

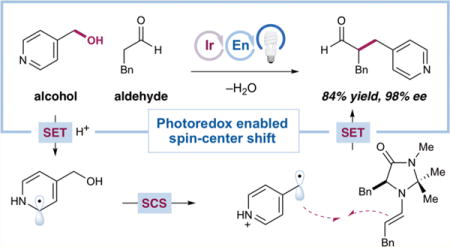
Abstract
Nature routinely engages alcohols as leaving groups, as DNA biosynthesis relies on the removal of water from ribonucleoside diphosphates by a radical-mediated “spin-center shift” (SCS) mechanism. Alcohols, however, remain underused as alkylating agents in synthetic chemistry due to their low reactivity in two-electron pathways. We report herein an enantioselective α-benzylation of aldehydes using alcohols as alkylating agents based on the mechanistic principle of spin-center shift. This strategy harnesses the dual activation modes of photoredox and organocatalysis, engaging the alcohol by SCS and capturing the resulting benzylic radical with a catalytically generated enamine. Mechanistic studies provide evidence for SCS as a key elementary step, identify the origins of competing reactions, and enable improvements in chemoselectivity by rational photocatalyst design.
Graphical abstract
INTRODUCTION
In DNA biosynthesis, deoxyribonucleoside diphosphate building blocks are procured from their corresponding ribonucleo-sides by the action of ribonucleotide reductase (RNR) enzymes.1 The key step in this deoxygenation occurs via a (3′,2′)-spin-center shift (SCS) event, which induces a β-C−O-scission and the net loss of water (Figure 1a).2 Despite this well-established open-shell mechanism that engages alcohols as leaving groups, alcohols remain underexploited as alkylating agents due to the substantial barrier to the displacement of the hydroxyl group by two-electron pathways.3 Nonetheless, the direct use of alcohols as electrophiles remains an important goal in synthetic organic chemistry due to their low genotoxicity, robustness, and ubiquity in naturally occurring molecules.4
Figure 1.
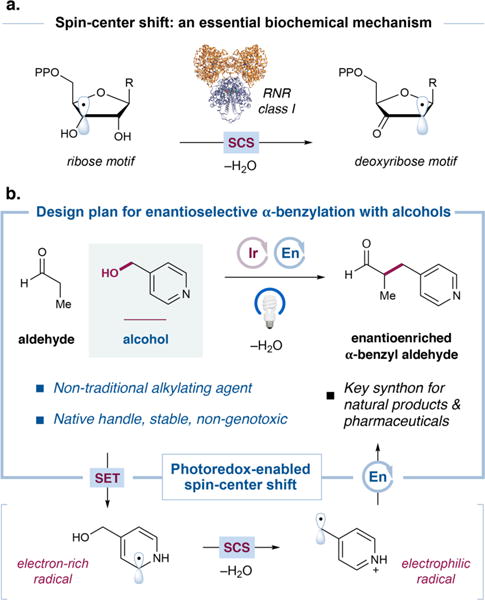
Spin-center shift (SCS) as a conceptual basis for the enantioselective α-benzylation of aldehydes with alcohols. (a) SCS in DNA biosynthesis. (b) Advantages of alcohols as alkylating agents and a possible mechanism in which benzylic alcohols may be engaged by SCS.
Inspired by nature’s spin-center shift process, our group recently reported the alkylation of heteroarenes with alcohols as latent alkylating agents, relying on dual photoredox and hydrogen atom transfer catalysis.5 Given that photoredox catalysis provides (1) mild access to open-shell radical intermediates and (2) a general platform to perform concurrent oxidation and reduction steps in the same vessel,6 we hypothesized that this activation mode, in concert with organocatalysis, could enable a direct, enantioselective α-benzylation of aldehydes with heterobenzylic alcohols as electrophiles by exploiting SCS (Figure 1b).
Pioneering work by Evans et al.,7 Oppolzer et al.,8 Seebach et al.,9 and Myers et al.10 has long established that the stereoselective α-benzylation of carbonyls can be readily accomplished using chiral auxiliaries. Surprisingly, however, catalytic enantioselective variants of this important transformation have been slower to develop, with the most notable examples being the phase transfer benzylation of glycine imines,11 chiral triamine ligation of ketone-derived lithium enolates,12 and Cr(salen) activation of preformed tin enolates.13 More recently, photoredox organocatalysis has emerged as a platform for the enantioselective construction of α-alkylated carbonyl motifs,14 including the α-benzylation of aldehydes using electron-deficient benzylic bromides.14b
A common feature of both catalytic and auxiliary-based strategies is the reliance on benzylic halide electrophiles or their equivalents (e.g., tosylates). Indeed, alkyl (pseudo)halides are archetypal alkylating agents due to the excellent leaving group ability of bromide, iodide, and sulfonate ions. This reactivity, however, also confers undesirable properties, such as genotoxicity and light-sensitivity, necessitating care in handling and storing these reagents. Furthermore, alkyl halides are frequently obtained by treatment of the corresponding alcohols with a stoichiometric activating agent,15 highlighting the appeal of engaging alcohols directly. In the context of asymmetric α-alkylation, the use of alcohols has been restricted to specialized cases where heterolytic C−O cleavage generates highly stabilized cations.16 In this paper, we report the design and application of an enantioselective α-benzylation of aldehydes with heterobenzylic alcohols as well as mechanistic studies that support the proposed SCS pathway, elucidate the major undesired reaction pathways, and enable improvements in chemoselectivity by photocatalyst modification.
DESIGN PLAN
Our design for the enantioselective α-benzylation of aldehydes with alcohols is outlined in Figure 1b. Single-electron reduction of a benzylic alcohol by a photoredox catalyst would initially give rise to an electron-rich radical. This intermediate would be poised to undergo SCS, whereby benzylic C–O bond cleavage and proton transfer would expel a molecule of water and reveal an electrophilic benzylic radical. This electron-deficient species would then react with a catalytically generated enamine, forming the desired C–C bond stereoselectively and ultimately leading to the enantioenriched α-benzyl aldehyde.
