

Abstract
Intercalation and surface modification of clays with surfactants are the essential process to tailor the clays’ surface chemistry for their extended applications. A full understanding of the interaction mechanism of surfactants with clay surfaces is crucial to engineer clay surfaces for meeting a particular requirement of industrial applications. In this study, the thermodynamic mechanism involved in the intercalation and surface modification of methanol preintercalated kaolinite by three representative alkane surfactants with different head groups, dodecylamine, cetyltrimethylammonium chloride (CTAC), and sodium stearate, were investigated using the adaptive biasing force accelerated molecular dynamics simulations. In addition, the interaction energies of surfactants with an interlayer environment (alumina surface, siloxane surface, and interlayer methanol) of methanol preintercalated kaolinite were also calculated. It was found that the intercalation free energy of CTAC with a cationic head group was relatively larger than that of stearate with an anionic head group and dodecylamine with a neutral head group. The attractive electrostatic and van der Waals interactions of surfactants with an interlayer environment contributed to the intercalation and surface modification process with the electrostatic force playing the significant role. This study revealed the underlying mechanism involved in the intercalation and surface modification process of methanol preintercalated kaolinite by surfactants, which can help in further design of kaolinite-based organic clays with desired properties for specific applications.
Graphical abstract
INTRODUCTION
Nature is an excellent source of abundant materials, among which clays have received a lot of attention due to their particular morphological and physicochemical properties and natural abundance. Clays belong to the layered aluminosilicate whose individual layers consist of a silica tetrahedral sheet and an alumina octahedral sheet. Among the clay families, the smectite clays have been widely used in the field of catalysts, environmental remediation, clay-based polymer nanocomposites, etc., due to their high cation exchange capacity, swelling behavior, and large surface area.1–3 Differing with smectite clays, kaolinite is a 1:1 dioctahedral clay mineral with the unit nanometer-thick layer formed by one alumina octahedral sheet linked with one silicon tetrahedral sheet by the sharing oxygen plane. The unit layers are stacked along the direction perpendicular to the basal surface, forming an interlayer space where the alumina surface covered by the hydroxyl groups faces the siloxane surface covered by basal oxygens. In the interlayer region, there are hydrogen bonds formed between the hydroxyl groups on the alumina surface and the basal oxygens on the siloxane surface. The clay surfaces show hydrophilic property that limits their applications especially when used as the reinforcing filler in the hydrophobic polymer matrix and adsorbent for particular organic contaminants. For instance, when kaolinite is directly used as the reinforcing filler for fabricating the clay-based polymer nanocomposites, the kaolinite platelets are difficult to disperse homogeneously in the hydrophobic polymer matrix due to their hydrophilic surfaces, which cannot bring the desired reinforcing effect for the kaolinite/polymer composites.4 In order to overcome their interfacial incompatibility, the organic modification of kaolinite surfaces by surfactants is introduced to render its surface chemistry. The commonly used surfactants for organic modification of clays are alkyl ammoniums consisting of hydrophobic alkyl chains and hydrophilic head groups. The latter can be fixed on the clay surfaces through electrostatic force, leading to the surfactants coated on the clay surfaces. Consequently, the surfactants can bridge the polymer matrix and clay platelets with their hydrophobic alkyl chains functioning as the cement. The interfacial compatibility of the polymer matrix and modified clay platelets with the help of surfactants makes a significant contribution to the improved mechanical, fire resistance, and gas barrier properties of the clay/polymer hybrid composites.4 Among the surface modification strategies of clays, the intercalation accompanied by surface modification has the excellent perspective, because intercalation of clays with surfactants can effectively expand the interlayer space and contribute to the subsequent exfoliation of clay platelets. Meanwhile, the surfactants adhered to the clay platelets significantly modify the topography and chemistry of clay surfaces. As a result, the delaminated and surface modified organoclays have a high aspect ratio and can be easily dispersed uniformly in the polymer matrix with a large contact area with the polymer matrix giving rise to a strong reinforcing effect for the hybrid composites.4,5 The swelling smectites can be easily intercalated and organically modified owing to their interlayer ion exchange capacity, and have been widely industrially utilized.6 However, for kaolinite, the cohesive forces between the adjacent layers lead to the difficult intercalation of compounds in its interlayer region. The surface or cleavage energy of kaolinite was reported to be 252.57 ± 2.75 mJ/m2.7 The kaolinite is generally considered as a nonswelling clay mineral with low chemistry reactivity. Nevertheless, kaolinite has two kinds of basal surfaces with different chemical reactivity in the interlayer environment due to its 1:1 dioctahedral structure, that is, alumina surface and siloxane surface. The alumina surface is covered by the hydroxyl groups, and the siloxane surface is covered by basal oxygens. The two kinds of surfaces can function as H-donors/acceptors inducing small polar molecules to enter the kaolinite interlayer via hydrogen bonding interaction to achieve the interlayer expansion.8 The surfactants with relatively large molecular size, however, are difficult to directly intercalate in the kaolinite interlayer due to the steric hindrance. Experimental studies have proven that the kaolinite–methanol intercalation complex can be used as the precursor to achieve the intercalation and surface modification of the kaolinite interlayer by surfactants.9–12 The particular interlayer environment of kaolinite makes it a strong candidate for fine chemistry if its interlayer is accessible and reacted with organic moieties to produce the functional kaolinite-based organic clays for the applications in clay/polymer nanocomposites and environmental remediation, etc.
