

Abstract
BACKGROUND:
Pediatric-onset systemic lupus erythematosus (pSLE) accounts for about 10%–20% of all patients with SLE. Deficiencies in early complement components of the classical pathway are the strong genetic risk factor for the development of SLE. In this study, clinical and laboratory manifestations of both complement-deficient and normal complement pSLE patients were compared.
MATERIALS AND METHODS:
To investigate clinical and immunological manifestations of pSLE in Iran, 36 consecutive pSLE patients (onset before 18 years) who were followed up over a period of 2 years, were studied. Complement C1q and C2 levels were measured using radial immunodifusion assay and complement C3 and C4 levels were measured using nephelometry. Medical records were retrospectively evaluated from patient database of Children Medical Center Hospital. Data were assessed through descriptive analysis (confidence interval = 95%), paired t-test, and Pearson correlation test.
RESULTS:
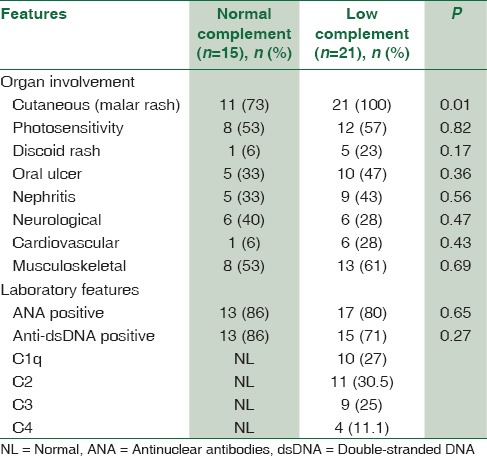
Twenty-one patients (58%) had at least one component of complement deficiency. Ten patients (27%) had low C1q level, 11 patients (30.5%) had low C2, nine patients (25%) had low C3, and four patients (11%) had low C4 level. Serum level of complement in pSLE was significantly lower than the control group, except C4 (P = 0.005). The low C1q patients had an earlier age of onset of disease (P < 0.0001). The cutaneous manifestations were more frequent and much more severe in pSLE with low complement (100% vs. 73%). The frequency of renal and musculoskeletal symptoms was equal, but renal morbidity was more common in pSLE with low complement. Positivity for anti-ds-DNA was less common in pSLE with low complement (71% vs. 86%).
CONCLUSION:
In pSLE patients with early disease onset and more aggressive SLE manifestations and negative anti-ds-DNA test, complement deficiency should be considered.
Keywords: C1q deficiency, childhood-onset, children, complement deficiency, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune rheumatic disorder characterized by multisystem involvement with periods of flare-ups and remissions.[1] Although SLE predominantly affects women of childbearing age, childhood-onset (<16 years of age) accounts for about 20% of all SLE cases.[2,3] The clinical features and immunological findings of SLE are similar in both pediatric-onset and adult SLE. However, in contrast to adults, pediatric-onset SLE (pSLE) patients appear to have a more aggressive organ involvement and poorer prognosis.[4] The exact etiology of SLE is not fully understood, and due to its complex nature, various mechanisms have been suggested to be responsible for its development. Previous studies have suggested that decreased levels of complement are associated with increased susceptibility to SLE.[5,6,7,8,9] Therefore, the objective of the present study was to assess the complement levels in pSLE patients and evaluate its prevalence and clinical characteristics.
Materials and Methods
Patients
The present study was a cross-sectional descriptive study conducted at Children's Medical Center (Pediatrics Center of Excellence affiliated to Tehran University of Medical Sciences, Tehran, Iran). A total of 36 SLE patients who had been referred to our Rheumatology Clinic were enrolled in this study. Age- and sex-matched healthy controls who were randomly selected from blood donors at Iranian blood transfusion organization were also enrolled in this study. This study was carried out between April 2013 and April 2015. The diagnosis of SLE was based on the revised criteria of the American College of Rheumatology for the classification of SLE.[10] Juvenile SLE onset was defined if the diagnosis was made at the age of 18 years or less. The clinical details of patients such as demographic characteristics, first clinical manifestation, and laboratory parameters were evaluated. Written informed consent was obtained from all the patients or through their legal guardians before blood sampling. The study was approved by the ethical committee of Tehran University of Medical Sciences.