A detailed mechanistic description of the proposed transformation is shown in Scheme 1. Excitation of an IrIII catalyst (1) with blue light would first generate a long-lived *IrIII excited state (2) [τ = 1.90 μs for Ir(ppy)3].17 This highly reducing species {E1/2red[IrIV/*III] = −1.81 V vs saturated calomel electrode (SCE) in CH3CN for Ir(ppy)3} should reduce a protonated heterobenzylic alcohol such as 4-(hydroxymethyl)pyridine (3, Ered = −1.29 V vs SCE in CH3CN for 3·HBr) to furnish electron-rich radical 4 and IrIV intermediate 5. Radical 4 would then undergo the key spin-center shift event to unveil electrophilic radical 6 and extrude a molecule of water after proton transfer. Within the same time frame, aldehyde 7 and an organocatalyst (8) would condense to form chiral enamine 9. The depicted DFT structure of 9 (with propionaldehyde as the aldehyde) illustrates that the benzyl substituent of the organocatalyst shields the Re-face of the enamine, leaving the Si-face exposed for reaction with electrophilic radical 6. The resulting α-amino radical 10 (E1/2ox = −1.12 to −0.92 V vs SCE in CH3CN for simple α-amino radicals)18 would be readily oxidized by the IrIV intermediate 5 {E1/2red[IrIV/III] = +0.77 V vs SCE in CH3CN for Ir(ppy)3} to regenerate ground-state IrIII photocatalyst 1 and iminium ion 11. Finally, hydrolysis of the latter species would liberate enantioenriched α-benzyl aldehyde 12 and organocatalyst 8.
Scheme 1.
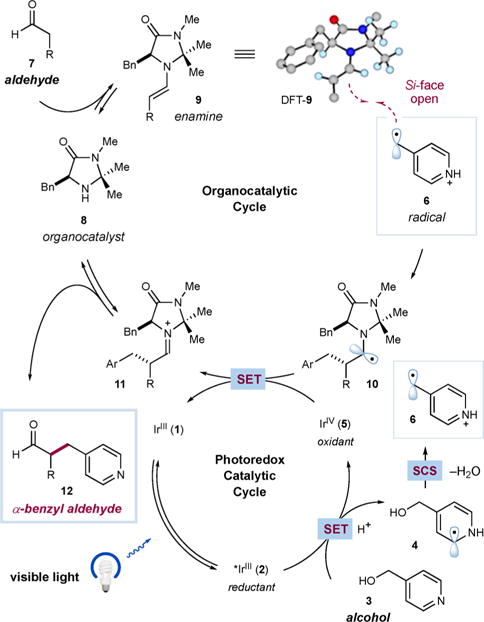
Proposed Mechanism for the Enantioselective α-Benzylation of Aldehydes with Alcohols
RESULTS
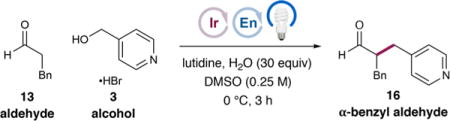
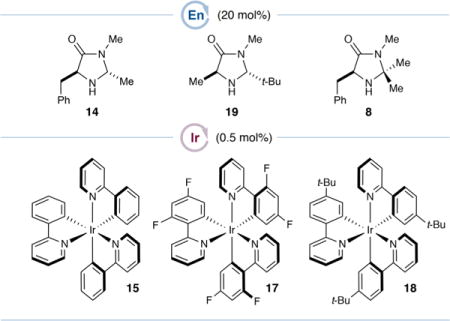
We first tested this hypothesis by subjecting hydrocinnamaldehyde (13) and alcohol 3, as its trifluoroacetic acid salt, to the reaction conditions that proved optimal in the enantioselective α-benzylation of aldehydes using benzylic bromides14b [20 mol % 14 as the organocatalyst, 0.5 mol % Ir(ppy)3 (15) as the photocatalyst, and 3 equiv lutidine in DMSO at rt] under blue-light irradiation (Table 1, entry 1). While none of the desired product was obtained, omitting the lutidine base (entry 2) gave rise to the desired α-benzyl aldehyde 16 in promising yield (37%) and enantioselectivity (62% ee). We postulate that a more acidic medium is necessary to facilitate both the reduction of alcohol 3 via protonation and ultimately the required spin-center shift event. Optimization of the reaction conditions [see Tables S1–S6, Supporting Information (SI)] revealed that employing a substoichiometric amount of lutidine (25 mol %) and HBr as the acid, a 2-fold dilution of the mixture, the addition of water (30 equiv), and cooling the mixture to 0 °C provided 16 in 48% yield and much improved 90% ee (entry 3). The modest efficiency was due primarily to the net reduction of alcohol 3 to 4-methylpyridine, rather than low consumption of 3, so we surmised that a less reducing photocatalyst such as fluorinated IrIII complex 1719 would minimize the production of the reduction byproduct. We were surprised, however, to observe a diminished 18% yield (entry 4). Instead, the more reducing photocatalyst 1817 improved efficiency without compromising enantioselectivity (entry 5, 78% yield, 92% ee; see later text for a detailed discussion). Further modification of stoichiometries and conducting the reaction in N,N-dimethylacetamide (DMA) gave optimal efficiency (90% yield, entry 6). Finally, while the sterically demanding tert-butyl organocatalyst 19 was unproductive (entry 7), catalyst 8, featuring a fully substituted aminal, provided 16 in 88% yield and 98% ee after 6 h (entry 8). Further photocatalyst modifications could improve the chemoselectivity and thus the yield (see Figure 3), but 18 proved optimal when considering alcohol conversion and synthetic accessibility (see the SI).
Table 1.
Optimization of the Enantioselective α-Benzylation of Aldehydes with Alcoholsa
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | lutidine | conditions | catalysts | yield | ee |
| 1b | 300 mol% | no H2O, 0.5 M, r.t. | 14 + 15 | 0% | n.d. |
| 2b | 0 mol% | no H2O, 0.5 M, r.t. | 14 + 15 | 37% | 62% |
| 3 | 25 mol% | as shown | 14 + 15 | 48% | 90% |
| 4 | 25 mol% | as shown | 14 + 17 | 18% | n.d. |
| 5 | 25 mol% | as shown | 14 + 18 | 78% | 92% |
| 6c | 50 mol% | DMA solvent | 14 + 18 | 90% | 92% |
| 7c | 50 mol% | DMA solvent | 19 + 18 | 6% | n.d. |
| 8c | 50 mol% | DMA solvent, 6 h | 8 + 18 | 88% | 98% |
|
| |||||
| |||||
Alcohol 3 (0.1 mmol), aldehyde 13 (1.5 equiv), lutidine, water, organocatalyst, and photocatalyst were irradiated in the indicated solvent with a 34 W blue LED lamp. Yields were determined by 1H NMR. Enantioselectivities were determined by chiral HPLC analysis following reduction of the crude aldehyde to the corresponding alcohol.
