

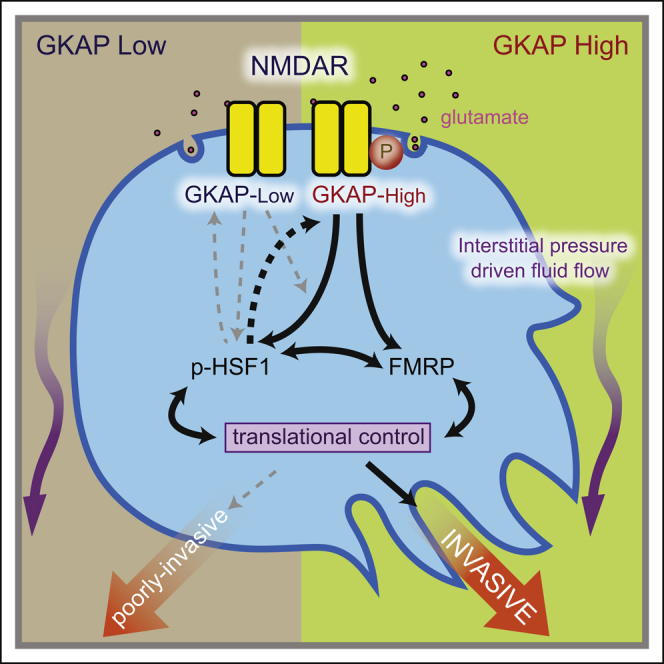
Summary
Genetic linkage analysis previously suggested that GKAP, a scaffold protein of the N-methyl-D-aspartate receptor (NMDAR), was a potential modifier of invasion in a mouse model of pancreatic neuroendocrine tumor (PanNET). Here, we establish that GKAP governs invasive growth and treatment response to NMDAR inhibitors of PanNET via its pivotal role in regulating NMDAR pathway activity. Combining genetic knockdown of GKAP and pharmacological inhibition of NMDAR, we implicate as downstream effectors FMRP and HSF1, which along with GKAP demonstrably support invasiveness of PanNET and pancreatic ductal adenocarcinoma cancer cells. Furthermore, we distilled genome-wide expression profiles orchestrated by the NMDAR-GKAP signaling axis, identifying transcriptome signatures in tumors with low/inhibited NMDAR activity that significantly associate with favorable patient prognosis in several cancer types.
Keywords: cancer modifier, GKAP/Dlgap1, glutamate receptor, FMRP, HSF1, NMDAR, GluN2b/NR2b/Grin2b, memantine, MK801, pancreatic ductal adenocarcinoma (PDAC), RIP1Tag2
Graphical Abstract
Highlights
-
•
GKAP governs invasive growth of pancreatic cancers via NMDAR signaling
-
•
HSF1 and FMRP are downstream effectors of the NMDAR-GKAP axis
-
•
A multigene signature of low/inhibited NMDAR-GKAP pathway activity is reported
-
•
A low-NMDAR-GKAP signature predicts better survival for various human cancers
Li et al. show that GKAP, a scaffold protein of NMDAR, and its downstream effectors FMRP and HSF1 play important roles in the invasive growth of pancreatic tumors. In several cancer types, low NMDAR activity, based on a transcriptomic signature, associates with favorable patient prognosis.
Significance
Genetic linkage analyses have associated multiple cancer modifier loci with disease susceptibility, malignant phenotypes, and responses to treatment. However, functional validation of cancer modifier genes has been challenging and insights into underlying mechanistic basis limited. In this proof-of-concept study, we used genetically engineered mouse models derived from distinct strain backgrounds to explore the validation process: we began with a candidate invasion modifier gene, Dlgap1 (which encodes GKAP protein), suggested by genetic linkage analysis, and went on to establish its role as a modifier of invasive growth orchestrated by NMDAR signaling. Using refined bioinformatics algorithms, we identified a signature for tumors lacking NMDAR-GKAP pathway activity that predicts better prognosis in various cancer types, suggestive of broader involvement of this pathway in malignancy.
Introduction
While distinct oncogenic “driver” genes are widely appreciated to be instrumental in cancer progression, the contributions of “modifier genes” have been less well studied. Modifier genes can alter the penetrance of specific driver oncogenes, exerting either protective or detrimental effects and affecting therapeutic outcomes. Numerous studies employing quantitative trait locus (QTL) mapping in mouse and genome-wide association analyses in humans have identified potential genetic modifier loci; however, few of these genetic modifiers have been validated mechanistically. Elucidating how genetic polymorphisms affect tumorigenesis at the molecular level is an important step toward appreciating individual variation in prognosis and in implementing personalized cancer therapies.
The RIP1Tag2 transgenic mouse model of pancreatic neuroendocrine tumor (PanNET) recapitulates the multi-stage nature of human cancer progression. As such, it has proved to be a valuable research tool for elucidating mechanisms of tumor invasion and growth. Interestingly, varying degrees of tumor invasiveness are observed at end stage depending on the genetic background in which the same transgene integration is resident, despite expressing similar levels of the driving oncoprotein (SV40 T-antigen) under control of the rat insulin promoter (RIP). In particular, the C57BL/6 (B6) background gives rise to highly invasive carcinomas, whereas mice in the C3HeB/Fe (C3H) background primarily develop well-defined, non-invasive islet tumors (Chun et al., 2010). Therefore, the RIP1Tag2 model may phenocopy a facet of the complexity of cancer progression in patients, where the same oncogenetic events can lead to varying outcomes in different patient populations.
Motivated by this observation, a classical linkage analysis was performed, identifying a QTL on mouse chromosome 17 that is highly associated with the invasive phenotype. As such, it was postulated to be a candidate “modifier locus” for mPanNET progression (Chun et al., 2010). This 13-Mb region harbors more than 50 genes; notably, there are no polymorphic differences in their coding regions, which led to a focus on differential expression. Among these genes, we became intrigued by Dlgap1, encoding GKAP, which serves as a key adaptor protein of the glutamate-activated N-methyl-D-aspartate (NMDA) receptors (NMDARs, also known as GluNRs). NMDAR is an important neuronal receptor involved in learning and memory, regulating synaptic plasticity in the central nervous system (Abbott and Nelson, 2000). As a first step, before evaluating GKAP itself, we asked in an earlier study whether NMDAR signaling was activated and, if so, was it influencing invasive tumor growth in the pro-invasive B6 background of this mouse model of PanNET, where GKAP was more highly expressed. Indeed, we established that the pathway was activated and that glutamate stimulated NMDAR activity and invasive tumor growth (Li and Hanahan, 2013). These findings prompted the current investigation to ascertain whether GKAP was a potential modifier gene whose differential expression contributes to genetic background-specific differences in pancreatic tumor invasiveness.
Results
Dlgap1 Is the Most Differentially Expressed Candidate Modifier Gene between the Invasive B6 and Non-invasive C3H Backgrounds
Initially, we further mined the expression data from Chun et al. (2010) and found Dlgap1 (encoding the GKAP protein) to be the most differentially expressed gene within the modifier locus, both in normal pancreatic islets and in fully developed β cell tumors (PanNETs) (Figure S1A). Interestingly, in wild-type animals, qRT-PCR also revealed elevated Dlgap1 expression in a number of B6 tissues when compared with C3H tissues (Figure S1B). In vitro, PanNET cell lines (βTC) derived from each of the two strains showed a clear difference in GKAP transcripts, with higher levels in βTC-B6 than in βTC-C3H (Figure 1A), consistent with the qRT-PCR analysis of fluorescence-activated cell sorting (FACS)-purified cells from primary PanNETs, which revealed β tumor cells to be the major Dlgap1-expressing cell population compared with associated stromal cells (Figures 1B and S1C).
Figure 1.

Differential GKAP Expression between the C57/BL6 and C3HeB/Fe Genetic Backgrounds Is Associated with a Differential NMDAR Pathway Activity In Vitro
(A) qRT-PCR of Dlgap1 mRNA (upper) and western blot for GKAP protein expression (lower) in mPanNET tumor-derived cancer cell lines (βTC-B6 and βTC-C3H) or primary tumors that arose in RIP1Tag2 transgenic mice inbred into the B6 and C3H backgrounds, respectively. ∗p < 0.05; ∗∗p < 0.01 (n = 3 individual tumors/genetic background; n = 3 independent RNA extraction/cell line).
(B) qRT-PCR analysis of FACS-sorted cell types from primary tumors derived from B6 mice. Cells were sorted from pools of multiple PanNETs isolated from two mice. One-way ANOVA, Dunnett multiple comparisons test was used when cancer cells were compared with all other populations (p < 0.0001 in all comparisons).
(C) Upper panel: a region within the Dlgap1 gene sequence containing a SNP site, as shown in red. Putative HSF1 binding domains (p < 0.004) are shown by the green circles. Lower panel: ChIP-qPCR for the Dlgap1 SNP site after immunoprecipitation with an anti-HSF1 antibody. The βmaj (β globin, Hbb-b1) promoter region was used as negative control. Mann-Whitney test: ∗p = 0.02 (n = 4, two batches of cell lysates per cell line, and two qPCR/batch).
(D) Western blot for HSF1 and GKAP in βTC-B6 cells. Expression levels were normalized to GAPDH and small interfering RNA (siRNA) control (n = 3 independent experiments).
(E) In vitro invasion assay of βTC-B6 and βTC-C3H cells, under either static or flow conditions. Two-way ANOVA, Bonferroni multiple comparisons test: n.s, not significant; ∗∗∗p < 0.001 (n = 4 independent assays for static condition; n = 6–9 for flow condition).
(F) Glutamate secretion by βTC-B6 and βTC-C3H cells under static and flow conditions, sampled from invasion assays. Two-way ANOVA, Bonferroni multiple comparisons test: ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; n.s., not significant (n = 3 invasion assay devices/condition/cell line).
All bar graphs represent the mean ± SEM. See also Figure S1.
