with 20 ng/ml BMP4 (SF+BMP4). GAPDH is used as a loading control. D, F, G, Representative IF images of human HtrA1 protein (green, arrows) in NSCs marked by vimentin (D, red), and BMP4-induced or BMP4/FGF1-induced astrocytes marked by GFAP or S100β (F, G, red). Scale bar, 25 μm; *p < 0.05.
To determine whether HtrA1 is also expressed in human astrocytes, we generated astrocytes from human embryonic stem cells and induced pluripotent stem cells using a previously reported differentiation protocol with some modifications (Duan et al., 2015). Briefly, LIF and BMP4 treatments were used to promote astroglial lineage commitment and cell-cycle exit, and further maturation of astrocytes was induced with the addition of FGF1 (Roybon et al., 2013). We did not detect HtrA1 in vimentin-expressing neural progenitor cells before astrocyte differentiation (Fig. 3E). In both BMP4-alone and BMP4/FGF1 conditions, HtrA1 expression is detected in GFAP-expressing and S100β-expressing astrocytes, where it shares similar protein distribution with its mouse ortholog (Fig. 3F,G). qRT-PCR analysis of HtrA1 mRNA in BMP4/FGF1-induced astrocytes and untreated NSC controls also supports the ICC observations (data not shown). These findings provide additional evidence that astrocytic expression of HtrA1 is both responsive to BMP signaling and evolutionarily conserved.
HtrA1-LacZ reporter confirms astrocyte-specific expression of HtrA1 in mouse forebrain
To further investigate cell-type specificity of HtrA1 expression in the mouse brain, we acquired and examined a mouse reporter line that expresses β-galactosidase (β-gal) under the control of endogenous HtrA1 regulatory elements (Brown and Moore, 2012). We found that in the cerebral cortex of adult mice, nearly all (98.7 ± 0.9%) β-gal-expressing cells also express the pan-astrocytic marker Aldh1L1 (Fig. 4A–A″). Conversely, nearly all (97.8 ± 1.14%) Aldh1L1+ cells express β-gal in the adult dorsal cortex, supporting the notion that HtrA1 is expressed in astrocytes. In addition to Aldh1L1, β-gal is also detected in 94.3 ± 3.1% of S100β+ cells (Fig. 4B–B″) and 88.6 ± 4.4% of layer 1 GFAP+ astrocytes (Fig. 4C–C″). Coimmunostaining with antibodies against NeuN (Fig. 4D–D″) and adenomatous polyposis coli (APC; Fig. 4E–E″) shows no β-gal expression in neurons (0 of 4640 NeuN+ cells) and oligodendrocytes (0 of 608 APC+ cells). Examination of β-gal expression in endothelial cells, pericytes, and microglia by coimmunostaining with CD31 (0 pf 523 CD31+ cells; Fig. 4F–F″), PDGFRβ (0 of 403 PDGFRβ+ cells; Fig. 4G–G″), and Iba1 (0 of 420 Iba1+ cells; Fig. 4H–H″) antibodies respectively also show no overlapping expression in the cerebral cortex. In addition, examination of β-gal in areas of adult neurogenesis with nestin costaining confirmed the absence of HtrA1 expression in NSCs, both in the SVZ (Fig. 4I–I″) and the SGZ (Fig. 4J–J″). Together, these findings support our mRNA expression analysis and provide further evidence for HtrA1 as an astrocyte-specific marker in the adult mouse cerebral cortex.
Figure 4.

HtrA1 expression is astrocyte specific. A–H″, Representative micrographs showing expression of HtrA1 β-gal reporter (green) with molecular markers of different neural and non-neural cell types in the dorsal cortex of adult mouse (n = 6, red). β-Gal is found to colocalize with known astrocyte markers, including Aldh1L1 (A–A″), S100β (B–B″), and GFAP (C–C″), where these markers are expressed. β-Gal expression does not colocalize with neuronal marker NeuN (D–D″), oligodendrocyte marker APC (E–E″), endothelial cell marker CD31 (F–F″), pericyte marker PDGFRβ (G–G″), or microglia marker Iba1 (H–H″). I–J″, IF imaging of β-gal (green) in germinal zones of the adult mouse brain did not show any colocalization with NSC marker nestin (red) in the anterior SVZ (I–I″) or the SGZ of the hippocampus (J–J″). DAPI is shown in blue in all panels. Scale bar, 20 μm.
We next sought to determine whether β-gal expression in HtrA1-LacZ mice reflects differential expression of HtrA1 observed at the mRNA level. We examined the level of β-gal expression in different forebrain regions of adult mice, including the cerebral cortex, hippocampus, corpus callosum, and thalamus, and found that not all astrocytes express β-gal at the same levels. Quantification of β-gal IF in cortical layers 1–3 (IF, 2344.57 ± 150.59 A.U.) versus layers 4–6 (IF, 1659.54 ± 99.3 A.U.) shows a significant difference in the integrated signal intensity (Fig. 5A,B,G; p = 0.003), suggesting that HtrA1 expression is differentially regulated in upper and deeper layers of the dorsal neocortex. In contrast, no difference in S100β IF (layers 1–3, 1780.38 ± 69.16 A.U.; layers 4–6, 1662.23 ± 95.39 A.U.; p = 0.223 A.U.) was detected in the same regions (Fig. 5A′,B′,G). GFAP IF is known to be stronger in the upper cortex, and our integrated intensity quantification confirms this finding (layers 1–3 IF, 1825.74 ± 105.76; layers 4–6 IF, 1103.85 ± 39.41; p = 8e-04; Fig. 5A″,B″,G). In addition to the neocortex, all three astrocyte markers were strongly expressed in the lateral dentate gyrus of the hippocampus (Fig. 5C–C″,G). However, β-gal IF was greatly reduced in the corpus callosum (832.99 ± 107.96) whereas GFAP, but not S100β, was strongly expressed (Fig. 5D–D″,G). In the diencephalon, only S100β is consistently expressed in the dorsal thalamus (Fig. 5E′,G) and the hypothalamus (Fig. 5F′,G), where GFAP is strongly expressed in select populations (Fig. 5E″,F″,G) and HtrA1-βGal is expressed at low levels (dorsal thalamus, 1063.4 ± 42.27; hypothalamus, 1232.77 ± 57.83; Fig. 5E–G).
Figure 5.

HtrA1 is differentially expressed by astrocytes in different forebrain regions. A–F″, IF images of β-gal (green), S100 β (red), and GFAP (magenta) expression in the forebrain of adult HtrA1-LacZ reporter mice (n = 3). Examination of multiple brain regions including upper (layers 1–3; A–A″) and deeper (layers 4–6; B–B″) neocortex, corpus callosum (C–C″), dentate gyrus of the hippocampus (D–D″), dorsal thalamus (E–E″), and ventral hypothalamus (F–F″) revealed varying levels of IF in multiple brain regions. G, Quantification of integrated signal density of IF for each protein in each brain region. Comparison of upper and deeper cortical layers reveals a significant difference (p = 0.003) in β-gal expression level, A.U., Arbitrary Unit. H, Quantification of the percentage of Aldh1L1-expressing cells that also express β-gal in different brain regions of adult HtrA1-LacZ reporter mice (n = 3). **p < 0.01. Scale bar, 50 μm.
