

Abstract
Mitochondria are well-known cellular organelles that play a vital role in cellular bioenergetics, heme biosynthesis, thermogenesis, calcium homeostasis, lipid catabolism, and other metabolic activities. Given the extensive role of mitochondria in cell function, mitochondrial dysfunction plays a part in many diseases, including diabetes and Alzheimer’s disease (AD). In most cases, there is overwhelming evidence that impaired mitochondrial function is a causative factor in these diseases. Studying mitochondrial function in diseased cells vs healthy cells may reveal the modified mechanisms and molecular components involved in specific disease states. In this chapter, we provide a concise overview of the major recent findings on mitochondrial abnormalities and their link to synaptic dysfunction relevant to neurodegeneration and cognitive decline in AD and diabetes. Our increased understanding of the role of mitochondrial perturbation indicates that the development of specific small molecules targeting aberrant mitochondrial function could provide therapeutic benefits for the brain in combating aging-related dementia and neurodegenerative diseases by powering up brain energy and improving synaptic function and transmission.
1. INTRODUCTION
Emerged evidence suggests that the deleterious and advanced cellular changes in aging and diabetes are linked to mitochondrial dysfunction.1,2 Brain aging is often characterized by neuronal loss and synaptic alteration, which are associated with mitochondrial abnormalities, energy failure, respiratory chain impairment, generation of reactive oxygen species (ROS), and neuronal perturbation.3 Further, various evidences suggest that mitochondrial dysfunction is a prominent and early oxidative stress-associated factor that produces neuronal abnormalities in aging and diabetes, resulting in susceptibility to aging-related neurodegenerative diseases.4 In the neurons, mitochondria are distributed throughout the length of the axons, presynaptic terminals, and dendrites. Mitochondria play active roles in regulating synaptogenesis and morphological/functional responses to synaptic activity; thus, mitochondrial dysfunction can lead to a stark neuronal energy deficit and, in the long run, to modifications in neuronal synapses and neurodegeneration in the aging brain.1
Alzheimer’s disease (AD) is a chronic aging-related disease with two pathological features: abnormal accumulations of amyloid beta peptide (Aβ) and phosphorylation of tau protein in the brain. Increased evidence indicates that mitochondrial and synaptic dysfunction is an early pathological feature of AD.5 Aβ has deleterious effects on mitochondrial function and structure and contributes to energy failure, respiratory chain impairment, ROS generation, induction of mitochondrial permeability transition pore (mPTP), imbalance of calcium homeostasis, disruption of mitochondrial dynamics, and mitochondrial DNA/RNA mutations.6 Although Aβ directly and indirectly causes abnormal mitochondrial and neuronal function, recent studies have highlighted the association between early mitochondrial dysfunction and the accumulation of Aβ in mitochondria, implicating mitochondrial Aβ in AD pathogenesis.7–28 These observations provide a better understanding of the relationship between mitochondria and AD pathogenesis.
Mitochondrial malfunction, synaptic damage, and the resultant impairment in cognitive function are pathological features of diabetes-affected brains.2 Diabetes adversely affects the brain and increases the risk for depression and dementia.29–39 In neurons, synaptic mitochondria are vital for the maintenance of synaptic function and transmission through normal mitochondrial dynamics, distribution, and trafficking as well as energy metabolism and synaptic calcium modulation. Imbalance of mitochondrial dynamics contributes to oxidative stress and hyperglycemia-induced alterations in mitochondrial morphology and function.38,40,41 Diabetes elicits AD-like brain changes linked with cognitive decline and neurodegeneration, such as elevated tau expression and phosphorylation and accumulation of Aβ,42–46 mitochondrial dysfunction, disruption of mitochondrial dynamics,37,38,41,47–51 oxidative stress,40,49 neuroinflammation, loss of synapses, impaired learning and memory, and synaptic plasticity deficits.29,35,36,44,52–55 The underlying mechanisms and strategies to rescue such injury and dysfunction are not well understood. Studies have identified several cellular and mitochondrial cofactors that are directly or indirectly involved in AD- and diabetes-mediated alterations in mitochondrial and synaptic structure and function. Such factors include cyclophilin D (CypD), presequence protease (PreP), Aβ, mPTP, N-methyl-D-aspartate, and the receptor for advanced glycation endproducts (RAGE).
This chapter addresses several aspects of AD- and diabetes-induced mitochondrial dysfunction with a special focus on mitochondrial molecular mechanisms underlying synaptic pathology and cognitive dysfunction.