Trifluoroacetic acid salt of the alcohol instead of the HBr salt.
Aldehyde 13 (2.0 equiv).
Figure 3.
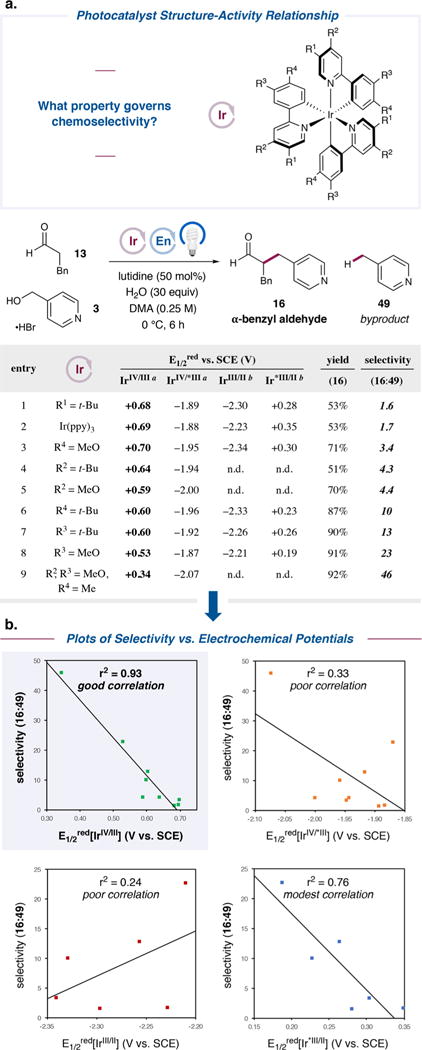
Impact of photocatalyst structure on chemoselectivity in the α-benzylation of aldehydes with alcohols. (a) Alcohol 3 (0.25 mmol), aldehyde 13 (2.0 equiv), lutidine (50 mol %), water (30 equiv), organocatalyst (±)-8 (20 mol %), and photocatalyst (0.5 mol %) were irradiated in DMA with a 34 W blue LED lamp. Yields of 16 and 49 were determined by 1H NMR. (b) Plots of selectivity vs each electrochemical potential. aMeasured in CH3CN. bMeasured in CH2Cl2..
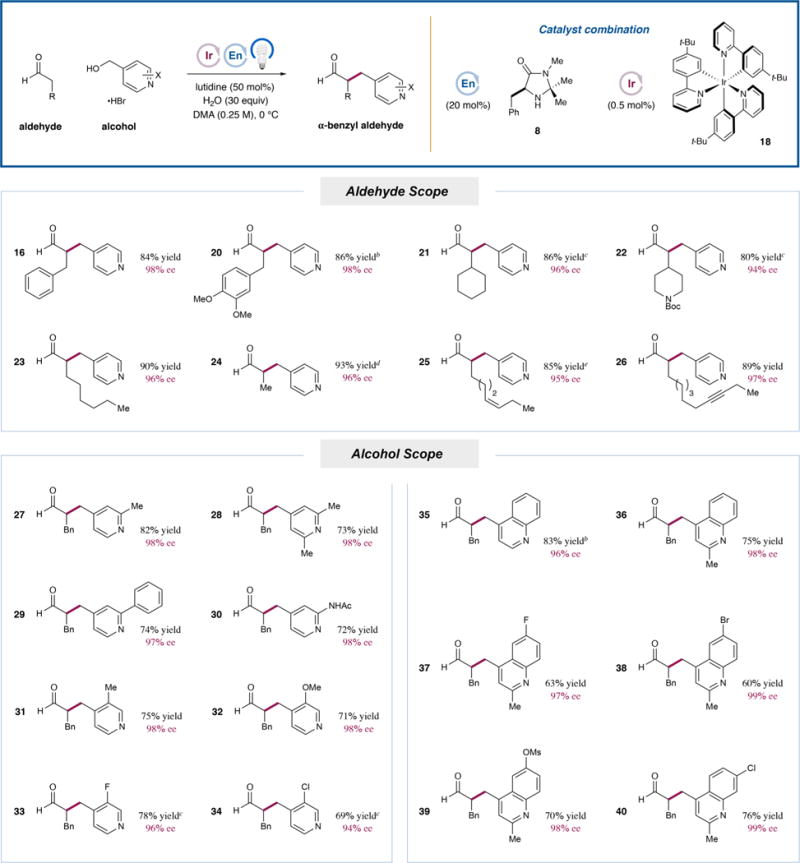
With this optimized set of conditions, we evaluated the scope of the enantioselective α-benzylation of aldehydes with alcohols (Table 2). First, a range of aldehydes undergo efficient and highly enantioselective benzylation with 4-(hydroxymethyl)-pyridine (3). Hydrocinnamaldehyde was alkylated to give 16 in 84% isolated yield and 98% ee, consistent with smaller-scale optimization studies. A dimethoxy-substituted analogue (20) was also obtained in excellent efficiency and selectivity (86% yield, 98% ee). β-Branched aldehydes are competent substrates, with cyclohexyl and piperidinyl products 21 and 22 obtained in good yields (86% and 80%, respectively) and enantioselectivities (96% and 94% ee, respectively). Simple alkanals such as octanal and propionaldehyde also reacted cleanly to give 23 (90% yield, 96% ee) and 24 (93% yield, 96% ee). Finally, unsaturation is tolerated, as shown by the production of alkene 25 (85% yield, 4.5:1 Z/E, 95% ee) and alkyne 26 (89% yield, 97% ee).
Table 2.
