

Abstract
Nucleic acid amplification of biomarkers is increasingly used to monitor microbial activity and assess remedial performance in contaminated aquifers. Previous studies described the use of filtration, elution, and direct isothermal amplification (i.e. no DNA extraction and purification) as a field-able means to quantify Dehalococcoides spp. in groundwater. This study expands previous work with direct loop mediated isothermal amplification (LAMP) for the detection and quantification of Dehalobacter spp. in groundwater. Experiments tested amplification of DNA with and without crude lysis and varying concentrations of humic acid. Three separate field-able methods of biomass concentration with eight aquifer samples were also tested, comparing direct LAMP with traditional DNA extraction and quantitative PCR (qPCR). A new technique was developed where filters were amplified directly within disposable Gene-Z chips. The direct filter amplification (DFA) method eliminated an elution step and provided a detection limit of 102 Dehalobacter cells per 100 mL. LAMP with crudely lysed Dehalobacter had a negligible effect on threshold time and sensitivity compared to lysed samples. The LAMP assay was more resilient than traditional qPCR to humic acid in sample, amplifying with up to 100 mg per L of humic acid per reaction compared to 1 mg per L for qPCR. Of the tested field-able concentrations methods, DFA had the lowest coefficient of variation among Dehalobacter spiked groundwater samples and lowest threshold time indicating high capture efficiency and low inhibition. While demonstrated with Dehalobacter, the DFA method can potentially be used for a number of applications requiring field-able, rapid (<60 min) and highly sensitive quantification of microorganisms in environmental water samples.
Keywords: Gene-Z, Dehalobacter, Real-time loop mediated isothermal amplification, Direct filter amplification, Field-able genetic diagnostics, Point-of-use, Groundwater
1. Introduction
The widespread contamination of groundwater aquifers with chlorinated solvents throughout the US and abroad has resulted in a number of treatment strategies (Moran et al., 2007). Metabolic reductive de-chlorination by Dehalobacter restrictus (Dehalobacter spp.) uniquely position its use for bioremediation of groundwater co-contaminated with tetrachloroethene, trichloroethene and dichloromethane (Grostern and Edwards, 2006a,b). The assessment of remedial performance increasingly relies on enumeration via nucleic acid amplification. Quantitative PCR (qPCR) is routinely used to quantify genes associated with reductive dehalogenation, however, this method can be inaccurate due to inhibition in environmental samples (Koloren et al., 2011; Kreader, 1996; Tsai and Olson, 1992) or target loss during DNA extraction (Findlay et al., 2016) and sample transport. Studies have also described an underestimation of reductive dehalogenase genes measured with qPCR compared to other methods (Matturro and Rossetti, 2015).
As previously reviewed (Craw and Balachandran, 2012), isothermal amplification techniques such as loop mediated isothermal amplification (LAMP) have many field-able and low cost (Plutzer and Karanis, 2009) advantages, including being less susceptible to environmental inhibition compared to qPCR (Koloren et al., 2011). In addition, LAMP analysis is faster than qPCR because amplification can be accomplished without DNA extraction or using a crude lysate (Kostic et al., 2015; Modak et al., 2016). Previous studies demonstrated LAMP assays for quantification of Dehalococcoides spp. in groundwater (Kanitkar et al., 2016).Our studies also examined a field-able means to concentrate bio-mass from groundwater via Sterivex cartridges and direct amplification from filtrate elution on a field-ready Gene-Z device (Stedtfeld et al., 2014). While a detection limit of 10 to 20 copies per reaction was observed for the Dehalococcoides LAMP assay, sensitivity in water samples varied with volume of concentrated water. For example, a detection limit of 105 cells per L was obtained with filtration of 4 L using the filter elution method and 108 cells per L were observed with no filtration. The lack of sensitivity, while suitable for some applications, was due to an elution step to remove concentrated biomass from filters, and the inability of the eluted sample to constitute over 10% of the amplification reaction volume.
For instances requiring higher sensitivity, a previously undescribed method was explored in which filters used to concentrate biomass were placed into reaction wells of disposable Gene-Z chips for direct filter amplification (DFA). By avoiding the elution step, this technique reduced time to results by 10 min and increased sensitivity over 100 fold. LAMP assays targeting the 16S rRNA gene specific to Dehalobacter spp. and the previously identified putative 1,2-DCA reductive dehalogenase gene (rdhA gene) (Grostern and Edwards, 2009) were tested. Experiments were performed to compare direct LAMP with qPCR including: i) spiking reactions with various concentrations of humic acid, ii) spiking multiple groundwater samples with Dehalobacter (from a commercially available mixed culture currently used for bioaugmentation, TCA-20) following a concentration step, and iii) comparing methods for field-able target enrichment followed by direct isothermal amplification or DFA.