To further analyze regional differences in HtrA1 expression, we quantified the percentage of Aldh1L1-expressing astrocytes that coexpress HtrA1 in the above-mentioned brain regions independent of expression levels. Consistent with the signal intensity analysis, we found that dorsal brain regions, including the hippocampus (99.35 ± 0.65%), cortical layers 1–3 (97.51 ± 0.86%), and cortical layers 4–6 (94.09 ± 3.24%), show a higher percentage of Aldh1L1+ cells expressing HtrA1 compared with ventral brain regions, including the thalamus (42.92 ± 6.58%) and hypothalamus (28.71 ± 2.67%; Fig. 5H). Similarly, cell counts in the corpus callosum also revealed low numbers of Aldh1L1+ astrocytes expressing HtrA1 (6.63 ± 3.19%; Fig. 5H). Similar results were obtained for the percentage of S100β cells that express HtrA1 in different forebrain regions (data not shown). Although there are some minor discrepancies between β-gal expression and ISH findings, possibly due to differences in β-gal protein and HtrA1 mRNA stability, the HtrA1-LacZ reporter analyses support our previous conclusion that HtrA1 is differentially expressed by astrocytes in different regions of the adult mouse forebrain. Furthermore, HtrA1 expression does not mirror expression of known astrocyte markers GFAP and S100β, and the combinatorial expression of the three markers may be used to identify subpopulations of forebrain astrocytes.
HtrA1 expression in NSCs inhibits postnatal astrogliogenesis
Developmentally, we observed HtrA1 mRNA first at embryonic day 14 enriched at the cortical hem (Fig. 6A), the most dorsal neurogenic region that receives high levels of BMP signaling (Furuta et al., 1997). Weaker signal is also detected in the ventral forebrain, which may be neuronal in origin (Launay et al., 2008). At P0, HtrA1 expression is found most strongly in the dorsolateral SVZ and weakly in the dorsal cortex (Fig. 6B). As more astrocytes populate the neocortex, the number of cortical HtrA1+ cells increases from P7 to P14 and the expression in the SVZ is lost (Fig. 6C,D). The evolving expression profile of HtrA1 during the perinatal period correlates well with the timeline of astrogliogenesis, leading us to ask whether HtrA1 has a role in astrocyte differentiation. We harvested and cultured postnatal NSCs from HtrA1-deleted mice and littermate controls, and differentiated the cells for 3 and 7 d in vitro (DIV) in the presence or the absence of BMP4 or Noggin. We first quantified the number of GFAP-expressing, nestin-expressing, or EdU-expressing cells in NSCs differentiated for 3 DIV without any cytokines and found a significant increase in the number of GFAP-expressing cells (Fig. 6E,E′,H; control, 14.3 ± 2.15%; mutant, 28.39 ± 1.97%; p = 0.0038) and a reduction in nestin-expressing cells when HtrA1 is ablated (Fig. 6E,E′,I; control, 69.64 ± 3.55%; mutant, 42.04 ± 8.16%; p = 0.037). In addition, the numbers of total EdU+ (control, 19.79 ± 3.9%; mutant, 4.99 ± 3%; p = 0.02) and nestin+EdU+ (control, 19.41 ± 3.89%; mutant, 4.28 ± 2.41%; p = 0.038) proliferating cells were also significantly reduced (Fig. 6J). Addition of BMP4 abolished the differences between control and mutant samples (Fig. 6F,F′,H–J), suggesting that sufficiently high levels of BMP4 overcome the inhibitory effects of HtrA1 on astrocyte differentiation. Conversely, blocking BMP signaling with Noggin prevented the increase of GFAP+ cells and the decrease of nestin+ stem cells observed in HtrA1 mutants (Fig. 6G,G′,H–J), suggesting that HtrA1 mediates astrocyte specification at least partly through inhibition of BMP signaling. Together, these findings suggest that HtrA1 expression in perinatal NSCs negatively regulates NSC differentiation into GFAP+ astrocytes.
Figure 6.

Deletion of HtrA1 in postnatal NSCs promotes astrocytic differentiation and maturation. A–D, Representative micrograph of HtrA1 mRNA expression by ISH during mouse forebrain development at embryonic day 14 (A) and at P0, P7, and P14 (B–D; n = 4 per age). E–G′, Representative micrographs of HtrA1 intact (HtrA1+/+; E–G; n = 5) or deleted (HtrA1−/−; E′, F′, G′; n = 4) NSCs after 3 DIV differentiation. IF stainings of GFAP (green), nestin (red), and EdU (white) illustrate that GFAP+ astrocytes are significantly increased while nestin+ NSCs and EdU+ cells are significantly reduced in HtrA1−/− mice. H–J, Quantification of the number of GFAP+ (H), nestin+ (I), and EdU+ (J) cells as a percentage of total DAPI+ cells at 3 DIV in NSC cultures from HtrA1+/+ and HtrA1−/− mice. Scale bars: A–D, 200 μm; E–G′, 50 μm. *p < 0.05.
To further validate these findings in vivo, we investigated whether HtrA1 deletion also alters astrogliogenesis in the postnatal mouse brain. Since GFAP only labels a subpopulation of astrocytes in the developing and adult mouse brain, we examined the number of Aldh1L1-expressing (Aldh1L1+) astrocytes in both the dorsal neocortex and the CA1 region of hippocampal primordium at P1. To identify dividing astrocyte progenitors, EdU was administered 4 h before death and the number of Aldh1L1+EdU+ cells was quantified. In both brain regions, more Aldh1L1+ astrocytes were found in HtrA1 mutants compared with the wild-type controls (Fig. 7A–B′, arrows, C). Interestingly, we also observed a significant increase in the number of Aldh1L1+EdU+ cells in the same brain regions (Fig. 7A–B′, arrowheads, D), supporting the idea that HtrA1 may regulate astrocyte differentiation through astrocyte precursor expansion that occurs away from germinal zones (Ge et al., 2012). However, no significant differences were observed at P7 in the total number of Aldh1L1+ astrocytes between HtrA1-null mice and the wild-type controls in either the cortex or the hippocampus (Fig. 7E–F′,G), suggesting that loss of HtrA1 does not lead to long-term changes in the total number of astrocytes. Quantification of Aldh1L1+EdU+ at P7 revealed significant reduction in cortical astrocyte precursors in HtrA1 mutants (Fig. 7E–F′, arrowheads, H), suggesting that earlier increases in precursor divisions may result in premature termination of cortical astrocyte generation. Together, these findings suggest that HtrA1 may regulate the tempo of cortical astrocyte differentiation in the postnatal forebrain.
Figure 7.

In vivo analysis of astrocyte differentiation in HtrA1-deleted mice reveal increase in astrogliogenesis at P1. A–B′, Representative IF staining of Aldh1L1 (green) and EdU (magenta) in P1 brains of HtrA1+/+ (n = 3) and HtrA1−/− (n = 5) mice. Areas of analysis include the dorsomedial neocortex (A, A′) and the CA1 region of the hippocampal primordium (B, B′; dotted line demarcates CA1 and dentate gyrus). Aldh1L1+ cells (arrows) and Aldh1L1+EdU+ precursors (arrowheads) are both significantly increased in both regions in HtrA1−/− mice. C, Quantification of Aldh1L1+ cells in the neocortex and the CA1 region of the hippocampus at P1. D, Quantification of Aldh1L1+EdU+ in the neocortex and the CA1 region of the hippocampus. A significant increase in both dividing and nondividing Aldh1L1+ cells were detected in HtrA1−/− mice. E–F′, Images of the neocortex (E, E′) and hippocampus (F, F′) stained with Aldh1L1 (green) and EdU (magenta) at P7 in HtrA1+/+ (n = 3) and HtrA1−/− (n = 3) mice. G, H, Quantification of Aldh1L1+ and Aldh1L1+EdU+ cells in P7 neocortex and hippocampus. A reduction in Aldh1L1+EdU+ cells (arrowheads) in the neocortex of P7 HtrA1−/− mice was observed. All data presented as mean ± SEM. Statistical significance was measured by unpaired Student's t test. Scale bar, 50 μm. *p < 0.05.