Scope of the Enantioselective α-Benzylation of Aldehydes with Alcoholsa
|
Alcohol (0.5 mmol), aldehyde (2.0 equiv), lutidine (50 mol %), water (30 equiv), organocatalyst 8 (20 mol %), and photocatalyst 18 (0.5 mol %) were irradiated in DMA with a 34 W blue LED lamp at 0 °C. Isolated yields are reported. Enantioselectivities were determined by chiral HPLC analysis following reduction of the crude aldehydes to the corresponding alcohols.
Characterized as the corresponding alcohol.
Aldehyde (5.0 equiv).
Yield determined by 1H NMR.
From the Z-starting material, 25 was obtained as a 4.5:1 mixture of Z and E isomers; chiral HPLC analysis was performed following reduction of the crude aldehyde to the corresponding alcohol and subsequent hydrogenation of the alkene.
With respect to the heterobenzylic alcohol, a variety of substituted pyridines are competent in the reaction with hydrocinnamaldehyde. Methyl substitution at the 2-position or disubstitution at the 2- and 6-positions were well-tolerated, as were 2-phenyl or protected 2-amino substituents (27–30, 72–82% yield, 97−98% ee). The 3-position can also be substituted, with methyl-, methoxy-, fluoro-, and chloro-containing products 31–34 obtained in good yields (69–78%) and excellent enantioselectivities (94–98% ee). Quinolines are also capable of inducing the requisite spin-center shift, and a variety of substitution patterns about this aromatic motif are accommodated in the α-benzylation of hydrocinnamaldehyde. Specifically, 4-(hydroxymethyl)quinoline served as a competent alkylating agent, furnishing product 35 in 83% yield and 96% ee. The 2-methyl analogue (36) was also cleanly isolated (75% yield, 98% ee). Alcohols bearing substituents at the 6-position of the quinoline system can be employed and gave rise to products 37–39 containing fluoro, bromo, and protected oxygen functionalities in synthetically useful yields (60–70%) without compromising enantioselectivity (97–99% ee). Finally, 7-chloroquinoline 40 was also isolated in 76% yield and 99% ee.
Products obtained by this enantioselective α-benzylation possess enantioenriched homobenzylic stereocenters and a versatile aldehyde functional handle and, thus, may serve as important synthons for the preparation of bioactive molecules. To demonstrate the utility of this protocol, we sought to prepare the stereoselective ligand of translocator protein (18 kDa), PK-14067 (44, Figure 2).20 To this end, propionaldehyde (41) was alkylated directly by alcohol 42. The crude aldehyde (not shown) was oxidized to the corresponding carboxylic acid, and subsequent HATU-mediated coupling with diethylamine provided amide 43 in 79% yield and 95% ee over three steps. Finally, the phenyl substituent was installed in modest efficiency via a Minisci-type arylation21 to afford the target (44) in 52% yield and without erosion of enantiopurity. It is noteworthy that this synthesis corroborates the assigned (R)-configuration of the active isomer. Previous studies of PK-14067 had obtained this compound by racemic synthesis, followed by resolution, and assigned the configuration of the bioactive enantiomer by comparison of its experimental VCD spectrum to the simulated spectrum of both enantiomers.22 The known stereochemical course of our α-benzylation (see the SI for a discussion) reliably delivered (R)-44, the optical rotation of which (95% ee, [α]D = −88°, c = 1.0, EtOH) matched the reported value for the active enantiomer (99% ee, [α]D = −90°, c = 2.86, EtOH).22
Figure 2.
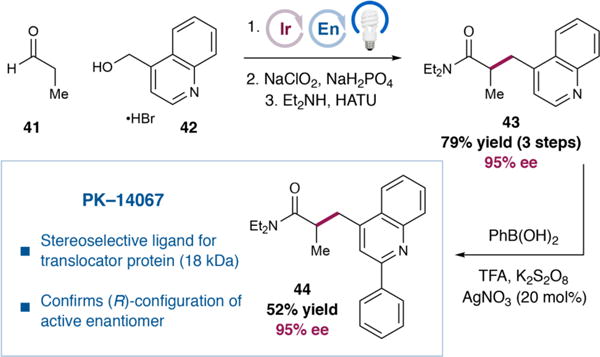
Enantioselective synthesis of stereoselective translocator protein (18 kDa) ligand PK-14067. The enantioselective α-benzylation procedure was conducted using alcohol 42 (2.0 mmol) and aldehyde 41 (5.0 equiv) under conditions listed in Table 2 for 42 h.
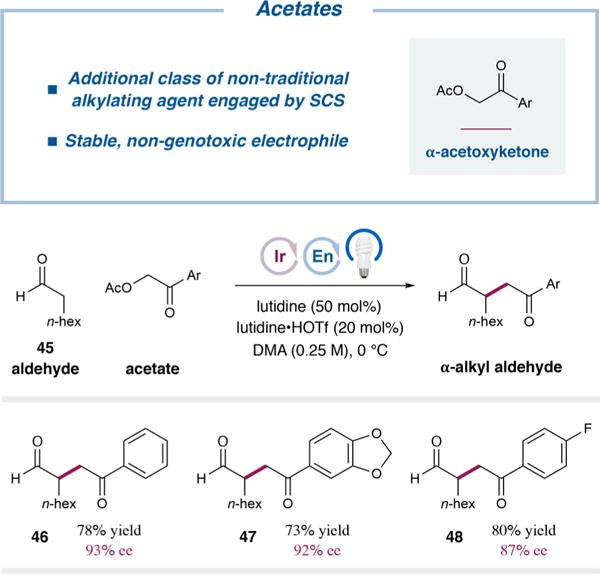
Finally, we sought further to broaden the utility of this spin-center shift paradigm for the enantioselective α-alkylation of aldehydes with unconventional electrophiles as latent alkylating agents. Beyond the heterobenzylic alcohols described above, work by Stephenson and co-workers23 suggested that alcohols such as α-hydroxy ketones or their derivatives may also be viable electrophiles in this alkylation manifold. While initial experiments demonstrated that free alcohols of this type are not competent alkylating agents, the corresponding acetates show excellent reactivity (Table 3). Therefore, under slightly modified conditions, octanal (45) was alkylated with α-acetoxyacetophenone, as well as the 3,4-(methylenedioxy) and 4-fluoro analogues, to procure the corresponding α-alkyl aldehydes in good yields and high enantioselectivities (46–48, 73–80% yield, 87–93% ee).
