

Abstract
CD8+ T cell immunosurveillance dynamics influence the outcome of intracellular infections and cancer. Here we used two-photon intravital microscopy to visualize the responses of CD8+ resident memory T cells (TRM cells) within the reproductive tracts of live female mice. We found that mucosal TRM cells were highly motile, but paused and underwent in situ division after local antigen challenge. TRM cell reactivation triggered the recruitment of recirculating memory T cells that underwent antigen-independent TRM cell differentiation in situ. However, the proliferation of pre-existing TRM cells dominated the local mucosal recall response and contributed most substantially to the boosted secondary TRM cell population. We observed similar results in skin. Thus, TRM cells can autonomously regulate the expansion of local immunosurveillance independently of central memory or proliferation in lymphoid tissue.
Naive T cells limit immunosurveillance to secondary lymphoid organs (SLOs) such as lymph nodes (LNs) through a restricted pattern of recirculation via blood and lymph vessels. Upon antigen encounter in LNs, naive T cells undergo rapid proliferation, giving rise to differentiated effector T cells and long-lived memory T cells that are distributed more broadly throughout the body. Memory T cells are grouped into subsets on the basis of the anatomic locations they patrol and their perceived functional role in the event of reinfection1,2. Central memory T cells (TCM cells), similarly to naive T cells, patrol LNs and seem to be specialized to proliferate in the event of reinfection, in this case when pathogen-derived antigens reach SLOs. TCM cells produce abundant secondary effector T cells that migrate to nonlymphoid sites of infection and also give rise to expanded populations of long-lived memory T cells2,3. Effector memory T cells (TEM cells) generally patrol regions outside of LNs and are typically thought of as terminally differentiated peripheral surveyors poised for rapid expression of effector functions, but not secondary expansion4–6. Both TCM cells and TEM cells recirculate, meaning they can be found in blood. TRM cells7, a third major population of memory T cells, are parked in tissues, where they accelerate clearance of local reinfections, and thus are absent from blood7,8.
Because TRM cells share properties with effector T cells and TEM cells, potentially including the expression of CD69 and granzyme B and the absence of LN homing receptors such as CD62L, the prevailing view is that TRM cells are also terminally differentiated, and thus are not responsible for expansion of their own population or boosting of local nonlymphoid tissue secondary memory T cell populations after reinfection. Rather, boosting of tissue memory is thought to require antigen trafficking to downstream lymphoid organs and proliferation and differentiation by the more ‘stem-cell-like’ TCM cells. Indeed, when TRM cells are restimulated outside of nonlymphoid tissues, expansion is poor compared with that of naive T cell or TCM cell populations, which suggests that T cell intrinsic differences impair proliferation potential9–11. However, TRM cells are difficult to study ex vivo because of their poor survival after being removed from tissues12–14.
TRM cells can trigger a tissue-wide state of pathogen resistance and immune stimulation, and precipitate the recruitment of recirculating lymphocytes to sites of TRM cell reactivation15,16. However, the fate of recruited cells is unclear. Moreover, formal descriptions of the relationship between CD8+ T cell magnitude, location and differentiation state and the efficiency of pathogen detection and clearance are in their infancy. Indeed, we lack in vivo data for mucosal TRM cell motility, which is intrinsically related to the scanning rate of potential target cells. Intravascular TRM cells that patrol liver sinusoids are motile, but it is unclear whether this is true of the numerous TRM cells that survey connective tissues and parenchymal barriers13. Indeed, TRM cell motility in skin epidermis is quite low (~2 μm min−1), in contrast to that of TCM cells surveilling LNs (~10 μm min−1)15,17–19. This suggests that resident populations of memory T cells not only do not migrate between tissues, but also may be relatively stationary within stromal or parenchymal tissues.
To address TRM cell immunosurveillance in the mucosa, we developed an intravital two-photon microscopy model to image mouse uterus after acute lymphocytic choriomeningitis virus (LCMV) infection and combined it with depletion strategies, parabiosis and dual-challenge models to test the relative contributions of TRM cells to secondary population expansion after local anamnestic antigen exposure. We found that compared with circulating memory T cells, TRM cells in both the female reproductive tract and the skin have the potential to dominate local recall responses and contribute most substantially to boosting of the secondary TRM cell population. These data indicate that TRM cells have the capacity to autonomously regulate the expansion of local immunosurveillance independently of TCM cells or proliferation in lymphoid tissue.
Results
T cell motility accelerates after antigen clearance in the female reproductive tract
To establish a model for observing dynamic T cell behavior in the female reproductive tract (FRT) in living mice, we transferred GFP+CD8+ P14 T cell receptor-transgenic (P14) T cells, which are specific for the immunodominant gp33 epitope of LCMV, into naive C57BL/6 mice and then infected the recipients with 2 × 105 plaque-forming units of LCMV Armstrong intraperitoneally (i.p.). This resulted in LCMV infection of the FRT (Fig. 1a), as well as establishment of GFP+ P14 TRM cells in all three layers of the uterine horn20–22 (Supplementary Fig. 1a,b) in the recipient mice (hereinafter referred to as GFP+ P14 immune chimeras). Mice are multiparous and have relatively long uterine horns, which could be sufficiently exteriorized to be imaged under laser-scanning intravital two-photon microscopy (IV-2PM; Methods) while remaining intact within the mouse. Using this approach, we visualized the motility of LCMV-specific GFP+ P14 TRM cells within the uterine mucosa at different stages of LCMV infection. We focused all live imaging on the most superficial 200 μm, which was the optimal depth for use with the instrumentation and tissue properties in this experiment. Thus, our analyses were restricted to the perimetrium and myometrium, and did not include the endometrium or luminal epithelium.
Fig. 1. Migration of CD8+ T cells in the FRT.
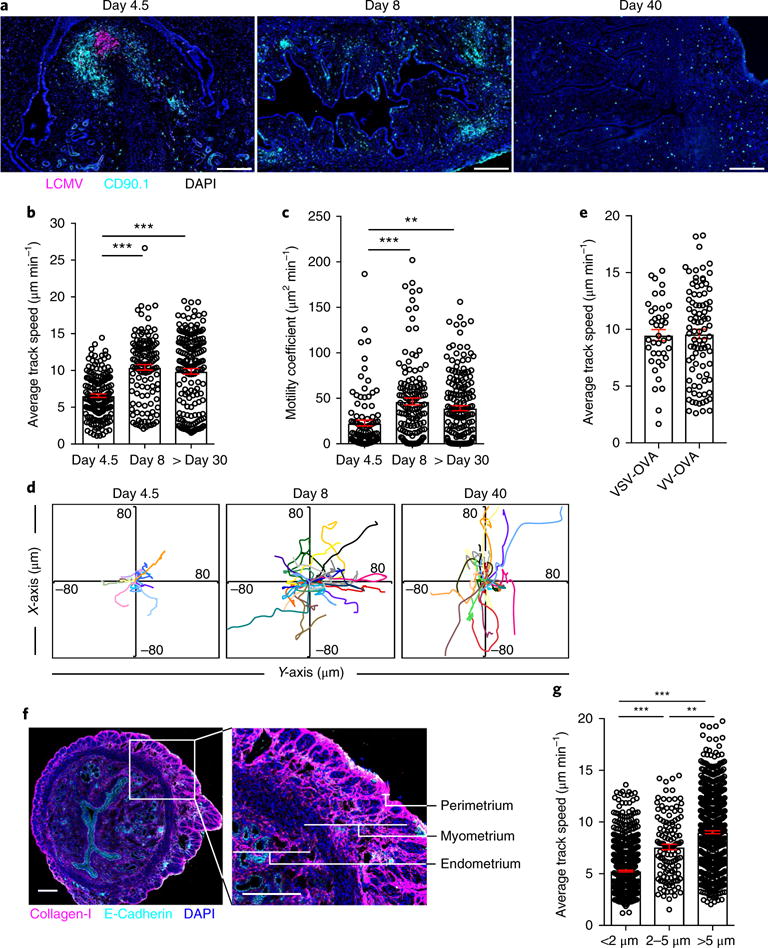
a, CD90.1+GFP+CD8+ P14 T cells were transferred to female C57BL/6J mice 1 d before Infection with LCMV Armstrong. We stained the FRT with DAPI (blue), anti-CD90.1 (to label P14; cyan) and anti-LCMV nucleoprotein (magenta) 4.5, 8 or 40 d later. Scale bars, 200 μm. b,c, The average track speed (b) and motility coefficients (c) of GFP+CD8+ P14 T cells in the FRT on the indicated days after LCMV infection, as determined by intravital microscopy. d, Flower plots of 2D tracks of several GFP+CD8+ P14 T cells, superimposed after normalization of their starting coordinates to the origin. e, GFP+CD8+ OT-I T cells were transferred to female C57BL/6J mice 1 d before infection with VSV-OVA or VV-OVA. The plot shows the average track speed of GFP+CD8+ OT-I T cells in the uterine horn 60 d after the respective infections. f, Transverse sections of mouse uterine horn stained for collagen-l (magenta), E-cadherin (cyan) and nuclei (DAPI; blue). Part of the section on the left (white rectangle) is shown at higher magnification on the right, depicting three layers of uterine horn with different collagen densities. Scale bars, 200 μm. g, CD8+ P14 memory T cell track speeds binned into three groups (<2 μm, 2–5 μm and >5 μm) on the basis of their average distance from the collagen (SHG) surface. Data in b,c,e,g are shown as mean ± s.e.m.; circles represent individual cells. **P<0.01, ***P< 0.001, Kruskal-Wallis one-way analysis of variance (ANOVA) with Dunn’s multiple comparison test. Data are representative of two independent experiments with n = 4 mice per infection (a,f) or three separate experiments with n = 3 mice per group per experiment (b,c,e,g).