2. Materials and methods
2.1. Design of LAMP primers
Three primer sets were initially designed targeting both the 16S rRNA gene (U84497.2) and Dehalobacter reductive dehalogenase genes. Sequences available in public databases were downloaded, aligned, and LAMP assays were designed from consensus sequences using Primer Explorer V4 software. Loop primers were also designed to decrease time to positive amplification (Tt). BLAST analysis was performed to determine coverage of each primer set. Degenerate bases were added to primer sets targeting the rdhA gene, which totaled 16 base variations (Table S1). LAMP primers were also designed from the luciferase (luc) gene to serve as a microbial internal positive control (Hatt et al., 2013). Previously described qPCR assays targeting the rdhA gene (Grostern and Edwards, 2009) and luc gene were compared with the LAMP assays.
2.2. qPCR and LAMP experiments
LAMP experiments were tested under isothermal conditions in conventional vials in the real-time cycler (Chromo4, BioRad) or in the Gene-Z device (Stedtfeld et al., 2012) with disposable chips. LAMP reactions consisted of 1× isothermal amplification buffer (New England Biolabs), 1.4 mM each dNTP (Invitrogen), 0.8 M Betaine solution (Sigma Aldrich), 6 mMMgSO4 (New England Biolabs), 8 U Bst Polymerase 2.0 WarmStart (New England Biolabs), 200 μM SYTO82 Orange Fluorescent Nucleic Acid Stain (ThermoFisher Scientific), template that constitutes 10% of the reaction volume (DNA, crudely lysed, or direct cells), and PCR grade water. All DNA extractions were performed using the PowerWater DNA Isolation Kit (12888-100, MoBio Laboratories, Inc.). For DFA experiments, cells were captured by the filter and thus template was not added to the reaction. All experiments were performed with an isothermal incubation at 63 °C for 60 min with plate reads at one minute intervals in the real-time thermal cycler, and every 16 s in the Gene-Z device. LAMP in the real-time cycler and Gene-Z device had 10 μL and 25 μL reaction volumes, respectively.
QPCR was performed in 25 μL volumes with the following constitutes: 500 nM forward and reverse primers, specified mass of gDNA, and reagents from the Power SYBR Green PCR Master Mix (Life Technologies). Real-time reactions were run using the real-time cycler, which included a 10-min enzyme activation at 95 °C followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. All experiments included a no-template control and triplicate reaction vials.
Experiments to test sensitivity were performed with dilutions of targeted rdhA gene amplicon and gDNA extracted from TCA-20, a mixed dehalogenating culture that contains Dehalobacter spp. among other organisms (ENV-TCA20™, CB&I). The quantity of Dehalobacter cells in TCA-20 was provided by CB&I and verified in house by qPCR of extracted DNA. Amplicons were generated by PCR of the F3 and B3 primers. Triplicate PCR reactions were pooled and purified using the Qiagen PCR purification kit. The mass of amplicon after purification was quantified using Qubit. Amplicons were diluted and tested with their respective primer sets. Triplicate reactions were performed for each experiment with positive and no template controls.
The rdhA gene and 16S rRNA gene assays used for subsequent experiments were selected based on initial sensitivity and specificity tests. Dilutions of gDNA extracted from TCA-20 and purified amplicons were tested with six designed LAMP assays. Experiments were performed in the real time thermal cycler. Primers selected for further analysis (Table S1) amplified down to 20 copies tested with purified amplicon and 100 genomic copies tested with gDNA extracted from TCA-20. Direct LAMP with the luc gene assay amplified in all dilutions down to 5 CFU per reaction. Lower dilutions did not amplify.
Specificity tests on the LAMP assays were performed with gDNA (5 ng) from 12 type strains. The 16S rRNA gene of selected type strains had varying levels of sequence similarity to Dehalobacter (Table S2). Specificity experiments were run in the real-time cycler. For the rdhA gene, gDNA from the non-targeted organisms did not amplify within the 60 minute LAMP reaction. However, the selected 16S rRNA gene assay amplified with gDNA from Syntrophobotulus glycolicus (DSM 8271). Thus, the 16S rRNA gene LAMP assay for Dehalobacter should only be used in parallel with rdhA gene primer as a secondary or redundant verification of presence.
2.3. Direct LAMP and qPCR in groundwater
For the experiments comparing heat lysed versus non-heat lysed Dehalobacter template,1 mL of TCA-20 (3 × 107 cells per mL) was spiked into 99 mL of phosphate buffered saline (PBS) and six dilutions were prepared yielding 3 × 105 to 3 cells per mL. Samples were passed through Sterivex filters (SVGPL10RC, Millipore) to remove extracellular DNA, eluted from filters using 0.9 mL of elution buffer as previously described (Stedtfeld et al., 2014). Briefly, samples were eluted from Sterivex filters by adding an elution buffer, vortexing Sterivex cartridges at minimum speed for 10 min, and collecting elution buffer with enriched biomass using a syringe. Following elution each dilution was separated into two vials and one of each vial was heat treated at 95 °C for 5 min to crudely lyse cells. Amplification reactions were performed in the real time cycler using the selected rdhA gene LAMP primer. Reactions were performed in triplicate with positive and no template controls. Using 1 μL of sample eluted from filters, and assuming 100% capture efficiency using Sterivex cartridges, LAMP reactions were run with dilutions of 3 × 104 to 0.3 cells per reaction.