Table 3.
Spin-Center Shift-Enabled Enantioselective α-Alkylation of Aldehydes with α-Acetoxy Ketonesa
|
Acetate (0.5 mmol), aldehyde 45 (2.0 equiv), lutidine (50 mol %), lutidine·HOTf (20 mol %), organocatalyst 8 (20 mol %), and photocatalyst 18 (0.5 mol %) were irradiated in DMA with a 34 W blue LED lamp at 0 °C. Isolated yields are reported. Enantioselectivities were determined by chiral HPLC analysis.
Notably, these preliminary results demonstrate that an additional class of nontraditional alkylating agents, α-acetoxy ketones, can be activated to this end by a spin-center shift. While acetates are activated leaving groups compared to alcohols (see Table 4 and the associated discussion), they are seldom employed directly in alkylation reactions, as they are still significantly less reactive than typical alkylating agents, such as alkyl bromides or iodides. Furthermore, like alcohols, they are less genotoxic and more stable than conventional electrophiles.
Table 4.
Leaving Group Scope in the Enantioselective Spin-Center Shift-Enabled α-Benzylation of Aldehydes and Its Impact on Reactivitya
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | X | Ered (V) | Ksv(mM−1) | pKa (XH) | yield (2 h) | yield [time] | 16:49 |
| 1 | OAc | −1.19 | 0.84 | 4.76 | 75% | 90% [3 h] | 18 |
| 2 | NMe3+Br− | n.d.b | 0.13 | 9.80 | 67% | 86% [5 h] | 14 |
| 3 | OH | −1.29 | 1.05 | 15.7 | 39% | 85% [5 h] | 7.7 |
| 4 | OMe | −1.29 | 1.15 | 15.2 | 29% | 71% [24 h] | 5.1 |
| 5 | OTBDPS | −1.30 | 0.82 | ⪝13.6 | 20% | 61% [48 h] | 3.1 |
|
| |||||||
| 6 | – | −1.30 | 1.30 | data for pyridine·HBr | |||
Electrophile (0.25 mmol), aldehyde 13 (2.0 equiv), lutidine (50 mol %), water (30 equiv), organocatalyst 8 (20 mol %), and photocatalyst 18 (0.5 mol %) were irradiated in DMA with a 34 W blue LED lamp. Yields of 16 and 49 were determined by 1H NMR. Enantioselectivities were determined by chiral HPLC analysis following reduction of the crude aldehyde to the corresponding alcohol. Acidity data in water from ref 24. See the SI for full experimental details.
Low solubility prevented electrochemical measurements in aprotic solvents.
MECHANISTIC STUDIES
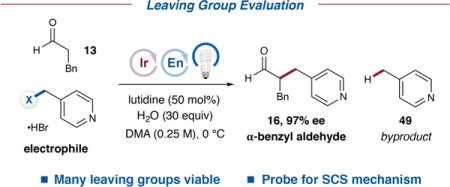
Further investigations were performed to gain a greater mechanistic understanding of the enantioselective α-benzylation of aldehydes with alcohols. Specifically, we sought to determine whether spin-center shift occurs as hypothesized, to elucidate the origin of the major byproduct [i.e., the formation of 4-methylpyridine (49) from 4-(hydroxymethyl)pyridine (3)] that initially complicated the optimization of this reaction, and to test the possibility of a radical chain mechanism. Thus, three investigations were performed: (1) an examination of how modifications to the leaving group in the electrophile impact reactivity and selectivity, (2) a photocatalyst structure–activity relationship (SAR) study, and (3) quantum yield measurements.
To begin, we investigated the impact of the leaving group X on reaction efficiency and chemoselectivity (Table 4). Thus, we prepared a series of (4-pyridyl)methyl electrophiles and subjected them to the standard reaction conditions with hydrocinnamaldehyde (13). First, several functional groups aside from alcohols can serve as leaving groups in this transformation. These electrophiles (acetate, trialkylammonium, alkyl ether, and silyl ether) all give rise to α-benzyl aldehyde 16 in respectable to excellent yields (61–90%) and with uniformly high enantioselectivity (97% ee). Further functional groups that do not generally confer alkylating ability can therefore be employed in the outlined enantioselective α-benzylation of aldehydes.
We then sought to account for the different reactivities observed among these electrophiles in order to evaluate our proposal that this reaction proceeds via SCS (Table 4, entries are sorted by decreasing yield and chemoselectivity, the latter parameter being the ratio between yields of desired product 16 and byproduct 49). Therefore, we measured their reduction potentials (Ered) and Stern–Volmer quenching constants (KSV) with photocatalyst 18, which, in this context, quantifies the relative rates at which the electrophile substrates receive an electron from the excited state of 18.
Notably, the reduction potentials of the electrophiles (entries 1–5, Ered = −1.19 to −1.30 V vs SCE in CH3CN) are nearly identical both to each other and to that of pyridine·HBr (entry 6, Ered = −1.30 V vs SCE in CH3CN). Such similar potentials suggest that the LUMOs of these compounds all reside primarily on their common structural feature, the protonated heteroaromatic system. If the leaving groups made a significant contribution to the LUMOs, we would expect a wider range of Ered values, given the appreciable stereoelectronic differences between these functionalities. As such, the photoredox activation of these electrophiles likely proceeds via initial SET to the aromatic core, followed by SCS, as proposed in Figure 1 and Scheme 1. For comparison, a conventional electrophile for this reaction, 4-(bromomethyl)pyridine (50; see Figure 4),14b is much more easily reduced (Ered = −0.88 V vs SCE in CH3CN for the HBr salt). We propose, therefore, that 50 is engaged by a photoredox catalyst for α-benzylation via direct SET to the C–Br σ* orbital, rather than by SCS.
Figure 4.
Quantum yield determination for the enantioselective α-benzylation of aldehydes with alcohols and bromides.
The KSV data, in comparison, exhibit appreciable variability among the electrophiles, although no clear relationship is evident between these values and reactivity. Since KSV directly reflects the relative rates of SET between the excited state photocatalyst and the electrophiles, we conclude that this SET is likely rapid, and a subsequent event, such as SCS, dictates reactivity. The measurement of nonzero KSV values also confirms that the excited state photocatalyst is quenched by these electrophiles, consistent with our proposal that the reaction is initiated by SET from the excited photocatalyst to the electrophile.