At day 4.5 after LCMV infection, GFP+ P14 T cell motility was approximately 6 μm min−1, with some GFP+ P14 T cells arrested (Fig. 1b–d and Supplementary Video 1). Motility increased to ~10 μm min−1 by day 8 after infection, which coincided with the time of observable antigen clearance after LCMV Armstrong infection in mice (Fig. 1a and Supplementary Video 2). At day 30 after LCMV infection, at which time T cells in the FRT are almost exclusively TRM cells12,21, GFP+ P14 T cell motility remained high at ~10 μm min−1 (Supplementary Video 3). Antigen clearance was also accompanied by an increase in the motility coefficient, expressed as the rate of GFP+ P14 T cell displacement from the starting position (Fig. 1c). These data suggest that CD8+ TRM cell motility in the stroma of the FRT is much faster than that reported in skin epidermis (2 μm min−1)17. The motility of GFP+ OT-I memory T cells, which recognize the ovalbumin (OVA)-derived peptide SIINFEKL, established in the FRT 60 d after infection with either vesicular stomatitis virus (VSV) or vaccinia virus (VV) expressing OVA (VSV-OVA or VV-OVA, respectively) was also ~10 μm min−1 (Fig. 1e).
The discrepancy in motility that we observed between the FRT and what has been reported for skin epidermis, which comprises compacted layers of flattened cells, could be due to the difference in tissue architecture. We found that the motility of GFP+ P14 T cells was lower (~6 μm min−1) in the perimetrium (the outer serosal lining of the uterus) and the subjacent thin layer of dense connective tissue than in the myometrium (the muscular layer of the uterus; ~10 μm min−1) (Supplementary Fig. 2). In fact, the average speed of GFP+ P14 T cells migrating in less collagen-rich areas (tracks > 2 μm away from second harmonic generation (SHG) signals) was significantly faster than that of cells close to collagen-dense regions (< 2 μm from SHG signals; Fig. 1f,g). In summary, both antigen and location have a role in CD8+ T cell migration within the uterine stroma.
Local reactivation results in motility arrest of TRM cells
Previous studies indicated that > 99% of LCMV-Armstrong-specific P14 CD8+ T cells within the FRT are resident at any time point 1–8 months after LCMV infection, 60–75% of TRM cells in the FRT express CD69, and only a minority express CD10312. To test whether TRM cell motility is affected by local antigen detection, which could influence subsequent immunosurveillance rates, we used mice that had received adoptively transferred GFP+ P14 T cells and been infected with LCMV Armstrong as described above, and we deposited LCMV-gp33 peptide into the lumen of the cervix (trans-cervical delivery) in these mice 60 d after primary LCMV infection. Corresponding with TRM cell reactivation16,21, we observed changes in dendritic cell morphology and a reduction in the average motility of GFP+CD8+ P14 T cells from 10.1 μm min−1 at steady state to 5.3 μm min−1 within 12 h (Fig. 2a,b and Supplementary Videos 4 and 5). These motility changes were transient, as GFP+CD8+ P14 T cells resumed their previous migration behavior with respect to both mean track speed and motility coefficient within 48 h after gp33 exposure (Fig. 2b).
Fig. 2. Motility arrest by TRM cells after cognate antigen interaction.
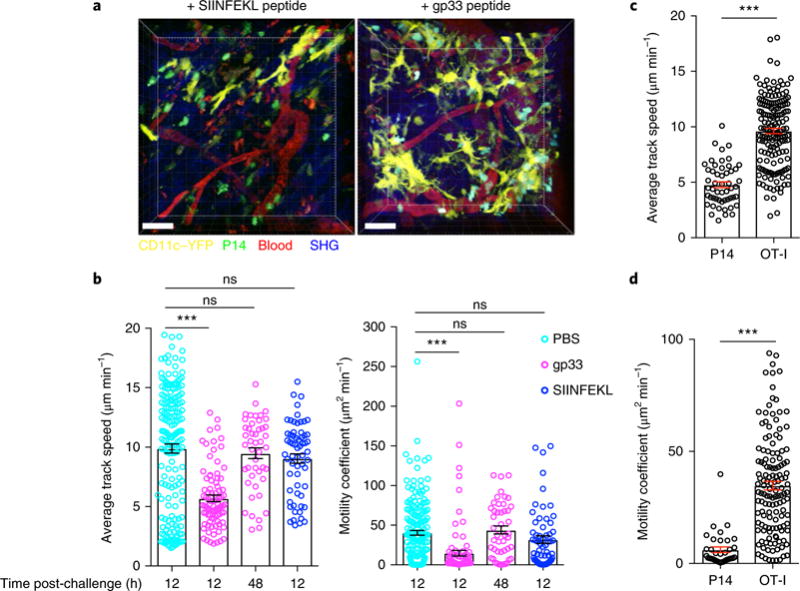
a, To assess in situ TRM cell reactivation, we transferred naive GFP+CD8+ P14 T cells into female C57BL/6J mice 1 d before infection with LCMV Armstrong, and 60 d later we challenged the mice trans-cervically with PBS, P14 reactivating peptide gp33 or control peptide SIINFEKL. Shown are representative snapshots, from Supplementary Videos 4 and 5, of the maximal projection of 3D z-stack images of P14 immune chimeras 12 h after trans-cervical exposure to peptide. Green, GFP+CD8+ P14 T cells; yellow, CD11c+ dendritic cells. Scale bars, 40 μm. b, Average track speed and motility coefficients of GFP+CD8+ P14 T cells 12 h or 48 h after trans-cervical challenge with the indicated peptide. c,d, To assess the motility of a bystander population of memory T cells, we transferred CFP+CD8+ P14 T cells into naive mice 1 d before LCMV infection. Thirty days later, we transferred GFP+CD8+ OT-I T cells into these LCMV immune chimeras, and then infected them with VSV-OVA. Thirty days after VSV-OVA infection (60 d after LCMV infection), we challenged these double-immune chimeras trans-cervically with gp33 peptide, and carried out intravital microscopy 12 h after challenge. The average track speed (c) and motility coefficients (d) of P14 and OT-I CD8+ T cells 12 h after gp33 peptide challenge are shown. Data are representative of two separate experiments with 4 mice per group per experiment. ***P< 0.001; ns, not significant; Kruskal-Wallis ANOVA with Dunn’s multiple comparison test (b) or Mann-Whitney U-test (c,d). Data in b-d are shown as the mean ± s.e.m.; circles represent individual cells.