Experiments included testing various concentrations of humic acid spiked into LAMP and qPCR assays with 500,000 copies of rdhA gene amplicon. In detail, humic acid (53680-10G, Aldrich) was spiked with concentrations of 0, 0.1, 0.3, 1, 3, 10, 30, 100, 300 mg per L. Based on results, subsequent experiments included testing six dilutions of rdhA gene amplicons (5 × 105 to 5 copies per reaction) with and without 30 mg per L of humic acid. Reactions were performed with isothermal conditions in a real time cycler in triplicate with positive and no template controls.
Experiments also examined the influence of seven enriched ground-water samples (from chlorinated solvent impacted sites) on direct LAMP and qPCR. In summary, 250 mL of sample was passed through a Sterivex filters (SVGPL10RC, Millipore) for testing direct amplification, and another 250 mL was concentrated via vacuum through a 47 mm diameter 0.22 μm filter (GSWG047S6, Millipore) for testing with DNA. Genomic DNA was extracted from vacuum filtered samples, and Sterivex filters were eluted as previously described (Stedtfeld et al., 2014). Direct LAMP and qPCR reactions were spiked with 1 μL of TCA-20 (3 × 107 Dehalobacter cells per mL) to yield 3 × 105 cells per reaction; and 0.4 μL of concentrated groundwater. For experiments with DNA, 5 ng of genomic DNA from TCA-20 was spiked into 2 ng of genomic DNA extracted from groundwater samples.
To test sensitivity of the DFA method, prepared dilutions of TCA-20 were spiked into 100 mL of sterile water yielding 104 to 0.1 cells per mL. Three replicate dilutions were passed through individual filters and all three filters along with a no template control were all run on a single Gene-Z chip (described below). The DFA method was performed with polycarbonate 13 mm diameter Isopore hydrophilic membrane filters with 0.45 μm pore size, track-etched black screen filter (HTBP01300, Merck Millipore Ltd.). The filters were placed into autoclaved semi-clear polycarbonate reusable holders with silicone gaskets (EW-29550-40, Cole-Parmer). Holders were attached to autoclaved 140 mL syringes with luer lock connections. Filter and syringe setup were sealed to a Büchner flask with parafilm to create a vacuum that pulled water samples through filters. After all water had passed, filters were removed from the reusable holders and placed into disposable Gene-Z chips. Reaction wells were sealed with clear optical film (MicroAmp, Applied Biosystems). LAMP reaction constitutes were dispensed into chips (300 μL per reaction well) via pipettes and run immediately following filtration. All DFA experiments were run with the LAMP assay targeting the rdhA gene.
For experiments testing various filtration methods, eight additional groundwater samples were collected from remediation sites. TCA-20 (1.25 mL) was spiked into each of five separate 200-mL groundwater samples to yield 2 × 105 Dehalobacter cells per mL. Samples were also spiked with a microbial internal amplification control (MIAC) (Hatt et al., 2013) to yield 2 × 106 CFU MIAC per mL. Cultures of Escherichia coli clones with the luciferase gene (luc gene), used for the MIAC, were provided by Dr. Frank Löffler (University of Tennessee, Knoxville). Cultures of MIAC were grown overnight at 37°CinLB medium amended with 50 mg per mL ampicillin, and counted by dilution plating. Spiked groundwater samples were concentrated using one of five methods including i) no enrichment (Direct), (ii) Sterivex filtration and elution as previously described (Ster.), iii) a two tiered filter train that included a 5 micron filter (SLSV025LS, Millipore) followed by Sterivex filtration and elution as previously described (5 + Ster.), iv) vacuum filtration and DNA extraction from filters (gDNA), and v) direct filter amplification after pre-filtration through 5 micron filter units (5 + DFA). Following enrichment, samples were tested for qPCR in a real time cycler or LAMP on the Gene-Z device. Extracted gDNA was diluted to 5 ng per μL concentrations, and all qPCR and LAMP experiments with non lysed samples or DNA were run with sample constituting 10% of amplification reaction volume.