Due to the strong cohesive force between kaolinite layers, the compounds used for intercalation and surface modification of kaolinite are mainly limited to some small molecules with high polarity and excellent ability to form hydrogen bonds, such as urea, dimethyl sulfoxide, hydrazine, etc.8,13–15 The studies on kaolinite intercalation by surfactants with relatively large molecular size are rare due to the challenge of the intercalation process. However, the methanol intercalated kaolinite has been proven to be a versatile precursor that can be used for further intercalation and surface modification by surfactants as mentioned above.16 Although the interfacial structures and interactions of kaolinite–surfactants complexes have been extensively characterized using X-ray diffraction (XRD), infrared spectroscopy (IR), scanning electron microscopy (SEM), and nuclear magnetic resonance (NMR),9–12 the thermodynamics associated with the intercalation and surface modification processes of kaolinite have been rarely addressed and it is still a challenging task to accomplish by the conventional experimental techniques. The atomistic molecular dynamics (MD) simulation has evolved into a powerful tool suitable for addressing the atomic scale interfacial structures in clay related materials and environment sciences.17–23 However, the simulations in order to accurately characterize the energetic properties of the dynamic intercalation process of clays by surfactants require the system size and simulation time scale beyond the traditional MD techniques. Recent advances in accelerated molecular dynamics such as the adaptive biasing force (ABF) method substantially speed up the MD simulations by enhancing the conformational space sampling and are capable of accurately describing the free energy surface of processes of large systems at long time scale.24,25 This technique has been successfully employed to calculate the free energies of the sorption process of PCBs from bulk water into clay minerals, agreeing well with the experimentally determined results.26 In the present work, the potential of mean force (PMF) or free energies for the intercalation of selected representative alkane surfactants with neutral and ionic head groups in methanol intercalated kaolinite were calculated using the ABF method. The driving force arisen from the enthalpic energy gain from their interactions with kaolinite interlayer surfaces as well as the interlayer methanol was quantified in terms of the electrostatic and van der Waals interaction energies. The contributions of the alkyl chain and head group segments of surfactants to the corresponding electrostatic and van der Waals interaction energies were also calculated. This research is significant to understand the underlying mechanism involved in the intercalation and surface modification process of the kaolinite interlayer as well as the interaction mechanism of different components of surfactants with a kaolinite interlayer environment, which can help in further design of kaolinite-based organic clays with desired properties for specific applications.
SIMULATION METHOD
The ABF molecular dynamics simulations were performed to explore the thermodynamic mechanism of intercalation and surface modification of the methanol preintercalated kaolinite interlayer by surfactants. The dodecylamine, CTAC, and sodium stearate were selected as the representative alkane surfactants with neutral, cationic, and anionic head groups whose molecular structures are shown in Figure 1. The unit cell of the kaolinite model was built following the procedure described in our previous study.10,14 The supercell with 8 × 5 × 2 unit cells of kaolinite possessing the size of 41.2 Å in the x direction and 44.67 Å in the y direction was created as the crystalline clay model. The direction perpendicular to the (001) crystal plane of kaolinite was chose as the z-axis (Figure S1 in the Supporting Information). One of the interlayers was placed at the center of the supercell to eliminate the electric dipole of the supercell in the direction perpendicular to the surface. This interlayer was artificially expanded in order to create an interlayer space of desired extension, where the behavior of solvent and adsorbate can be modeled. The other interlayer remained intact. Since we are interested in the initial stage of the intercalation process with relatively low loading of surfactant, such a partial intercalation system is sufficient for structural analysis and PMF calculations. Similar clay models have been successfully adopted in previous studies.27,28 In the interlayer region of the kaolinite model, the methanol molecules were randomly placed to make the methanol preintercalated interlayer environment. In order to calculate the free energies of surfactants transferring between the interlayer micropore of methanol preintercalated kaolinite and the surrounding solution phase (for CTAC and dodecylamine, the solvent in solution phase is methanol; for sodium stearate, the solvent is water), the methanol preintercalated kaolinite interlayer–bulk methanol/water two-phase periodic systems were created. For constructing the two kinds of systems, the kaolinite model with the methanol intercalated interlayer was cleaved along the (010) plane where the dangling bonds were saturated with OH groups. The Al atoms on the termination of the (010) edge surface adopt a 5-fold coordination state according to the study by Liu et al.29 Then, a vacuum slab with a width of 30 Å was added above the (010) edge surface. For calculation of free energies of dodecylamine and CTAC, the vacuum slab was packed with the bulk methanol to create the methanol preintercalated kaolinite interlayer–bulk methanol model to match the real environmental condition where the methanol was used to dissolve dodecylamine and CTAC. For sodium stearate, the vacuum slab was packed with the bulk water to make the methanol preintercalated kaolinite interlayer–bulk water model. Then, one molecule of surfactants was placed in the middle position of the kaolinite interlayer. The representative initial configuration of the methanol preintercalated kaolinite interlayer–bulk methanol two-phase systems with intercalated CTAC is shown in Figure S2. A different axis system was used for the two-phase periodic models in which the orientations of x-, y-, and z-axes of the two-phase periodic systems were reset, and the z-axis was set perpendicular to the (010) edge surface as shown in Figure S2. The free energies of surfactants transferring between the methanol preintercalated kaolinite interlayer and the contacting solution phase were calculated with the potential of mean force using the adaptive biasing force accelerated MD method (ABF).30 This method has been successfully used for computing the free energy profiles of the sorption process of PCBs from bulk water into clay minerals, and the calculated free energies were reasonably consistent with experimentally determined adsorption free energies.26 All the MD simulations were carried out using the LAMMPS package.31 The INTERFACE force field32 developed specifically for clays, silicates, metal oxides, and other inorganic minerals was used to describe the kaolinite, and OPLS-AA force field33 for surfactants and methanol. The compatibility of the two force fields and their applicability for inorganic–organic interfaces have been validated in refs 32, 34, and 35. The water molecules were described using the simple point charge (SPC) water model.36 The force field parameters for methanol and selected surfactants were directly adopted from the OPLS-AA force field except that the atomic charges for CTAC were taken from the work of Lopes and Pádua.37 The MD simulation details are provided in the text of the Supporting Information.