2. MITOCHONDRIAL FUNCTION
Mitochondria are essential organelles for cell survival, playing a crucial role in calcium homeostasis, energy metabolism, detoxification of ROS generation, and induction of cell death, including apoptosis and necrosis. Mitochondria in different types of cells or in different subcompartments of one cell differ significantly in their morphology and function and can be divided into multiple subgroups within one cell.56 The recent recognition of mitochondrial heterogeneity facilitates our understanding of mitochondrial biology.
Mitochondria are the major site of ATP synthesis and are also the site of amino acid biosynthesis, fatty acid oxidation, steroid metabolism, calcium homeostasis, and ROS production and detoxification. The inner mitochondrial membrane is largely impermeable and contains a variety of enzymes, including those responsible for making ATP, and forms the major barrier between the cytosol and the mitochondrial matrix. The five complexes of the respiratory chain [complex I (NADH ubiquinone oxidoreductase), complex II (succinate ubiquinone oxidoreductase), complex III (ubiquinone-cytochrome c reductase), complex IV (cytochrome oxidase), and complex V (ATP synthase)] are embedded in the inner mitochondrial membrane. The transmission of electrons along the respiratory chain provides the energy to pump protons from the matrix into the intermembrane space, thereby generating the electrochemical gradient required to drive ATP synthesis.56
3. SYNAPTIC MITOCHONDRIAL PATHOLOGY IN AD
Synapses are the neuronal contact sites through which neurons receive and send information.57,58 Energy provision and calcium fluctuation in synapses are prerequisite for interneuronal communication.59 To meet the high energy demands and to cope with constant calcium flux, synapses are enriched with mitochondria for on-site energy provision and calcium modulation.60
Although the detrimental impacts of Aβ on synapses and synaptic function are extensive, multiple studies demonstrate that mitochondrial structure and function are particularly susceptible to the effects of mitochondrial Aβ accumulation.7–28 Further, synaptic mitochondria serve as a reservoir for Aβ accumulation in aging and AD1,5,61–64; thus mitochondrial dysfunction is a major player in the synaptic alterations seen in AD and diabetes.3,56,65
First, mitochondrial and neuronal malfunction in AD is linked to the progress accumulation of Aβ in the mitochondria of both human AD and transgenic AD mouse brains.1,7–9,11,15–18,66–68 Aβ can directly import into mitochondria via the translocase of the outer membrane machinery,67 RAGE,69 or other unknown mechanisms. Aβ may also be locally produced in mitochondria via gamma-secretase that is localized in mitochondria.70–72 Notably, accumulation of mitochondrial Aβ precedes extracellular Aβ deposition in AD brains, increases with age, and associates with early onset synaptic loss, synaptic damage, and mitochondrial oxidative damage,5,7,10,11,22,73–83 suggesting that early accumulation of Aβ in mitochondria may be an initiating pathological event, leading to mitochondrial and neuronal perturbation. Second, interaction of Aβ with mitochondrial matrix proteins such as amyloid-binding alcohol dehydrogenase (ABAD)7,10,11,84 and CypD23,85–87 exacerbates Aβ-induced mitochondrial and neuronal stress. Increasing PreP activity by antagonizing the Aβ-ABAD interaction decreases mitochondrial and cerebral Aβ accumulation in AD mice overexpressing Aβ and improves mitochondrial function.88 Third, increasing neuronal PreP expression and activity in Aβ-enriched synaptic mitochondria of mAPP mice greatly reduces mitochondrial accumulation. Accordingly, synaptic function and learning and memory are significantly improved in PreP-overexpressed mitochondria.28 These data strongly indicate that PreP is critical for maintaining mitochondrial integrity and function by clearance of mitochondrial Aβ. Strategies that reduce Aβ levels in mitochondria in addition to the brain by increasing PreP expression and activity are critical to consider as new avenues for both preventing and halting AD progression at the early stage. One such therapeutic strategy involves the development of a small-molecule agonist of PreP in order to safely decrease mitochondrial and cerebral Aβ accumulation by accelerating Aβ clearance.
These recent studies highlight the significant role of Aβ in synaptic mitochondrial pathology and significantly advance our understanding of the mechanisms underlying mitochondrial dysfunction in AD, especially in the early stage when the presence of Aβ has not yet set in motion the devastating cognitive impairments often associated with AD. Ameliorating alterations in mitochondrial function could improve synaptic function and reverse cognitive decline in AD.