We next sought to investigate the basis for the elevated expression of GKAP in B6 tissues, focusing on potential regulatory polymorphisms, given that there were no differences in the coding region of Dlgap1 between B6 and C3H. Genomic analysis for single nucleotide polymorphisms (SNP) in putative transcription factor-binding sites within 5Kb upstream and downstream of Dlgap1 identified a SNP (rs33397766) that maps to a potential heat shock factor (HSF)-binding element (Figure 1C, upper panel). While the consensus HSF1 binding site is TTCnnGAAnnTTC (http://hocomoco11.autosome.ru/motif/HSF1_HUMAN.H11MO.0.A), some studies have shown that, while the TTC repeats at each end are highly conserved, the internal GAA site can be variable (http://stormo.wustl.edu/ScerTF/details/HSF1/). Notably, the B6 allele of Dlgap1 (ATTCTCTTCTCTTCT) has TTC repeats at each end of this putative HSF1 site and is therefore predicted to enable HSF1 binding (p < 0.004). In contrast, the C3H allele of this site in Dlgap1 lacks the second TCC repeat due to the SNP variation, which can be predicted to impair HSF1 binding. Indeed, chromatin immunoprecipitation (ChIP) qPCR in βTC-B6 and βTC-C3H mPanNET cell lines showed enrichment of HSF1 binding at the proposed site within Dlgap1 in B6 compared with C3H (Figure 1C, lower panel). Furthermore, GKAP expression was decreased when we knocked down HSF1 in βTC-B6 cells (Figure 1D), suggesting that HSF1 is an upstream regulator of GKAP.
Cancer Cells from the Invasive B6 Background Have Higher NMDAR Pathway Activity than Those from the Non-invasive C3H Background
We previously established an invasion assay mimicking interstitial pressure-driven fluid flow that activates glutamate secretion and NMDAR signaling, thereby enhancing invasiveness (Li and Hanahan, 2013). Using this assay, we found that B6 cancer cells (βTC-B6) were more invasive than C3H cancer cells (βTC-C3H) (Figure 1E). Moreover, while βTC-B6 and βTC-C3H had similar levels of glutamate secretion under static conditions, flow conditions selectively enhanced glutamate secretion by βTC-B6 compared with βTC-C3H (Figure 1F).
In its capacity as a transmembrane calcium channel, activation of NMDAR by glutamate leads to calcium influx into the cell. Therefore, we used a fluorescent calcium indicator to assess the functionality of NMDAR in βTC-B6 and βTC-C3H cultures (Figure 2A). As the name implies, the synthetic amino acid NMDA is a highly specific and potent ligand for NMDAR, and NMDAR is its only known receptor; as such, a response to NMDA is indicative of a functional NMDAR. When we applied NMDA to cultured cancer cells in puffs from a micropipette, a strong signal for calcium influx was observed in βTC-B6 cells, whereas no signal was detectable in βTC-C3H (Figure 2B), indicating the presence of functional NMDA-responding calcium channels (i.e., NMDAR) in βTC-B6 but not in βTC-C3H. This finding was further confirmed with whole-cell patch-clamp recording, in which application with a micropipette of the synthetic ligand NMDA or of glutamate resulted in NMDAR activation only in βTC-B6 but not in βTC-C3H (Figures 2C and 2D). Furthermore, intracellular perfusion of glutamate through a membrane-sealed micropipette in the whole-cell patch-clamp configuration activated the surface-localized NMDAR in βTC-B6, suggestive of autocrine glutamate secretion from the perfused βTC-B6 cells (Figures 2E and 2F), congruent with a previous report (Robinson and Li, 2017). Notably, besides being distinct in NMDAR activity, the electrophysiology and passive membrane properties in these two cell lines were similar (Figure 2G), and consistent with previous analyses of normal β cells.
Figure 2.

Intracellular Calcium Responses and Electrophysiology Reveals Functional NMDAR in βTC-B6 but Not in βTC-C3H Cells
(A) Oregon Green-labeled calcium indicator BAPTA-AM was applied to βTC-B6 and βTC-C3H cancer cells bathed in a Mg-free Ringer solution; puffing an NMDA solution (1 mM, 1 s, through perfusion pipette at left) induced calcium influx into the cells, thereby producing an increased fluorescence signal (ΔF) compared with the background fluorescence signal (F). The top left image shows βTC-B6 in phase-contrast, whereas the lower left image shows a green-fluorescence signal overlaid with a phase-contrast image. The graph at the right shows time-resolved fluorescence signals (sampling frequency/frame rate = 12.5 Hz), where each trace represents one recorded cell. The y axis indicates the change in fluorescence intensity.
(B) Using the fluorescence reporter assay in (A), the number of βTC cells with active NMDAR signaling was determined following puff application of 1 mM NMDA. The ΔF/F measurements refer to the normalized difference in each cell's signal measured immediately before the application of agonist compared with the peak of the response after the puff. For βTC-B6, 263 cells from 15 different regions of three independent culture dishes were recorded. Light green bar indicates cells with no response; dark green bars indicate cells with ΔF. For βTC-C3H, 155 cells from eight regions of two different dishes were analyzed. Wilcoxon rank-sum test, p < 4.24e−16.
(C) Left: exogenous application of 1 mM NMDA to βTC-B6 cells, using a puffer pipette pressure application during the period shown by gray bar. (Low-noise whole-cell recording, holding at −90 mV.) Right: exogenous application of 500 μM NMDA (two cells), 1 mM NMDA (seven cells), or glutamate (50 μM, four cells) to βTC-C3H cells.
(D) L-Glutamate application to βTC-B6 cancer cells. Three successive membrane current responses (in voltage-clamp mode) are shown, using Mg-free Ringer solution, with a membrane potential of −80 mV (upper panel); voltage-response, including action potentials, was measured in current-clamp (i.e., voltage recording) mode (lower panel).
(E) Intracellular glutamate perfusion during low-noise whole-cell recordings to assess autocrine activation of NMDARs in βTC cells. Left: βTC-B6 cells, n = 12 cells. A segment at higher time resolution is shown at bottom, as indicated. Right: βTC-C3H cells, n = 9 cells. (a–c) Representative segments of recording from one of three different cells.
(F) Current amplitude histogram of autocrine-activated NMDARs in a βTC-B6 cell, showing peaks corresponding with single and double openings of channels (indicated by arrows) with a chord conductance of 48 pS, assuming reversal at 0 mV.
(G) Current-clamp recordings with step current injection in βTC-B6 (left) and βTC-C3H (right).
GKAP Expression Is Associated with Differential NMDAR Activity and Sensitivity to Pharmacological Inhibition In Vivo
We went on to evaluate the NMDAR pathway in PanNETs that had arisen de novo in RIP1Tag2 mice from the two strain backgrounds. The NMDAR includes both GluN1 and GluN2 subunits, of which GluN2b interacts with GKAP. We found that both GluN1 (encoded by Grin1) and GluN2b (encoded by Grin2b) were expressed in B6 and C3H PanNETs at comparable levels, yet GKAP was significantly lower in the C3H background (Figures 3A and B). Notably, while GluN2b was expressed at similar levels in PanNETs from both strain backgrounds (Figures 3B and 3C), p-GluN2b (at Y1252), an indicator of NMDAR activation, was only detectable in B6 but not in C3H PanNETs (Figure 3C). Consistently, C3H RIP1Tag2 mice were largely non-responsive in a pharmacological “intervention” trial using the NMDAR inhibitor MK801, in contrast with B6 RIP1Tag2 mice (Figure 3D). The result establishes that, despite similar expression levels of NMDAR mRNA and protein in PanNETs from both genetic backgrounds, comparatively elevated expression of the scaffold protein GKAP in B6 PanNETs was associated with higher signaling activity, and hence increased sensitivity to pharmacological inhibition. Notably, given that C3H RIP1Tag2 mice have appreciably higher tumor burden compared with B6 RIP1Tag2 mice and that no QTL locus associated with tumor burden (Chun et al., 2010), we conclude that alternative pathways are activated in C3H tumors to promote tumor growth and proliferation independent of the NMDAR-GKAP pathway.
Figure 3.

High GKAP Expression Is Associated with Increased NMDAR Pathway Activity In Vivo
(A) qRT-PCR evaluation for the NMDAR subunits GluN1 (Grin1), GluN2b (Grin2b), and the scaffold protein GKAP (Dlgap1) in PanNET tumors from the two genetic backgrounds. Mean ± SEM. Mann-Whitney test was used to compare the expression of each gene in the B6 and C3H tumors. ∗p = 0.0175 (qRT-PCR: n = 7 tumors/7 mice/background).
(B) Western blot of GluN2b and GKAP in PanNETs from B6 and C3H backgrounds. After normalization, the one-column t test was used for comparison, hypothetical value = 1; ∗p = 0.01; n.s., not significant (mean ± SEM; n = 4 tumors/4 mice).
(C) Immunohistochemistry (IHC) analysis of large T oncoprotein, GluN2b, and p-GluN2b in PanNET tumor tissue sections. Images are representative of >50 tumors from >10 RIP1Tag2 mice/background. S, spleen; LN, lymph node.
(D) MK801 treatment in RIP1Tag2 mice. Cohorts of seven to nine mice were used for control (saline treated) and MK801 treatment in each genetic background; mean ± SEM. Mann-Whitney test: ∗∗p < 0.01; n.s., not significant.
GKAP Knockdown Phenocopies the “Low NMDAR Activity” Status and Reduces Invasiveness of mPanNET Cells In Vitro
The qRT-PCR data implied that the comparatively higher levels of GKAP in B6 cancer cells might be required for activation of glutamate secretion and NMDAR signaling, as GKAP was the only differentially expressed gene from the core NMDAR signaling axis (Figure 3A). We therefore evaluated the impact of GKAP knockdown on NMDAR pathway activity and cell invasiveness in PanNET cancer cells (βTC-3) (Figures 4A and S2A). Indeed, the GKAP-knockdown cells showed decreased GluN2b phosphorylation, indicative of reduced NMDAR signaling activity (Figure 4A). Furthermore, GKAP-knockdown cells exhibited a significantly reduced response to the NMDA ligand in the fluorescent calcium indicator assay (Figure 4B) and decreased invasiveness under flow conditions (Figure 4C). Thus, the GKAP knockdown functionally phenocopied the C3H phenotype, in relation to NMDAR activity and cancer cell invasiveness.