Figure 1.
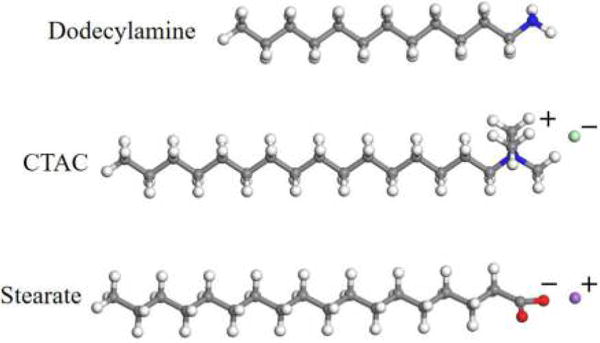
Molecular structures of selected alkane surfactants with neutral and ionic head groups. The ball color scheme for the molecular structure is O (red), N (blue), C (gray), H (white), Cl− (green), and Na+ (purple).
RESULTS AND DISCUSSION
Free Energies Calculated by ABF Method
The intercalation free energies of methanol preintercalated kaolinite by surfactants were directly calculated with the ABF MD simulations. The energy variation related to the surfactants transferring between the kaolinite interlayer domain and the surrounding solution phase gives a direct insight into the reaction trend pertaining to the intercalation process of surfactants. Figure 2 shows that the calculated free energies for all surfactants in the kaolinite interlayer domain are always lower than that in solution phase, indicating that the interlayer environment of methanol preintercalated kaolinite is energetically favorable for the intercalation of surfactants. In this study, the intercalation free energy ΔG for the intercalation process of surfactants was defined as the energy difference between the stationary stage of the surfactant molecule located at the center of the interlayer and the interface of the clay edge and solution phase, that is, the point −15 Å and 2.5 Å along the reaction coordinate of PMF. The intercalation free energies of three surfactants are −38, −15, and −10 kcal/mol for CTAC, stearate, and dodecylamine, respectively. The methanol preintercalated kaolinite interlayer shows relatively stronger affinity to CTAC with a cationic head group compared with dodecylamine with a neutral head group and stearate with an anionic head group. The magnitude of calculated intercalation free energies for the three surfactants agrees with the experimentally observed intercalation trend that the CTAC can more easily achieve the intercalation and surface modification of methanol preintercalated kaolinite compared with stearate and dodecylamine.10,12,38 It is interesting to note that the PMF of CTAC (Figure 2) has the bumpy feature before completely entering the kaolinite interlayer. The local maximums of the free energies along the reaction coordinate represent the energy barrier when CTAC continually move into the interlayer, which are related with the transfer of the head group N(CH3)3+ of CTAC above the adjacent ditrigonal cavities on the siloxane surface in the kaolinite interlayer. The ditrigonal cavities surrounded by six negatively charged oxygen atoms are the dominant adsorption sites where the cationic head group of CTAC can be firmly anchored through strong electrostatic interaction. This point can be verified by the atom distribution map of the head group of CTAC on the slioxane surface in Figure 5b, which directly reveals that the head groups of CTAC are strongly adsorbed at the ditrigonal cavity. Thus, CTAC needs to overcome the energy barrier for moving its head group above the adjacent ditrigonal cavities of the kaolinite siloxane surface.
Figure 2.
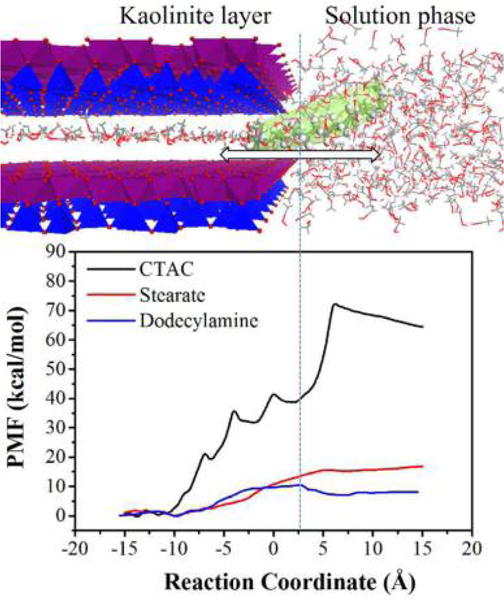
Simulation snapshot (upper panel) and calculated free energy profiles of CTAC, stearate, and dodecylamine transferring between the kaolinite interlayer region and the solution phase by the ABF method (lower panel). For CTAC and dodecylamine, the solvent in the solution phase is methanol; for stearate, the solvent is water. CTAC (in light green color) between the kaolinite interlayer and the bulk methanol along the reaction coordinate is shown in the snapshot; methanol molecules are displayed in line style; and kaolinite layers are displayed in the polyhedron.
Figure 5.