Serology evaluation
All individuals underwent serologic testing in the Clinical Immunology Laboratory at Children's Medical Center. Blood samples were obtained at each visit. Baseline measurements of routine laboratory analysis such as hemoglobin, white blood cell counts, lymphocytes, neutrophils, platelets, erythrocyte sedimentation rate, creatinine, C-reactive protein, and antinuclear antibodies were evaluated. Anticardiolipin antibodies and anti-double-stranded DNA antibodies (were measured using a “plasmid ELISA” as described previously.[11]
Determination of circulating levels of complement components
Blood samples were obtained from cases and controls into ethylenediaminetetraacetic acid (EDTA) anticoagulated tubes for the preparation of EDTA plasma. Then, EDTA tubes were immediately centrifuged at room temperature with at 2000 g for 10 min, and the aliquots of plasma were stored at −80°C until the analyses. Serum C1q and C2 concentrations were assessed by single radial immunodiffusion (normal C1q range >33 mg/dl and normal C2 range >14 mg/dl) on commercially available plates (Endoplates; Chaska, Minnesota) according to the method of Mancini et al.[12] Levels of serum complement components C3 and C4 were determined by nephelometry (normal C3 range >90 mg/dl and normal range for C4 >10 mg/dl).
Statistical analysis
Continuous variables were expressed as the mean ± standard deviation (SD) or median and range, and categorical variables were reported as absolute values and percentages. Means and SDs were compared using an unpaired t-test. Statistical comparisons were performed using independent t-test (for quantitative variables) and the Chi-square test (for qualitative variables). A Student's t-test was used for comparison of continuous variables, and Chi-square and Fisher's exact tests were used to compare categorical data. The value of P < 0.05 was considered statistically significant in all tests. The analysis of data was performed using SPSS version 19.0 software (SPSS Inc., Chicago, IL, USA).
Results
Demographic data
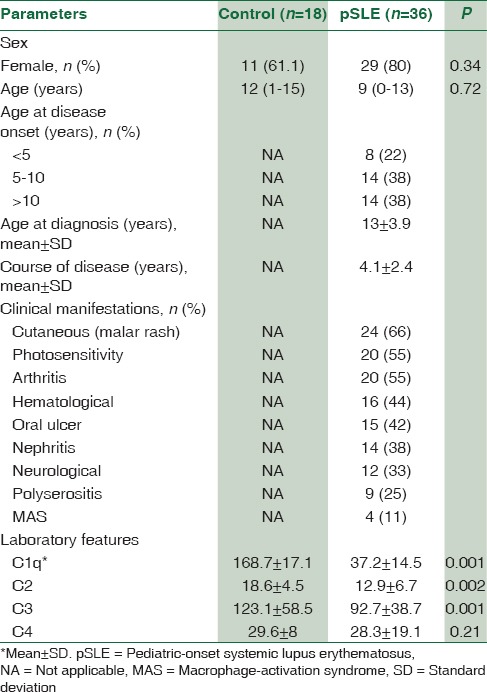
The anthropometric parameters and biochemical characteristics of patients and controls are presented in Table 1. A total of 36 patients were included for analysis. Among these patients, 8 (22%) cases were younger than 5, 14 (38%) were 5–10 years, and 14 (38%) older than 10 years. Female-to-male ratio was 4.1:1 (29 females and 7 males). The mean (SD) age at the time of diagnosis was 13 (3.9) years. Mean follow-up period was 50 ± 29 (range: 5–144) months. A positive family history of rheumatologic disorders was found in 8 (20%) families.
Table 1.
The demographic data and laboratory features of children with systemic lupus erythematosus and control group
Clinical manifestations
The initial manifestations are summarized in Table 2. The most common symptoms at presentation were fatigue and weight loss. Joint and mucocutaneous manifestations were the most frequent clinical features (20% and 24%, respectively). Six patients had a history of infectious disease including pneumonia, sepsis, meningitis, and pansinusitis. There were three cases of deep venous thrombosis, two cases of seizure, two cases of Reynaud, a case of cerebellitis, and 3 cases of myocarditis. Renal biopsies were performed in 12 out of 14 patients with renal involvement, in which the most common finding included class IV lupus nephritis (diffuse proliferative) and glomerulonephritis type V, in 6 and 3 patients, respectively.
Table 2.
Comparison of demographic and clinical features of normal versus low complement component levels in pediatric-onset systemic lupus erythematosus patients
Complement determination
SLE patients had significantly lower levels of C1q, C2, C3, and C4 compared with healthy controls. As shown in Table 2, 21 (58%) out of 36 SLE patients demonstrated low complement levels during initial evaluations. Of these patients, 10 (27%) had a decreased Clq level, with normal C4 and C3 levels, 11 (30.5%) had a decreased C2, with normal Clq and C4 levels, 9 (25%) had a decreased C3 levels and four patients had decreased C4 levels with normal C1q and C2 levels [Figure 1]. Among SLE patients, the levels of C1q, C2 and C3 was lower than the control group (P < 0.001).