Figure 4.

GKAP Regulates Cancer Cell Invasion through NMDAR Activity and Downstream Effectors FMRP and HSF1
(A) Western blot for GKAP and p-GluN2b in control and GKAP-knockdown βTC-3 cells under unstimulated cell culture conditions. The numbers below indicate levels of p-GluN2b and GKAP normalized to untransfected βTC-3.
(B) Fluorescence reporter assay was performed in βTC-3 small hairpin RNA (shRNA) control and GKAP-knockdown (KD) cells lines, comparing calcium transients induced by puffing an NMDA solution onto the cells. Clusters of cultured control βTC-3 cells or GKAP-KD βTC-3 cells were analyzed under a bright-field microscope after an NMDA solution (1 mM, 1 s) was puffed through perfusion pipette at left (upper panels). The arrows indicate the direction of puffing. βTC-3 shRNA control KD cells: 78 of 284 cells examined showed a response (orange bars). βTC-3 shRNA-GKAP KD cells: 5 of 178 cells showed minor NMDA responses (light blue bars). p < 10−11, Wilcoxon rank-sum test. Histogram of transient amplitude, denoting time-resolved fluorescence signals (sampling frequency/frame rate = 12.5 Hz). Light orange bar and blue bar indicate cells with no response.
(C) Invasion assay in control and GKAP-KD βTC-3 cells. Two-way ANOVA, Bonferroni multiple comparisons test: ∗∗p < 0.01; n.s., not significant (mean ± SEM, n = 3 invasion assay devices/condition in one experiment; two independent experiments were performed with consistent results).
(D) IHC staining of FMRP in B6 PanNETs. Similarly sized invasive versus non-invasive primary tumors on the same section were used for comparison (tumor borders marked by yellow dashed line in the representative images). Rare, multiple metastatic lesions in the liver from one mouse (indicated by the red arrowheads, tumor borders marked by yellow dashed line). Images shown are representative of an analysis of >50 PanNETs from >10 B6 RIP1Tag2 mice, one section per mouse, and all staining was performed in the same experiment. Magnified lesion is representative of >100 metastases from one liver.
(E) Western blot shows the efficiency of FMRP knockdown in βTC-3 cells. The numbers below indicate levels of FMRP normalized to GAPDH. Bar graph: invasion assay. Unpaired t test: ∗p < 0.05; ∗∗p < 0.01. FMRP #1 and #2 indicate two different siRNA constructs used (mean ± SD, n = 3 invasion assay devices per condition in one experiment; two independent experiments).
(F) Western blots comparing FMRP and p-HSF1 expression in βTC-3 cancer cells infected with control shRNA or shRNA-GKAP lentiviral vectors (left), and comparing expression of p-GluN2b, FMRP, and p-HSF1 in βTC-3 cancer cells treated with either vehicle or MK801 (right). GAPDH was used as a loading control and the numbers below indicate levels of p-GluN2b, FMRP, and p-HSF1 normalized to GAPDH. (n = 3).
(G) Left: tissue immunostaining shows expression of total and active GluN2b, FMRP, and total and active HSF1 in tumors from saline- and MK801-treated B6 RIP1Tag2 mice. Data shown are representative of 9–21 random pictures from >15 PanNETs from three mice per group. Scale bar in the blow-up picture represents 25 μm. Right: quantification of FMRP and pHSF1 expression in saline- and MK801-treated tumors. Mean ± SEM. FMRP, n = 9 pictures in saline treated groups, n = 21 in MK801-treated group; p-HSF1, n = 15 pictures in saline treated groups, n = 20 in MK801-treated group. Mann-Whitney test: ∗p < 0.05; ∗∗p < 0.01.
See also Figure S2.
NMDAR Signaling Modulates FMRP and HSF1
In neurons, NMDAR signaling governs synaptic plasticity through regulating local protein translation, for which the mRNA-binding protein FMRP (fragile X mental retardation protein) is critical (Hoeffer and Klann, 2009). Moreover, in neurons, NMDAR activity has been shown to increase FMRP protein expression (Gabel et al., 2004, Todd et al., 2003). Interestingly, a previous study demonstrated that FMRP promoted invasiveness and metastasis of breast cancer cells (Lucá et al., 2013), although the upstream signaling events leading to FMRP activation were unexplored. Therefore, we took a candidate gene approach to investigate FMRP as a potential downstream target of NMDAR signaling in mPanNET.
We found higher FMRP protein expression in B6 tumors compared with C3H tumors (Figure S2B); moreover, in B6 mPanNETs, FMRP expression was particularly elevated in more invasive tumors (Figures 4D, S2C, and S2D) and in liver metastases (Figure 4D). Consistently, knocking down FMRP decreased βTC-3 invasion in vitro (Figure 4E). Interestingly, while FMRP mRNA showed modest stepwise upregulation during PanNET tumorigenesis according to data from a published dataset (Sadanandam et al., 2015) (Figure S2E), the upregulation of FMRP protein in liver metastases when compared with primary tumors was far more impressive (Figures 4D and S2F). This result is consistent with observations in neurons that upregulation of FMRP by NMDAR signaling is largely post-transcriptional (Gabel et al., 2004, Todd et al., 2003).
Intriguingly, previously reported RIP-chip (ribonucleoprotein immunoprecipitation followed by microarray analysis) data showed that FMRP binds to HSF1 mRNA (Ascano et al., 2012). Moreover, both HSF1 and FMRP are known to play important roles in translational regulation (Darnell and Klann, 2013, Santagata et al., 2013). Therefore, we asked whether targeting the NMDAR pathway might also affect HSF1 activation. Indeed, inhibition of NMDAR either by MK801 treatment or by GKAP knockdown decreased both FMRP expression and HSF1 phosphorylation at S326, a marker for its activation (Figure 4F), which was recapitulated by MK801 treatment in vivo (Figure 4G). Notably, mRNA levels for FMRP (Fmr1) remain unchanged after GKAP knockdown or MK801 treatment (Figures S2G and S2H), in contrast to the evident reduction in levels of the protein. Collectively, the data presented above establish GKAP as a genetically polymorphic modifier that differentially regulates an invasive growth program mediated by NMDAR signaling as a function of differential expression governed by genetic background, evidently involving HSF1 and FMRP.
NMDAR Activity in PDAC: a Similar Signaling Axis Uncovered
Next we sought to investigate whether NMDAR activation through GKAP and the consequent upregulation of FMRP/HSF1 could be important for other cancer types. We focused on an appraisal of pancreatic ductal adenocarcinoma (PDAC), in which NMDAR was previously implicated (Li and Hanahan, 2013). We selected two mPDAC cell lines to represent high and low GKAP protein expression. Western blots revealed that the GKAPhigh line (mPDAC-4361) had elevated p-GluN2b compared with the GKAPlow line (mPDAC-2263) (Figure 5A), which was correlated with increased invasiveness, especially under flow conditions (Figure 5A), as well as with increased sensitivity to MK801 inhibition (Figure 5B). Concordantly, knocking down GKAP diminished GluN2b phosphorylation and dampened invasion of the GKAPhigh cell line (Figure 5C). Moreover, p-HSF1 was significantly decreased by GKAP knockdown, while the effect on FMRP levels was marginal (Figure 5C), perhaps reflective of the incomplete knockdown. Congruent results were observed in two hPDAC cell lines, DanG and SUIT2, in which GKAP knockdown and MK801 treatment both decreased hPDAC invasiveness (Figure 5D). MK801 treatment also decreased HSF1 phosphorylation and FMRP expression (Figure 5E). Finally, knocking down either FMRP or HSF1 in hPDAC cells decreased hPDAC invasiveness in the flow-stimulated invasion assay (Figure 5F).
Figure 5.

NMDAR Signaling through GKAP Promotes Invasion in Both Mouse and Human PDAC Cell Lines
(A) Left panels: western blot analysis of GKAP and p-GluN2b levels in mPDAC-4361 and mPDAC-2263 cell lines. The numbers indicate quantification (n = 3). Right panel: invasion assay in static and flow-stimulated conditions. Two-way ANOVA, Bonferroni's multiple comparisons test (right panel): ∗∗∗∗p < 0.0001 (mean ± SEM, n = 3 invasion assay devices per condition per cell line in one experiment; two independent experiments).
(B) MK801 treatment of mPDAC cell lines in static and flow-stimulated invasion assays. The data were normalized to each corresponding “control” in the same static/flow conditions. Two-way ANOVA, Bonferroni's multiple comparisons test: n.s., not significant; ∗p < 0.05 (mean ± SEM, n = 3 invasion assay devices per condition per cell line in one experiment; two independent experiments).
(C and D) GKAP mRNA was knocked-down in mPDAC-4361 (C) and in two hPDAC cell lines, DanG and SUIT2 (D). The knockdown efficiency was assessed by western blot analysis; numbers below indicate levels of GKAP normalized to GAPDH. Cell invasiveness in the invasion assays is shown in bar graphs. Representative images of DAPI-stained nuclei from the invasion assay illustrate the cells that reached the other side of the membrane of a Boyden chamber (scale bar, 100 μm) (C). Two-way ANOVA, Bonferroni's multiple comparisons test (C) or unpaired t test (D): ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. For all invasion assays: n = 3 invasion assay devices per condition in one experiment. Mean ± SEM. Two independent experiments.
(E) Western blot analysis of p-HSF1 and FMRP levels in control and MK801-treated hPDAC cells. The numbers below indicate levels of p-HSF1 and FMRP normalized to GAPDH (n = 3).
(F) Western blot analysis assessing siRNA-mediated HSF1 and FMRP knockdown in DanG cells and SUIT2 cells are shown in the left panels. The numbers below indicate levels of HSF1 and FMRP normalized to GAPDH. Flow-guided-invasion assays of hPDAC cells after HSF1 or FMRP knockdown are shown in bar graphs on the right). Mean ± SEM, unpaired t test: ∗p < 0.05; ∗∗p < 0.01 (n = 3 invasion assay devices/condition in one experiment). Two independent experiments were performed with consistent results.