In AD and non-AD cell and animal models, treatment of mitochondria-targeted molecules mitoQ and SS31 significantly reverses Aβ-induced CypD elevation, mitochondrial fusion/fission proteins imbalance, and neurite growth.89 MitoQ and SS31 also reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage.90 Additionally, antioxidants attenuate mitochondrial transport and function in cybrid cells containing AD-derived mitochondria.4,91 These results suggest a close relationship between neuronal mitochondrial dysfunction and synaptic perturbation and the value of eliminating neuronal mitochondrial oxidative stress in the treatment of neuronal/synaptic alterations in AD.1,90
4. IMPACT OF CypD-DEPENDENT mPTP ON MITOCHONDRIAL DEFECTS
CypD is a crucial component of the mPTP. CypD released from matrix can bind to the adenine nucleotide translocase in the inner mitochondrial membrane and the voltage-dependent anion channel in the outer mitochondrial membrane to trigger the opening of mPTP, a nonselective, high conductance pore allowing the transport not only calcium but any solute below the pore size. The opening of mPTP results in osmotic swelling, dissipation of the mitochondrial membrane potential, reduced mitochondrial calcium retention capacity, decreased membrane potential, increased ROS production, and eventually, cell death (Fig. 1).92 Increased expression of CypD occurs in neurodegenerative diseases including AD, Parkinson’s disease (PD), Huntington’s disease (HD),23,87,93–97 and diabetes, and con tributes to mitochondrial perturbation.5,23,65,98
Fig. 1.
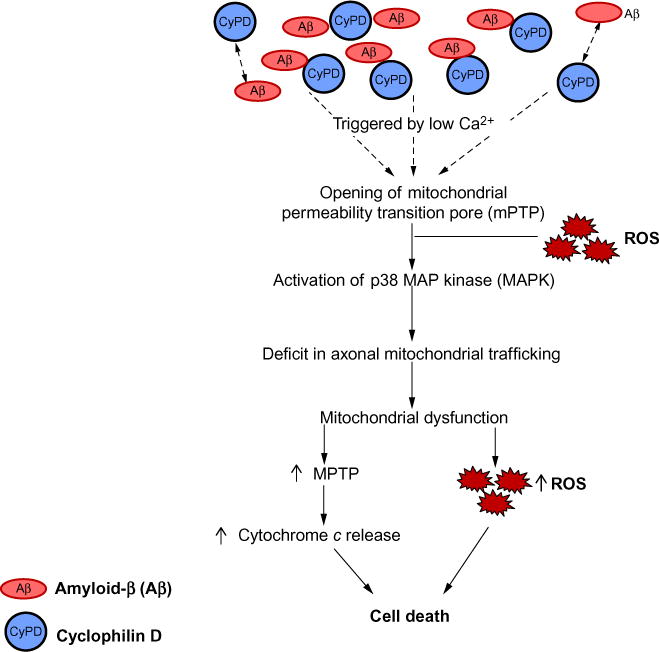
Effect of Aβ on CypD-involved mPTP formation. Aβ-cyclophilin D interaction mediates impairments in axonal mitochondrial transport due to an increase in the opening of CypD-mediated mitochondrial permeability transition pore (mPTP). This leads to the disruption of Ca2+ balance and increases the production/accumulation of reactive oxygen species (ROS). Elevation of Ca2+ and oxidative stress activates the downstream p38 MAP kinase signaling pathway, thus contributing to mitochondrial dysfunction.
Studies from in vitro cellular and in vivo animal models have demonstrated that blockade of CypD significantly attenuates mPTP-related mitochondrial dysfunction and cell death, which are relevant to the pathogenesis of stroke, AD, and diabetes.23,65,87,98–102 Furthermore, blockade of mPTP by genetic depletion or pharmacological inhibition of CypD rescues axonal mitochondrial trafficking and protects synapses from Aβ toxicity. The potential mechanisms underlying the protective effect of CypD deficiency on axonal mitochondrial trafficking include the reduction of Aβ-induced calcium perturbation, the suppression of axonal ROS accumulation, and the activation of the downstream P38/MAPK signaling pathway.