We next tested whether TRM cell motility arrest is dependent on stimulation by cognate antigen or is a result of bystander effects of local peptide exposure and/or neighboring TRM cell activation. To generate mice with traceable TRM cells with discrete antigenic specificities, we adoptively transferred GFP+ OT-I T cells into cyan fluorescent protein (CFP)-expressing P14 immune chimeras 30 d after LCMV infection, and then infected the chimeras with VSV-OVA. Thirty days after VSV-OVA infection, we exposed the CFP+ P14−GFP+ OT-I immune chimeric mice to gp33 peptide locally through trans-cervical rechallenge, and monitored the mean track speeds of CFP+ P14 T cells and GFP+CD8+ OT-I T cells simultaneously (Supplementary Video 6). Only the migration of CFP+ P14 T cells was reduced at 12 h compared with that of resting OT-I CD8+ memory T cells that did not recognize recall antigen (Fig. 2c,d), which indicated that TRM cell motility arrest depends on cognate-antigen-specific T-cell-receptor-mediated signals, rather than bystander effects.
In situ division in the female reproductive tract
Next, we assessed the dynamics of the response in the FRT after local antigen rechallenge by infecting mice carrying adoptively transferred P14 CD8+ T cells with LCMV Armstrong and then subjecting them to transcervical rechallenge with gp33 60 d after LCMV infection. IV-2PM monitoring of the recall response indicated the accumulation of GFP+ P14 T cells in the uterine mucosa 36 and 48 h after gp33 peptide inoculation (Fig. 3a,b). We observed hundreds of GFP+ P14 T cells undergoing in situ division at 36 h after gp33 peptide inoculation, with rare examples as early as 32 h (Fig. 3c and Supplementary Video 7). Consistent with this observation, P14 CD8+ T cells in the FRT were Ki67+ 48 h after local rechallenge, which indicates that they were undergoing proliferation (Fig. 3d,f and Supplementary Fig. 1c,d). The mice were also i.p. pulsed with 5-bromo-2′-deoxy-uridine (BrdU) 2 h before they were killed. By 2 h after BrdU treatment, ~25% of P14 CD8+ T cells had incorporated BrdU (Fig. 3d), which indicated that they were undergoing active DNA synthesis and thus confirmed that they were proliferating.
Fig. 3. In situ division of CD8+ T cells in the FRT.
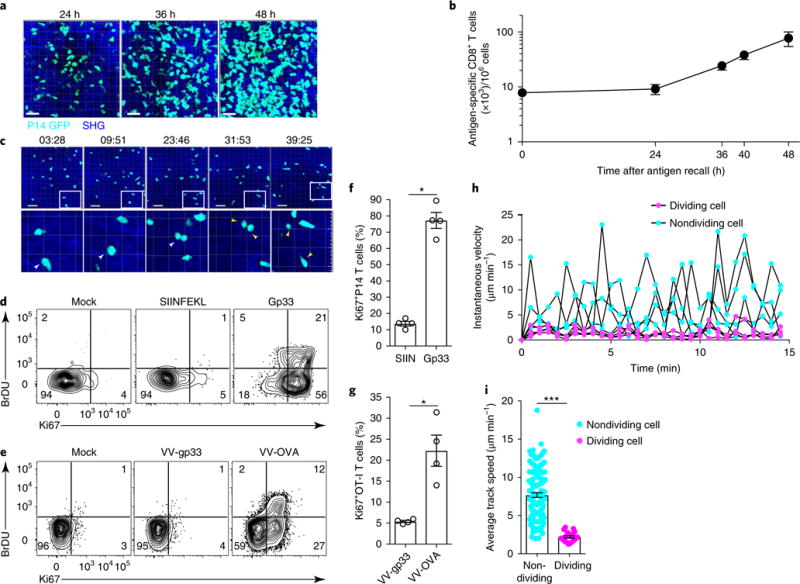
a,b, Accumulation of P14 CD8+ T cells after local TRM cell reactivation in FRT. a, Representative maximal projections of 3D z-stack images of GFP+CD8+ P14 T cells (cyan) after trans-cervical challenge with gp33. Scale bars, 30 μm. b, The abundance of P14 CD8+ T cells enumerated by QIM in 7-μm sections of FRT. c, Top, representative time-lapse images (time stamps in minutes and seconds are shown above the images) of GFP+CD8+ P14 T cells (cyan) undergoing division beginning ~36 h after gp33 challenge. Scale bars, 40 μm. Bottom, magnified views (4× magnification compared with the images above) of dividing cells, indicated by white arrowheads (or two yellow arrowheads for cells after cytokinesis). Magnified cells are outlined by white rectangles in the images above. d, P14 immune chimeras were challenged with peptides (gp33 or SIINFEKL) or mock-challenged (with PBS), and tissues were harvested 48 h after challenge. Animals received 2 mg of BrdU i.p. before cell collection. e, OT-I immune chimeras were challenged with viruses (VV-OVA and VV-gp33) or mock-challenged, and tissues were harvested 48 h after recall. In d and e, numbers in corners indicate the percentage of cells in each quadrant. f,g, The frequency of Ki67+ cells in FRT of the mice described in d (see f) and e (see g). SIIN, SIINFEKL. h, Instantaneous track speed versus time, beginning 15min before cytokinesis, for four representative dividing and four nondividing P14 CD8+ T cells. i, The mean track speed of dividing and nondividing cells 36h after recall. *P< 0.05, ***P< 0.001, Mann-Whitney U-test. Data in b,f-i are shown as the mean ± s.e.m. Data are representative of two separate experiments with 3 (a,b) or 4 (f,g) mice per group per experiment or are pooled from 6 individual movies and 3 animals (i).
Transgenic fluorophore-expressing CD8+ T cells are necessary for IV-2PM visualization of antigen-specific responses. To determine whether the division of GFP+ P14 T cells was an artifact of adoptive transfer, we immunized C57BL/6 J (B6; ‘wild-type’) mice with LCMV, rechallenged the mice with gp33 peptide trans-cervically 90 d after infection, and measured Ki67 expression and BrdU incorporation in endogenous LCMV-specific CD8+ T cells detected with H-2Db/gp33 MHC I tetramers (Supplementary Fig. 3). Tetramer-positive cells became Ki67+ and BrdU+, which indicates that antigen-specific TRM cells divide in the FRT after local rechallenge. To test whether in situ division of TRM cells occurs in response to live replicating virus, we transferred naive CD45.1+CD8+ OT-I T cells into naive B6 recipients and then immunized them with VSV-OVA. Ninety days later, we challenged the mice trans-cervically with VV-OVA or VV expressing gp33 epitope (VV-gp33). CD45.1+CD8+ OT-I T cells became Ki67+ and/or BrdU+ in response to cognate-antigenbearing viral rechallenge, which indicates that live replicating virus induced division by antigen-specific CD8+ T cells within the FRT (Fig. 3e,g). Lastly, we assessed the relationship between motility and division. Two months after primary LCMV infection, we challenged GFP+ P14 immune chimeras with LCMV-gp33 peptide trans-cervically, and 36 h after gp33 recall we used intravital microscopy to monitor the motility of GFP+ P14 T cells undergoing active division compared with that of GFP+ P14 T cells that were not dividing in a 15-min period. Dividing GFP+ P14 T cells underwent a transient reduction in motility, then regained rapid motility after cytokinesis was completed (Fig. 3h,i and Supplementary Video 7). In summary, these data indicate that antigen-specific memory T cells in the FRT undergo pausing and cytokinesis after local antigen rechallenge.
TRM cells divide in the female reproductive tract after reactivation
We next investigated whether TRM cells in the FRT undergo division in situ, or whether antigen rechallenge induces the division of only antigen-specific T cells newly recruited from the blood, such as TCM cells. Using mice that had previously received adoptively transferred GFP+ P14 T cells and been infected with LCMV Armstrong as described above, we administered anti-CD90.1 titrated to induce depletion of blood P14 CD8+ T cells, but insufficient to deplete P14 CD8+ T cells confined within the FRT. A single dose of anti-CD90.1 caused P14 CD8+ T cells to be almost completely eliminated from blood, LNs and spleen while remaining abundant in the FRT (Fig. 4a). We additionally blocked the migration of circulating P14 CD8+ T cells into the FRT by injecting anti-VCAM-1 and anti-CD49d 24 h before and 24 h after trans-cervical challenge with gp3316. For proliferation monitoring, we administered BrdU i.p. 2 h before we collected P14 CD8+ T cells from the FRT. We observed that 24 and 36 h after gp33 challenge, P14 CD8+ T cells in the FRT of mice treated with anti-CD90.1, anti-VCAM-1 and anti-CD49d showed equivalent BrdU incorporation and Ki67 upregulation compared with T cells in challenged mice that did not receive antibodies (Fig. 4b,c). In addition, intravital microscopy showed GFP+ P14 T cell proliferation in situ in GFP+ P14 immune chimeras treated with these antibodies (Supplementary Video 8). These results indicate that P14 CD8+ TRM cells rapidly initiate in situ proliferation in the FRT mucosa after anamnestic challenge.