2.4. Gene-Z device and chips
The previously described Gene-Z device (Stedtfeld et al., 2012) was used for real-time LAMP of Dehalobacter spiked into groundwater samples. Disposable chips were laser etched (3rd Gen 40W laser, Full Spectrum Laser) into 1/16 inch black acrylic sheets (24112-07, Inventables), cleaned with distilled water, soaked in 70% ethanol for 5 min, dried with compressed air, and enclosed on one side using clear optical film (MicroAmp, Applied Biosystems). Primers were dispensed using a pipette, dried at 70 °C for 10 min, enclosed on the top-side using MicroAmp tape, and chips were stored as previously described (Stedtfeld et al., 2015). The chip configuration consisted of eight reaction wells per sample lane, and four sample lanes per chip (i.e. 32 reaction wells per chip, each with 20 μL reaction volume), as previously described (Kanitkar et al., 2016). Primers targeting rdhA gene and 16S rRNA gene were each dispensed into three separate reaction wells per sample lane, and primers targeting the MIAC were dispensed into two reaction wells.
For DFA experiments, 1/16 inch black acrylic plastic was cut to have 4 reaction wells per chip. Wells were cut with 18 mm diameter for placement of 13 mm diameter DFA filters. The size of each well permitted six sensors (LEDs/optical fibers) within the Gene-Z device to monitor reactions in real-time (Fig. 1). Reaction wells had a tear-drop shape on both the entrance and exit for loading with minimal bubble formation (Fig. 1C). DFA chips were designed so that three replicates and one negative control could be tested per chip for a single primer set. Each reaction well had a loading port and air vent that were sealed with clear optical film (MicroAmp, Applied Biosystems) after reaction mixes were dispensed.
Fig. 1.

Pictures of filter apparatus used for DFA: A) placement of 140 mL syringe into Büchner flask, B) close up picture shows reusable filter holder with vacuum pressure retained with parafilm wrapped between the syringe and Büchner flask, and C) disposable Gene-Z chips with 13 mm filters placed into four individual reaction wells, enclosed with optical adhesive, and loaded with LAMP reagents.
Data analysis was performed as previously described (Stedtfeld et al., 2015) to calculate the threshold time (Tt) of LAMP, akin to threshold cycle (Ct). The signal to noise ratio (SNR) was calculated as the signal at a given time minus the median signal at the start of the reaction, divided by the standard deviation of the signal at the start of the reaction. For DFA experiments, the average Tt of all six sensors monitoring a single re-action well was used for analysis. A student t-test was used to test for significant differences between tested methods (p <0.05).
3. Results and discussion
3.1. Direct amplification of Dehalobacter
Heat treatment of Dehalobacter prior to LAMP had no observable influence in Tt or sensitivity (Fig. 2A) compared to direct amplification without lysis. Regardless of lysis, amplification was only observed in the highest tested dilutions (3 × 104 to 3 × 102 cells per reaction) and no amplification was observed in lower dilutions. Results showed no significant difference (p <0.05) in sensitivity or Tt between crudely heat lysed and non-lysed dilutions of Dehalobacter.
Fig. 2.
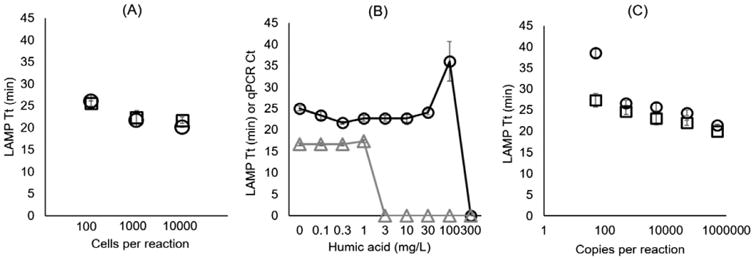
Testing direct amplification and inhibition with rdhA gene LAMP assay. A) Tt for dilution series of TCA-20 tested with heat lysis at 95 °C for 5 min (open circles) and without lysis (open squares). X-axis is theoretical number of cells per reaction after filtering 100 mL of dilutions through Sterivex cartridges (to eliminate extracellular gDNA) and 0.9 mL of elution. B) 500,000 copies of rdhA gene amplicons with LAMP (circles, Tt) and qPCR (triangles, Ct) assays spiked with varying concentrations of humic acid. Points at 0 indicate less than two of three technical replicates amplified with the specified concentration. C) Six dilutions of rdhA gene amplicon with (circles) and without (squares) humic acid spiked at 30 mg per L in the LAMP reaction. Dilutions lower than 50 rdhA gene copies per reaction did not amplify with or without humic acid. Error bars represent standard error of three reaction vials.
The LAMP assays were less susceptible to inhibition by humic acid than qPCR, amplifying with up to 100 mg per L of humic acid per LAMP reaction compared to only 1 mg per L for qPCR (Fig. 2B). Further-more, the LAMP assay did not show a significant change in Tt for humic acid concentrations below 100 mg per L in the reaction. A dilution series of rdhA gene amplicon was also spiked with and without 30 mg per L of humic acid to test for inhibition with lower target concentrations. Results showed that there was a slight shift in Tt, but only with the lowest number of copies showing amplification (50 copies per reaction). All other dilutions had no significant difference between reactions with and without humic acid (Fig. 2C). Dilutions below 50 copies per reaction did not amplify with or without humic acid.