The protein kinase A/cAMP regulatory element-binding (PKA/CREB) signaling pathway, a crucial regulator of synaptic plasticity and learning memory, is adversely affected by an Aβ-rich environment, leading to dendritic spine architecture changes in an AD mouse model.102 Aβ reduces phosphorylation of PKA, thus disrupting PKA/CREB signal transduction and causing synaptic and cognitive dysfunction.100,102 Notably, neurons lacking CypD reverse Aβ-induced synaptic dysfunction and are protected against Aβ-induced alterations in PKA and CREB phosphorylation. These results indicate the involvement of CypD in Aβ-induced abnormalities in signal transduction including PKA/CREB signaling. Sustained CypD-induced neuronal/synaptic mitochondrial stress is a potential mechanism underlying synaptic failure in the pathogenesis of AD.
Recently, Wang et al. demonstrated that CypD expression levels were significantly elevated in the hippocampi of streptozotocin-induced diabetic mice.56 The CypD expression levels are further elevated in Aβ-enriched diabetic brain compared to nondiabetic mAPP mice.56 These results suggest that CypD expression is increased in diabetes mellitus and further enhanced in an Aβ-rich environment. Increased levels of CypD in mitochondria trigger/enhance the mPTP opening, leading to colloidal osmotic swelling of the mitochondrial matrix, dissipation of the inner membrane potential, generation of ROS, and release of many proapoptogenic proteins and procaspases.99 Hence, blockade of CypD may be a potential therapeutic strategy for preventing and halting synaptic and mitochondrial pathology in AD. Specifically, the development of small-molecule CypD inhibitors could hold therapeutic potential for the treatment of neurodegenerative diseases including AD and diabetes.103,104
5. EFFECT OF NEURONAL PreP ACTIVITY AND RAGE SIGNALING ON MITOCHONDRIAL DYSFUNCTION
PreP is a mitochondrial peptidasome that is localized in the mammalian mitochondrial matrix.105 It is the key for maintenance of mitochondrial health and integrity. PreP proteolytic activity is significantly reduced in AD-affected brain mitochondria and transgenic AD mouse models106 and is negatively correlated to mitochondrial Aβ accumulation. Du et al. demonstrated that increased expression and activity of neuronal PreP significantly reduced mitochondrial Aβ load and the production of proinflammatory mediators, improved mitochondrial function and synaptic plasticity, and attenuated cognitive decline in AD mice.28 Furthermore, PreP proteolytic activity is required for degradation and clearance of mitochondrial Aβ. Mitochondrial Aβ accumulation may interfere with normal mitophagy and release of mitochondria-derived damage-associated molecular patterns from the injured neurons, leading to increased production of TNF-α, IL-1β, and MCP1, the cytokines known to be involved in the inflammatory process of AD.107 Thus, dysfunctional or damaged mitochondria can produce excessive inflammation and tissue damage possibly via overproduction of cytokines and ROS.
RAGE-dependent signal transduction via Aβ-RAGE interaction plays an important role in mitochondrial dysfunction. RAGE serves as an important cell-surface receptor mediating chemotactic and inflammatory reaction to Aβ and other proinflammatory ligands.69,108–113 RAGE signaling in neurons and microglia is known to promote induction of proinflammatory mediators, including cytokines and chemokines, and activation of microglia by increased expression of microglial markers (CD4 and CD11).107,110 Additionally, over-expression of neuronal PreP in mAPP mice not only reduces Aβ accumulation in the brain but also remarkably suppresses RAGE expression as compared with mAPP mice,69 suggesting a possible connection between mitochondrial defects and RAGE signaling relevant to the activation of transcription and the proinflammatory response.28,69,107,109,110,112 Further investigation is required to elucidate the role of RAGE in mitochondrial dysfunction relevant to the pathogenesis of AD and diabetes.
6. EFFECTS OF METHIONINE SULFOXIDE REDUCTASE ON Aβ SOLUBILITY AND MITOCHONDRIAL FUNCTION
Accumulation of oxidized proteins, especially Aβ, is thought to be one of the common causes of AD. Induced ROS generation is one of the earliest consequences of toxic insults mediated by soluble Aβ oligomers.81 Mitochondria are particularly sensitive to ROS, and reduced metabolic activity resulting from oxidative damage to vital mitochondrial components has been demonstrated in AD.10
Methionine (Met) is highly susceptible to oxidation in vivo, particularly under conditions of oxidative stress. The sulfoxide form comprises 10%–50% of Aβ in amyloid plaques of AD brain.114 Oxidation of Met to Met(O) is reversible and the reverse reaction is catalyzed in vivo by the methionine sulfoxide reductase (Msr) system, composed of peptide-methionine (S)-S-oxide reductase (MsrA) and peptide-methionine (R)-S-oxide reductase (MsrB), which, respectively, reduce the S and R enantiomers of the sulfoxide group. These enzymes provide both an efficient repair mechanism for oxidative damage to Met residues and general protection against oxidative stress by scavenging ROS through the recycling of Met.