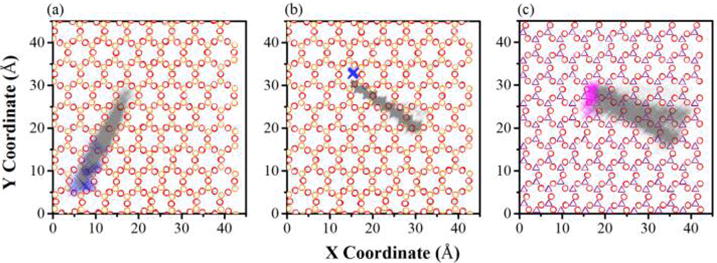
Atom distribution map of head group and alkyl chain segments of (a) dodecylamine, (b) CTAC, and (c) stearate on the kaolinite interlayer surfaces. For (a) dodecylamine and (b) CTAC, the substrate is the siloxane surface where the orange and red hollow circles refer to Si and O on the siloxane surface, the blue cross represents N in the head group NH2 in dodecylamine and N(CH3)3+ in CTAC, and the gray cross represents C in the alkyl chain backbone. For (c) stearate, the substrate is the alumina surface where the purple hollow triangle and red hollow circle represent Al and O on the alumina surface, respectively; the magenta and gray crosses represent O in the head group COO− of stearate and C in the alkyl chain backbone, respectively.
Interaction Energies of Surfactants with Interlayer Environment
The intercalation and surface modification of methanol preintercalated kaolinite by surfactants is favored by gaining the enthalpic energy through their interactions with the kaolinite interlayer surfaces as well as the interlayer methanol, and the entropic gain by partitioning of less hydrated surfactants with long alkyl chains from bulk polar solvents into the interlayer also plays an important role.39 Quantifying the enthalpy contribution to such a process can facilitate the understanding of the thermodynamic driving force for the intercalation and surface modification process.40,41 In order to evaluate the enthalpic energy gain through the interactions of surfactants with the interlayer environment of methanol preintercalated kaolinite including the two opposing interlayer surfaces, viz. alumina surface and siloxane surface, and the interlayer methanol, the interaction energies between them were calculated. The method of interaction energy calculation is detailed in the Supporting Information.
Figure 3 shows the interaction energies of surfactants with the interlayer environment of methanol preintercalated kaolinite, and the contributions of electrostatic and van der Waals energies to the corresponding total interaction energies along with the standard deviations. The decomposition of the interaction energy provides a direct insight into what interaction dominates the ehthalpic energy gain for the intercalation and surface modification process. For the energy values, a negative value represents attractive force while a positive value represents repulsive force. The results clearly show that the intercalation and surface modification of the kaolinite interlayer is driven by both electrostatic and van der Waals forces. The interaction energies of neutral dodecylamine with the kaolinite alumina surface, siloxane surface, and interlayer methanol are −4.64, −3.41, and −7.31 kcal/mol, respectively (Figure 3 (1)), indicating that the kaolinite interlayer surfaces and interlayer methanol all interact favorably with the dodecylamine. The electrostatic energy component to the total interaction energy of dodecylamine with the alumina surface accounts for 62% (−2.88 kcal/mol), indicating a slightly prevailing role of electrostatic interaction. In contrast, the van der Waals energy component dominates slightly for the interaction with the siloxane surface with a 65% (−2.21 kcal/mol) contribution to the total interaction energy. The interlayer methanol has a relatively larger interaction energy with the dodecylamine compared with the kaolinite surfaces, of which the electrostatic component accounts for 64% (−4.71 kcal/mol) and the van der Waals component is 36% (−2.60 kcal/mol), respectively. The interaction energies of CTAC and stearate possessing ionic head groups with the interlayer environment of methanol preintercalated kaolinite show different features to dodecylamine. The interaction energies of CTAC with the alumina surface, siloxane surface, and interlayer methanol were calculated as 38.39, −13.47, and −48.15 kcal/mol, respectively. For stearate, the corresponding interaction energies were −43.43, 7.79, and −2.07 kcal/mol, respectively. It shows that not all the components of the interlayer environment show attractive interactions with the charged surfactants. The alumina surface and siloxane surface demonstrate the repulsive interaction with the CTAC and stearate, respectively. This is attributed to the repulsive force between like charges since the alumina surface and siloxane surface carry local positive and negative charge, respectively. Therefore, they show a rational repulsive force with the positively charged head group N(CH3)3+ in CTAC and the negatively charged head group COO− in stearate, respectively. In spite of the repulsive force, the opposing kaolinite surfaces that have opposite charge show attractive interaction with the surfactants. It is shown that the interaction energy of stearate with the alumina surface is −43.43 kcal/mol (electrostatic component is −39.74 kcal/mol, and van der Waals component is −3.69 kcal/mol) much larger in magnitude than that with the siloxane surface with 7.79 kcal/mol, of which the electrostatic and van der Waals components are 11.84 and −4.05 kcal/mol, respectively. For the CTAC, although the repulsive interaction energy with the alumina surface is 38.39 kcal/mol much larger in magnitude than the attractive interaction energy with the siloxane surface (−13.47 kcal/mol), the attractive interaction with interlayer methanol with the value of −48.15 kcal/mol is sufficient to compensate the repulsive force from the alumina surface. Recall that the interlayer methanol also demonstrated a considerable attractive interaction with dodecylamine, agreeing with the experimental observation that the interlayer methanol plays the significant role in the intercalation and surface modification of methanol preintercalated kaolinite by quaternary ammonium salt and alkyl amine.9,10,12 Similar to the stearate, the interaction energies of CTAC with the interlayer environment are also dominated by the electrostatic component as shown in Figure 3 (2). The results also show that, even if the van der Waals energy components occupy a smaller proportion than the electrostatic energy component for all surfactants, they demonstrate attractive interaction with all components of the interlayer environment. Overall, the interlayer environment of methanol preintercalated kaolinite shows relatively strong affinity to the surfactants having ionic head groups, which can gain more enthalpic energy via electrostatic interaction with the interlayer environment to easily achieve the intercalation and surface modification process.