The influence of humic acid on qPCR and LAMP assays targeting the amoA gene was previously reported by Tani et al. (2007). Samples were tested with 0–8 mg per L of humic acid, which had a concentration dependent influence on Tt with turbidity based LAMP. qPCR reactions tested in Tani's study failed to amplify with humic acid concentrations above 4 mg per L. Experiments described here were to examine inhibition with reagents selected/optimized for genetic testing of Dehalobacter with the Gene-Z device.
3.2. qPCR and LAMP in groundwater samples spiked into filtered samples
Direct LAMP of Dehalobacter assays was less influenced by inhibition in groundwater samples (collected from remediation sites) compared to qPCR. In detail,TCA-20 culture was added to seven groundwater samples following elution from Sterivex cartridges, yielding 3 × 105 Dehalobacter cells per reaction. Of note, amplification of the rdhA gene or Dehalobacter specific 16S rRNA gene was not observed in qPCR or LAMP reactions in non-spiked groundwater samples (genomic DNA, or direct cells). Four of the groundwater samples spiked with TCA-20 inhibited qPCR while none of the samples inhibited LAMP to the extent that amplification did not occur (Fig. 3A). Six out of seven spiked groundwater samples had a Tt similar to the LAMP reaction run without groundwater. Sample MI315, which contained the highest concentration of suspended solids (Fig. S2) had a shifted Tt, compared to the other six groundwater samples tested with direct LAMP. Excluding the MI315 sample, the Tt coefficient of variation (CV) was 9% among the seven groundwater samples tested with LAMP rdhA gene and 16S rRNA gene assays. The CV among qPCR-based Ct was also 9% between samples that amplified with qPCR. Subsequent experiments (described in next section) tested the use of a 5 micron filter prior to enrichment and DFA to reduce suspended solids in groundwater samples (e.g. MW315).
Fig.3.
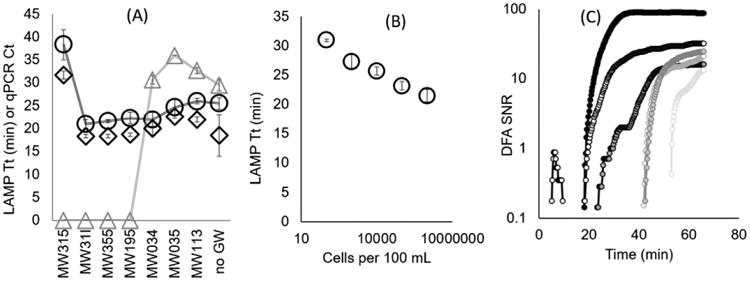
Direct LAMP in groundwater samples and testing DFA method. A) Sterivex enriched groundwater samples spiked with 3 × 105 Dehalobacter cells per reaction from TCA-20 following elution from cartridges; and tested for LAMP assays targeting rdhA gene (circles), 16S rRNA gene (diamond), and qPCR assay targeting rdhA gene (triangles). Points at 0 indicate less than two of three technical replicates amplified with the specified sample. B) Dilutions of TCA-20 spiked into 100 mL of sterile water and tested using the DFA method with the Gene-Z device. C) Signal to noise ratio (SNR) observed throughout a DFA reaction for all six sensors monitoring a single reaction well loaded with a filter used to concentrate 102 cells per 100 mL. This figure demonstrates how amplified product diffuses across the surface of the filter during the reaction. Error bars in (A–B) represent standard error of three technical replicates.
All groundwater samples spiked with gDNA extracted from TCA-20 amplified with qPCR and LAMP assays. Inhibition was not observed, as the Ct and Tt CV between all seven samples was 2%, 3%, and 6% for qPCR (rdhA gene), LAMP (rdhA gene), and LAMP (16S rRNA gene) assays, respectively (Fig. S1).
3.3. Direct filter amplification (DFA) with Gene-Z
For DFA experiments, 100 μL of serially diluted TCA-20 was spiked into 100 mL of sterile distilled water, filtered, and analyzed on the Gene-Z device for the rdhA gene. Amplification was observed down to 102 Dehalobacter cells spiked into 100 mL. Amplification was not observed in controls run without TCA-20 spikes or lower tested dilutions (Fig. 3B). At lower dilutions (e.g. 102 cells spiked into 100 mL), increased fluorescence due to an amplification event was initially observed on one side of the filter, and this fluorescence diffused across the reaction well temporally, varying the Tt among the six sensors used to monitor each reaction well (Fig. 3C). As such, the mean Tt among the six sensing spots was reported for all DFA chips (Figs.3B, 4A, D).While not transparent, filters were translucent allowing a mean value of 13 ± 5% of green LED light to pass through the reaction (captured by the Gene-Z photodiode). Lower fluorescent signals caused by the filters did not influence Tt. More translucent filters that allow for greater transmission of light are currently being explored.