HtrA1 alters astrocyte morphology and ECM protein expression
Since the expression of HtrA1 is heterogeneous among astrocytes in different brain regions, we next used an established cultured astrocyte model to discover whether HtrA1 plays any role in astrocyte physiology and function. We first generated a GFP-tagged lentivirus expressing an shRNA to specifically knockdown HtrA1, and validated the reduction of protein production by Western blots (Fig. 8A). We found that HtrA1 expression is clearly detected in cultured astrocytes derived and maintained in media containing FBS, and the level of expression is reduced significantly when transduced with shRNA-expressing lentivirus (Fig. 8A). We transduced cultured mouse cortical astrocytes with shRNA-expressing lentivirus and examined their morphology at 7 d and the effects on neurite outgrowth in cortical neuron cocultures at 14 d. Morphologically, we found that astrocytes with reduced HtrA1 exhibited more stellate GFAP-expressing processes compared with control astrocytes, which acquired a more fibroblast-like morphology as expected from serum-containing astrocyte growth conditions (Fig. 8B). Quantification of cell size, shape variance, and process number validated this observation (Fig. 8C). When HtrA1-knockdown astrocytes were cultured with cortical neurons, reduced neurite length (40.1 ± 4.1 μm) was observed compared with neurons cultured with control astrocytes (59.5 ± 4.7 μm, p = 0.0145; Fig. 8B,D). These findings suggest that HtrA1 in astrocytes regulates cellular morphology and modulates neuronal–glial interactions.
Figure 8.

Loss of HtrA1 leads to morphological, biochemical, and functional changes in astrocytes. A, Validation of HtrA1 shRNA lentivirus efficiency by Western blot analysis. B, Representative micrograph of astrocyte (GFAP; top, red) and cortical neuron (βIII tubulin; bottom) coculture studies with GFP-tagged scrambled or HtrA1 shRNAs (top, green). C, Quantification of morphological changes of HtrA1 knockdown (HtrA1 KD, n = 179) and control (scrambled, n = 78) cultured astrocytes. Significant changes were observed in compactness (p = 0.008), cell area (p = 0.042), and process number (p = 3.4E-6). D, Quantification of cortical neurite length under culture conditions without an astrocyte monolayer (No AML) or with astrocytes transduced with scrambled (Control AML) or HtrA1 shRNA (HtrA1 KD AML). Significant reduction of neurite outgrowth (p = 0.0145) was observed when HtrA1 expression is reduced in astrocytes (n = 4). E, Western blot for neurocan in cultured astrocytes transduced with either scrambled control, HtrA1 shRNA #1, or HtrA1 shRNA #1 and #2 (n = 3). Lower panel graph represents quantification of relative signal intensity to the scrambled shRNA control condition, demonstrating that HtrA1-deficient astrocytes express significantly higher neurocan (p = 0.003). F, IF staining of GFAP-expressing (green) and Aldh1L1-expressing (red) astrocytes in cortical layer 1 of 6-month-old wild-type control (n = 4) and HtrA1-null mutant (n = 5) mice. G, Quantifications of GFAP and CSPG signal intensity, GFAP-expressing cells, and Aldh1L1-expressing cells in cortical layer 1 of HtrA1 control and mutant mice. H, IF staining of CSPG (green, CS-56 antibody) and collagen IV (red) in HtrA1 mutant mice shows an increase in CSPG expression while collagen IV expression remains unchanged. I, Western blots of BMP4 and TGF-β from HtrA1+/+ and HtrA1−/− astrocytes after 7 d of differentiation. Two bands were detected that correspond to the preprocessing form (∼200 kDa for BMP4; 150 kDa for TGF-β) and the mature dimer (∼65 kDa for BMP4; 50 kDa for TGF-β). Deletion of HtrA1 led to a near twofold but not statistically significant increase in pro-BMP4 and mature BMP4, while pro-TGF-β and mature TGF-β are significantly increased and reduced respectively. All data presented as mean ± SEM. Statistical significance measured by one-way ANOVA with Tukey's post hoc test for A, D, and E, and Student's t test for C, G, and H. Scale bar: B, 25 μm; F, H, 50 μm; *p < 0.05; **p < 0.01.
Previous studies on the function of HtrA1 as a protease have reported the cleavage of multiple ECM proteins by HtrA1, including members of the CSPG family (Chamberland et al., 2009). Elevated expression of CSPGs after injury is known to inhibit neurite outgrowth in the CNS (Inatani et al., 2001; Jones et al., 2003), which led us to investigate whether changes in CSPG expression contribute to the reduced cortical neurite length observed in cocultures with HtrA1-reduced astrocytes. We examined the protein expression of the CNS-enriched CSPG neurocan in cultured astrocytes with and without HtrA1 knockdown, and found that the level of neurocan protein is significantly increased in astrocytes with reduced HtrA1 (251.67 ± 8.01% compared with control, p = 0.00013; Fig. 8E). Conversely, incubating cultured wild-type astrocytes with recombinant HtrA1 protein leads to a reduction in the level of CSPGs, detectable by either pan-CSPG or neurocan-specific antibodies (data not shown). These findings support the idea that altered ECM, including increased levels of CSPGs, such as neurocan, may contribute to inhibition of neurite outgrowth observed in coculture with HtrA1-deficient astrocytes.
We next sought to validate our in vitro findings by examining astrocytes in adult HtrA1 mutant mice, focusing on the GFAP+ cells in layer 1 of the dorsomedial cortex. At 6 months, HtrA1 mutant mice show increased GFAP intensity (control, 754.3 ± 91.79; mutant, 1124.05 ± 77.2) and an enlarged, hypertrophic cellular morphology (Fig. 8F,G). However, quantification of GFAP-expressing cells in layer 1 revealed similar numbers in HtrA1 mutants and control mice (Fig. 8G). To distinguish the role of HtrA1 in astrocyte differentiation from its role in regulation of GFAP expression, we examined the same brain region for changes in the expression of the pan-astrocytic marker Aldh1L1 and found no significant difference between HtrA1 mutant and control mice (Fig. 8F,G). Similar to our findings in vitro, examination of CSPG expression in HtrA1−/− cortical layer 1 revealed an increase in signal intensity (Fig. 8G,H; control, 989.32 ± 60.9; mutant, 1290.04 ± 54.45), while the basement membrane-enriched collagen IV expression surrounding the vasculature appeared unaffected (Fig. 8H). Finally, since HtrA1 has been shown to regulate expression of TGFβ family proteins in other organs (Oka et al., 2004), we asked whether astrocytes lacking HtrA1 exhibit changes in both BMP4 and TGFβ protein expression. Western blot analyses using protein harvested from HtrA1-null cultured astrocytes revealed significant increases in both BMP4 and TGFβ precursor proteins (Fig. 8I; pro-BMP4 and pro-TGFβ), suggesting that HtrA1 participates in the cleavage of the immature forms of BMP4 and TGFβ proteins. Interestingly, we observed a marked reduction in the mature TGFβ but a trend toward increased levels of mature BMP4 in HtrA1-deleted astrocytes (Fig. 8I), suggesting that HtrA1 may differentially regulate the levels of mature BMP and TGFβ family members. These observations suggest that HtrA1 may be functionally important for the regulation of ECM and TGFβ family proteins in astrocytes.