Fig. 4. TRM cells initiate division in mucosae after reactivation.
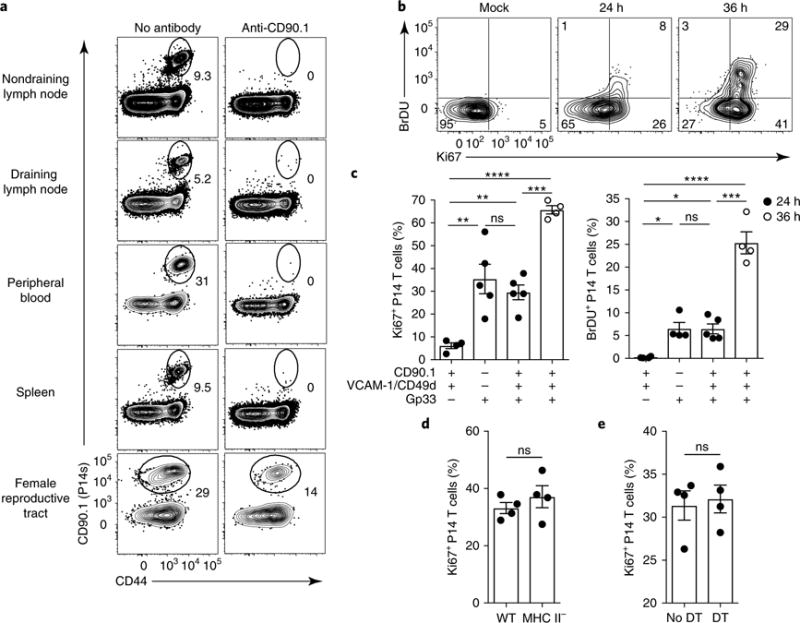
a, The presence of remaining P14 memory CD8+ T cells in the indicated tissues 4 d after i.p. anti-CD90.1 treatment in P14 LCMV immune chimeric mice. Numbers indicate the percentage of cells in the gate. b, P14 immune chimeras were treated as in a. Their blood was assessed for successful depletion of P14 CD8+ T cells 4 d later, and they received anti-VCAM-1 and anti-CD49d i.p. on day 4. On day 5, mice were rechallenged trans-cervically with gp33 peptide or PBS (Mock), and they were assessed 24h or 36 h after rechallenge. Mice received 2 mg of BrdU i.p. 2 h before they were killed, and P14 CD8+ T cells isolated from FRT were stained for Ki67 and BrdU incorporation by flow cyotmetry. Numbers in corners indicate the percentage of cells in the gate. c, The percentage of Ki67+ or BrdU+ cells among P14 CD8+ T cells isolated from the FRT of mice treated with anti-CD90.1, anti-VCAM-1, anti-CD49d and gp33 as indicated. Data points represent individual mice. d, Ki67+CD8+ P14 T cell frequency as enumerated by flow cytometry 24h after challenge in wild-type (WT) and MHC-II-deficient P14 immune chimeric mice treated as in b. e, P14 immune chimeras were generated in CD11c-DTR chimeric mice. Sixty days later, mice were either treated with diphtheria toxin (every 48 h starting 2 d before recall) or left untreated. The mice were also treated with antibodies to CD90.1, VCAM-1, CD49d and gp33 peptide as in b and c. The plot shows the percentage of Ki67+CD8+ P14 T cells isolated from FRT as determined by flow cytometry 24 h after challenge. ns, not significant; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001; Kruskal-Wallis ANOVA (c) or Mann-Whitney U-test (d,e). Data in c–e are shown as the mean ± s.e.m. Data are representative of two separate experiments with 3 (a) or 4 (d,e) mice per group per experiment, or three separate experiments with 4 mice per group per experiment (b,c).
It was previously shown that help from CD4+ T cells is required to promote CD8+ T cell migration to the mouse FRT and to establish local CD8+ T cell memory after infection with a thymidine-kinase-deficient strain of herpes simplex virus (HSV) type 223. However, CD4+ T cell help is dispensable for the establishment of skin CD8+ TRM cells after exposure to VV24. Moreover, CD4+ T cell help has been reported to be important for priming memory CD8+ T cells that retain proliferative potential, although this has not been addressed for TRM cells25·26. Thus we addressed whether the establishment of TRM cells in the FRT requires help from CD4+ T cells after infection with LCMV, by transferring naive P14 CD8+ T cells into MHC-II-deficient recipients and then infecting them with LCMV Armstrong 1 d after cell transfer. We collected P14 CD8+ T cells from the FRT of MHC-II-deficient mice and from wild-type B6 mice 40 d after infection, and found that the cell numbers were similar in both groups of mice (Supplementary Fig. 4). We observed similar results at 7 d after LCMV infection and in Cd4−/− mice (data not shown and Supplementary Fig. 4). Next, we depleted the circulating P14 CD8+ T cells with anti-CD90.1 (administered i.p. 96 h before rechallenge) and blocked de novo migration into the FRT with anti-VCAM-1 and anti-CD49d (administered i.p. 1 d before the rechallenge) in wild-type and MHC-II-deficient P14 immune chimeras, and then rechallenged the mice with gp33 trans-cervically. Upregulation of Ki67 by CD90.1+CD8+ P14 T cells was equivalent (~40%) in wild-type and MHC-II-deficient mice (Fig. 4d), which suggests that CD90.1+CD8+ P14 T cells did not require CD4+ T cell help during either priming or reactivation to initiate entry into the cell cycle. We also tested the requirement for CD11c+ cells, which include dendritic cells, for TRM cell reactivaion. We irradiated B6 mice and then transplanted bone marrow from CD11c-DTR mice, in which CD11c+ cells express the diphtheria toxin (DT) receptor. Two months later, we delivered adoptively transferred CD90.1+CD8+ P14 T cells into the CD11c-DTR chimeric mice and infected them with LCMV Armstrong. Sixty days after LCMV infection, we began treating the mice with DT, with administration every 2 d starting at 48 h before trans-cervical rechallenge with gp33, which resulted in depletion of ~90% of CD11c+ cells in the FRT (Supplementary Fig. 4d). Twenty-four hours after gp33 challenge, ~35% of P14 CD8+ T cells upregulated Ki67 at similar levels in DT-treated and untreated control mice (Fig. 4e). These results indicate that bona fide TRM cells were triggered to undergo proliferation in situ after mucosal tissue rechallenge, and that this process did not depend on CD4+ T cell help and was probably independent of CD11c+ host cells.
Recruited circulating bystanders convert to TRM cells
TRM cell reactivation in the mucosa induces rapid upregulation of inflammatory homing molecules, which results in local recruitment of circulating memory T cells in a chemokine- and integrin-dependent process21. However, the long-term fate of these newly recruited bystander CD8+ T cells is unknown. To address this issue, we isolated memory OT-I CD8+ T cells from the spleen and LNs of mice that had been infected with VSV-OVA at least 60 d prior and transferred them i.v. into mice that had received adoptively transferred P14 CD8+ T cells and been infected with LCMV Armstrong at least 60 d prior. We then subjected these mice to trans-cervical rechallenge with gp33 1 d after memory OT-I CD8+ T cell transfer. At 45 and 75 d after gp33 rechallenge, we detected the accumulation of CD45.1+CD8+ OT-I T cells in the FRT of recipient CD90.1+ P14 immune chimeric mice that were challenged with LCMV-gp33 trans-cervically, compared with cell numbers in PBS-challenged mice (Fig. 5a). A substantive fraction of the CD45.1+CD8+ OT-I T cells were CD69+, although 60% were CD69−, and very few expressed CD103 (Fig. 5b,c). Despite this phenotypic heterogeneity, we assessed whether the recruited OT-I CD8+ T cells were resident by transferring circulating memory CD45.1+CD8+ OT-I T cells into CD90.1+ P14 immune chimeric mice and then challenging the mice with gp33 peptide trans-cervically to recruit the OT-I CD8+ T cells to the FRT. Forty days after gp33 challenge, we surgically joined these mice with naive B6 mice (Fig. 5d). At 35 d after parabiosis, we could not detect any CD45.1+CD8+ OT-I T cells in the FRTs of the naive parabiotic partners (Fig. 5e), which indicated that almost all bystander memory OT-I T cells retained were resident. These data suggest that induction of the TRM cell program within the FRT does not absolutely require local cognate antigen recognition, that recirculating memory T cells retain the developmental potential to differentiate into TRM cells, and that periodic reinfection events may induce bystander memory T cell redistribution out of circulation and into mucosal tissues.