Next, immunostaining of tumor sections from the PDAC genetically-engineered mouse model (GEMM) revealed that p-GluN2b, GKAP, HSF1, and FMRP were all highly expressed both in primary tumors and in liver metastases (Figure 6A). Furthermore, in human tissue microarrays (TMAs) of PDAC, the expressions of p-GluN2b, GKAP, HSF1, and FRMP all showed a trend of progressive elevation from primary PDAC to lymph node metastases (Figure 6B). Moreover, p-GluN2b was positively associated with GKAP expression (Figure 6C), in further support of the conclusion that variable GKAP expression modulates NMDAR signaling activities in hPDAC. Notably, p-GluN2b also correlated with HSF1 and FMRP expression, with larger tumor size (Figure 6C), and with vascular invasion (Figure 6D). In contrast to PanNET, where GKAP was constitutively expressed at similar levels in the normal pancreatic islets and throughout multistep tumorigenesis (Sadanandam et al., 2015), GKAP was evidently upregulated during the malignant progression in PDAC (Figure 6B), suggesting a distinctive mode of gene regulation.
Figure 6.

The NMDAR/GKAP/FMRP/HSF1 Signaling Axis Is Active in PDAC Tumors
(A) IHC staining of GKAP, FMRP, and HSF1 in both primary tumors (upper panels) and liver metastases (lower panels) from a PDAC GEMM. The primary tumor panels are representative of >5 tumor fields per pancreas from >20 mice. The liver metastasis panels are representative of two liver macro-metastases (∼1 cm in diameter).
(B) IHC staining of p-GluN2b, GKAP, FMRP, and HSF1 in hPDAC tumors displayed in a tissue microarray. Lower table: quantification of immunostaining (percentage) in each tissue section. Wilcoxon rank-sum test.
(C) Correlation between GKAP, FMRP, HSF1, and tumor size with p-GluN2b. Spearman's correlation coefficient.
(D) Correlation between vascular invasion by cancer cells, classified as absent (V0) or present (V1) with p-GluN2b. Wilcoxon rank-sum test.
Transcriptome Profiling Reveals Gene Signatures for Inhibition of NMDAR Signaling
Collectively, our data revealed extensive regulations of the NMDAR signaling axis that are independent of mRNA expression, including phosphorylation (GluNR2b, HSF1), translation (FMRP), in addition to differential binding to DNA (HSF1). While the results from the TMA analysis were informative, immunostaining of patient samples to ascertain NMDAR activity is both time consuming and labor intensive, and often limited by the paucity of appropriate clinical samples. Therefore, we sought to identify gene expression signatures that might be reflective of differential NMDAR activity, by analyzing RNA sequencing (RNA-seq) data from B6 mPanNETs, B6 MK801-treated mPanNETs, and C3H mPanNETs (Figure 7A).
Figure 7.

Identification of Gene Expression Signatures for mPanNETs
(A) Schematic presentation for gene expression signatures associated with tumor phenotypes. Red and blue boxes mark the samples used in the signature analysis; red suggests enrichment of the gene expression signature, while blue suggests depletion.
(B) Mouse strain signature. The heatmap shows major drivers of the strain signature (fold change >2; |Z| > 9) and the boxplot illustrates the standardized signature scores of sample groups. The box marks the 25th to 75th percentiles, and the whiskers show minimum to maximum. The line in the middle of the box indicates the median. No data point is beyond the limit of lines. Genes with |Z| > 9 in the signature are shown. Red, upregulated in B6; blue, upregulated in C3H.
(C) MK801 treatment signature. The heatmap shows the 330 MK801 treatment signature genes, which segregated MK801-treated B6 tumor samples from B6 control tumors, and the boxplot illustrates the standardized signature scores of sample groups. The box marks the 25th to 75th percentiles, and the whiskers show minimum to maximum. The line in the middle of the box indicates the median. No data point is beyond the limit of lines. Genes with |Z| > 9 in the signature are shown. Red, upregulated in MK801-treated B6 tumors; blue, upregulated in B6 control tumors.
(D) GSEA analysis revealed that the MK801 treatment signature showed high enrichment when compared with the strain signature. p = 0 for both up- and downregulated gene sets.
(E) Leading edge analysis from (D) identified 148 common driver genes in both MK801 treatment signature and mouse strain signature, representing NMDAR-pathwaylow signature. Shown here are the “core” common driver genes, which have |Z| > 3 in both signatures. Red, upregulated in MK801-treated B6 tumors and C3H tumors; blue, upregulated in B6 controls.
First, we identified a “strain signature” that distinguished untreated B6 tumor samples from C3H tumor samples (Figure 7B and Table S1). Signature correlation values were represented as Z scores. The higher a |Z| score is, the more likely that the gene is a major factor (driver) in the signature. In our analysis, genes with |Z| > 2.5 were considered to be significantly associated with their corresponding signature. Importantly, Dlgap1 (|Z| = 9.6) was the second most poorly expressed gene in C3H (Figure 7B), consistent with Dlgap1 being a potential phenotype-defining gene distinguishing B6 versus C3H PanNETs. Notably, Dlgap1 has nine differentially spiced protein-coding isoforms, of which three were detected by RNA-seq, and all three were differentially expressed between B6 and C3H PanNETs (Figure S3A). Next, we identified a set of 330 genes that distinguished MK801-treated B6 tumors from untreated B6 tumors (“MK801 treatment signature”, Figure 7C, Tables S2, and S3). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) between the “strain signature” and the “MK801 treatment signature” revealed striking similarity between the two signatures (Figure 7D), suggesting that C3H tumors were similar to MK801-treated B6 tumors when compared with B6 control tumors, which further supported our hypothesis that differences in NMDAR pathway activity are indeed among the defining characteristics distinguishing B6 and C3H PanNETs.
The similarity between these two datasets led us to perform GSEA leading edge analysis (Subramanian et al., 2005) to identify an “NMDAR-pathwaylow signature”, which encompassed common driver genes in both strain signature and MK801 signature (i.e., high in B6 control, low in both MK801-treated and C3H samples, or vice versa) (Figure 7E and Table S4). Pathway analyses of the NMDAR-pathwaylow signature highlighted neurogenesis and synaptogenesis (Figure S3B), consistent with an expectation for genes regulated by NMDAR signaling. Notably, one of the major phenotypes of MK801-treated B6 tumors was decreased proliferation and hence lower tumor burden, in contrast to highly proliferative C3H tumors (Figure 7A). Therefore, selecting for genes that were similarly expressed in both C3H and MK801-treated B6 tumors allowed us to filter out genes commonly associated with cell proliferation and focus on genes more specifically reflecting NMDAR pathway activation.
Gene Expression Signatures for Low NMDAR Activity Predict Better Patient Survival in Multiple Cancer Types
As a prelude to assessing prognostic associations, we performed survival trials in the PDAC GEMM using MK801 and a clinically approved, albeit less potent and less toxic, NMDAR inhibitor, memantine. Both inhibitors prolonged survival in treated animals (Figure 8A), establishing that NMDAR inhibition is therapeutically beneficial for treating PDAC in a mouse model, as previously shown for PanNET (Li and Hanahan, 2013). We then employed the MK801 treatment signature to query The Cancer Genome Atlas (TCGA) database for associated survival differences. A significant survival benefit was associated with PDAC patients whose tumors correlated with the MK801 treatment signature, as if tumors from these patients had been treated with MK801, compared with the rest of the patients (Figure 8B). Moreover, lower-grade tumors (T1/T2) were better associated with the MK801 treatment signature compared with higher-grade tumors (T3/T4) (Figure S4A). Cox proportional hazards regression analysis identified the MK801 treatment signature as a significant, independent prognostic factor, while controlling for other covariates (including T and N stage classification) in a multivariable model (Figure 8C).
Figure 8.

Activity of the NMDAR Signaling Pathway is Associated with Poor Prognosis in Human Cancer Types as Assessed in the TCGA Patient Cohort
(A) MK801 (left panel) and memantine (right panel) treatments in PDAC GEMM. (Left) Control group: 28 mice; median survival, 23 days after enrollment. MK801 group: 25 mice; median survival, 36 days after enrollment. p = 0.0206, log rank test. (Right) Control group: 35 mice; median survival, 13.4 weeks. Memantine group, 33 mice; median survival, 15.4 weeks. Log rank test, ∗p < 0.05.
(B) Survival analysis employing the mPanNET MK801 treatment signature in PDAC patients (n = 13 for associated, n = 165 for not associated). Kaplan-Meier analysis with log rank p value shown.
(C) Cox regression analysis in PDAC patients, both in univariate and multivariable analyses while controlling for other clinical covariates. HR, hazard ratio; CI, confidence interval; T score, primary tumor size/invasiveness; N score, lymph node metastasis; Pinteraction, p value of interaction between significant covariates (model comparison; likelihood ratio test).
(D) Survival analysis employing MK801 treatment signature in patients from several cancer types in addition to PDAC, including glioma (combining low-grade glioma and glioblastoma) and kidney cancers (combining three major subtypes of kidney cancer: chromophobe renal cell carcinoma, clear cell renal carcinoma, and papillary kidney carcinoma). All patients were included in each cancer type shown, regardless of treatment and staging. Brain cancer, associated, n = 566; not associated, n = 93. Kidney cancers, associated, n = 684; not associated, n = 197.
(E) Empirical cumulative distribution function (CDF) plot demonstrating the association of low-grade gliomas (LGG; marked in blue) compared with high-grade glioblastomas (GBM; marked in red) with the (pathway-low) MK801-treatment signature (p < 2.22e-16; Kolmogorov-Smirnov test).
(F) Survival analysis employing NMDAR-pathwaylow signature in PDAC patients (n = 13 for associated, n = 164 for not associated). Kaplan-Meier analysis with log rank p value shown.
(G) Survival analysis employing the NMDAR-pathwaylow signature in patients from several cancer types. All patients were included in each cancer type shown, regardless of treatment and staging. Brain cancer, associated, n = 572; not associated, n = 88. Kidney cancers, associated, n = 501; not associated, n = 378. Uveal melanoma, associated, n = 55; not associated, n = 25.