Figure 1.
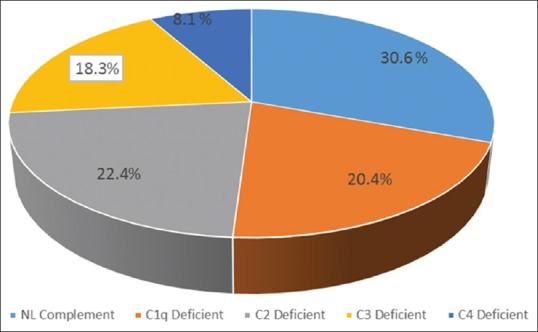
Frequency of complement components subtype among pediatric-onset systemic lupus erythematosus subjects
Low versus normal complement levels among patients with systemic lupus erythematosus
We evaluated the relationship between SLE patients with low complement levels compared to those with normal complement levels. Six out of eight patients who had developed SLE before the age of 6 years had low levels of complement, of which 5 had decreased levels of C1q and one patient had low levels of C2 and C4. Furthermore, an inverse correlation was observed between plasma levels of C1q and age of onset of SLE symptoms (r = −0.231; P < 0.0001). Six patients with a positive family history of rheumatologic disorder had low levels of complement. Among patients with decreased levels of complement components, 4 patients (19%) had a history of developing infections including meningitis, sepsis, recurrent pneumonia, and pansinusitis.
Complement determinations in relation to different organ involvement
Figure 2 presents the frequency of the complement levels and different organ involvement.
Figure 2.
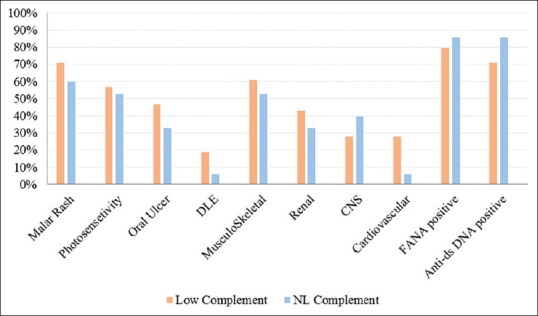
The frequency of different organ involvement in pediatric-onset systemic lupus erythematosus subjects with normal versus low complement component levels
Of the 9 SLE patients with decreased levels of complement component, 6 (66%) presented with grade V and VI lupus nephritis, and only two patients presented with a grade II nephritis with mild proteinuria. In addition to renal involvement, central nervous system involvement was seen in 6 patients with decreased levels of complement. These neurological manifestations included a headache (4 patients), aseptic meningitis (one patient), and seizure (one patient).
Discussion
SLE is a prototype of an autoimmune disease characterized by fluctuating disease activity, production of autoantibodies, and immune-complex deposition which eventually leads to multi-organ damage. While the etiology of SLE is not yet fully elucidated, numerous dysregulations of the immune system have been observed among these patients. Despite numerous reports on the clinical and laboratory features of the adult population, less is known about the immunological dysregulation occurring in the pediatric population affected by SLE. Recent evidence has demonstrated that homozygous deficiency of any of the early components of the classical pathway of complement activation (C1q, C1r, C1s, C4, and C2) predisposes individuals to the development of SLE.[13,14] The results of this study, support the finding that decreased levels of complement are prevalent in children with SLE and highlights the importance of dysregulation of this part of the immune system in the development and progression of this disease in a younger population.
Previous studies have shown the younger age of onset of SLE symptoms among patients with lower complement levels.[15] In the present study, the median age at onset was 5.5 years among patients with deficiency in one of the complement components, which is similar to prior reports. In addition, patients who demonstrated lower levels of C1q were found to present at even a younger age than other patients in our cohort, further emphasizing the role of this component of the complement system in the development of pediatric SLE. Previous studies had shown that deficiency of the early complement components predisposes individuals to the development of SLE.[16,17] C1q deficiency has been shown to be among the strongest genetic risk factor for the SLE development with more than 90% of C1q deficient individuals developing SLE later in life.[16,18] While the mechanism of this association has not been fully elucidated, abnormalities of immune complex processing and ineffective clearance of immune complexes apoptotic cells could be a direct effect of C1q deficiency leading to tissue injury autoantibody production.[19,20]
Conclusion
The results of the present study showed that among SLE patients with reduced complement levels, the severity of disease was higher than other patients. High-grade lupus nephritis and neurological manifestations were only observed among patients with reduced complement levels. This finding suggests that measuring complement levels may be considered as a marker of severity of disease and can help identify a group of patients who are more susceptible to disease manifestation.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
This study was a part of a dissertation (of Dr. P. Afzali) and was approved and supported by Vice-chancellor for Research of School of Medicine, Tehran University of Medical Sciences (Grant No: 9211165009).