Figure 3.
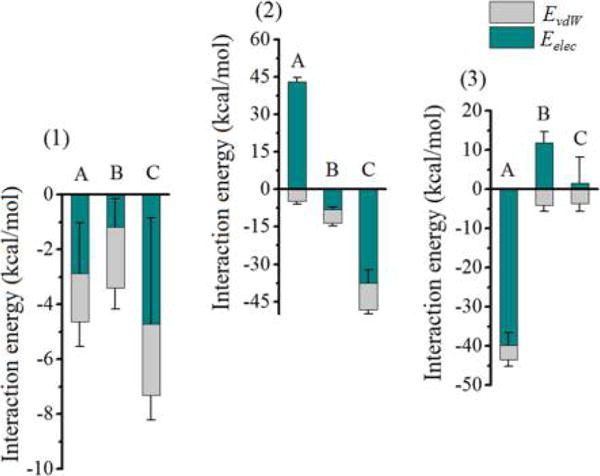
Interaction energies of (1) dodecylamine, (2) CTAC, and (3) stearate with the interlayer environment of methanol preintercalated kaolinite, and the proportions of electrostatic energies Eelec and van der Waals energies EvdW accounting for the interaction energies, where A, B, and C represent the interlayer alumina surface, siloxane surface, and interlayer methanol, respectively.
The proportions of electrostatic and van der Waals energies that originated from the head groups and alkyl chains accounting for the electrostatic and van der Waals interactions of surfactants with the interlayer environment of methanol preintercalated kaolinite were also calculated to quantify how different segments of surfactants make the contributions to the electrostatic and van der Waals interactions. The details of how to split the energy contributions of head groups and alkyl chains are provided in the Supporting Information. Figure 4 (2) and (3) indicate that the charged head groups of CTAC and stearate that have big charges on them in spite of the small atom numbers dominate the corresponding electrostatic interactions of surfactants with all components of the interlayer environment. The proportions of electrostatic energy that stemmed from the charged head groups account for more than 80% of the corresponding total electrostatic interaction energies; the alkyl chains, although having a large number of atoms, only make an insignificant contribution to the electrostatic interactions since the atoms of alkyl chains have smaller charge on them. In the case of neutral dodecylamine (Figure 4 (1)), the alkyl chain and head group dominate the attractive electrostatic interactions with kaolinite interlayer surfaces and interlayer methanol, respectively. In contrast to the proportions of charged head groups of surfactants accounting for the electrostatic interactions, the alkyl chain segments of all surfactants play a central role in the van der Waals interaction energies as displayed in Figure 4. The proportions of van der Waals energy that originated from alkyl chains for all surfactants occupy more than 85% of the corresponding total van der Waals interaction energies, especially for the neutral dodecylamine with a percentage of almost 100%. This indicates that the alkyl chain moiety of surfactants prefers the hydrophobic environment. The hydrophobic interaction is also a driving force for the continuous intercalation and surface modification of methanol preintercalated kaolinite by surfactants. Once the surfactants penetrate into the kaolinite interlayer, it can make the interlayer environment more hydrophobic. The hydrophobic interactions between alkyl chains can aid in inducing more surfactants to enter the kaolinite interlayer. When the first layer of surfactants is densely packed in the interlayer, the additional intercalated surfactants lead to the further interlayer expansion of kaolinite for making more space to accommodate the surfactants. The process of interlayer filling of methanol preintercalated kaolinite by dodecylamine and CTAC can increase the kaolinite basal spacing from 7.2 Å to around 40 Å.10,12 As a result, the original cohesive force between kaolinite adjacent layers is weakened considerably; then the slacked kaolinite layers can be easily exfoliated to the platelets with organically modified surfaces, which has excellent prospect in kaolinite-based clay/polymer nanocomposites. The kaolinite stacks can even be exfoliated to platelets with very few kaolinite unit layers by the intercalation with CTAC, which can automatically form the nanoscrolls due to the structural mismatch between the alumina octahedral sheet and the silica tetrahedral sheet of the kaolinite unit layer.12
Figure 4.
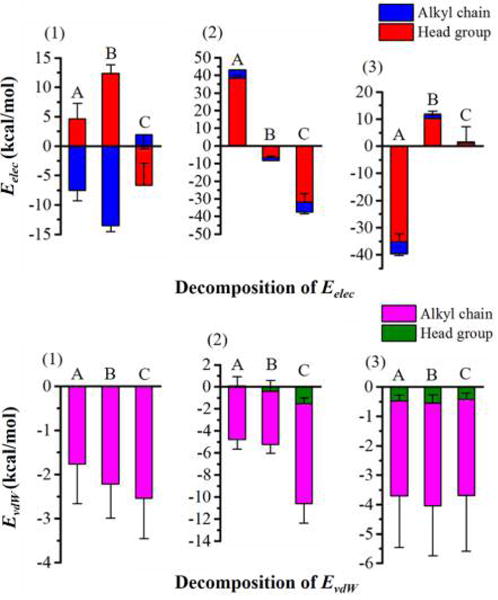
Decomposition of electrostatic interaction energies (Eelec) and van der Waals interaction energies (EvdW) of (1) dodecylamine, (2) CTAC, and (3) stearate with interlayer environment of methanol preintercalated kaolinite into the head groups and alkyl chains, where A, B, and C represent the alumina surface, siloxane surface, and interlayer methanol, respectively.