Fig.4.
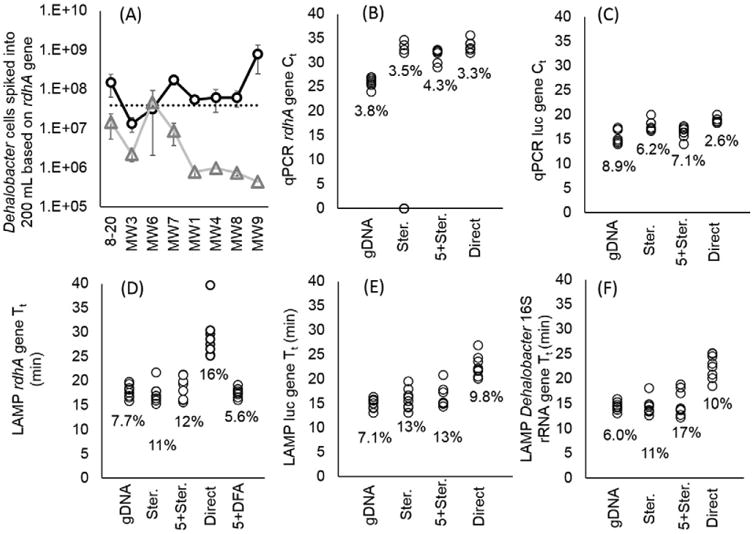
Comparing methods of biomass concentration followed by LAMP on Gene-Z or qPCR in real-time cycler with TCA-20 spiked into 200 mL of eight separate groundwater samples. A) Estimated quantity of Dehalobacter cells based on calibration curves and Tt and Ct of DFA (circle) and qPCR of gDNA (triangle), respectively. The dotted line represents total number of cells spiked into 200 mL of groundwater samples prior to sample processing. Error bars represent standard error of Tt between three replicates. B–F) Graphs showing Tt and Ct of different assays measured with MIAC and TCA-20 spiked into 200 mL of eight separate groundwater samples prior to sample processing including filtration with Sterivex and direct LAMP (Ster.), a filter train with 5 micron filter prior to Sterivex (5 + Ster.), no filtration prior to amplification (Direct), conventional sample preparation using vacuum filtration followed by DNA extraction (gDNA), and direct filter amplification (DFA). Percentage indicates the Tt or Ct coefficient of variation (CV) among the eight groundwater samples.
The DFA method was also tested to enrich for low target concentrations in environmental waters. During our previous study with Sterivex filters, a minimum detection limit of 105 Dehalococcoides cells per L (Stedtfeld et al., 2014) was observed with filtration of 4 L of water. Demonstrated with filtration of 100 mL in this current study, the lowest sensitivity observed with DFA exceeds this 100 fold. While tested with different microbial targets (e.g. Dehalococcoides versus Dehalobacter), LAMP assays for both targets had similar sensitivity (10 to 100 gene copies per reaction depending on the assay tested). Placement of filters directly onto Gene-Z chips also reduced the time required for an elution step (10–15 min), which required an elution buffer stored at 4 °C and a means to vortex the Sterivex cartridges. Thus, DFA may provide a field-able alternative for biomass enrichment of water for genetic diagnostics.
3.4. Sample concentration methods
Five methods of biomass concentration prior to LAMP on the Gene-Z device or qPCR in a conventional real-time cycler were tested in eight groundwater samples spiked with 2 × 106 CFU per mL of MIAC and 2 × 105 Dehalobacter per mL from TCA-20. Since the same amount of bacterial target was added to all eight groundwater samples, the ideal method of sample processing will have a minimum Tt and Ct CV among samples. Overall, the 5 + DFA method tested on the Gene-Z device and gDNA tested with qPCR both had the lowest Tt and Ct CV among the eight different groundwater samples. A CVof 5.6% was measured for DFA and 3.8% for qPCR of gDNA, both tested with the rdhA gene assay (Fig. 4B–F). However, Dehalobacter genomic copies measured with qPCR of gDNA was underestimated in all but one of the groundwater samples (24 ± 40% estimated to actual rdhA gene copies). The DFA method appeared to be less influenced by inhibition since all groundwater had been removed, providing estimates that were closer to the actual amount of spiked Dehalobacter cells. Overestimation was observed in groundwater sample MW9 run with DFA. Compared to all other methods, the DFA method tested with rdhA gene assay provided the lowest Tt, higher estimated yield compared to qPCR, and lowest CV among the eight groundwater samples.