See also Figure S4.
In addition to PDAC, this MK801 treatment signature was also significantly associated with favorable prognosis in patients with several other cancer types (Figures 8D and S4B). Furthermore, among glial brain cancers, low-grade gliomas were significantly more correlated with the MK801 treatment signature when compared with advanced (more invasive and aggressive) glioblastomas (Figure 8E).
We described above (Figure 7E) a sub-signature of the MK801 treatment signature, the NMDAR-pathwaylow signature, which encompassed driver genes common between the C3H strain signature and the B6-MK801 treatment signature but lacked common proliferation-associated genes that were asynchronous between highly proliferative C3H and poorly proliferative MK801-treated tumors. Analysis of the same patient tumor datasets revealed that this 148 gene NMDAR-pathwaylow signature could also predict survival in PDAC patients (Figure 8F) and other cancer types (Figures 8G and S4C), comparable with the associations revealed by the 330 genes in the full MK801 treatment signature. The results suggest that our approach for identifying phenotype-distinguishing signatures can bypass the obstacle imposed by the extensive post-transcriptional modification of the core NMDAR signaling components and their minimal transcriptional variation, thus presenting a robust and effective method for identifying tumors with high versus low NMDAR pathway activity.
In sum, these data begin to broaden the association of NMDAR signaling (via GKAP, HSF1, and FMRP) with invasive tumor growth and malignancy. The results additionally suggest that NMDAR antagonists may be therapeutically beneficial in PDAC and other cancer patients whose tumors are inferred to have elevated NMDAR signaling by virtue of lacking this favorable “NMDARlow” MK801 treatment signature.
Discussion
It is well recognized that genetic background can have significant impact on cancer susceptibility, progression, and response to treatment, both in humans and in mice. Here, we present evidence that expression of the intracellular signal transducer GKAP modulates activity of the glutamate-to-NMDAR signaling pathway in the genetically engineered RIP1Tag2 mouse model of cancer, producing tumors that are invasive or not, as a function of polymorphic variation in constitutional levels of GKAP expression in the cell of origin. We further demonstrate via GKAP knockdown that activation of NMDAR signaling, governed by GKAP and mediated through FMRP/HSF1, promotes invasiveness in mouse neuroendocrine (PanNET) and in both mouse and human ductal (PDAC) cancer cell lines. We also identify gene expression signatures associated with GKAP-mediated NMDAR signaling and its pharmacological inhibition that reveal striking prognostic associations in various cancer types.
As part of a multi-protein scaffold involved in transmitting ligand-induced signals from NMDAR, GKAP protein interacts with a number of other synaptic scaffold proteins. Thus, GKAP is considered to be an adaptor between the core synaptic NMDAR complex and subsynaptic signaling molecules in neurons. We envision that GKAP similarly orchestrates diverse functional effects in cancer cells as a result of connecting downstream effectors with the NMDAR, thereby affecting multiple signaling circuits. The finding that a SNP in the HSF1 binding site of Dlgap1 might contribute to the differential expression of GKAP between the B6 and C3H backgrounds echoes a previous study in which a SNP within an intron of a gene resident in a super enhancer region affected binding of a transcription factor and hence gene expression, and in turn susceptibility to neuroblastoma (Oldridge et al., 2015). While the Dlgap1 SNP we identified is not conserved in humans, bioinformatic analysis has found a number of SNP sites in the human DLGAP1 promoter/enhancer regions that overlap with potential HSF1 binding sites (data not shown). Whether these SNPs might be associated with differential GKAP expression and/or prognosis in cancer patients is worthy of future investigation.
NMDAR is instrumental in learning and memory through regulating synaptic plasticity, which involves extensive de novo protein synthesis (Hoeffer and Klann, 2009). NMDAR activation has been shown to increase translation of certain proteins without affecting their mRNA levels. While protein translation is frequently implicated in proliferation, several lines of evidence have demonstrated an additional role in cancer invasion and metastasis (Hsieh et al., 2012). Moreover, localized translation contributes to invadopodia formation (Klemke, 2012), and FMRP has been shown to localize mRNA to invadopodia (Mili et al., 2008), consistent with the important role FMRP plays in localized translational activities in neurons. As the name implies, FMRP is best known for its association with the fragile X mental retardation syndrome (FXS), where its loss of expression leads to abnormal synaptic function and resultant impairment in learning and memory. FMRP regulates mRNA translation of several components of the NMDAR interactome (Darnell and Klann, 2013), and decreased NMDAR-dependent synaptic activity has been reported in Fmrp knockout mice (Bostrom et al., 2015). Intriguingly, FXS patients, who have reduced FMRP protein expression, were previously reported to have a reduced overall cancer incidence (Schultz-Pedersen et al., 2001), and FMRP was also implicated in aggressiveness of breast cancer (Lucá et al., 2013). Now our report further establishes a mechanistic foundation for FMRP in cancer. We showed that FMRP activation is downstream of the NMDAR/GKAP signaling complex and is functionally involved in the invasive phenotype. Interestingly, while we identified FMRP in mouse neuroendocrine tumors, FMRP overexpression is also evident in certain other cancer types, including PDAC (our results), cervical cancer (Jeon et al., 2011), and breast cancer (Lucá et al., 2013), suggesting a broader role in epithelial tumors.
The transcription factor HSF1 orchestrates malignant phenotypes of various tumors (Mendillo et al., 2012). Here, we further highlight the importance of HSF1 activity in invasion via its interconnection with the NMDAR pathway. Interestingly, HSF1 may sit both upstream and downstream of NMDAR/GKAP: on the one hand its activity is upregulated by NMDAR/GKAP signaling, while on the other it is implicated in a positive feedback loop that regulates GKAP expression via the HSF1 binding sites within the GKAP gene. Similarly, we show that NMDAR/GKAP signaling increases expression of FMRP protein, whereas other reports have shown that FMRP regulates translation of GluN2b and additional components of the NMDAR signaling apparatus in neurons (Bostrom et al., 2015), suggestive of a feedback circuitry. In addition, the regulation and interaction between HSF1 and FMRP (and related family proteins) might be reciprocal and complex as well (Ascano et al., 2012, Santagata et al., 2013); it will be of interest in future studies to further characterize the functional roles and regulatory control of HSF1 and FMRP (and the details of their interconnections).
It is notable that the NMDAR signaling axis is highly complex, regulated at multiple levels to allow for fine-tuning of neuronal activities. In addition to the GKAP-dependent GluN2b (NR2b) subunit, which we have implicated in this study, there are several other NMDAR subunits that can alternatively assemble into the heterotetrameric receptor. The obligatory subunit NR1/GluN1 can pair with three other NR2/GluN2 subunits (GluN2a, GluN2c, GluN2d) and two more NR3/GluN3 subunits (GluN3a, GluN3b). Among these, GluN2a and GluN2b are the most commonly expressed in neurons, but the relative levels and potential functional roles of each subunit in cancer remain to be investigated. These NMDAR subunits have different intracellular C-terminal domains, allowing for preferential association with different downstream scaffold proteins (Paoletti et al., 2013). Additionally, three of nine differentially spiced isoforms of GKAP are (differentially) expressed in the mPanNETs. Interestingly, it has been shown that splice variants of DAP102, another scaffold protein of NMDAR, differentially regulate NMDAR trafficking in a subunit-dependent manner (Wei et al., 2015). As such, it remains to be explored whether some or all GKAP isoforms specifically contribute to invasive tumor growth in this and other cancer types. Moreover, Dlgap1 is one of the five members of the Dlgap gene family (Dlgap1–Dlgap5); whether others can contribute to NMDAR activity in tumors also remains to be elucidated.
Given this complexity, single-gene-based assessment of GKAP or of the core NMDAR signaling components in tumor samples is not certain to be informative about pathway activity and its association with prognosis. Rather, it will be auspicious to begin prognostic assessment with identification of NMDAR pathway-high versus pathway-low tumors using the MK801-treatment/NMDAR-pathwaylow signatures described here. Then, for tumors showing a pathway-high association, individual genes in the NMDAR signaling axis could be profiled, noting the proviso that some components are regulated post-transcriptionally, by translational control, by phosphorylation, and potentially by alternative mRNA spicing.
In conclusion, we have presented several lines of evidence that reveal how differential expression of GKAP promotes cancer invasion through modulating the NMDAR pathway. The results conceptually expand and extend previous studies documenting activity of NMDAR signaling in cancer and the benefits of its pharmacological inhibition, suggesting that co-option of this neuronal signaling pathway to promote malignant growth may prove to be a relatively common phenomenon.