Interfacial Structure
The interfacial structure can help to explore the interfacial interactions between particular segments (head group and alkyl chain) of surfactants and the kaolinite interlayer surfaces at the atomic scale, which can be visualized from the MD simulations. Figure 5 shows the atom distribution maps of surfactant segments on the kaolinite interlayer surfaces plotted based on the atom coordinates recorded during the 1 ns production runs after the 10 ns equilibrium runs, as stated in the Simulation Protocol in the Supporting Information. Here, the N represents the NH2 of dodecylamine and N(CH3)3+ of CTAC, and O represents the COO− of stearate; the C in the alkyl chain segments refers to the backbone CH2 and tail CH3 of alkyl chains. The atom distribution maps provide clear information on the preferred adsorption sites for different segments of surfactants on the interlayer surfaces. The head group N(CH3)3+ of CTAC was strongly anchored at the ditrigonal cavity on the siloxane surface and did not change its position during the 1 ns production runs as shown in Figure 5b. The ditrigonal cavity surrounded by six negatively charged basal oxygens relaxing outward slightly with respect to the silicon atoms is the main adsorption site for N(CH3)3+ of CTAC through electrostatic force. This is also an important finding in this study. It explains why the quaternary ammoniums are commonly used as the cationic collectors for the flotation of kaolinite from mineral deposits that contain kaolinite as the gangue mineral.42 The strong adhesion of cationic head groups of quaternary ammoniums on the kaolinite siloxane surface can effectively separate kaolinite from other minerals. In addition, the steric effect resulting from the hindrance of the bulky N(CH3)3+ group by the ditrigonal cavity with a similar diameter (Figure 6a) also contributes to its movement limitation on the siloxane surface. For the dodecylamine, its neutral head group NH2 exhibits greater lateral mobility (Figure 5a) compared with N(CH3)3+ of CTAC due to the weaker interactions with kaolinite interlayer surfaces. The anionic head group of stearate is preferentially adsorbed at the alumina octahedral vacancy surrounded by hydroxyl groups as shown in Figures 5c and 6b. Similar to the ditrigonal cavity on the siloxane surface, the octahedral vacancy surrounded by hydroxyl groups on the alumina surface can anchor the anionic head group COO− of stearate through electrostatic force and H-bonds formed between COO− and hydroxyl groups (Figure 6b). On the basis of the CH2 distribution of alkyl chains of surfactants on the kaolinite interlayer surfaces, it is concluded that the alkyl chains of all surfactants keep a linear structure due to the confinement of the kaolinite interlayer.
Figure 6.
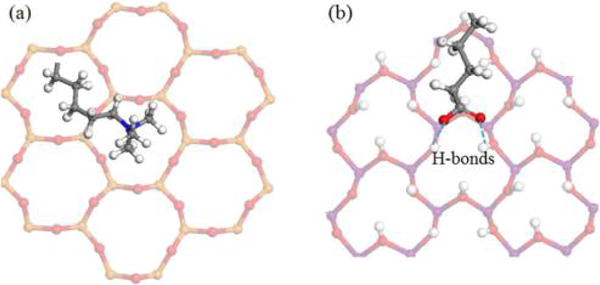
Snapshots of (a) CTAC head group N(CH3)3+–siloxane surface and (b) stearate head group COO−–alumina surface. The ball color scheme is O (red), Si (orange), Al (purple), H (white), N (blue), and C (gray).
CONCLUSION
In this work, the adaptive biasing force accelerated molecular dynamics simulations were employed to calculate the potential of mean force of alkane surfactants with neutral, cationic, and anionic head groups transferring between the methanol preintercalated kaolinite interlayer region and the surrounding solution phase. By doing the thermodynamics analyses, it was found that the intercalation free energies were −38, −15, and −10 kcal/mol for CTAC, stearate, and dodecylamine, respectively. This indicates that the interlayer environment of methanol preintercalated kaolinite shows relatively stronger affinity to CTAC with a cationic head group compared with dodecylamine with a neutral head group and stearate with an anionic head group. The driving force for the intercalation and surface modification process contributed from the enthalpic energy gain via the interactions of surfactants with the interlayer environment was controlled by both electrostatic and van der Waals interactions with the electrostatic interaction playing the major role. The preintercalated methanol played the important role for inducing the CTAC and dodecylamine to enter the kaolinite interlayer through attractive electrostatic and van der Waals forces. The strong electrostatic interactions of ionic head groups of CTAC and stearate with the interlayer environment can help in gaining more enthalpic energies for their intercalation and surface modification process. The ditrigonal cavity on the siloxane surface and alumina octahedral vacancy on the alumina surface were the dominated adsorption sites, where the head groups N(CH3)3+ of CTAC and COO− of stearate can be firmly anchored, respectively. The present study provided an insight into the thermodynamic mechanism of intercalation and surface modification of methanol preintercalated kaolinite by alkane surfactants, which could serve as a guidance to engineer modification of kaolinite surfaces for its further applications in clay-based nanocomposites and environmental remediation.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Natural Science Foundation of China (51034006), (41602171), (41301535), and the National Institute of Environmental Health Sciences of the National Institutes of Health, United States (P42 ES004911). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health, United States. Support from the China Scholarship Council (CSC) for S. Zhang’s study at Michigan State University is also gratefully acknowledged. The computation resources and time provided by the High Performance Computing Center (HPCC) at Michigan State University is also acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.6b12919.