Sterivex with LAMP assays also had lower Tt, however, the higher CV compared to DFA method indicates more variability in biomass loss. In all three LAMP assays, prefiltration with the 5 micron filter slightly increased the average Tt (18.5 ± 2.3 min for the rdhA gene) compared to sole filtration with Sterivex (17.7 ± 0.9 min for the rdhA gene). This indicates a slight loss of Dehalobacter cells using the 5 micron filters. Interestingly, Ct values were lower for qPCR experiments run with the 5 micron pore filter, which we surmise is due to higher level of inhibition in samples without pre-filtration. The level of suspended solids was lower when the 5 micron filtration step was implemented (Fig. S2B, C). Both Ct and Tt values were highest in non-enriched (Direct) samples as biomass was not concentrated.
To our knowledge, no previous study has described a method similar to DFA with a field-able device such as the Gene-Z for remediation applications. A manuscript is also in preparation by our group describing real-time DFA for detection of Legionella pneumophila in water samples, monitored in real-time using a camera and the Gene-Z device (Samhan et al., 2016). A handful of studies have described lab on chip devices in which samples are automatically processed and crude or purified lysate is amplified isothermally (see reviews by Mauk et al., 2015 and Notomi et al., 2015). However, the DFA method is better suited for 1) genetic diagnostics applications that require greater sensitivity when organism/gene concentrations are low, and 2) potential use outside of the laboratory via direct amplification without DNA extraction and purification. The DFA method could also potentially be used in other field-able devices, in which filters are placed directly into amplification vials.
Limitations of the DFA method include the inability to test multiple assays in parallel, which would require a setup for enriching and handling of numerous smaller filters. Used with Gene-Z chips, the current configuration also requires 300 μL per reaction well; however, chips etched into thinner pieces of acrylic would reduce volumes. The smaller 13 mm diameter filter may also have reduced filtration capacity compared to Sterivex and 47 mm filters, thus requiring the prefiltration step with 5 micron syringe filters. For some organisms and targets, direct LAMP amplification without DNA lyses may cause reduction in quantitative capacity or sensitivity. For example, previous studies with Staphylococcus aureus observed a 1 min difference between crudely lysed and non lysed cells (Kostic et al., 2015). Perhaps one of the greatest limitations of LAMP is increased possibility of contamination to subsequent reactions, due to the large concentration of generated amplicons. As such, proper handling of chips and vials is critical to ensure vials or chips remained unopened following anamplification event.
Genetic techniques such as qPCR have proven extremely useful in accessing remedial performance in contaminated sites. The development of LAMP assay targeting Dehalobacter will also allow for specific detection of samples from bioaugmented sites. Minimal sample processing associated with the DFA method can potentially reduce costs and time for detection, and compliments the use of the field-able real-time tools such as the Gene-Z device. The DFA technique facilitates detection of Dehalobacter far below the accepted limit for natural attenuation (107 gene copies per L).
Supplementary Material
Acknowledgments
We would like to thank Simon Vainberg (CB&I) for supplying the TCA-20 culture and groundwater samples, and Dr. Frank Löffler (UTK) for providing cultures of the microbial internal control with the luciferase gene. This research was funded by the United States Department of Defense Strategic Environmental Research and Development Program (SERDP) grant ER-2309. Farag Samhan was supported by the Environmental Protection Agency Great Lakes Restoration Initiative (GL-00E01127-0), and Robert Stedtfeld was partially supported by the Superfund Research Program (2P42 ES004911-22A1) from the National Institute for Environmental Health Sciences.
Footnotes
Appendix A. Supplementary data: Supplementary data to this article can be found online at http://dx. doi.org/10.1016/j.mimet.2016.09.025.
References
- Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip. 2012;12:2469–2486. doi: 10.1039/c2lc40100b. [DOI] [PubMed] [Google Scholar]
- Findlay M, Smoler DF, Fogel S, Mattes TE. Aerobic vinyl chloride metabolism in groundwater microcosms by methanotrophic and etheneotrophic bacteria. Environ Sci Technol. 2016;50:3617–3625. doi: 10.1021/acs.est.5b05798. [DOI] [PubMed] [Google Scholar]