Fig. 5. Recruited circulating bystander memory CD8+ T cells adopt a TRM cell phenotype.
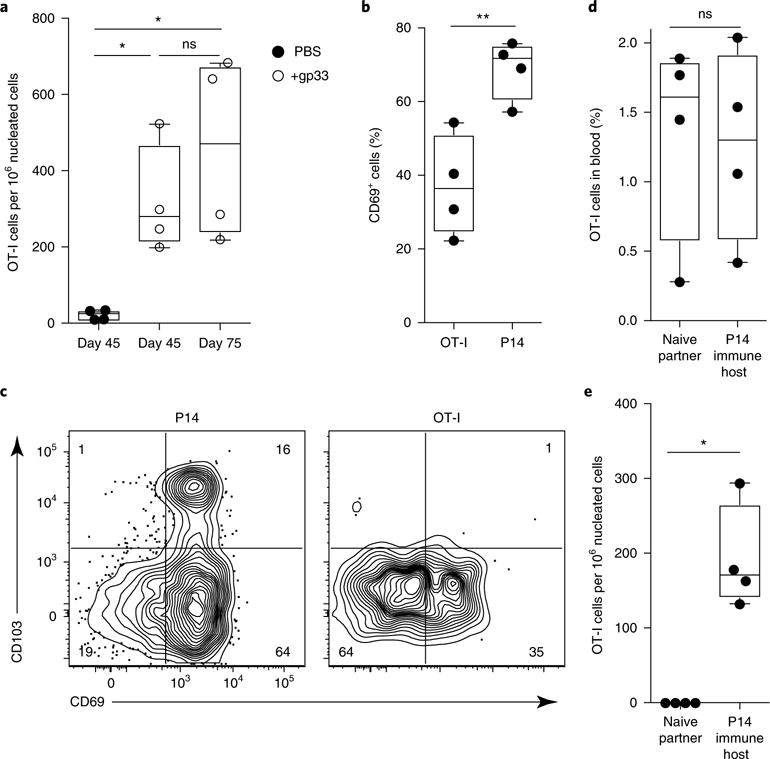
a, To pull non-antigen-specific recirculating OT-I memory cells to the FRT, we transferred OT-I memory CD8+ T cells i.v. into P14 immune chimeras. The next day, we challenged recipients trans-cervically with PBS (mock) or gp33 peptide. Mice were rested until day 45 or day 75, at which time we used QIM to enumerate OT-I CD8+ T cells in the FRT. b, The frequency of FRT OT-I or P14 CD8+ T cells that were CD69+, as evaluated by flow cytometry. c, Representative flow cytometry plots of CD69 and CD103 staining on P14 and OT-I CD8+ T cells 45 d after recall. Numbers in corners indicate the percentage of cells in each quadrant. d,e, To confirm that recruited OT-I CD8+ T cells were resident, we carried out parabiotic surgery to conjoin mice treated 40 d prior with trans-cervical gp33 peptide as described above (P14 immune hosts) with naive B6 partner mice. We examined each parabiont for the presence of OT-I CD8+ T cells in blood and FRT 35 d after surgery. *P<0.05, **P< 0.01, Kruskal-Wallis ANOVA (a) or Mann-Whitney U-test (b,d,e). Box plots indicate medians (center lines), 25th and 75th percentiles (bottom and top box edges, respectively), minima and maxima (whiskers), and individual data points (circles). Data are representative of three separate experiments with 4 mice per group per experiment (a–c) or two separate experiments with four parabiont pairs per experiment (d,e).
TRM cell proliferation dominates expansion of local secondary memory
Local exposure to anamnestic antigen resulted in rapid and profound population expansion of mucosal antigen-specific T cells (Fig. 3). This recall response could have been dominated by non-TRM cells (for example, TCM cells that were reactivated in the draining LN or were newly recruited to the FRT) or in situ expansion of preformed TRM cells. To distinguish among these possibilities, we adoptively transferred naive CD8+ T cells from CD45.1+ P14 mice or CD90.1+ P14 mice into CD45.2+CD90.2+ C57BL/6 recipient mice and infected those recipients with LCMV the following day. Sixty days after LCMV infection, we carried out parabiotic surgery to join CD45.1+ P14 mice and CD90.1+ P14 immune chimeric mice, and we allowed 14–30 d after surgery to induce equilibration of circulating P14 CD8+ T cells before proceeding to trans-cervical challenge with gp33 in both mice of the parabiotic pair (Supplementary Fig. 5). As shown by both flow cytometry and quantitative immunofluorescence microscopy (QIM), the ratios of CD45.1+CD8+ P14 T cells to CD90.1+CD8+ P14 T cells were similar in the spleens of both mice in the parabiotic pair 2 d after gp33 challenge (Fig. 6a,b). In contrast, in the FRT compartment, host P14 CD8+ T cells dominated over parabiotic-partner-derived P14 CD8+ T cells (Fig. 6b,c), even though the total number of P14 CD8+ T cells in the FRT had increased tenfold (Fig. 6d), which indicated that 2 d after local rechallenge most of the accumulating antigen-specific T cells were derived from TRM cells. Consistent with this interpretation, at this early time point, most host-derived P14 CD8+ T cells in the FRT expressed markers of TRM cells (particularly CD69), in contrast to parabiont-partner-derived P14 CD8+ T cells (Fig. 6a). The disequilibrium between host and donor-derived P14 CD8+ T cells in the FRT was more pronounced 30 d after local challenge (the host/parabiont-partner-derived P14 CD8+ T cell ratio was ~10:1 at day 2, compared with ~24:1 at day 30) (Fig. 6c,e–g), which suggests that TRM cells locally expanding during the effector phase of the response had a greater propensity to differentiate into secondary memory TRM cells compared with recirculating memory T cells. We observed an approximately threefold increase in the number of total memory T cells (combined host and parabiont-partner-derived P14 CD8+ T cells) in the FRT 30 d after rechallenge (Fig. 6d), with the host/parabiont-partner P14 CD8+ T cell ratio highly biased in favor of the host cells (Fig. 6g). These data demonstrate that at least in this reductionist setting, TRM cells can contribute substantively to secondary effector accumulation and local anamnestic memory.
Fig. 6. TRM cell proliferation dominates expansion of local secondary memory T cell populations.
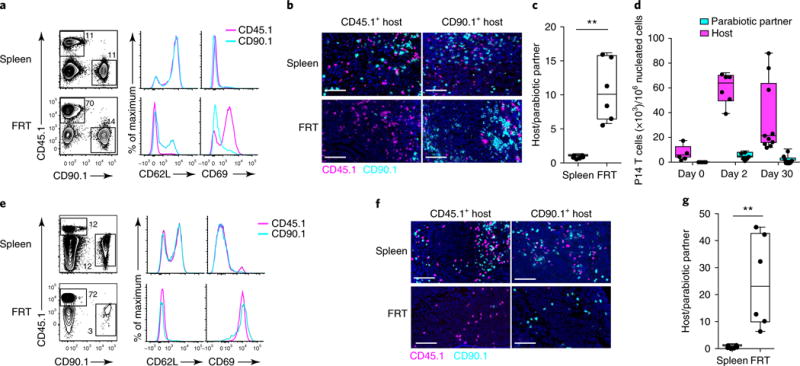
a, Equilibration of recirculating memory CD8+ T cells in parabiotically conjoined CD45.1+ and CD90.1+ P14 immune chimeras challenged trans-cervically with gp33 peptide 14–30 d after surgery. Scale bars, 50 μm. a–g, We analyzed spleen and FRT by flow cytometry 2 d (a) or 30 d (e) after gp33 challenge to evaluate the relative contributions of host and donor P14 CD8+ T cells to the secondary response. Tissues from a CD45.1+ immune chimera are shown, and histograms indicate phenotypes of gated CD45.1+ and CD90.1+ P14 CD8+ T cells at day 2 (a) or day 30 (e) post-recall. CD45.1+ and CD90.1+ P14 CD8+ T cells were also analyzed in each parabiont by immunofluorescence staining 2 d (b) and 30 d (f) after t.c. gp33 challenge. d, Host and parabiotic partner P14 CD8+ T cells enumerated by QIM on the indicated days after trans-cervical gp33 challenge. c,g, The ratio of host and parabiotic partner P14 CD8+ T cells in each parabiont 2 d (c) and 30 d (g) after trans-cervical peptide recall. All data are representative of two separate experiments with at least four parabiont pairs/time points, with a total of > 16 individual mice in individual groups. **P<0.01, Mann-Whitney U-test. In a and e, numbers adjacent to outlines indicate the percentage of cells in the gate. Box plots indicate medians (center lines), 25th and 75th percentiles (bottom and top box edges, respectively), minima and maxima (whiskers), and individual data points (circles).