The observed reactivity trends are best explained by the acid–base properties of the leaving groups. A comparison of the literature pKa values for the parent acids (XH) of the leaving groups (X−) with reaction rates and yields in Table 4 suggests that the electrophiles sort into two classes. First, the more reactive electrophiles possess weakly basic leaving groups (entry 1, X = OAc, and entry 2, X = NMe3+). The protonation states of these leaving groups following simple heterolytic C–X scission (anionic carboxylate and neutral trialkylamine, respectively) should be stable in the pyridine/pyridinium reaction buffer. For these electrophiles, therefore, SCS directly follows single-electron reduction of the pyridinium moiety. In contrast, the less reactive electrophiles possess strongly basic leaving groups (entries 3–5, X = OH, OMe, OTBDPS), the corresponding anions of which should be unstable in the reaction medium. These leaving groups must be activated by protonation or hydrogen-bonding before C–X cleavage, and this additional barrier slows the α-benzylation reaction. This clear dependence of reactivity on leaving group acidity suggests that the rate of C–X bond breaking contributes to the overall rate of reaction. A discussion of reactivity trends within the two classes of electrophiles is provided in the SI.
On balance, the data in Table 4 suggest that a rapid SET from the excited state photocatalyst to the electrophile initiates the reaction, followed by slow C–X cleavage (SCS). This step impacts the rate of α-benzylation and is the rate-determining step (RDS) in the linear reaction between the electrophile and the enamine. While these experiments do not assess the kinetics of C–C bond-formation, as they all involve a common electrophilic radical and enamine that would participate in this step, (a) an examination of reactivities among the different alcohol electrophiles employed in Table 2 is consistent with SCS being slower than C–C bond formation, and (b) initial rate studies suggest that the enamine is not involved the RDS of its photoredox-mediated alkylation by the electrophile (see the SI). Higher loadings of either aldehyde or organocatalyst lead to increased rates beyond this initial period, however, as the organocatalytic cycle must turn over to provide further enamine and preliminary experiments suggest that iminium ion hydrolysis is turnover-limiting (also see the SI). The chemoselectivity between desired product 16 and byproduct 49 (Table 4, final column) is addressed in the following discussion of Figure 3.
Next, we conducted a photocatalyst SAR study. We systematically prepared a series of tert-butyl- and methoxy-substituted derivatives of Ir(ppy)3 and measured their photophysical and electrochemical properties (Figure 3a; also see Figures S3–S35, SI). We then evaluated their performance in the α-benzylation of hydrocinnamaldehyde (13) with 4-(hydroxymethyl)pyridine (3) and focused on the selectivity between the yields of desired α-benzyl aldehyde 16 and undesired 4-methylpyridine (49).
Figure 3a tabulates these results, which are sorted from least selective to most selective (final column). Preliminary examinations of two potentially important properties of these photocatalysts, their excited state lifetimes17 and Stern–Volmer quenching rates with 4-(hydroxymethyl)pyridine (3) (see Figure S53, SI), showed negligible differences. Instead, their electrochemical potentials (E1/2red) seemed to play a primary role in determining selectivity. Since there are four such values to consider (ground-state oxidation by IrIV, ground-state reduction by IrII, and excited-state oxidation or reduction by *IrIII), we plotted the observed chemoselectivity as a function of each E1/2red series, as shown in Figure 3b. The strongest correlation (r2 = 0.93) was found between selectivity and E1/2red[IrIV/III], the oxidizing power of the IrIV ground state, with lower oxidation potentials leading to higher selectivity. A modest correlation was also found between selectivity and E1/2red[Ir*III/II], the oxidizing ability of the *IrIII excited state (r2 = 0.76). We postulate that this correlation is incidental, however, as the lower oxidation potentials of *IrIII excited states compared to IrIV ground states suggest that the former species are not responsible for the oxidation leading to 49 (see below). The remaining potentials, E1/2red[IrIV/*III] and E1/2red[IrIII/II], clearly do not explain the observed trends in chemoselectivity (r2 = 0.33 and 0.24, respectively).
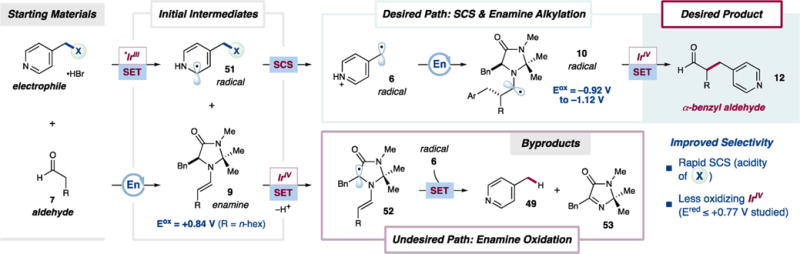
From the preceding studies, a detailed mechanistic understanding of chemoselectivity emerges, which is outlined in Scheme 2. The electrophile starting material is first reduced by the excited state *IrIII photocatalyst to give radical 51 and an IrIV species. At this stage, the relative reactivities of radical 51, enamine 9, and the IrIV intermediate dictate the final chemoselectivity. Desired α-benzyl aldehyde 12 is formed (Scheme 2, top) when a spin-center shift occurs to give electrophilic radical 6, which alkylates enamine 9. The resulting α-amino radical 10 is oxidized by the IrIV species to produce iminium ion 11 (see Scheme 1), which is hydrolyzed to deliver 12. Major byproduct 49 arises (Scheme 2, bottom) when the IrIV species oxidizes enamine 9 directly. This SET presumably leads to radical 52, which formally reduces electrophilic radical 6, likely with the assistance of the photocatalyst. The resulting byproducts are thus the previously discussed 49, from net alcohol reduction, and oxidized organocatalyst 53,25 which we have also isolated from several reaction mixtures.26
Scheme 2.