References
- 1.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–39. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 2.Ståhl-Hallengren C, Jönsen A, Nived O, Sturfelt G. Incidence studies of systemic lupus erythematosus in Southern Sweden: Increasing age, decreasing frequency of renal manifestations and good prognosis. J Rheumatol. 2000;27:685–91. [PubMed] [Google Scholar]
- 3.Kaufman DB, Laxer RM, Silverman ED, Stein L. Systemic lupus erythematosus in childhood and adolescence-the problem, epidemiology, incidence, susceptibility, genetics, and prognosis. Curr Probl Pediatr. 1986;16:545–625. doi: 10.1016/0045-9380(86)90033-2. [DOI] [PubMed] [Google Scholar]
- 4.Taddio A, Rossetto E, Rosé CD, Brescia AM, Bracaglia C, Cortis E, et al. Prognostic impact of atypical presentation in pediatric systemic lupus erythematosus: Results from a multicenter study. J Pediatr. 2010;156:972–7. doi: 10.1016/j.jpeds.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 5.Bowness P, Davies KA, Norsworthy PJ, Athanassiou P, Taylor-Wiedeman J, Borysiewicz LK, et al. Hereditary C1q deficiency and systemic lupus erythematosus. QJM. 1994;87:455–64. [PubMed] [Google Scholar]
- 6.Holers VM. Complement deficiency states, disease susceptibility, and infection risk in systemic lupus erythematosus. Arthritis Rheum. 1999;42:2023–5. doi: 10.1002/1529-0131(199910)42:10<2023::AID-ANR1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Bryan AR, Wu EY. Complement deficiencies in systemic lupus erythematosus. Curr Allergy Asthma Rep. 2014;14:448. doi: 10.1007/s11882-014-0448-2. [DOI] [PubMed] [Google Scholar]
- 8.Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. 2016;7:55. doi: 10.3389/fimmu.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–6. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 10.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 11.Radice A, Sinico RA. A new oligonucleotide-based ELISA for the detection of anti-double-stranded DNA antibodies. Autoimmunity. 2006;39:113–9. doi: 10.1080/08916930500448422. [DOI] [PubMed] [Google Scholar]
- 12.Mancini G, Carbonara AO, Heremans JF. Immunochemical quantitation of antigens by single radial immunodiffusion. Immunochemistry. 1965;2:235–54. doi: 10.1016/0019-2791(65)90004-2. [DOI] [PubMed] [Google Scholar]
- 13.Jesus AA, Liphaus BL, Silva CA, Bando SY, Andrade LE, Coutinho A, et al. Complement and antibody primary immunodeficiency in juvenile systemic lupus erythematosus patients. Lupus. 2011;20:1275–84. doi: 10.1177/0961203311411598. [DOI] [PubMed] [Google Scholar]
- 14.Tahernia L, Namazi S, Rezaei N, Ziaee V. Cytokines in systemic lupus erythematosus: Their role in pathogenesis of disease and possible therapeutic opportunities. Rheumatol Res J. 2016;1:e7556. [Google Scholar]
- 15.Bhattad S, Rawat A, Gupta A, Suri D, Garg R, de Boer M, et al. Early complement component deficiency in a single-centre cohort of pediatric onset lupus. J Clin Immunol. 2015;35:777–85. doi: 10.1007/s10875-015-0212-y. [DOI] [PubMed] [Google Scholar]
- 16.Al-Mayouf SM, Abanomi H, Eldali A. Impact of C1q deficiency on the severity and outcome of childhood systemic lupus erythematosus. Int J Rheum Dis. 2011;14:81–5. doi: 10.1111/j.1756-185X.2010.01574.x. [DOI] [PubMed] [Google Scholar]
- 17.Karp DR. Complement and systemic lupus erythematosus. Curr Opin Rheumatol. 2005;17:538–42. doi: 10.1097/01.bor.0000172799.03379.86. [DOI] [PubMed] [Google Scholar]
- 18.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 19.Trendelenburg M. Antibodies against C1q in patients with systemic lupus erythematosus. Springer Semin Immunopathol. 2005;27:276–85. doi: 10.1007/s00281-005-0007-y. [DOI] [PubMed] [Google Scholar]
- 20.Böhm I. Quantification of absolute peripheral white blood cells and their subsets in patients with lupus erythematosus: Comparison with other inflammatory diseases with and without autoimmune background. Biomed Pharmacother. 2006;60:92–5. doi: 10.1016/j.biopha.2005.06.007. [DOI] [PubMed] [Google Scholar]