- Grostern A, Edwards EA. A 1,1,1-trichloroethane-degrading anaerobic mixed microbial culture enhances biotransformation of mixtures of chlorinated ethenes and ethanes. Appl Environ Microbiol. 2006a;72:7849–7856. doi: 10.1128/AEM.01269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grostern A, Edwards EA. Growth of Dehalobacter and Dehalococcoides spp. during degradation of chlorinated ethanes. Appl Environ Microbiol. 2006b;72:428–436. doi: 10.1128/AEM.72.1.428-436.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grostern A, Edwards EA. Characterization of a Dehalobacter coculture that dechlorinates 1,2-dichloroethane to ethene and identification of the putative reductive dehalogenase gene. Appl Environ Microbiol. 2009;75:2684–2693. doi: 10.1128/AEM.02037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatt JK, Ritalahti KM, Ogles DM, Lebron CA, Loffler FE. Design and application of an internal amplification control to improve Dehalococcoides mccartyi 16S rRNA gene enumeration by qPCR. Environ Sci Technol. 2013;47:11131–11138. doi: 10.1021/es4019817. [DOI] [PubMed] [Google Scholar]
- Kanitkar Y, Stedtfeld RD, Steffan RJ, Hashsham SA, Cupples AM. Development of loop mediated isothermal amplification (LAMP) for rapid detection and quantification of Dehalococcoides spp. biomarker genes in commercial reductive dechlorinating cultures KB-1 and SDC-9. Appl Environ Microbiol. 2016;82:1799–1806. doi: 10.1128/AEM.03660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koloren Z, Sotiriadou I, Karanis P. Investigations and comparative detection of Cryptosporidium species by microscopy, nested PCR and LAMP in water supplies of Ordu, Middle Black Sea, Turkey. Ann Trop Med Parasitol. 2011;105:607–615. doi: 10.1179/2047773211Y.0000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic T, Ellis M, Williams MR, Stedtfeld TM, Kaneene JB, Stedtfeld RD, Hashsham SA. Thirty-minute screening of antibiotic resistance genes in bacterial isolates with minimal sample preparation in static self-dispensing 64 and 384 assay cards. Appl Microbiol Biotechnol. 2015;99:7711–7722. doi: 10.1007/s00253-015-6774-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreader CA. Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein. Appl Environ Microbiol. 1996;62:1102–1106. doi: 10.1128/aem.62.3.1102-1106.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matturro B, Rossetti S. GeneCARD-FISH: detection of tceA and vcrA reductive dehalogenase genes in Dehalococcoides mccartyi by fluorescence in situ hybridization. J Microbiol Methods. 2015;110:27–32. doi: 10.1016/j.mimet.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Mauk MG, Liu C, Song J, Bau HH. Integrated microfluidic nucleic acid isolation, isothermal amplification, and amplicon quantification. Microarrays. 2015;4:474–489. doi: 10.3390/microarrays4040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak SS, Barbar CA, Geva E, Abrams WR, Malamud D, Ongagna YSY. Rapid point-of-care isothermal amplification assay for the detection of malaria without nucleic acid purification. Infect Dis. 2016;9:1. doi: 10.4137/IDRT.S32162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MJ, Zogorski JS, Squillace PJ. Chlorinated solvents in groundwater of the United States. Environ Sci Technol. 2007;41:74–81. doi: 10.1021/es061553y. [DOI] [PubMed] [Google Scholar]
- Notomi T, Mori Y, Tomita N, Kanda H. Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J Microbiol. 2015;53:1–5. doi: 10.1007/s12275-015-4656-9. [DOI] [PubMed] [Google Scholar]
- Plutzer J, Karanis P. Rapid identification of Giardia duodenalis by loop-mediated isothermal amplification (LAMP) from faecal and environmental samples and comparative findings by PCR and real-time PCR methods. Parasitol Res. 2009;104:1527–1533. doi: 10.1007/s00436-009-1391-3. [DOI] [PubMed] [Google Scholar]
- Samhan FA, Stedtfeld RD, Stedtfeld TM, Williams K. 2016 In preparation. [Google Scholar]
- Stedtfeld RD, Tourlousse DM, Seyrig G, Stedtfeld TM, Kronlein M, Price S, Ahmad F, Gulari E, Tiedje JM, Hashsham SA. Gene-Z: a device for point of care genetic testing using a smartphone. Lab Chip. 2012;12:1454–1462. doi: 10.1039/c2lc21226a. [DOI] [PubMed] [Google Scholar]
- Stedtfeld RD, Stedtfeld TM, Kronlein M, Seyrig G, Steffan RJ, Cupples AM, Hashsham SA. DNA extraction-free quantification of Dehalococcoides spp. in groundwater using a hand-held device. Environ Sci Technol. 2014;48:13855–13863. doi: 10.1021/es503472h. [DOI] [PubMed] [Google Scholar]
- Stedtfeld RD, Liu YC, Stedtfeld TM, Kostic T, Kronlein M, Srivannavit O, Khalife WT, Tiedje JM, Gulari E, Hughes M, Etchebarne B, Hashsham SA. Static self-directed sample dispensing into a series of reaction wells on a microfluidic card for parallel genetic detection of microbial pathogens. Biomed Microdevices. 2015;17:1–12. doi: 10.1007/s10544-015-9994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Teramura T, Adachi K, Tsuneda S, Kurata S, Nakamura K, Kanagawa T, Noda N. Technique for quantitative detection of specific DNA sequences using alternately binding quenching probe competitive assay combined with loop-mediated isothermal amplification. Anal Chem. 2007;79:5608–5613. doi: 10.1021/ac070041e. [DOI] [PubMed] [Google Scholar]
- Tsai YL, Olson BH. Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl Environ Microbiol. 1992;58:2292–2295. doi: 10.1128/aem.58.7.2292-2295.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.