Preclinical trials in the mouse model of PDAC with NMDAR inhibitors, one of which (memantine) is clinically tractable, suggest that therapeutic targeting of NMDAR signaling may have benefit in this highly malignant form of human cancer. Notably, the extended survival plateau in PDAC patients with an MK801 treatment signature indicative of non-induced/repressed glutamate-NMDAR signaling is very striking, considering that the overall 5-year survival rate is only 6% in PDAC patients; similar associations in other tumor types are also provocative. Therapeutic targeting of NMDAR signaling may therefore have promise in a precision medicine strategy focused on patient subsets whose tumors express the core NMDAR pathway genes, including GKAP, but lack the MK801 treatment signature, which predicts impaired NMDAR signaling (and hence therapeutic non-responsiveness and better prognosis), so as to focus on potentially responsive at risk cohorts. Finally, we have begun in this study to chart the downstream pathways that orchestrate the invasive growth phenotype driven by GKAP-modulated glutamate-NMDAR signaling, implicating two effectors, FMRP and HSF1, in distinctive forms of pancreatic cancer.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NMDAR2B Polyclonal Antibody | Pierce | #PA3-105; RRID: AB_2112445 |
| Anti-SAPAP1 antibody produced in rabbit | Sigma-Aldrich | #PRS4623; RRID: AB_1850605 |
| Human GKAP/DLGAP1 MAb (Clone 727425) antibody | R&D | #MAB7296; RRID:AB_10972459 |
| Rabbit Anti-FMRP Polyclonal Antibody, Unconjugated | Cell Signaling | #4317; RRID: AB_1903978 |
| Phospho-NMDAR2B (Tyr1252) Polyclonal Antibody | Life technology | #485200; RRID: AB_2533846 |
| HSF1 Antibody | Cell Signaling | #4356; RRID: AB_2120258 |
| HSF1 (phospho S326) antibody [EP1713Y] | Abcam | #ab76076, RRID:AB_1310328 |
| Biological Samples | ||
| Human Pancreatic Tissue Microarray | Institute of Pathology, Universität Bern, Bern, Switzerland http://www.ngtma.com/Home.10.html |
N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK801 | Sigma-Aldrich | #M107-50MG |
| Memantine | Tocris | #0773/50 |
| Critical Commercial Assays | ||
| Amplex Red glutamine acid/glutamate oxidase assay kit | Thermo scientific | #A12221 |
| Deposited Data | ||
| RNA-seq | GEO | GSE102598 |
| Experimental Models: Cell Lines | ||
| βTC-B6 (βTC-PO1) | This paper. | N/A |
| βTC-C3H | This paper. | N/A |
| βTC-3 | (Efrat et al., 1988) | RRID: CVCL_0172 |
| mPDAC-2263 | This paper. | N/A |
| mPDAC-4361 | This paper. | N/A |
| DanG | DSMZ | RRID: CVCL_0243 |
| SUIT2 | JCRB | RRID:CVCL_3172 |
| Experimental Models: Organisms/Strains | ||
| C57/Bl6N | Charle river laboratories | Strain Code: 027 |
| C3HeB/FeJ | Jackson laboratories | SN : 658 |
| RIP1-Tag2 in C57/Bl6 strain background | (Chun et al., 2010) | N/A |
| RIP1-Tag2 in C3Heb/Fe strain background | (Chun et al., 2010) | N/A |
| Ptf1a-Cre lox-stop-lox(LSL)-KrasG12D p53R172H/+ in pure FBV/n strain background | (Hingorani et al., 2005). Backcrossed into pure FBV/n background in our laboratory. | N/A |
| Oligonucleotides | ||
| GKAP/dlgap1 shRNA | Openbiosystem | Clone ID: TRCN0000088935 |
| GKAP/dlgap1 (human) siRNA #1 | Thermo Fisher Scientific | #4392420, Assay ID s17644 |
| GKAP/dlgap1 (human) siRNA #2 | Thermo Fisher Scientific | #4392420, Assay ID s52141 |
| FMRP/fmr1 (human) siRNA #1 | Thermo Fisher Scientific | #4392420, Assay ID s5316 |
| FMRP/fmr1 (mouse) siRNA #1 | Thermo Fisher Scientific | #4390771, Assay ID s66176 |
| FMRP/fmr1 (mouse) siRNA #2 | Thermo Fisher Scientific | #4390771, Assay ID s66177 |
| HSF1 (human) siRNA #1 | Thermo Fisher Scientific | #4392420, Assay ID s6951 |
| HSF1 (human) siRNA #2 | Thermo Fisher Scientific | #4392420, Assay ID s6952 |
| HSF1 (mouse) siRNA #1 | Thermo Fisher Scientific | #4390771, Assay ID s67870 |
| HSF1 (mouse) siRNA #2 | Thermo Fisher Scientific | #4390771, Assay ID s67871 |
| FMR1 (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm01339582_m1 |
| DLGAP1 (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm00510688_m1 |
| HSF1 (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm01201402_m1 |
| Rpl19 (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm02601633_m1 |
| GluN1/grin1 (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm00433790_m1 |
| GluN2B/grin2b (mouse) Taqman Probe | Thermo Fisher Scientific | # 4331182, Assay ID Mm00433820_m1 |
| Software and Algorithms | ||
| Graph Pad Prism 7.0 | Graphpad | RRID:SCR_002798 |
| MetaCore | Thomson Reuters https://portal.genego.com/ |
RRID:SCR_008125 |
| Independent Component Analysis for RNA-seq data | This paper. | N/A |
| Other | ||
| Millicell-PCF 8.0μm insert 12mm diameter | Millipore | #PI8P01250 |
| Growth factor reduced matrigel, phenol red free 10 ml | BD Bioscience | #356231 |
| Rat tail collagen type I ABOVE 4.0 ug/ml 100 ml | BD Bioscience | #354236 |
| Vectastain Elite ABC kit | Vector Labs | #PK-6100 |
| DAB | Sigma-Aldrich | #D5637-1G |
| Signal Stain Antibody Diluent | Cell Signaling | #8112 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas Hanahan (douglas.hanahan@epfl.ch).
Experimental Model and Subject Details
Mice
All studies involving mice were approved by the committee for animal research of Canton Vaud, Switzerland, in accordance with the Swiss regulation of experimental animal welfare. All mice used in this study were maintained in the Center of Phenogenomics at EPFL.
Cell Culture
βTC-B6 (βTC-PO1) and βTC-C3H were derived by former members (Peter Olson and Matthew G. Chun, respectively) in the Hanahan lab, from B6 and C3H PanNETs, respectively. Both lines were cultured in DMEM (Life Technology #41965) with 10 % FBS and pan-strep. The βTC-C3H line was maintained at low passage number, particularly for the flow-based invasion assay, preferably less than 20 passages. All the cell lines described in this paper were cultured using the same culture conditions.
Intervention Trial with MK801 in RIP1Tag2 Mice
Intervention trials were performed as described previously (Li and Hanahan, 2013). Only male mice were included in the trial, as the tumor burden is significantly different between male and female RIP1Tag2 mice (Chun et al., 2010). The trials start at 10.5 weeks and end at 14 weeks. Saline or MK801 (1 mg/kg) was injected intraperitoneally daily for 5 days a week. At end point, the mice were cardiac-perfused with normal saline followed by zinc formalin, and the tissues were harvested. The PanNETs were measured by caliber, and the volume of each tumor was calculated by the following formula: (short axis)2 x (long axis) x 0.52. The total tumor burden represents the cumulative tumor burden from all tumors in the pancreas.
Survival Trial with MK801 and Memantine in PDAC Mice
A mouse model of pancreatic ductal adenocarcinoma (LSL-KrasG12D; p53LSLR172H; p48cre (Hingorani et al., 2005)) was used for the survival analysis. Different treatment schemes have been utilized for PDAC preclinical trials (Gopinathan et al., 2015): for early intervention/survival trials, PDAC mice were typically enrolled from 10-12 weeks of age (Miller et al., 2015, Olson et al., 2011); for later intervention/survival trials, PDAC mice were monitored by ultrasound regularly, until the enrolment sizes were reached. We employed the early intervention/survival trial scheme for MK801 trial. Both male and female mice were included. Whenever possible, gender- and weight-matched littermates were evenly distributed into control (saline) and MK801/Memantine treatment groups. The treatment started from 10.5 weeks onwards. MK801 was diluted in normal saline, and administrated at 1 mg/kg, once daily, i.p., 5 days a week. For the MK801 trial, when the mice reached 14 weeks old, they were given a one-week drug break in order to mitigate drug-related toxicity. Thereafter, the mice were dosed for 3 days a week from 15 weeks old until the end stage. Memantine was diluted in normal saline, and administered at 10 mg/kg, once daily, i.p., 5 days a week, continuously from 10.5 weeks until the animal reached end point. Weight loss of more than 15 % of initial body weight, or the development of severe ascites, was considered to dictate end points.
Method Details
Invasion Assay
As described previously (Li and Hanahan, 2013). A gel composite of 1.2 mg/ml rat tail collagen type I and 10 % growth factor reduced matrigel was prepared. The final gel/cell mixture will be 500 μl for each set of technical triplicates, with 275 μl of gel, 125 μl of cell suspension (at 2 million/ml), and either 100 μl of medium or 100 μl of 500 μM MK801 diluted in medium (so that the final concentration of MK801 will be 100 μM). Ice-cold pipette tips were used whenever they need to be in contact with the gel, and the gel was kept on ice until adding to the insert. 150 μl gel/cell mixture per well was placed onto transwell inserts. The gel/cell mixture was added to the side of the transwell, avoiding direct contact of the membrane. The inserts were placed into 24 well plates, then put into the 37°C incubator for ∼40 min - 1 hr until the gel solidified. After incubation, plates were removed and for static conditions: medium was added (150 μl) to the top of the gel and underneath the transwell (650 μl); for flow conditions: medium was added (650 μl) to the top of the gel and underneath the transwell (150 μl). Only serum-free medium (DMEM) was used in the whole device. After overnight incubation (∼16-18 hr), inserts and medium were removed, and the gels were wiped with cotton tips while avoid disturbing the bottom side of the membrane followed by fixation with ice-cold methanol and washes with PBS for three times. DAPI staining was used for visualization of nuclei under upright microscope. The results were quantified using Fiji.
Immunohistochemical Staining
As described previously (Li and Hanahan, 2013). Zinc-formalin-fixed, paraffin-embedded tissue sections were used. Mice were intracardially-perfused with PBS and zinc-formalin before tissue harvest. Slides were immersed in citrate buffer (pH = 6.0) for antigen retrieval for 20 min at 95°C. Endogenous peroxidase activity was blocked with 3 % H2O2 in PBS for 10 min at room temperature. Protein blocking was done 1 hr at room temperature with 5 % normal donkey serum in 0.1 %TBST. Primary antibodies were incubated in the cold room overnight in blocking buffer, followed by biotinylated secondary antibodies incubation at room temperature for 30 min, then by incubation with avidin/biotinylated complex for 30 min at room temperature. 0.1 % TBST was used as wash buffer. DAB was used for visualization for 8 min at room temperature. After washes, stained slides were counterstained with Meyer’s hematoxylin. For HSF1 IHC, Signal Stain Antibody Diluent was used as suggested on the antibody datasheet.