To extend our analysis to a different rechallenge model and a different anatomic site, we adoptively transferred either CD45.1+CD8+ OT-I T cells or CD90.1+CD8+ OT-I T cells into C57BL/6 mice and then infected the mice i.v. with VSV-OVA, which induced the generation of OT-I memory CD8+ T cells in both the FRT and the skin (Supplementary Fig. 6). Thirty days after VSV-OVA infection, we established parabiotic pairs, each consisting of one mouse that had received transferred CD45.1+ OT-I T cells and one that had received CD90.1+ OT-I T cells, and allowed equilibration of circulating OT-I CD8+ T cells for 14–30 d. We then rechallenged each conjoined parabiont locally with modified VV Ankara expressing the immunodominant SIINFEKL epitope derived from OVA (MVA-OVA) via either the trans-cervical (Fig. 7a–c) or the epicutaneous route27 (Fig. 7d–f). One month later, we used QIM to examine the expansion and origin of OT-I CD8+ T cell populations in each mouse’s FRT or skin. There were approximately ninefold more host-derived OT-I CD8+ T cells than parabiotic-partner-derived OT-I CD8+ T cells in both FRT (Fig. 7b) and skin (Fig. 7e), which indicated that local FRT or skin challenge with MVA-OVA resulted in local amplification of host-derived TRM cell populations. These data suggest that TRM cells may autonomously amplify local immune surveillance and memory in various barrier sites in response to a range of stimuli.
Fig. 7. TRM cell proliferation dominates expansion of local secondary memory T cell populations after FRT or skin rechallenge.
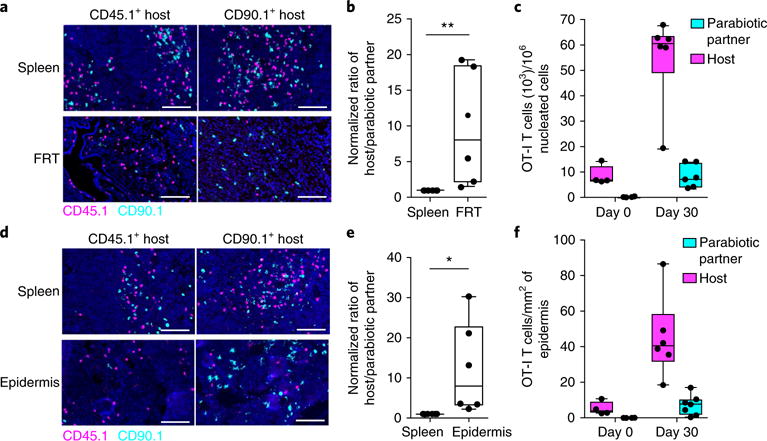
a, Tissues from VSV-OVA-immunized CD45.1+ and CD90.1+ OT-I immune chimeras that were surgically conjoined by parabiosis to achieve equilibration of recirculating memory CD8+ T cells. a–f, Both parabionts were challenged with MVA-OVA 14–30 d after surgery via either the (a–c) trans-cervical or (d–f) epicutaneous route. OT-I CD8+ T cells were analyzed by immunofluorescence staining and enumerated by QIM in either FRT (b,c) or skin epidermis (e,f) and in the spleen 30 d after rechallenge. Magenta, CD45.1; cyan, CD90.1; blue, DAPI (nuclei). Scale bars, 50 μm. Ratios of host and donor-derived OT-I CD8+ T cells were calculated after normalization to the respective ratios in spleen. Absolute numbers of OT-I CD8+ T cells in FRT (c) and skin epidermis (f) in unchallenged and MVA-OVA-challenged (30 d post-recall) mice are shown. All data are representative of two separate experiments with three parabiont pairs per experiment, with a total of 12 individual mice in individual groups. *P< 0.05, **P<0.01, Mann-Whitney U-test (b) or unpaired t-test (e). Box plots indicate medians (center lines), 25th and 75th percentiles (bottom and top box edges, respectively), minima and maxima (whiskers), and individual data points (circles).
Discussion
This study demonstrates that CD8+ TRM cells within the stroma of the FRT exhibit motility rates that are similar to those of circulating lymphocytes in LNs. In response to local re-encounter with cognate antigen, TRM cells pause, then proliferate even in the absence of CD4+ T cell help. In the contexts examined here, FRT and skin TRM cells dominated the local recall response and made a greater contribution than did circulating memory T cell subsets, including TCM cells, to the expansion of a long-lived anamnestic TRM cell population. These data highlight nonlymphoid-tissue-autonomous amplification of adaptive immune responses.
We observed that TRM cells patrolled with a speed of 10 μm min−1 within the FRT myometrium, similar to the motility rate observed in SLOs. Unlike uterine myometrium and SLOs, the epidermis is densely populated with stationary keratinocytes, which suggests that motility may be partly regulated by tissue architecture28,29. Indeed, we found that motility was substantially reduced (~6 μm min−1) in the matrix-dense region of the uterine perimetrium. These observations argue against the concept that TRM cells are necessarily stationary within tissues and reveal the marked heterogeneity of TRM cell immunosurveillance patterns, which has important implications for modeling the requirements for immunity at frontline tissues.
It has been reported that after HSV infection in mice, effector CD4+ T cells are required to migrate to the FRT before CD8+ T cells can enter23. This important observation, if generalized, suggests that cytotoxic T lymphocyte responses to nonlymphoid tissues are intrinsically delayed, and thus are likely to impede the rapidity of viral control. It is therefore important to note that we observed no impairment in the migration of CD8+ effector T cells (or the establishment of CD8+ TRM cells) to the FRT after LCMV infection in mice that lacked CD4+ T cells, similar to what has been shown recently in an intracerebral LCMV infection model30.
Our observations confirmed previous reports that TRM cell reactivation induces the recruitment of bystander (non-antigen-specific) memory T cells to sites of ongoing infection21,31,32. In the current study, we found that some recruited bystander CD8+ T cells persisted for more than 75 d in the nonlymphoid site of recruitment. This suggests that T cell migration from recirculating to resident compartments could be more dynamic in organisms that experience frequent recall infections (unlike laboratory mice that live in specific-pathogen-free facilities). Antigen encounter in non-lymphoid tissues has a role in shaping TRM cell development, but the rules are unclear and seem to be tissue specific20,33–35, although this issue has not been well addressed in the FRT. We found that a fraction of recruited bystander memory CD8+ T cells upregulated CD69, but not CD103. Thus, cognate antigen recognition in tissue was clearly not required for CD69 induction. It remains unclear whether bystander recruited T cells, which include TCM cells and TEM cells, had lost the developmental capacity to fully acquire TRM cell differentiation markers, or distribute differently, or whether local infection was required to enhance CD69 and CD103 induction signals36. The lineage relationship between TRM cells and other memory T cell subsets is an area of active investigation.