Since mutation in HtrA1 in humans is linked to cerebral vascular disease CARASIL, we asked whether loss of HtrA1 changes the overall morphology and distribution of the cerebral vasculature in 6-month-old adult mouse brains. Despite a significant increase in GFAP immunoreactivity in the HtrA1 mutant cortex (Fig. 9A,B), no change in the overall expression pattern and signal density of CD31, an endothelial cell marker, was observed in the dorsal cortex (Fig. 9A,C). There were no changes in distribution or density of the pericyte marker PDGFR-β (Fig. 9A,D). Based on CD31 expression, we next performed a series quantitative analyses of cerebral vasculature properties that included vessel density (Fig. 9E, vessel percentage area), vessel branching frequency (Fig. 9E, junctions per area), number of vessel endpoints (Fig. 9E, endpoints per area), and total and average vessel length, as well as vessel pattern uniformity (Fig. 9E, lacunarity). We did not observe significant differences between controls and HtrA1 mutants in any of these measures. Last, to address whether increased GFAP immunoreactivity in the HtrA1 mutant cortex is the result of altered neuroinflammatory responses, we examined the morphology and number of microglia by CD11b and Iba1 IHC. Again, no differences were detected between HtrA1 mutants and controls (Fig. 9F). These findings suggest that the overall distribution and morphology of the neurovascular and neuroimmune cells were not significantly affected by the loss of HtrA1 in young-adult mice.
Figure 9.
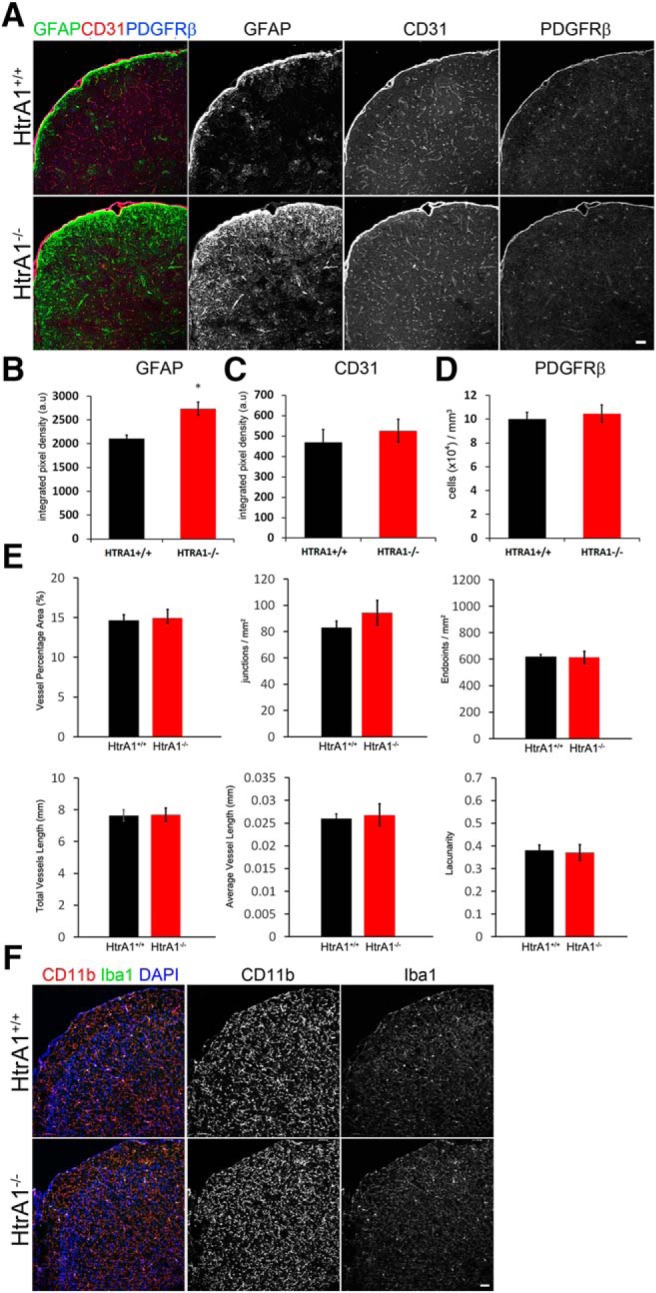
Deletion of HtrA1 does not alter vascular or immune cell morphology or distribution in the young-adult mouse neocortex. A, Representative micrographs illustrating the overall distribution and density of neurovascular unit components marked by GFAP (green), CD31 (red), and PDGFRβ (blue) in 6-month-old HtrA1 control (n = 6) and null mutant cortex (n = 4). B–D, Graphs showing the integrated density quantification of astrocytes (B; GFAP+ cells) and endothelial cells (C; CD31+ cells), as well as pericyte numbers (D; PDGFRβ+ cells) in HtrA1 control and null mutant cortex. E, Quantification of blood vessel properties in the cerebral cortex of 6-month-old adult HtrA1+/+ and HtrA1−/− mice. CD31 IHC signal was used to measure vessel properties listed on the y-axis of each graph. No significant differences were detected between HtrA1 mutant (n = 6) and control (n = 4) mice. F, Representative images of microglia markers CD11b (red) and Iba1 (green) in HtrA1 control and mutant mouse cortex. No significant differences were observed, suggesting HtrA1 deletion does not induce inflammatory responses in the uninjured adult mouse forebrain. Scale bar, 50 μm. *p ≤ 0.05.
HtrA1 in reactive astrogliosis regulates lesion area size and proliferation after injury
The morphology of HtrA1-deficient astrocytes is reminiscent of reactive astrocytes that surround lesions after injury, leading us to investigate whether HtrA1 is involved in the astrocytic response to injury. We first performed cortical stab wound injury in the dorsal-medial cortex of wild-type mice, and examined the expression of HtrA1 by mRNA ISH at 3 d after injury. We found that reactive astrocytes surrounding the lesion area expressed high levels of HtrA1 (Fig. 10A), including deeper layer astrocytes that normally have low GFAP and HtrA1 expression (Fig. 10A, inset). The change in HtrA1 expression was accompanied by elevated BMP4 expression, which was strongly expressed by cells surrounding the lesion after injury, in contrast to the gradient in uninjured controls (Fig. 10B). Western blot analysis of injured cultured astrocytes also confirmed increased levels of HtrA1 protein, along with increased levels of neurocan protein (Fig. 10C).
Figure 10.