Mechanistic Description of Chemoselectivity in the Enantioselective SCS-Enabled α-Benzylation of Aldehydes
This description accounts for the chemoselectivity trends outlined in Table 4 and Figure 3 in terms of two competing pathways for the IrIV intermediate. Desired α-benzyl aldehyde 12 is obtained when the IrIV species oxidizes the strongly reducing α-amino radical 10 (E1/2ox = −0.92 to −1.12 V vs SCE in CH3CN for simple α-amino radicals),18 an SET which should be rapid and irreversible for all photocatalysts employed in this investigation (E1/2red[IrIV/III] = +0.34 V to +0.70 V vs SCE in CH2Cl2; see Figure 3). Conversely, undesired byproducts 49 and 53 form when the IrIV species oxidizes enamine 9 (Eox = +0.84 vs SCE in CH3CN for R = n-hex). This SET is feasible, albeit endergonic, for all the above-listed IrIV oxidation potentials (see above for data in CH2Cl2; for photocatalysts soluble in CH3CN, E1/2red[IrIV/III] ≤ +0.77 V vs SCE in this solvent; see the SI). Furthermore, appreciable concentrations of enamine 9 are present throughout the reaction, whereas radical 10, which must be oxidized to obtain the desired product, should only be present in trace amounts.
With respect to the electrophile, the least basic leaving groups give the highest chemoselectivities (see Table 4) due to the corresponding acceleration of the SCS step. While SCS must occur to form both desired α-benzyl aldehyde 12 and byproduct 49, the rate of this elementary step has a different impact on the pathways leading to each product. In the limiting case when SCS is fast, α-amino radical 10 forms rapidly, and this strong reductant reacts preferentially with the IrIV intermediate to close the photocatalytic cycle and generate desired product 12. Conversely, when SCS is slow, 10 is unavailable to reduce the IrIV species. Instead, enamine 9 can be oxidized by the IrIV intermediate, giving 52 (or a related species) after proton transfer. Radicals such as 52 should be modest reducing agents, and upon the eventual formation of electrophilic radical 6, its formal reduction by 52 (likely mediated by a photocatalyst) competes with C–C bond formation, ultimately producing 49 and 53.
With respect to the photocatalyst, selectivity for desired product 12 increases straightforwardly with decreasing IrIV oxidation potentials. Lower IrIV potentials render undesired enamine oxidation increasingly endergonic, while oxidation of the strongly reducing radical 10 remains highly exergonic and ensures that the desired product can still be accessed without complication. Indeed, as shown in Figure 3, chemoselectivity rises from modest levels when using photocatalysts with the most oxidizing IrIV states (entries 1–3, 16:49 = 1.6:1 to 3.4:1 for E1/2red[IrIV/III] = +0.68 to +0.70 V vs SCE in CH2Cl2) to excellent when using the least oxidizing derivative, which we designed explicitly for this purpose (entry 9, 16:49 = 46:1 for E1/2red[IrIV/III] = +0.34 V vs SCE in CH2Cl2).
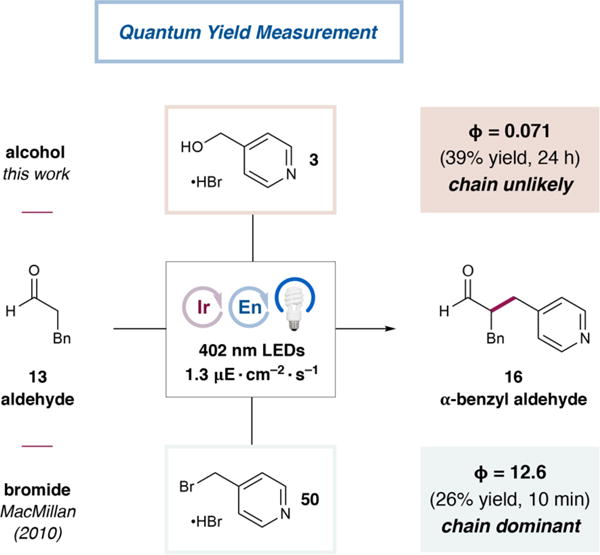
Finally, we questioned whether a radical chain propagation mechanism could be occurring in this transformation, as work by Yoon et al. has demonstrated that such pathways operate in a range of photoredox-catalyzed transformations,27 including the enantioselective α-alkylation of aldehydes with alkyl bromides.14a With the present alcohol electrophiles, however, we hypothesized that the relatively difficult reduction of the model substrate (3, Ered = −1.29 V vs SCE in CH3CN for the HBr salt) would prohibit its reduction by any organic intermediates formed during the reaction (the most likely candidate would be α-amino radical 10, depicted in Schemes 1 and 2, but the literature data suggest that the potentials of simple analogues, E1/2ox = −0.92 to −1.12 V vs SCE in CH3CN, are still insufficiently reducing).18 As shown in Figure 4, the quantum yield for the reaction of alcohol 3 with hydrocinnamaldehyde (13) is 0.071. While this observation does not rule out propagation events conclusively, the relatively low value is consistent with our mechanistic hypothesis that each photon absorbed by the photocatalyst should lead, at most, to a single product molecule. In contrast, we surmised that the formation of reducing intermediates such as 10 would enable radical chain propagation events when a more easily reduced electrophile, such as the corresponding benzylic bromide (50), is employed (Ered = −0.88 V vs SCE in CH3CN for the HBr salt). Indeed, for the α-benzylation of hydrocinnamaldehyde (13) with 50, under the optimal conditions for benzylic bromide electrophiles reported in 2010, we measured a quantum yield of 12.6. In this reaction, therefore, the photocatalyst serves primarily as an initiator for a self-propagating chain responsible for the majority of product formation.
CONCLUSIONS
We have developed a strategy based on spin-center shift that enables the enantioselective α-benzylation of aldehydes with electron-deficient heterobenzylic alcohols. To our knowledge, this work represents the first example of a direct enantioselective α-alkylation of carbonyl compounds with alcohols where the electrophile does not contain specialized cation-stabilizing features to promote SN1-type activation. Additional nontraditional leaving groups, such as acetates and ethers, are also competent in this reaction, and α-acetoxy ketone electrophiles can be employed to access a further aldehyde α-alkylation motif via SCS. Mechanistic studies are consistent with spin-center shift as a key elementary step and elucidate the impact of electrophile and photocatalyst structures on reactivity. Finally, enamine oxidation was identified as the origin of the major side reaction, enabling optimal yields to be obtained by rational photocatalyst design.