Transcription Factor Prediction
SNPs were retrieved from the SNP query tool of MGI at http://www.informatics.jax.org/javawi2/servlet/WIFetch?page=snpQF. Within the Dlgap1 gene and its 2Kb upstream, “different” SNPs between C57BL/6 (reference) and C3H/HeJ strains were selected (205 dbSNP from Build 137 entries). These SNP sites were subjected to further analysis using FIMO (Grant et al., 2011) with Transfac 2012 Position Weight Matrices (PWM) to predict transcription factor binding.
Chromosome Immune-Precipitation (ChIP) Assay
ChIP was performed as described previously (Mendillo et al., 2012), with several modifications. After immunoprecipitation, enrichments were determined using SYBRSelect Master Mix for CFX (Life Technologies) on the CFX96 Real-Time PCR Detection System (Bio-Rad). Enrichments were verified using two biological replicates, each with two ChIP experimental replicates, followed by ChIP-qPCR in duplicate. The primer pairs used were: HSP70 (Hspa1a) (forward: 5’-TCCAGCAGTTTCGCGTCTG-3’; reverse: 5’-TCCGTACTCTCCAGTGAACCC-3’); potential HSF1 binding element on the GKAP gene (forward: 5’-CATTTTAAAACTAGGCCCAAACTCAGC-3’; reverse: 5’-AGAGAATATTTCTTCCCCAGGTAGGAT-3’); β-maj (Hbb-b1) (forward: 5’-GGGAGAAATATGCTTGTCATC-3’; reverse: 5’-CAACTGATCCTACCTCACCTT-3’). In brief, for cell lines, cell numbers were counted after trypsinization, and crosslinked with 1 % formaldehyde solution (Sigma) in 10 % FBS/PBS. The cell pellets were resuspended in sonication buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1 % Na-Deoxycholate, 0.5 % N-lauroylsarcosine, 1× protease inhibitors), and sonicated with a Bioruptor Next Gen (Diagenode). After sonication, TritonX was added to a final concentration of 1 %. DNA concentration was measured with Qubit dsDNA BR Assay kit (Life Technologies, #Q32850) on the Qubit Fluorometer (LifeTechnologies). Sonicated DNA was incubated with anti-HSF1 antibody (Santa Cruz, #9144) at 4°C overnight with rotation. On the next day, 25 μl Pierce Protein A/G Magnetic beads (Thermo Scientific, #88803) were added to each sample, and rotated at 4°C for 1.5 hour. The beads were then washed with the following buffers: 6X with low salt washing buffer (Noordermeer et al., 2011), 2X with high salt washing buffer (Noordermeer et al., 2011), 2X with LiCl washing buffer (Noordermeer et al., 2011), and 2X with TE + 50 mM NaCl. Finally, the beads were eluted with 210 μl elution buffer (Lee et al., 2006) at 65°C for 30 minutes, with vortexing every 5 minutes. The crosslink was reversed by incubating samples at 65°C overnight. DNA purification was done according to previous protocol (Lee et al., 2006), and DNA concentration was measured with the Qubit dsDNA HS Assay kit (Life Technologies).
Western Blotting
Transfer was performed using nitrocellulose membrane (GE Healthcare), and blocked with 5 % BSA in 0.1 % TBST. Primary antibody was diluted in the same blocking buffer and incubated at either room temperature for two hours or 4°C overnight, and secondary antibody was incubated for 1 hour at room temperature in blocking buffer, followed by visualization with SuperSignal West Pico Chemiluminescent Substrate (Pierce, #34077). Imaging was done on the Fusion Fx7 imaging platform for fluorescence and chemiluminescence (PEQLAB, Germany), and quantification was performed using build-in software and Fiji. Background subtraction was performed using the rolling-ball algorithm. After quantification, images were inverted to black signal/white background, and adjusted for better visualization using Photoshop.
Electrophysiology
Cells were bathed in a magnesium-free Ringer solution, containing (mM): 140 NaCl, 2.5 KCl, 2 CaCl2, 10 glucose, 0.01 glycine, 10 HEPES/Na, pH adjusted to 7.4 with NaOH. For whole-cell recording, patch pipettes were filled with a solution containing 105 K gluconate, 30 KCl, 10 HEPES/KOH, 4 ATP / Mg, 0.3 GTP Na2, 10 creatine phosphate / Na, pH adjusted to 7.3 with NaOH. Membrane potentials were corrected for liquid junction potential nulled before seal formation. Whole-cell recordings were established using an Axopatch 200A patch-clamp amplifier (Axon Instruments) in capacitative feedback mode for low noise, low-pass filtered at 5 kHz (Bessel, 8-pole) and sampled at 20 kHz with 16-bit resolution using a National Instruments X-series board. Further low-pass Gaussian filtering at corner frequencies between 500 Hz – 1 kHz was applied offline. Glutamate and NMDA responses were elicited by pressure ejection of agonist dissolved in the Ringer solution, through pipettes with tip diameters of 10-20 μm, and pressures steps of 5-10 mbar.
Intracellular Calcium Measurement
For recording calcium signals, cells were loaded with the fluorescent indicator Oregon Green 488 BAPTA-1 AM (Life Technologies) at 5 μM for 1 hour, and imaged using epifluorescence (Olympus IX71 microscope, UMPlan FI 10X objective, X-Cite 120 light source, EXFO Photonic Solutions), and a sCMOS camera (Zyla 4.2P, Andor). Using the Matlab Image Processing toolbox (Mathworks), cell regions were selected, and the average signals across pixels in each region were analysed as the change in fluorescence (ΔF) relative to the baseline level (F), i.e. ΔF/F.
RNA-Seq Sample Collection (B6/C3H/MK801)
mPanNETs were collected from RIP1Tag2 mice ranging from 14-16 week old (end stage). Control mice were either not treated, or treated with normal saline (“sal”) for 3 days prior to sacrifice; MK801-treated mice were treated for 3 days prior to sacrifice. Harvested tumors were snap-frozen in liquid nitrogen, before being processed for RNA-extraction. The miRNeasy kit (QIAGEN) was used for RNA extraction. Tissue homogenization was done on TIssueLyser II (QIAGEN) in pre-cooled cassettes, with disposable beads in 700 μl Qiazol (from the miRNeasy kit)/ tumor. On-column DNase digestion (QIAGEN) were performed for all samples during RNA extraction according to manufacturer’s protocol.
Antibodies
GluN2b (Pierce #PA3-105) for IP, WB and IHC, (NeuroMab #75-097, 75-101) for WB; GKAP (Sigma # PRS4623) for IHC, IP, and WB 1:1000 in mouse, (R&D #MAB7296) for IHC in human PDAC; FMRP (Cell Signaling, #4317); HSF1 (Santa Cruz, #sc-9144) for ChIP, (Cell Signaling, #4356) for IHC 1:50.
siRNA Knockdown of Human PDAC Cell Line
100 pmol commercially available siRNA (Life Technologies) was transfected with 5 μl Lipofectamine 2000/well in 6 well plates. Data was analyzed 30 hours post transfection.
Quantification and Statistical Analysis
RNA-Seq Analyses
Illumina HiSeq 2500 101-nt single-ended reads were mapped to the UCSC mm9 mouse genome build (http://genome.ucsc.edu/) using RSEM (Li and Dewey, 2011). Raw estimated expression counts were upper-quartile normalized to a count of 1000 (Bullard et al., 2010). Given the complexity of the dataset in terms of a mixture of different biological conditions, a high-resolution signature discovery approach was employed to characterize global gene expression profiles. Independent Component Analysis (ICA), an unsupervised blind source separation technique, was used on this discrete count-based expression dataset to elucidate statistically independent and biologically relevant signatures (Dimitrova et al., 2016, Li et al., 2015). ICA is a signal processing and multivariate data analysis technique in the category of unsupervised matrix factorization methods.
Conceptually, ICA decomposes the overall expression dataset into independent signals (gene expression patterns) that represent distinct signatures. High-ranking positively and negatively correlated genes in each signature represent gene sets that drive the corresponding expression pattern (in either direction). Multi-sample signatures were visualized using relative signature profile boxplots. Biologically relevant and statistically significant signatures were identified using a Mann-Whitney U test. Each signature is two-sided, allowing for identification of up-regulated and down-regulated genes for each signature within each sample.
Formally, utilizing input data consisting of a genes and samples matrix, ICA uses higher order moments to characterize the dataset as a linear combination of statistically independent latent variables. These latent variables represent independent components based on maximizing non-gaussianity, and can be interpreted as independent source signals that have been mixed together to form the dataset under consideration. Each component includes a weight assignment for each gene that quantifies its contribution to that component. Additionally, ICA derives a mixing matrix that describes the contribution of each sample towards the signal embodied in each component. This mixing matrix can be used to select biologically relevant signatures among components with distinct gene expression profiles across the set of samples. The R implementation of the core JADE algorithm (Joint Approximate Diagonalization of Eigenmatrices) (Biton et al., 2013, Miettinen et al., 2017, Rutledge and Jouan-Rimbaud Bouveresse, 2013) was used along with custom R utilities. All RNA-seq analyses were conducted in the R Statistical Programming language (http://www.r-project.org/). Isoform-level differential analyses were performed using EBSeq (Leng et al., 2013). Gene set enrichment analysis (GSEA) was carried out using the pre-ranked mode with default settings (Subramanian et al., 2005). Enrichment maps were generated using the EM (Merico et al., 2010) plugin for Cytoscape (Shannon et al., 2003). Heatmaps were generated using the Heatplus package in R. For the derivation of common driver genes in the strain and the MK801 treatment signatures, GSEA leading edge analyses (Subramanian et al., 2005) were performed by using the ranked gene list from the strain signature to interrogate against the driver genes (defined by |Z|>3) in the MK801 treatment signature, and vice versa. The union of these two analyses was taken as the common driver genes.