HtrA1 expression in reactive astrocytes regulates lesion size and proliferation after injury. A, Main panels illustrate ISH detection of HtrA1 mRNA (without GFAP IHC) in P60 wild-type control (left, n = 6) and injured (right, n = 6) mice, which revealed an increase in HtrA1 expression surrounding the injury site 3 d after cortical stab wound injury. Insets shows HtrA1 mRNA in cortical layer 4 where it is detected in low GFAP-expressing astrocytes in the control brain but becomes strongly expressed in high GFAP-expressing reactive astrocytes in the injured brain. B, ISH of BMP4 mRNA in sections adjacent to A. The increase in HtrA1 in reactive astrocytes correlates with an increase in BMP4 surrounding the lesion. C, Western blot of HtrA1 and neurocan (Ncan) protein in cultured cortical astrocytes with (+, n = 3) and without injury (−, n = 3). An increase in both HtrA1 and neurocan was found in injured astrocytes relative to the control astrocytes. D, Representative micrographs of HtrA1 mRNA and GFAP protein expression in GFAP-Cre; BMPR1a1fx/− mutant mice without (n = 5) and with (n = 4) cortical stab wound injury. Deletion of BMP receptor 1a in astrocytes diminishes layer 1 HtrA1 expression but fails to reduce HtrA1 expression in reactive astrocytes after injury. E, IF colabeling of GFAP (green), CD31 (red), and EdU (blue) in the lesion area of HtrA1 wild-type (n = 4) and null mutant mice (n = 3). Uninjured brains do not show significant differences in cerebral vasculature between wild-type and HtrA1-deleted neocortex. F, Quantification of lesion area size in HtrA1 wild-type and null mutant mice. G, Quantification of total proliferating cells (EdU+), proliferating endothelial cells (CD31+EdU+), and proliferating astrocytes (GFAP+EdU+) in the lesion area of HtrA1 wild-type and null mutant mice. H, Representative images of EdU-incorporating microglia (Iba1), fibroblasts (FN), pericytes (PDGFR-β), and oligodendrocytes (CNPase) in injured HtrA1+/+ and HtrA1−/− mice. I, Quantification of EdU+ cell types presented in J. All data presented as mean ± SEM. Statistical significance was measured by unpaired Student's t test. Scale bar, 50 μm. *p < 0.05; ***p < 0.001.
We next asked whether BMP signaling is necessary for HtrA1 expression in normal and injured astrocytes in vivo. We examined HtrA1 expression in mice lacking BMP receptor 1a (BMPR1a) in GFAP-expressing cells (GFAP-Cre; BMPR1afx/−), and observed a loss of HtrA1 expression in GFAP-expressing astrocytes in cortical layer 1 (Fig. 10D, inset). However, deletion of BMPR1a did not alter HtrA1 upregulation in reactive astrocytes (Fig. 10D, inset), suggesting that molecular signals other than BMPs contribute to increased HtrA1 expression after injury.
Since astrocytic responses after injury are critical for blood–brain barrier repair and lesion reduction, we next asked whether HtrA1 ablation affects the lesion area size or vascular repair at 3 d after injury (Fig. 10E–G). Measurement of the lesion area surrounded by GFAP immunostaining revealed a significant increase in HtrA1 mutants compared with the controls (Fig. 10E,F; control, 118,943 ± 18,953 μm3; mutant, 260,831 ± 13,745 μm3; p = 8.7E-7). The number of EdU+ dividing cells near the lesion area was also significantly increased (Fig. 10E,G; control, 30.46 ± 6.62; mutant, 224.95 ± 30.9; p = 2.4E-7). Colabeling with EdU, CD31, and GFAP revealed that proliferation was increased in endothelial cells (Fig. 10G; control, 18.19 ± 4.79; mutant, 69.22 ± 9.04; p = 0.02) but not in GFAP+ astrocytes (Fig. 10G; control, 0.40 ± 0.47; mutant, 6.61 ± 2.26; p = 0.08). Further analysis of EdU-incorporating cells surrounding the lesion revealed that Iba1-expressing microglia constitute most EdU+ cells, indicating that HtrA1 deletion leads to elevated inflammatory responses (Fig. 10H,I). These findings suggest that HtrA1 expression in astrocytes regulates brain injury recovery by limiting vascular and immune cell proliferation as well as controlling lesion size.
Discussion
One of the goals of this study was to identify novel molecular markers of differentiated astrocytes. A common complication associated with studying astrocyte differentiation and diversity is that classic astroglial markers, such as GFAP and astrocyte-specific glutamate transporter (GLAST), are also expressed by adult NSCs. S100β and glutamate synthetase, two well accepted astrocyte markers not found in NSCs, are expressed in ependymal cells, neurons, and oligodendrocytes in the CNS (Vives et al., 2003; Carlén et al., 2009; Bernstein et al., 2014). Even Aldh1L1, a more recent addition to the list of pan-astrocytic markers, is also found in both differentiated astrocytes and postnatal NSCs (Foo and Dougherty, 2013). In short, the identification of differentiated astrocytes requires a combination of currently existing markers. We have demonstrated here that HtrA1 is expressed by mature astrocytes but not by adult NSCs. This unique property will allow future investigations to distinguish two functionally distinct cell types that share significant molecular similarities in the adult mouse brain.
We have also demonstrated that HtrA1 mRNA is not found in neurons or cells in the oligodendroglial lineage, and coexpression analysis of additional cell-type markers in HtrA1-LacZ mice validated this finding and confirms astrocyte specificity of HtrA1 expression. Two recent RNAseq studies that examined neural and non-neural cell types in the mouse brain identified HtrA1 as astrocyte enriched (Zhang et al., 2014; Zeisel et al., 2015). However, one of the studies suggested that HtrA1 may also be expressed at low levels in oligodendrocyte precursors (OPCs) and in endothelial cells during postnatal development (Zhang et al., 2014). Our HtrA1 mRNA in situ with OPC marker Olig2 at P14 did not show any Olig2+ cells expressing HtrA1, and we did not detect β-gal expression in endothelial cells in the adult neocortex. This discrepancy may be due to detectability of expression in our method or limitations in cell-type isolation in the published report. Regardless, the ability to identify select astrocyte populations will be beneficial in studying astrocyte diversity, particularly when combined with a pan-astrocyte-specific marker, such as Aldh1L1, or with other existing astrocyte subtype markers, such as GFAP.
Based on combinatorial expression of HtrA1, S100β, and GFAP, we observed astrocytes that are HtrA1hiGFAPhiS100βhi (cortical layers 1–3 and hippocampus), HtrA1lowGFAPlowS100βhi (cortical layers 4–6), HtrA1lowGFAPhiS100βlow (corpus callosum), and HtrA1lowGFAPlowS100βhi (dorsal thalamus). Whether these molecularly distinct astrocytes have a unique developmental origin is an intriguing question. Transgenic and adenovirus-based fate-mapping of perinatal NSCs from different forebrain regions has demonstrated contributions of astrocytes from regionally specific progenitors in a radial fashion (Tsai et al., 2012). The broad nature of HtrA1 expression suggests that multiple regional progenitors can give rise to HtrA1+ astrocytes, and that heterogeneity of HtrA1 expression may reflect differences in local signals. The increase in expression in deeper-layer astrocytes after injury suggests that HtrA1 can be modulated by changes in the local environment. Another example of such a phenomenon is the expression of Gli1 in astrocytes of the deeper cortical layers. Ablation of Sonic hedgehog (Shh) signaling from neighboring neurons significantly reduces Gli1 expression in deep-layer astrocytes without affecting total astrocyte numbers (Garcia et al., 2010). In view of the antagonistic roles of Shh and BMP during neuronal specification (Jessell, 2000), it is also intriguing that there is a reciprocal pattern of expression between BMP-regulated HtrA1 and Shh-regulated Gli1 in cortical astrocytes. Future studies that examine how Shh signaling affects HtrA1 expression may provide new insights into the establishment of cortical astrocyte diversity.