The simulation protocol of PMF calculation using the adaptive biasing force accelerated molecular dynamics simulations and the method of interaction energies calculation. The initial crystalline kaolinite structure and initial configuration of methanol preintercalated kaolinite interlayer–bulk methanol two-phase system with intercalated CTAC. The scheme of interaction energies calculation in the case of dodecylamine with interlayer environment of methanol preintercalated kaolinite (PDF)
ORCID
Qinfu Liu: 0000-0001-8992-7571
Notes
The authors declare no competing financial interest.
References
- 1.de Paiva LB, Morales AR, Valenzuela Díaz FR. Organoclays: Properties, Preparation and Applications. Appl Clay Sci. 2008;42:8–24. [Google Scholar]
- 2.Kotal M, Bhowmick AK. Polymer Nanocomposites from Modified Clays: Recent Advances and Challenges. Prog Polym Sci. 2015;51:127–187. [Google Scholar]
- 3.Boyd SA, Sheng G, Teppen BJ, Johnston CT. Mechanisms for the Adsorption of Substituted Nitrobenzenes by Smectite Clays. Environ Sci Technol. 2001;35:4227–4234. doi: 10.1021/es010663w. [DOI] [PubMed] [Google Scholar]
- 4.Dai JC, Huang JT. Surface Modification of Clays and Clay–Rubber Composite. Appl Clay Sci. 1999;15:51–65. [Google Scholar]
- 5.Omara SS, Abdel Rehim MH, Ghoneim A, Madkour S, Thünemann AF, Turky G, Schönhals A. Structure–Property Relationships of Hyperbranched Polymer/Kaolinite Nanocomposites. Macromolecules. 2015;48:6562–6573. [Google Scholar]
- 6.Chiu C-W, Huang T-K, Wang Y-C, Alamani BG, Lin J-J. Intercalation Strategies in Clay/Polymer Hybrids. Prog Polym Sci. 2014;39:443–485. [Google Scholar]
- 7.Helmy AK, Ferreiro EA, de Bussetti SG. The Surface Energy of Kaolinite. Colloid Polym Sci. 2004;283:225–228. [Google Scholar]
- 8.Ledoux RL, White JL. Infrared Studies of Hydrogen Bonding Interaction between Kaolinite Surfaces and Intercalated Potassium Acetate, Hydrazine, Formamide, and Urea. J Colloid Interface Sci. 1966;21:127–152. [Google Scholar]
- 9.Komori Y, Sugahara Y, Kuroda K. Intercalation of Alkylamines and Water into Kaolinite with Methanol Kaolinite as an Intermediate. Appl Clay Sci. 1999;15:241–252. [Google Scholar]
- 10.Zhang S, Liu Q, Cheng H, Li X, Zeng F, Frost RL. Intercalation of Dodecylamine into Kaolinite and Its Layering Structure Investigated by Molecular Dynamics Simulation. J Colloid Interface Sci. 2014;430:345–350. doi: 10.1016/j.jcis.2014.05.059. [DOI] [PubMed] [Google Scholar]
- 11.Matusik J, Kłapyta Z, Olejniczak Z. NMR and IR Study of Kaolinite Intercalation Compounds with Benzylalkylammonium Chlorides. Appl Clay Sci. 2013;83–84:426–432. [Google Scholar]
- 12.Liu Q, Li X, Cheng H. Insight into the Self-Adaptive Deformation of Kaolinite Layers into Nanoscrolls. Appl Clay Sci. 2016;124–125:175–182. [Google Scholar]
- 13.Cheng H, Liu Q, Yang J, Ma S, Frost RL. The Thermal Behavior of Kaolinite Intercalation Complexes-a Review. Thermochim Acta. 2012;545:1–13. [Google Scholar]
- 14.Zhang S, Liu Q, Cheng H, Zeng F. Combined Experimental and Theoretical Investigation of Interactions between Kaolinite Inner Surface and Intercalated Dimethyl Sulfoxide. Appl Surf Sci. 2015;331:234–240. [Google Scholar]
- 15.Dedzo GK, Detellier C. Functional Nanohybrid Materials Derived from Kaolinite. Appl Clay Sci. 2016;130:33–39. [Google Scholar]
- 16.Komori Y, Sugahara Y, Kuroda K. A Kaolinite-NMF-Methanol Intercalation Compound as a Versatile Intermediate for Further Intercalation Reaction of Kaolinite. J Mater Res. 1998;13:930–934. [Google Scholar]
- 17.Heinz H, Ramezani-Dakhel H. Simulations of Inorganic-Bioorganic Interfaces to Discover New Materials: Insights, Comparisons to Experiment, Challenges, and Opportunities. Chem Soc Rev. 2016;45:412–448. doi: 10.1039/c5cs00890e. [DOI] [PubMed] [Google Scholar]
- 18.Suter JL, Groen D, Coveney PV. Chemically Specific Multiscale Modeling of Clay–Polymer Nanocomposites Reveals Intercalation Dynamics, Tactoid Self-Assembly and Emergent Materials Properties. Adv Mater. 2015;27:966–984. doi: 10.1002/adma.201403361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Li H, Johnston CT, Boyd SA, Teppen BJ. Relating Clay Structural Factors to Dioxin Adsorption by Smectites: Molecular Dynamics Simulations. Soil Sci Soc Am J. 2012;76:110–120. [Google Scholar]
- 20.Zhou J, Boek ES, Zhu J, Lu X, Sprik M, He H. Molecular Simulation Study of Hydrated Na-Rectorite. Langmuir. 2015;31:2008–2013. doi: 10.1021/la503900h. [DOI] [PubMed] [Google Scholar]