Supplementary Material
Acknowledgments
Financial support was provided by the NIHGMS (R01 GM-103558-05) and kind gifts were from Merck, Abbvie, BMS, and Janssen. E.D.N. acknowledges the NIH for a postdoctoral fellowship (F32 GM119362-01A1).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12768.
Experimental procedures and compound characterization data (PDF)
ORCID
David W. C. MacMillan: 0000-0001-6447-0587
Notes
The authors declare no competing financial interest.
References
- 1.Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P. Prog Biophys Mol Biol. 2001;77:177. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- 2.Wessig P, Muehling O. Eur J Org Chem. 2007;2007:2219. [Google Scholar]
- 3.Dryzhakov M, Richmond E, Moran J. Synthesis. 2016;48:935. [Google Scholar]
- 4.Henkel T, Brunne RM, Müller H, Reichel F. Angew Chem, Int Ed. 1999;38:643. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 5.Jin J, MacMillan DWC. Nature. 2015;525:87. doi: 10.1038/nature14885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (b) Xuan J, Xiao WJ. Angew Chem, Int Ed. 2012;51:6828. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]; (c) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]
- 8.Oppolzer W, Moretti R, Thomi S. Tetrahedron Lett. 1989;30:5603. [Google Scholar]
- 9.Seebach D, Wasmuth D. Helv Chim Acta. 1980;63:197. [Google Scholar]
- 10.Myers AG, Yang BH, Chen H, Gleason JL. J Am Chem Soc. 1994;116:9361. [Google Scholar]
- 11.Maruoka K, Ooi T. Chem Rev. 2003;103:3013. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]
- 12.Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J Am Chem Soc. 1994;116:8829. [Google Scholar]
- 13.Doyle AG, Jacobsen EN. J Am Chem Soc. 2005;127:62. doi: 10.1021/ja043601p. [DOI] [PubMed] [Google Scholar]
- 14.(a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shih HW, Vander Wal MN, Grange RL, MacMillan DWC. J Am Chem Soc. 2010;132:13600. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu Y, Zhang L, Luo S. J Am Chem Soc. 2014;136:14642. doi: 10.1021/ja508605a. [DOI] [PubMed] [Google Scholar]; (d) Welin ER, Warkentin AA, Conrad JC, MacMillan DWC. Angew Chem, Int Ed. 2015;54:9668. doi: 10.1002/anie.201503789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohlmann R. Synthesis of Halides. In: Trost BM, editor. Comprehensive Organic Synthesis. Vol. 6. Pergamon: Oxford; 1991. pp. 203–220. [Google Scholar]
- 16.(a) Cozzi PG, Benfatti F, Zoli L. Angew Chem, Int Ed. 2009;48:1313. doi: 10.1002/anie.200805423. [DOI] [PubMed] [Google Scholar]; (b) Guo QX, Peng YG, Zhang JW, Song L, Feng Z, Gong LZ. Org Lett. 2009;11:4620. doi: 10.1021/ol901892s. [DOI] [PubMed] [Google Scholar]; (c) Capdevila MG, Benfatti F, Zoli L, Stenta M, Cozzi PG. Chem - Eur J. 2010;16:11237. doi: 10.1002/chem.201001693. [DOI] [PubMed] [Google Scholar]; (d) Ikeda M, Miyake Y, Nishibayashi Y. Angew Chem, Int Ed. 2010;49:7289. doi: 10.1002/anie.201002591. [DOI] [PubMed] [Google Scholar]; (e) Song L, Guo QX, Li XC, Tian J, Peng YG. Angew Chem, Int Ed. 2012;51:1899. doi: 10.1002/anie.201106275. [DOI] [PubMed] [Google Scholar]; (f) Mo X, Hall DG. J Am Chem Soc. 2016;138:10762. doi: 10.1021/jacs.6b06101. [DOI] [PubMed] [Google Scholar]
- 17.Dedeian K, Djurovich PI, Garces FO, Carlson G, Watts RJ. Inorg Chem. 1991;30:1685. [Google Scholar]
- 18.Wayner DDM, Dannenberg JJ, Griller D. Chem Phys Lett. 1986;131:189. [Google Scholar]
- 19.Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubroeucq MC, Bénavidès J, Doble A, Guilloux F, Allam D, Vaucher N, Bertrand P, Guérémy C, Renault C, Uzan A, Le Fur G. Eur J Pharmacol. 1986;128:269. doi: 10.1016/0014-2999(86)90776-4. [DOI] [PubMed] [Google Scholar]
- 21.Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J Am Chem Soc. 2010;132:13194. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brouwer C, Jenko K, Zoghbi SS, Innis RB, Pike VW. J Med Chem. 2014;57:6240. doi: 10.1021/jm5007947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen JD, Matsuura BS, Stephenson CRJ. J Am Chem Soc. 2014;136:1218. doi: 10.1021/ja4113462. [DOI] [PubMed] [Google Scholar]
- 24.(a) Bacarella AL, Grunwald E, Marshall HP, Purlee EL. J Org Chem. 1955;20:747. [Google Scholar]; (b) Berg U, Jencks WP. J Am Chem Soc. 1991;113:6997. [Google Scholar]; (c) Reeve W, Erikson CM, Aluotto PF. Can J Chem. 1979;57:2747. [Google Scholar]; (d) Arm H, Hochstrasser K, Schindler PW. Chimia. 1974;28:237. [Google Scholar]
- 25.Walaszek DJ, Rybicka-Jasińska K, Smoleń S, Karczewski M, Gryko D. Adv Synth Catal. 2015;357:2061. [Google Scholar]
- 26.Imine 53 is the only oxidative byproduct we have isolated, though it does not account fully for the generation of 49. A small amount of nonspecific aldehyde decomposition also occurs that could account for the formation of 49, and we can detect trace quantities of cinnamaldehyde in situ in reactions employing aldehyde 13, but the full array of byproducts resulting from 52 has yet to be elucidated. H atom abstraction from solvent by 6 does not occur, as no D-incorporation into 49 is observed when conducting the reaction in DMF-d7.
- 27.Cismesia MA, Yoon TP. Chem Sci. 2015;6:5426. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.