The degree to which TRM cells have the capacity for recall expansion in tissues also remains unclear8. Although some reports indicate that neither effector nor memory T cells divide within nonlymphoid tissues, an elegant study in the brain parenchyma challenged this view37. It showed that effector T cells could complete a division in situ, although those cells were pre-committed to divide (perhaps in the lymphoid tissue) prior to entry into the central nervous system37. Naive T cells can be primed in low-shear-flow vascular compartments, including the liver38. T cells can also be induced to divide within nonlymphoid tissue39 by transplantation of the dorsal root ganglia of HSV-infected mice under the kidney capsule of naive mice, which induces viral reactivation. In this scenario, division depends on CD4+ T cells and dendritic cells, which suggests that the rules for tissue division may be context dependent39. Here we found that TRM cells in the FRT could be induced to undergo cell division in situ and complete antigen-induced cytokinesis within only 36 h of local stimulation. This was independent of CD4+ T cell help, and was not impeded when 90% of CDl1c+ cells were depleted. In addition, in situ TRM cell proliferation dominated secondary expansion.
Residence provides a mechanism for regionalizing immunosurveillance by retaining cells in relevant locations, rather than relying on a tissue-restricted recirculation program. Our study extends this paradigm by revealing tissue-autonomous amplification of local memory, and suggests that iterative stimulation at a local site could enrich memory T cell immunosurveillance at very specific sites. One important implication is that these recall responses would remain undetected if analysis were limited to blood. The clinical relevance of local self-amplifying T cell responses may extend beyond infectious disease40,41, perhaps to include T-cell-driven autoimmune and allergic responses7,42.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at https://doi.org/10.1038/s41590-017-0029-3.
Methods
Mice
C57BL/6 J (B6), C57BL/6-Tg(UBC-GFP)30Scha/J, B6.129(ICR)-Tg (CAG-ECFP)CK6Nagy/J, B6.129S2-H2dlAb1-Ed/J (MHC II−), B6.FVB-Tg(Itgax-DTR/EGFP)57Lan/J (CD11c-DTR), B6.Cg-Tg(Itgax-Venus)1Mnz/J (CD11c-YFP) and B6.SJL-PtprcaPepcb/BoyJ (CD45.1+) female mice were from The Jackson Laboratory and were maintained in specific-pathogen-free conditions at the University of Minnesota. CD90.1+ P14, CD45.1+ P14 and CD45.1+ OT-I mice were fully backcrossed to C57BL/6 J mice and maintained in our animal colony. We generated P14 and OT-I reporter mice by crossing the mice with C57BL/6-Tg(UBC-GFP)30Scha/J or B6.129(ICR)-Tg(CAG-ECFP)CK6Nagy/J mice. Sample size was chosen on the basis of previous experience. No sample exclusion criteria were applied. No method of randomization was used during group allocation, and investigators were not blinded. All mice used in experiments were 6–14 weeks of age. All mice were used in accordance with the Institutional Animal Care and Use Committees guidelines at the University of Minnesota.
Adoptive transfers and infections
We generated P14 immune chimeras by transferring 5 × 104 P14 CD8+ T cells into naive C57BL/6 J mice and then infecting those mice with 2 × 105 plaque-forming units (PFU) of LCMV Armstrong i.p. 1 d later. For endogenous CD8+ T cell studies, we infected naive C57BL/6 J mice with 2 × 105 PFU of LCMV Armstrong i.p., and we stained lymphocytes with H-2Db/gp33 MHC I tetramers. We generated OT-I immune chimeras by transferring 5 × 104 naive OT-I CD8+ T cells into C57BL/6 mice. We infected mice with 1 × 106 PFU of vesicular stomatitis virus expressing chicken ovalbumin (VSV-OVA) i.v. We generated double-immune chimeras by seeding P14 immune chimeras with 5 × 104 naive OT-I CD8+ T cells and then infecting them with VSV-OVA. For intravital imaging experiments, we seeded C57BL/6 or CD11c-YFP mice with 5 × 104 GFP+CD8+ or CFP+CD8+ P14 T cells and then infected the mice with LCMV, with the following exception: to aid analysis, mice for use in experiments on days 4.5 and 8 received a mixture of 4.5 × 104 P14 CD8+ T cells and 0.5 × 104 GFP+CD8+ P14 T cells. To investigate bystander memory CD8+ T cell recruitment and acquisition of the TRM cell phenotype, we purified CD45.1+CD8+ OT-I memory T cells from OT-I memory donors (generated by infection with VSV-OVA at least 60 d prior) with a CD8+ T cell isolation kit (StemCell Technologies) and injected 1.5 × 106 OT-I CD8+ T cells i.v. into a single CD90.1+ P14 immune host.
Intravascular staining, lymphocyte isolation and phenotyping
We used an intravascular staining method to discriminate cells present in the vasculature from cells in the tissue parenchyma, as described43. We injected mice i.v. with biotin/fluorochrome-conjugated anti-CD8α through the tail vein. Three minutes after the injection, we killed the animals and harvested tissues as described44. Isolated mouse cells were surface-stained with antibodies to CD3 (145-2C11), CD45 (30F-11), CD11b (M1/70), CD11c (N418), CD90.1 (OX-7), CD45.1 (A20), MHC II (Ia-Ie) (M5/114.15.2), CD8α (53-6.7), CD8β (YTS156.7.7), CD45.2 (104), CD4 (RM4-5), CD62L (MEL-14), CD44 (IM7), CD69 (H1.2F3) and CD103 (M290), all from BD Biosciences, Biolegend or Affymetrix eBiosciences. Cell viability was determined with Ghost Dye 780 (Tonbo Biosciences). The stained samples were acquired with LSRII or LSR Fortessa flow cytometers (BD) and analyzed with FlowJo software (Treestar).
In vivo BrdU assay
For studies investigating BrdU incorporation, animals were injected i.p. with 2 mg of BrdU (Sigma-Aldrich) 2 h before they were killed for cell isolation. Intracellular staining of cells for BrdU and Ki67 was done with the BD BrdU flow kit (BD Biosciences) with antibodies to BrdU (Bu20a) and Ki67 (SolA15) according to the manufacturer’s guidelines.
Tissue freezing, immunofluorescence and microscopy
Harvested mouse tissues were fixed in 2% paraformaldehyde for 2 h before being treated with 30% sucrose overnight for cryoprotection. The sucrose-treated tissue was embedded in OCT tissue-freezing medium and frozen in an isopentane liquid bath. Frozen blocks were processed, stained, imaged, and enumerated by QIM as described12, including staining with antibodies to the following markers: CD8-β (YTS156.7.7; BD Biosciences), CD90.1 (OX-7; BD Biosciences), CD45.1 (A20; Biolegend), E-cadherin (DECMA-1; Abcam), collagen-IV (goat anti-mouse polyclonal; Millipore), collagen-I (rabbit anti-mouse polyclonal; Millipore), Ki67 (TEC3; Dako), and LCMV nucleoprotein (VL-4; Bioxcell). We also counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) to detect nuclei. The following secondary antibodies were from Jackson Immunoresearch: donkey anti-rabbit (polyclonal), bovine anti-goat (polyclonal), and donkey anti-rat (polyclonal).
Preparation of epidermal whole mounts
We prepared epidermal whole mounts for immunofluorescence as described45. We prepared epidermal sheets from back skin by affixing the epidermis side to slides with an optically clear doublesided adhesive (Thorlabs). Slides were incubated in 2.5 mg/ml Dispase II (Roche Diagnostics) in PBS for 1 h at 37°C, and then the dermis was physically removed. Epidermal mounts were fixed in chilled acetone for 5 min. They were subsequently stained, and images were captured as described above. Antigen-specific CD8+ T cells in epidermis were counted manually in Photoshop. The area of the epidermis was measured with the ruler tool in Photoshop.
In vivo antibody treatment and local TRM cell reactivation
We depleted circulating CD90.1+CD8+ P14 T cells by injecting 0.6–1.5 μg of titrated anti-CD90.1 (HIS51) i.p. as described16. To block the migration of peripheral memory CD8+ T cells to the site of reactivation, we injected 500 μg of blocking anti-VCAM-1 (M/K-2.7) or anti-CD49d (PS/2) (BioXcell) i.p. 24 h before and 24 h after trans-cervical peptide challenge as described16. For local FRT TRM cell rechallenge experiments involving peptides, 50 μg of the indicated peptides (New England Peptides) was delivered trans-cervically as described21 in a volume of 30 μl delivered by modified gel loading pipet. For reactivation of skin TRM cells, 3 × 106 PFU of modified vaccinia virus Ankara expressing the SIINFEKL peptide of ovalbumin (MVA-OVA) was delivered epicutaneously into the back skin by 10–15 pokes with a bifurcated needle as described27. Reactivation of OT-I TRM cells in FRT was achieved by trans-cervical delivery of either 4 × 106 PFU of VV-OVA or 3 × 106 PFU of MVA-OVA as indicated.