- 21.Rao Q, Leng Y. Molecular Understanding of CO2 and H2O in a Montmorillonite Clay Interlayer under CO2 Geological Sequestration Conditions. J Phys Chem C. 2016;120:2642–2654. [Google Scholar]
- 22.Krishnan M, Saharay M, Kirkpatrick RJ. Molecular Dynamics Modeling of CO2 and Poly(Ethylene Glycol) in Montmorillonite: The Structure of Clay–Polymer Composites and the Incorporation of CO2. J Phys Chem C. 2013;117:20592–20609. [Google Scholar]
- 23.Shapley TV, Molinari M, Zhu R, Parker SC. Atomistic Modeling of the Sorption Free Energy of Dioxins at Clay–Water Interfaces. J Phys Chem C. 2013;117:24975–24984. [Google Scholar]
- 24.Laio A, Gervasio FL. Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep Prog Phys. 2008;71:126601. [Google Scholar]
- 25.Darve E, Rodríguez-Gómez D, Pohorille A. Adaptive Biasing Force Method for Scalar and Vector Free Energy Calculations. J Chem Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- 26.Liu C, Gu C, Yu K, Li H, Teppen BJ, Johnston CT, Boyd SA, Zhou D. Integrating Structural and Thermodynamic Mechanisms for Sorption of PCBs by Montmorillonite. Environ Sci Technol. 2015;49:2796–2805. doi: 10.1021/es505205p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greathouse JA, Hart DB, Bowers GM, Kirkpatrick RJ, Cygan RT. Molecular Simulation of Structure and Diffusion at Smectite–Water Interfaces: Using Expanded Clay Interlayers as Model Nanopores. J Phys Chem C. 2015;119:17126–17136. [Google Scholar]
- 28.Haria NR, Grest GS, Lorenz CD. Viscosity of Nanoconfined Water between Hydroxyl Basal Surfaces of Kaolinite: Classical Simulation Results. J Phys Chem C. 2013;117:6096–6104. [Google Scholar]
- 29.Liu X, Lu X, Wang R, Meijer EJ, Zhou H, He H. Atomic Scale Structures of Interfaces between Kaolinite Edges and Water. Geochim Cosmochim Acta. 2012;92:233–242. [Google Scholar]
- 30.Fiorin G, Klein ML, Hénin J. Using Collective Variables to Drive Molecular Dynamics Simulations. Mol Phys. 2013;111:3345–3362. [Google Scholar]
- 31.Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J Comput Phys. 1995;117:1–19. [Google Scholar]
- 32.Heinz H, Lin T-J, Kishore Mishra R, Emami FS. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The Interface Force Field. Langmuir. 2013;29:1754–1765. doi: 10.1021/la3038846. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 34.Mishra RK, Flatt RJ, Heinz H. Force Field for Tricalcium Silicate and Insight into Nanoscale Properties: Cleavage, Initial Hydration, and Adsorption of Organic Molecules. J Phys Chem C. 2013;117:10417–10432. [Google Scholar]
- 35.Zhang S, Liu Q, Gao F, Li X, Liu C, Li H, Boyd SA, Johnston CT, Teppen BJ. Mechanism Associated with Kaolinite Intercalation with Urea: Combination of Infrared Spectroscopy and Molecular Dynamics Simulation Studies. J Phys Chem C. 2017;121:402–409. doi: 10.1021/acs.jpcc.6b10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction Models for Water in Relation to Protein Hydration. In: Pullman B, editor. Intermolecular Forces. Springer Netherlands; Dordrecht: 1981. pp. 331–342. [Google Scholar]
- 37.Canongia Lopes JN, Pádua AAH. Molecular Force Field for Ionic Liquids Composed of Triflate or Bistriflylimide Anions. J Phys Chem B. 2004;108:16893–16898. [Google Scholar]
- 38.Wang S, Zuo X, Cheng H, Yang Y, Liu Q. Structural Model and De-Intercalation Kinetics of Kaolinite-Methanol-Sodium Stearate Intercalation Compound. J Braz Chem Soc. 2016;27:1311–1318. [Google Scholar]
- 39.Teppen BJ, Miller DM. Hydration Energy Determines Isovalent Cation Exchange Selectivity by Clay Minerals. Soil Sci Soc Am J. 2006;70:31–40. [Google Scholar]
- 40.Yu T, Lee O-S, Schatz GC. Steered Molecular Dynamics Studies of the Potential of Mean Force for Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J Phys Chem A. 2013;117:7453–7460. doi: 10.1021/jp401508w. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Wohlert J, Berglund LA, Tu Y, Ågren H. Molecular Dynamics Simulation of Strong Interaction Mechanisms at Wet Interfaces in Clay–Polysaccharide Nanocomposites. J Mater Chem A. 2014;2:9541–9547. [Google Scholar]
- 42.Rodrigues OMS, Peres AEC, Martins AH, Pereira CA. Kaolinite and Hematite Flotation Separation Using Etheramine and Ammonium Quaternary Salts. Miner Eng. 2013;40:12–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.