Clinical Data Analysis
Genes with standardized signature correlation scores |z| > 3 were used as gene sets to score TCGA (https://tcga-data.nci.nih.gov/tcga/) patient datasets using ssGSEA (Barbie et al., 2009). Patient tumors were stratified using standardized scores and Kaplan-Meier survival analyses were conducted to compare the top scoring patients (within 3.5 of the top Z score) with the low scoring patients (within 3.5 of the bottom Z score) to assess differences in survival time. P values from the log-rank test are reported. Cox proportional hazards analysis was conducted across all patients in the TCGA PDAC cohort (with survival greater than 30 days to eliminate possible complications arising from surgical intervention) to asssess the prognostic significance of the signature while controlling for various clinical covariates. Hazard ratio proportionality assumptions for the Cox regression model fit were validated by testing for all interactions simultaneously (p=0.422). All survival analyses were conducted using the survival package in R.
Statistics
Statistics was performed using GraphPad Prism. In general, when technical replicates were shown for in vitro experiments, student-t-test was used for statistic analysis, and the same experiment was at least repeated once with similar trend observed. When data from multiple experiments was merged into one figure, Mann-Whitney test was performed. If each experiment was normalized to its internal control, then one-column t-test was performed. For in vivo experiments, Mann-Whitney test was used. ANOVA with post-hoc test was performed when analyzing experiments with multiple variants. Detailed statistic methods for each figure can be found in corresponding figure legends. The Kolmogorov-Smirnov test was used to assess statistical significance in ECDF plots.
Data and Software Availability
The RNA-seq data for mPanNETs has been deposited in the GEO database under ID codes GSE102598.
Acknowledgments
We thank Tyler E. Jacks (Massachusetts Institute of Technology [MIT]) for his gracious support of L.L. and A.B.; Ehud Drori for technical support; the Duboule lab (Swiss Institute for Experimental Cancer Research/École Polytechnique Fédérale de Lausanne [EPFL]) for assistance with ChIP experiments; A. Necsulea (EPFL and Université de Lyon) for bioinformatics analysis; J. Delafontaine and J. Rougement (EPFL) for transcription factor analysis; the De Palma lab (EPFL) for help with lentiviral infection; M.G.H. Chun (University of California, San Francisco [UCSF] and Salk Institute), P. Olson (UCSF), and K. Shchors (EPFL) for cell line and reagents; S. Saqafi (EPFL) for bioinformatic advice; A. Jaegger (MIT), A. Balmain (UCSF), G. Ciriello (University of Lausanne [UNIL]), and I. Michael (EPFL) for constructive comments on the manuscript; and the Genome Core at UNIL and the Histology and Animal Care Facilities at EPFL for technical services. The PDAC TMA was made available through Tissue Biobank Bern (TBB, University of Bern). This research was supported by an Advanced Grant from the European Research Council (grant numbers: 587516/322491).
Author Contributions
L.L., M.L.I.-A., and D.H. conceived the study. H.R. performed all NMDAR activity analyses (fluorescence reporter assay and electrophysiology studies). J.A.G., E.K., A. Perren, and I.Z. performed hPDAC TMA analysis. L.L., Q.Z., and M.W.P. performed all other experiments. A.B. conducted bioinformatics analyses. A. Piersigilli. and A. Perren consulted on mouse pathology. D.N. helped with the ChIP experiment. L.L., M.L.I.-A., and D.H. wrote the paper with input from all authors.
Declaration of Interests
Three of the co-authors (L.L., A.B., and D.H.) are in the process of filing a patent application for the MK801 treatment signature and the NMDAR-low signature.
Published: March 29, 2018
Footnotes
Supplemental Information includes four figures and four tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.02.011.
Supplemental Information
References
- Abbott L.F., Nelson S.B. Synaptic plasticity: taming the beast. Nat. Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Ascano M., Jr., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton, A., Zinovyev, A., Barillot, E., and Radvanyi, F. (2013). MineICA: independent component analysis of transcriptomic data. https://www.bioconductor.org/packages/devel/bioc/vignettes/MineICA/inst/doc/MineICA.pdf
- Bostrom C.A., Majaess N.M., Morch K., White E., Eadie B.D., Christie B.R. Rescue of NMDAR-dependent synaptic plasticity in Fmr1 knock-out mice. Cereb. Cortex. 2015;25:271–279. doi: 10.1093/cercor/bht237. [DOI] [PubMed] [Google Scholar]
- Bullard J.H., Purdom E., Hansen K.D., Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun M.G., Mao J.H., Chiu C.W., Balmain A., Hanahan D. Polymorphic genetic control of tumor invasion in a mouse model of pancreatic neuroendocrine carcinogenesis. Proc. Natl. Acad. Sci. USA. 2010;107:17268–17273. doi: 10.1073/pnas.1012705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.C., Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N., Gocheva V., Bhutkar A., Resnick R., Jong R.M., Miller K.M., Bendor J., Jacks T. Stromal expression of miR-143/145 promotes neoangiogenesis in lung cancer development. Cancer Discov. 2016;6:188–201. doi: 10.1158/2159-8290.CD-15-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrat S., Linde S., Kofod H., Spector D., Delannoy M., Grant S., Hanahan D., Baekkeskov S. β cell lines derived from transgenic mice expressing a hybrid insulin oncogene. Proc. Natl. Acad. Sci. USA. 1988;85:9037–9041. doi: 10.1073/pnas.85.23.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel L.A., Won S., Kawai H., McKinney M., Tartakoff A.M., Fallon J.R. Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J. Neurosci. 2004;24:10579–10583. doi: 10.1523/JNEUROSCI.2185-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinathan A., Morton J.P., Jodrell D.I., Sansom O.J. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model Mech. 2015;8:1185–1200. doi: 10.1242/dmm.021055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant C.E., Bailey T.L., Noble W.S. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hoeffer C.A., Klann E. NMDA receptors and translational control. In: Van Dongen A.M., editor. Biology of the NMDA Receptor. CRC Press/Taylor & Francis; 2009. Chapter 6. [PubMed] [Google Scholar]
- Hsieh A.C., Liu Y., Edlind M.P., Ingolia N.T., Janes M.R., Sher A., Shi E.Y., Stumpf C.R., Christensen C., Bonham M.J. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S.J., Seo J.E., Yang S.I., Choi J.W., Wells D., Shin C.Y., Ko K.H. Cellular stress-induced up-regulation of FMRP promotes cell survival by modulating PI3K-Akt phosphorylation cascades. J. Biomed. Sci. 2011;18:17. doi: 10.1186/1423-0127-18-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R.L. Trespassing cancer cells: ‘fingerprinting’ invasive protrusions reveals metastatic culprits. Curr. Opin. Cell Biol. 2012;24:662–669. doi: 10.1016/j.ceb.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Johnstone S.E., Young R.A. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M., Haag J.D., Gould M.N., Stewart R.M., Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.M., Gocheva V., Oudin M.J., Bhutkar A., Wang S.Y., Date S.R., Ng S.R., Whittaker C.A., Bronson R.T., Snyder E.L. Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Genes Dev. 2015;29:1850–1862. doi: 10.1101/gad.267393.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Hanahan D. Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell. 2013;153:86–100. doi: 10.1016/j.cell.2013.02.051. [DOI] [PubMed] [Google Scholar]
- Lucá R., Averna M., Zalfa F., Vecchi M., Bianchi F., La Fata G., Del Nonno F., Nardacci R., Bianchi M., Nuciforo P. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol. Med. 2013;5:1523–1536. doi: 10.1002/emmm.201302847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo M.L., Santagata S., Koeva M., Bell G.W., Hu R., Tamimi R.M., Fraenkel E., Ince T.A., Whitesell L., Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen J., Nordhausen K., Taskinen S. Blind source separation based on joint diagonalization in R : The packages JADE and BSSasymp. J. Stat. Softw. 2017;76 [Google Scholar]
- Mili S., Moissoglu K., Macara I.G. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453:115–119. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.W., Morton J.P., Pinese M., Saturno G., Jamieson N.B., McGhee E., Timpson P., Leach J., McGarry L., Shanks E. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer D., Leleu M., Splinter E., Rougemont J., De Laat W., Duboule D. The dynamic architecture of Hox gene clusters. Science. 2011;334:222–225. doi: 10.1126/science.1207194. [DOI] [PubMed] [Google Scholar]
- Oldridge D.A., Wood A.C., Weichert-Leahey N., Crimmins I., Sussman R., Winter C., McDaniel L.D., Diamond M., Hart L.S., Zhu S. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528:418–421. doi: 10.1038/nature15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson P., Chu G.C., Perry S.R., Nolan-Stevaux O., Hanahan D. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc. Natl. Acad. Sci. USA. 2011;108:E1275–E1284. doi: 10.1073/pnas.1111079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P., Bellone C., Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Robinson H.P.C., Li L. Autocrine, paracrine and necrotic NMDA receptor signalling in mouse pancreatic neuroendocrine tumour cells. Open Biol. 2017;7 doi: 10.1098/rsob.170221. 170221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge D.N., Jouan-Rimbaud Bouveresse D. Independent components analysis with the JADE algorithm. TrAC Trends Analyt. Chem. 2013;50:22–32. [Google Scholar]
- Sadanandam A., Wullschleger S., Lyssiotis C.A., Grötzinger C., Barbi S., Bersani S., Körner J., Wafy I., Mafficini A., Lawlor R.T. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, metastatic, developmental, and metabolic characteristics. Cancer Discov. 2015;5:1296–1313. doi: 10.1158/2159-8290.CD-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S., Mendillo M.L., Tang Y.C., Subramanian A., Perley C.C., Roche S.P., Wong B., Narayan R., Kwon H., Koeva M. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science. 2013;341:1238303. doi: 10.1126/science.1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Pedersen S., Hasle H., Olsen J.H., Friedrich U. Evidence of decreased risk of cancer in individuals with fragile X. Am. J. Med. Genet. 2001;103:226–230. [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd P.K., Malter J.S., Mack K.J. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res. Mol. Brain Res. 2003;110:267–278. doi: 10.1016/s0169-328x(02)00657-5. [DOI] [PubMed] [Google Scholar]
- Wei Z., Behrman B., Wu W.H., Chen B.S. Subunit-specific regulation of N-methyl-D-aspartate (NMDA) receptor trafficking by SAP102 protein splice variants. J. Biol. Chem. 2015;290:5105–5116. doi: 10.1074/jbc.M114.599969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.