HtrA1 was also selected for further analysis because of its known inhibitory role in TGF-β family protein signaling (Oka et al., 2004). Our microarray and qRT-PCR results demonstrated that HtrA1 expression is a target of BMP4 activation, suggesting that increases in HtrA1 expression in astrocytes may act as an autoregulatory response to modulate the levels of BMP and/or TGF-β ligands through proteolysis. The increase of BMP4 in HtrA1-null astrocytes, as well as the loss of HtrA1 in BMPR1a-deleted layer 1 astrocytes, support this hypothesis. The intriguing loss of HtrA1 expression in cortical layer 1 after BMPR1a deletion likely reflects loss of a region-specific source of BMP ligand expression; for example, meningeal fibroblasts express high levels of BMP ligands, including BMP7 (Choe et al., 2014). However, the maintenance of HtrA1 expression in deeper cortical layer reactive astrocytes in BMPR1a-deficient mice suggests that BMPs are not the sole regulator of HtrA1 expression. TGF-β proteins, which are also known cleavage targets of HtrA1, were shown to be elevated in HtrA1-null astrocytes in our study. In many systems, BMP4 and TGF-β signaling are often viewed as antagonistic in nature (Zeisberg et al., 2003; Zode et al., 2009; Oshimori and Fuchs, 2012). How HtrA1 is regulated by these cytokines and vice versa in different cellular contexts is central to elucidating the role of HtrA1 in astrocytes.
HtrA1 also is known for its cleavage of ECM proteins (Chamberland et al., 2009; Jones et al., 2011). This is consistent with our observation of increased neurocan expression in HtrA1-knockdown astrocytes, suggesting that astrocytic expression of HtrA1 inhibits neurocan accumulation in astrocytes. Neurocan is also a known target of TGF-β family signaling (Asher et al., 2000; Fuller et al., 2007), making it unclear whether the increase in neurocan levels in HtrA1-deficient astrocytes is a direct effect from reduced ECM protein cleavage or the result of increased BMP signaling. Regardless, coculture studies suggest that astrocyte expression of HtrA1 is permissive for cortical neuron outgrowth, consistent with influences of astrocytic HtrA1 on neighboring cells through ECM modification. This finding also suggests that increased HtrA1 in reactive astrocytes after injury may play a positive role in the regeneration of neuronal processes.
In the context of brain injury, we found that loss of HtrA1 leads to an increase in lesion area size and higher proliferation in CD31+ and Iba1+ cells. As we only detected increased HtrA1 in reactive astrocytes after injury, these findings suggest astrocytic HtrA1 noncell-autonomously modulates the injury milieu. Our ISH of BMP4 and HtrA1 show concomitant increases in mRNA levels 3 d after injury, raising the possibility of astrocytic HtrA1 acting as a negative feedback regulator of elevated TGF-β family proteins associated with the early inflammatory response (Schachtrup et al., 2010; Simon et al., 2017). We hypothesize that in the absence of HtrA1, unchecked levels of TGF-β family proteins in the injury milieu further simulates the immune response and angiogenesis, resulting in increased proliferation of microglia and endothelial cells in injured HtrA1-null mice. Alternatively, ECM modifications necessary for proper neurovascular unit remodeling may be defective in the absence of HtrA1. Due to the feedback-inhibitory relationship between TGF-β/BMP and HtrA1, deletion of HtrA1 likely results in prolonged elevation of TGF-β/BMP signaling in the injury site, which may have long-term beneficial or detrimental effects on the recovery process. Further analyses of injury responses at additional time points following brain injury will be critical in understanding how astrocytic HtrA1 modulates cytokine-mediated and ECM-mediated recovery responses after injury.
Mutation in HtrA1 is linked to CARASIL, a rare small blood vessel disease characterized by endothelial cell mineralization and associated with high risks of stroke and cognitive impairments (Hara et al., 2009). Several studies have hypothesized that the pathology of CARASIL is the result of dysregulation of TGF-β signaling (Hara et al., 2009; Shiga et al., 2011; Beaufort et al., 2014), although a consensus on how HtrA1 modulates TGF-β signaling has yet to be reached. Our study demonstrated that HtrA1 in the adult mouse brain is only expressed by astrocytes and is not detected in endothelial cells or pericytes surrounding the vasculature. We also showed that HtrA1 deletion increases the expression of astrocyte-produced TGF-β family proteins, which is consistent with changes observed in CARASIL patients (Hara et al., 2009). Dysfunction of astrocytic proteins has been previously linked to neurodegenerative diseases with oligodendroglial and neuronal pathology (Marignier et al., 2010; Haidet-Phillips et al., 2011). Our findings raise the possibility that CARASIL may be astrocytic in origin. Our findings suggest deletion of HtrA1 in astrocytes may increase TGF-β levels and altered the ECM environment at the blood–brain barrier, which in turn leads to the vascular pathology. In addition, heterogeneity of HtrA1 expression among forebrain astrocytes may reflect, or result in, differential TGF-β/BMP signaling in different regions. A recent report demonstrated that molecularly distinct astrocyte subtypes respond differently to inflammatory stimuli from microglia, and this differential response can be modulated by TGF-β signaling (Liddelow et al., 2017). Future studies that isolate and examine the interactions of HtrA1-expressing and HtrA1-nonexpressing astrocytes with microglia or vascular endothelial cells will provide insights into functional astrocyte diversity and may contribute to the treatment of brain injury and cerebral vascular diseases.
Footnotes
This work was supported by the National Institute of Health Grant R01 NS 20778 to J.A.K. and National Institutes of Health/National Cancer Institute Training Grant T32 CA080621 to S.V.G. We thank Amy Lu, Murtaza Ahktar, and Joshua Leyenson for their excellent technical assistance, and are grateful for the helpful discussions with Dr. Michael Bonaguidi. We also thank Drs. Nadereh Jafari, Simon Lin, and Pan Du from the Northwestern University Genomics and Bioinformatics Core Facility for their assistance with microarray sample processing and data analysis.
The authors declare no competing financial interests.