Studies from primary hippocampal and cortical neurons show increased total Msr activity, ascribed to increased activity in both MsrA and MsrB, in conjunction with protection against cell death induced by the sulfoxide forms of Aβ40 or Aβ42. Exposure of wild-type and MsrA knockout mouse cortical neurons to Aβ42 and Met(O)-Aβ demonstrated that lack of MsrA abolishes the protective effect induced by Met(O)-Aβ.115 Furthermore, lack of MsrA promotes a shift from aggregated forms of Aβ toward soluble oligomers. Given that soluble oligomer Aβ are thought to be more toxic to neurons and synapses than aggregated Aβ forms,115 enhancing MsrA activity by regulating transcription may have therapeutic applications. Alterations in MsrA expression levels and Aβ structure during normal aging might be a cofactor in AD-related mitochondrial malfunction.115
7. IMPACT OF MITOCHONDRIAL DYNAMICS IN MCI AND AD
Mitochondria are highly dynamic organelles that undergo continuous fission and fusion, which are regulated by the GTPase hydrolysis activity mitochondrial fission proteins (DLP1 and Fis1) and mitochondrial fusion protein [mitofusin 1 and 2 (Mfn1 and 2) and optic atrophy (Opa1)]. Mitochondrial dynamics are important for the proper distribution of mitochondria within cells, which is particularly critical for morphologically complex cells such as neurons.116 Alterations in mitochondrial dynamics significantly impact almost all aspects of mitochondrial function including energy metabolism, calcium buffering, ROS generation, and apoptosis regulation.117,118 Unbalanced fusion and fission lead, respectively, to mitochondrial elongation and excessive mitochondrial fragmentation, both of which impair the function of mitochondria. It has been shown that exchange of mitochondrial contents is important for mitochondrial function as well as organelle distribution in neurons. Mitochondrial fusion, in particular that mediated by Mfn2, is required for proper development and maintenance of the cerebellum.119 Mutations in the Mfn2 gene cause neurodegenerative diseases, such as Charcot–Marie–Tooth type 2A, and mutations in OPA1 cause dominantly inherited optic atrophy. Increasing evidence implicates altered mitochondrial trafficking and fusion–fission dynamics in aging-related AD, PD, HD, and amyotrophic lateral sclerosis.
7.1 Effect of Mfn2 on Mitochondrial Function
Mitofusins Mfn1 and Mfn2 are outer membrane GTPases that mediate outer mitochondrial membrane fusion. Mfn2 expression is crucial for maintaining the morphology and operation of the mitochondrial network and mitochondrial metabolism. Recent studies demonstrate that markedly reduced mitochondrial mass and transport may contribute to neuronal loss due to the specific loss of Mfn2 but not Mfn1.120 Du et al. examined the role of Mfn2 in the human-induced pluripotent stem cells (hiPSCs) differentiation system and reported that knockdown of Mfn2 results in mitochondrial dysfunctions and defects in neurogenesis and synapse formation.119 By contrast, Mfn2 overexpression in neural progenitor cells directs differentiation and maturation into neurons with enhanced mitochondrial functions, suggesting that Mfn2 is crucial for mitochondrial development, and thereby essential to hiPSCs differentiation. Importantly, this also provides a novel neurophysiologic model of mitochondrial development in neurogenesis, which enhances our understanding of the involvement of dysfunctional mitochondria in aging and neurodegenerative diseases.119 Under pathological conditions, Mfn2 expression levels are increased such as mild cognitive impairment (MCI)-derived mitochondria, leading to aberrant mitochondrial fusion and fission event evidenced by abnormal mitochondrial morphology and function.