Parabiotic surgery
Parabiotic surgery was done as described21 with the following modification. Lateral skin incisions on both pairs were conjoined with surgical wound clips. Parabionts were then allowed to rest for 14–30 d before experiments. Equilibration was confirmed in the peripheral blood before TRM cell reactivation.
Two-photon microscopy and analysis
Mice were anesthetized with 2,2,2-tribromoethanol (Avertin), and uterine horns were located through a midline incision along the linea alba. One uterine horn was gently exteriorized and positioned on a custom-made polystyrene platform, where it was stabilized with cyanoacrylate tissue adhesive. The platform allowed constant bathing of the organ in warm oxygenated buffer and permitted continuous temperature monitoring. The mouse was then transferred onto a microscope stage fully enclosed within an environmental chamber that helped maintain body temperature at 36.5 ± 1°C. Mice were injected with 50 μl of rhodamine isothiocyanate, 70 Kd (20 mg/ml stock), where indicated to visualize blood vessels.
Movies were acquired with an MP SP5 two-photon microscope TCS (Leica) equipped with Mai Tai HP DeepSee lasers (SpectraPhysics), an 8,000-Hz resonant scanner, a 25 × /0.95-NA (numerical aperture) objective, and two NDD and two HyD photomultiplier tubes. Samples were excited at 860 or 890 nm, and multiple fluorophores were imaged with the custom dichroic mirrors with the following collections: SHG, < 440 nm; CFP, 435–485 nm; GFP, 500–520 nm; YFP, 520–550 nm; and rhodamine dextran, 565–605 nm. All imaging studies focused on the perimetrium and myometrium regions of the uterus. Imaging data were processed with Imaris software (versions 7.6.4 and 8.0.1). For the calculation in Fig. 1g, a surface was created for SHG signal and then converted to a distance transformation channel to determine spot distance to the SHG surface at all time points. Determination of mean track velocities (in micrometers per minute) and motility coefficients and other migrational analyses were carried out with MotilityLab (http://www.motilitylab.net/) or a macro46. The drift correct function in Imaris and MotilityLab was used to correct for uterine tissue contractions during imaging. In cases where uterine tissue contractions were severe, we injected terbutaline hemisulfate (Sigma-Aldrich), a β-adrenergic agonist, i.p. at 50 mg/kg body weight to achieve transient uterine muscle relaxation. No adverse effect on CD8+ T cell surveillance behavior was noted during this short-term terbutaline treatment.
Bone marrow chimeras and diphtheria toxin treatment
Bone marrow was harvested from femurs and tibias of CD11c-DTR mice, and 1–2 × 106 bone marrow cells were injected i.v. into recipient mice that had received a lethal irradiation dose of 1,100 cGy. Mice received antibiotics in drinking water for 4 weeks after irradiation and were allowed 8 weeks for full reconstitution of bone marrow, which was confirmed by analysis of donor peripheral cells. We depleted CD11c+ cells in the chimeric mice by treating them with an initial dose of 1 μg of DT i.p. and then 0.2 μg of DT i.p. every 2 d until the end of the experiment.
Statistics
If the samples followed a Gaussian distribution, then parametric tests (unpaired two-tailed Student’s t-test for two groups and one-way ANOVA with Tukey’s multiple comparison test for more than two groups) were used. If the samples deviated from a Gaussian distribution, non-parametric tests (Mann-Whitney U-test for two groups, Kruskal-Wallis with Dunn’s multiple comparison test for more than two groups) were used unless otherwise stated. The DAgostino and Pearson omnibus normality test was used to determine whether samples adhered to a Gaussian distribution. Variances between groups were compared by F test and found to be equal. All statistical analysis was done in GraphPad Prism (GraphPad Software Inc.). P < 0.05 was considered significant.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank the members of the Masopust laboratory for helpful discussions. This work was funded by the Howard Hughes Medical Institute Faculty Scholars program (D.M.) and the US National Institutes of Health (grants R01AI111671 and R01AI084913 to D.M.; grant R21AI123600 to B.J.B.). H.D.H. was funded by the Intramural Research Program of the US National Institute of Allergy and Infectious Diseases.
Footnotes
Author contributions
L.K.B., J.S.M., E.A.T., J.M.S., J.M., S.W, R.F. and B.J.B. performed the experiments; H.D.H., V.V. and B.T.F. contributed critical reagents and experimental help; L.K.B., J.S.M. and E.A.T. analyzed intravital microscopy data; L.K.B. and D.M. wrote the manuscript; and D.M. was responsible for research supervision, coordination and strategy.
Competing interests
The authors declare no competing financial interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41590-017-0029-3.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 2.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 3.Stemberger C, et al. Stem cell-like plasticity of naive and distinct memory CD8+ T cell subsets. Semin Immunol. 2009;21:62–68. doi: 10.1016/j.smim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 5.Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 7.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 9.Mowat AM, McInnes IB, Parrott DMV. Functional properties of intra-epithelial lymphocytes from mouse small intestine. IV Investigation of the proliferative capacity of IEL using phorbol ester and calcium ionophore. Immunology. 1989;66:398–403. [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert EC, Roberts AI, Brolin RE, Raska K. Examination of the low proliferative capacity of human jejunal intraepithelial lymphocytes. Clin Exp Immunol. 1986;65:148–157. [PMC free article] [PubMed] [Google Scholar]
- 11.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. 2006;176:2079–2083. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 12.Steinert EM, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell. 2015;161:737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez-Ruiz D, et al. Liver-resident memory CD8+ T cells form a frontline defense against malaria liver-stage infection. Immunity. 2016;45:889–902. doi: 10.1016/j.immuni.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Wakim LM, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. 2012;189:3462–3471. doi: 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariotti S, et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346:101–105. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- 16.Schenkel JM, et al. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gebhardt T, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 18.Zaid A, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA. 2014;111:5307–5312. doi: 10.1073/pnas.1322292111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- 20.Casey KA, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8+ T cells. Nat Immunol. 2013;14:509–513. doi: 10.1038/ni.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beura LK, et al. Lymphocytic choriomeningitis virus persistence promotes effector-like memory differentiation and enhances mucosal T cell distribution. J Leukoc Biol. 2015;97:217–225. doi: 10.1189/jlb.1HI0314-154R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature. 2009;462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang X, et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 27.Hickman HD, et al. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe. 2013;13:155–168. doi: 10.1016/j.chom.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaylo A, Schrock DC, Fernandes NRJ, Fowell DJ. T cell interstitial migration: motility cues from the inflamed tissue for micro- and macro-positioning. Front Immunol. 2016;7:428. doi: 10.3389/fimmu.2016.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- 30.Steinbach K, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med. 2016;213:1571–1587. doi: 10.1084/jem.20151916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glennie ND, et al. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212:1405–1414. doi: 10.1084/jem.20142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stary G, et al. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science. 2015;348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med. 2016;213:951–966. doi: 10.1084/jem.20151855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackay LK, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu T, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95:215–224. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Çuburu N, et al. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J Clin Invest. 2012;122:4606–4620. doi: 10.1172/JCI63287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang SS, et al. Migration of cytotoxic lymphocytes in cell cycle permits local MHC I-dependent control of division at sites of viral infection. J Exp Med. 2011;208:747–759. doi: 10.1084/jem.20101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein I, Crispe IN. Complete differentiation of CD8+ T cells activated locally within the transplanted liver. J Exp Med. 2006;203:437–447. doi: 10.1084/jem.20051775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319:198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- 40.Posavad CM, et al. Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal Immunol. 2017;10:1259–1269. doi: 10.1038/mi.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu J, et al. Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature. 2013;497:494–497. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. 2015;7:269rv1. doi: 10.1126/scitranslmed.3010641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson KG, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson EA, Beura LK, Nelson CE, Anderson KG, Vezys V. Shortened intervals during heterologous boosting preserve memory CD8 T cell function but compromise longevity. J Immunol. 2016;196:3054–3063. doi: 10.4049/jimmunol.1501797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammed J, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat Immunol. 2016;17:414–421. doi: 10.1038/ni.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fife BT, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.