References
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW (2000) Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci 20:2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C, Rowitch DH, Wong WH, DePinho RA (2004) Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci U S A 101:8384–8389. 10.1073/pnas.0402140101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batter DK, Kessler JA (1991) Region-specific regulation of preproenkephalin mRNA in cultured astrocytes. Brain Res Mol Brain Res 11:65–69. 10.1016/0169-328X(91)90022-P [DOI] [PubMed] [Google Scholar]
- Beaufort N, Scharrer E, Kremmer E, Lux V, Ehrmann M, Huber R, Houlden H, Werring D, Haffner C, Dichgans M (2014) Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling. Proc Natl Acad Sci U S A 111:16496–16501. 10.1073/pnas.1418087111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Rowitch DH (2017) Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18:31–41. 10.1038/nrn.2016.159 [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Bannier J, Meyer-Lotz G, Steiner J, Keilhoff G, Dobrowolny H, Walter M, Bogerts B (2014) Distribution of immunoreactive glutamine synthetase in the adult human and mouse brain. Qualitative and quantitative observations with special emphasis on extra-astroglial protein localization. J Chem Neuroanat 61–62:33–50. 10.1016/j.jchemneu.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Bonaguidi MA, McGuire T, Hu M, Kan L, Samanta J, Kessler JA (2005) LIF and BMP signaling generate separate and discrete types of GFAP-expressing cells. Development 132:5503–5514. 10.1242/dev.02166 [DOI] [PubMed] [Google Scholar]
- Bond AM, Peng CY, Meyers EA, McGuire T, Ewaleifoh O, Kessler JA (2014) BMP signaling regulates the tempo of adult hippocampal progenitor maturation at multiple stages of the lineage. Stem Cells 32:2201–2214. 10.1002/stem.1688 [DOI] [PubMed] [Google Scholar]
- Brown SD, Moore MW (2012) The international mouse phenotyping consortium: past and future perspectives on mouse phenotyping. Mamm Genome 23:632–640. 10.1007/s00335-012-9427-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Göritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabé-Heider F, Yeung MS, Naldini L, Honjo T, Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisén J (2009) Forebrain ependymal cells are notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci 12:259–267. 10.1038/nn.2268 [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberland A, Wang E, Jones AR, Collins-Racie LA, LaVallie ER, Huang Y, Liu L, Morris EA, Flannery CR, Yang Z (2009) Identification of a novel HtrA1-susceptible cleavage site in human aggrecan: evidence for the involvement of HtrA1 in aggrecan proteolysis in vivo. J Biol Chem 284:27352–27359. 10.1074/jbc.M109.037051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y, Huynh T, Pleasure SJ (2014) Migration of oligodendrocyte progenitor cells is controlled by transforming growth factor beta family proteins during corticogenesis. J Neurosci 34:14973–14983. 10.1523/JNEUROSCI.1156-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvatal A, Pastor A, Mauch M, Sykova E, Kettenmann H (1995) Distinct populations of identified glial cells in the developing rat spinal cord slice: ion channel properties and cell morphology. Eur J Neurosci 7:129–142. 10.1111/j.1460-9568.1995.tb01027.x [DOI] [PubMed] [Google Scholar]
- Davies JE, Pröschel C, Zhang N, Noble M, Mayer-Pröschel M, Davies SJ (2008) Transplanted astrocytes derived from BMP- or CNTF-treated glial-restricted precursors have opposite effects on recovery and allodynia after spinal cord injury. J Biol 7:24. 10.1186/jbiol85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Peng CY, Pan L, Kessler JA (2015) Human pluripotent stem cell-derived radial glia recapitulate developmental events and provide real-time access to cortical neurons and astrocytes. Stem Cells Transl Med 4:437–447. 10.5966/sctm.2014-0137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Dougherty JD (2013) Aldh1L1 is expressed by postnatal neural stem cells in vivo. Glia 61:1533–1541. 10.1002/glia.22539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, Miller RH (2007) Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann Neurol 62:288–300. 10.1002/ana.21179 [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL (1997) Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124:2203–2212. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV (2004) GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7:1233–1241. 10.1038/nn1340 [DOI] [PubMed] [Google Scholar]
- Garcia AD, Petrova R, Eng L, Joyner AL (2010) Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci 30:13597–13608. 10.1523/JNEUROSCI.0830-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY (2012) Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484:376–380. 10.1038/nature10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golgi S. (1885) Sulla fina anatomia degli organi centrali del sistema nervosa (in Italian). Riv Sper Fremiat Med Leg Alienazione Ment 11:51. [Google Scholar]
- Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA (1996) Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron 17:595–606. 10.1016/S0896-6273(00)80193-2 [DOI] [PubMed] [Google Scholar]
- Hadfield KD, Rock CF, Inkson CA, Dallas SL, Sudre L, Wallis GA, Boot-Handford RP, Canfield AE (2008) HtrA1 inhibits mineral deposition by osteoblasts: requirement for the protease and PDZ domains. J Biol Chem 283:5928–5938. 10.1074/jbc.M709299200 [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. 10.1038/nbt.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, Kawata H, Koyama A, Arima K, Takahashi T, Ikeda M, Shiota H, Tamura M, Shimoe Y, Hirayama M, Arisato T, Yanagawa S, Tanaka A, Nakano I, Ikeda S, et al. (2009) Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med 360:1729–1739. 10.1056/NEJMoa0801560 [DOI] [PubMed] [Google Scholar]
- Herrera J, Yang H, Zhang SC, Proschel C, Tresco P, Duncan ID, Luskin M, Mayer-Proschel M (2001) Embryonic-derived glial-restricted precursor cells (GRP cells) can differentiate into astrocytes and oligodendrocytes in vivo. Exp Neurol 171:11–21. 10.1006/exnr.2001.7729 [DOI] [PubMed] [Google Scholar]
- Inatani M, Honjo M, Otori Y, Oohira A, Kido N, Tano Y, Honda Y, Tanihara H (2001) Inhibitory effects of neurocan and phosphacan on neurite outgrowth from retinal ganglion cells in culture. Invest Ophthalmol Vis Sci 42:1930–1938. [PubMed] [Google Scholar]
- Jessell TM. (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet 1:20–29. 10.1038/35049541 [DOI] [PubMed] [Google Scholar]
- Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD (1996) Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev 10:3129–3140. 10.1101/gad.10.24.3129 [DOI] [PubMed] [Google Scholar]
- John Lin CC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, Mohila CA, Ahmed N, Patel AJ, Arenkiel BR, Noebels JL, Creighton CJ, Deneen B (2017) Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20:396–405. 10.1038/nn.4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, Anderson J, Amrita, Fillerup H, McCloskey M, Luo L, Yang Z, Ambati B, Marc R, Oka C, Zhang K, Fu Y (2011) Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U S A 108:14578–14583. 10.1073/pnas.1102853108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182:399–411. 10.1016/S0014-4886(03)00087-6 [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. (2009) Astrocyte heterogeneity or homogeneity? In: Astrocytes in (patho) physiology of the nervous system (Haydon P, Parpura V., eds). Boston, MA: Springer; 10.1007/978-0-387-79492-1_1 [DOI] [Google Scholar]
- Launay S, Maubert E, Lebeurrier N, Tennstaedt A, Campioni M, Docagne F, Gabriel C, Dauphinot L, Potier MC, Ehrmann M, Baldi A, Vivien D (2008) HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ 15:1408–1416. 10.1038/cdd.2008.82 [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillien LE, Sendtner M, Rohrer H, Hughes SM, Raff MC (1988) Type-2 astrocyte development in rat brain cultures is initiated by a CNTF-like protein produced by type-1 astrocytes. Neuron 1:485–494. 10.1016/0896-6273(88)90179-1 [DOI] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker JC, Doody J, Cárcamo J, Harland RM, Massagué J (1996) A human mad protein acting as a BMP-regulated transcriptional activator. Nature 381:620–623. 10.1038/381620a0 [DOI] [PubMed] [Google Scholar]
- Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin-Doyer M, Cavillon G, Rogemond V, Confavreux C, Honnorat J, Giraudon P (2010) Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 133:2578–2591. 10.1093/brain/awq177 [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902. 10.1083/jcb.85.3.890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, Miyazono K, Taga T (1999) Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284:479–482. 10.1126/science.284.5413.479 [DOI] [PubMed] [Google Scholar]
- Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, Tsuchiya A, Ueta Y, Soma A, Kanda H, Matsumoto M, Kawaichi M (2004) HtrA1 serine protease inhibits signaling mediated by tgfbeta family proteins. Development 131:1041–1053. 10.1242/dev.00999 [DOI] [PubMed] [Google Scholar]
- Oshimori N, Fuchs E (2012) Paracrine TGF-beta signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell 10:63–75. 10.1016/j.stem.2011.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Abney ER, Miller RH (1984) Two glial cell lineages diverge prenatally in rat optic nerve. Dev Biol 106:53–60. 10.1016/0012-1606(84)90060-5 [DOI] [PubMed] [Google Scholar]
- Rao MS, Noble M, Mayer-Pröschel M (1998) A tripotential glial precursor cell is present in the developing spinal cord. Proc Natl Acad Sci U S A 95:3996–4001. 10.1073/pnas.95.7.3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards LJ, Kilpatrick TJ, Dutton R, Tan SS, Gearing DP, Bartlett PF, Murphy M (1996) Leukaemia inhibitory factor or related factors promote the differentiation of neuronal and astrocytic precursors within the developing murine spinal cord. Eur J Neurosci 8:291–299. 10.1111/j.1460-9568.1996.tb01213.x [DOI] [PubMed] [Google Scholar]
- Roybon L, Lamas NJ, Garcia AD, Yang EJ, Sattler R, Lewis VJ, Kim YA, Kachel CA, Rothstein JD, Przedborski S, Wichterle H, Henderson CE (2013) Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep 4:1035–1048. 10.1016/j.celrep.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni V, Mukhopadhyay A, Tysseling V, Hebert A, Birch D, Mcguire TL, Stupp SI, Kessler JA (2010) BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J Neurosci 30:1839–1855. 10.1523/JNEUROSCI.4459-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J Neurosci 30:5843–5854. 10.1523/JNEUROSCI.0137-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga A, Nozaki H, Yokoseki A, Nihonmatsu M, Kawata H, Kato T, Koyama A, Arima K, Ikeda M, Katada S, Toyoshima Y, Takahashi H, Tanaka A, Nakano I, Ikeuchi T, Nishizawa M, Onodera O (2011) Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-β1 via cleavage of proTGF-β1. Hum Mol Genet 20:1800–1810. 10.1093/hmg/ddr063 [DOI] [PubMed] [Google Scholar]
- Simon DW, McGeachy MJ, Bayır H, Clark RS, Loane DJ, Kochanek PM (2017) The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13:171–191. 10.1038/nrneurol.2017.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov AA, Wu X, Tsankova NM, Guilfoyle E, McKhann GM 2nd, Goldman JE (2014) Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J Neurosci 34:2285–2298. 10.1523/JNEUROSCI.4037-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G, Pellegrini S (1994) Association and activation of jak-tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 263:92–95. 10.1126/science.8272873 [DOI] [PubMed] [Google Scholar]
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME (2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104:365–376. 10.1016/S0092-8674(01)00224-0 [DOI] [PubMed] [Google Scholar]
- Supanji, Shimomachi M, Hasan MZ, Kawaichi M, Oka C (2013) HtrA1 is induced by oxidative stress and enhances cell senescence through p38 MAPK pathway. Exp Eye Res 112:79–92. 10.1016/j.exer.2013.04.013 [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T (2001) DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell 1:749–758. 10.1016/S1534-5807(01)00101-0 [DOI] [PubMed] [Google Scholar]
- Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SP, Merkle F, Kessaris N, Alvarez-Buylla A, Richardson WD, Rowitch DH (2012) Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 337:358–362. 10.1126/science.1222381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives V, Alonso G, Solal AC, Joubert D, Legraverend C (2003) Visualization of S100B-positive neurons and glia in the central nervous system of EGFP transgenic mice. J Comp Neurol 457:404–419. 10.1002/cne.10552 [DOI] [PubMed] [Google Scholar]
- Wang N, Eckert KA, Zomorrodi AR, Xin P, Pan W, Shearer DA, Weisz J, Maranus CD, Clawson GA (2012) Down-regulation of HtrA1 activates the epithelial-mesenchymal transition and ATM DNA damage response pathways. PloS One 7:e39446. 10.1371/journal.pone.0039446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TH, Lee DY, Gianino SM, Gutmann DH (2009) Microarray analyses reveal regional astrocyte heterogeneity with implications for neurofibromatosis type 1 (NF1)-regulated glial proliferation. Glia 57:1239–1249. 10.1002/glia.20845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968. 10.1038/nm888 [DOI] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347:1138–1142. 10.1126/science.aaa1934 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Schools GP, Kimelberg HK (2006) Development of GLAST(+) astrocytes and NG2(+) glia in rat hippocampus CA1: mature astrocytes are electrophysiologically passive. J Neurophysiol 95:134–143. 10.1152/jn.00570.2005 [DOI] [PubMed] [Google Scholar]
- Zode GS, Clark AF, Wordinger RJ (2009) Bone morphogenetic protein 4 inhibits TGF-beta2 stimulation of extracellular matrix proteins in optic nerve head cells: role of gremlin in ECM modulation. Glia 57:755–766. 10.1002/glia.20803 [DOI] [PubMed] [Google Scholar]
- Zudaire E, Gambardella L, Kurcz C, Vermeren S (2011) A computational tool for quantitative analysis of vascular networks. PloS One 6:e27385. 10.1371/journal.pone.0027385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the number of differentially expressed genes identified by the microarray. 21217 probes that were identified as “present” were compared among experimental groups in pairwise fashion using listed criteria (N1, N2, N3). The number of differential expressed genes (N) was determined for each experimental group, which included genes that were induced (up) and suppressed (down). Download Figure 1-1, XLSX file (10.1KB, xlsx)
Expression of known astrocyte markers in Neurospheres, BMP induced, and LIF induced astrocytes. A summary of gene expression levels for fourteen commonly used astrocytes markers. While every marker is expressed in all three experimental groups, majority of the genes show strongest expression in BMP induced astrocytes. Download Figure 1-2, XLSX file (10.5KB, xlsx)
A list of differentially expressed genes identified by the comparison of BMP-induced astrocytes and neurosphere. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 830 genes are differentially expressed. 455 genes are expressed more highly in BMP-induced astrocytes compared to neurospheres. Conversely, 375 genes are expressed more highly in neurospheres compared to BMP-induced astrocytes. Download Figure 1-3, XLSX file (62.2KB, xlsx)
A list of differentially expressed genes identified by the comparison of BMP- and LIF-induced astrocytes. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 383 genes are differentially expressed. 257 genes are expressed more highly in BMP-induced astrocytes compared to LIF-induced astrocytes. Conversely, 124 genes are expressed more highly in LIF-induced astrocytes compared to BMP-induced astrocytes. Download Figure 1-4, XLSX file (34.8KB, xlsx)
A list of differentially expressed genes identified by the comparison of LIF-induced astrocytes and neurosphere. Genes are ordered based on largest to smallest fold difference. Genes that show less than two-fold change are excluded. A total of 69 genes are differentially expressed. 57 genes are expressed more highly in LIF-induced astrocytes compared to neurospheres. Conversely, 12 genes are expressed more highly in neurospheres compared to LIF-induced astrocytes. Download Figure 1-5, XLSX file (16.5KB, xlsx)