7.2 Oxidative Stress and MCI- and AD-Related Mitochondrial Dynamics
MCI is characterized by a decline in cognitive abilities that is noticeable yet not severe enough to completely disrupt an individual’s daily activity. MCI is generally considered to be a transitional phase between normal aging and early dementing disorders, especially AD.121
In cybrid model, MCI-induced mitochondrial defects manifest as alterations in mitochondrial dynamics, function, and morphology. These dysfunctional MCI cybrid mitochondria exhibit impaired fission/fusion events, impaired mitochondrial respiratory chain enzyme activity, decreased membrane potential, increased mitochondrial and intracellular ROS, and impairment in energy metabolism with decreased ATP levels when compared to non-MCI cybrid mitochondria. Given that mitochondrial Mfn2 is involved in mitochondrial fusion,119 increased mitochondrial Mfn2 levels in MCI cybrids suggest that altered Mfn2 expression likely contributes to enhanced mitochondrial fusion. Accordingly, changes in MCI mitochondrial morphology display as elongated mitochondria. Interestingly, suppression of Mfn2 overexpression by inhibiting oxidative stress-mediated activation of extracellular signal-regulated kinases (ERK) reverses abnormalities in mitochondrial structure and function.122 Thus, generation of Mfn2 antagonist may hold potential for prevention and treatment at the early stage of AD.123
In contrast to MCI-derived mitochondria, AD mitochondria exhibit fragmentation as shown by overabundant fission, elongate, and aggregated mitochondria, compared to cybrid cells containing mitochondria from normal age-matched subjects with the relatively normal cognitive function. DLP1, which plays a key role in balancing mitochondrial dynamics by regulating mitochondrial fission, was significantly increased in AD mitochondria.123 Additionally, the abnormal interaction of DLP1 with hyperphosphorylated tau was found in AD neurons.124 Interaction of DLP1 with glycogen synthase kinase-3 (GSK3β) mediates changes in mitochondrial morphology and dynamics.125–127 Mitochondrial dynamics modulates the induction of proinflammatory mediators in microglial cells.128,129 ROS-induced activation of the mitogen-activated protein (MAP) kinase family appears to play a key role in mediating cellular responses to multiple stresses. ERK signaling is involved in mitochondrial function and neuronal stress.123,130 Taken together, this suggests that oxidative stress-induced activation of MAP kinase via upregulation of DLP1 or Mfn2 expression contributes to mitochondrial dysfunction and abnormal mitochondrial dynamics122,123 by disrupting the balance of mitochondrial fission and fusion and promoting translocation of DLP1 to mitochondria, leading to mitochondrial fragmentation in AD. Most importantly, suppression of ERK signaling and inhibition of mitochondrial fission or fusion pathways rescues defective mitochondrial morphology and function induced by AD or MCI123 (Fig. 2). Antioxidant treatment attenuates AD mitochondrial defects, leading to improvements in axonal mitochondrial transport and mitochondrial bioenergy and function.4,91
Fig. 2.
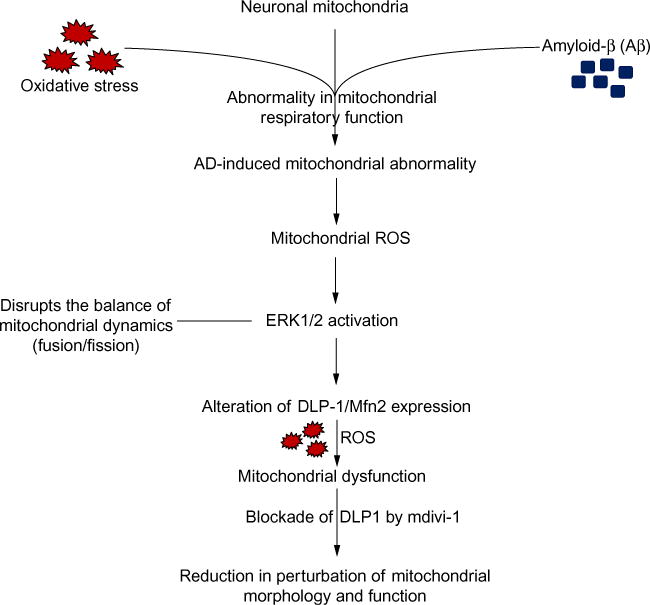
Effect of AD on mitochondrial dynamics. AD-induced mitochondrial respiratory function abnormality orchestrates ROS generation and accumulation and subsequently activates ERK signal transduction. Activation of ERK signaling disrupts mitochondrial dynamics and results in altered DLP1 and Mfn2 expression, which eventually leads to mitochondrial dysfunction. Inhibition of DLP1 or Mfn2 expression attenuates AD- or MCI-derived mitochondrial and neuronal dysfunction (Mdivi-1, an inhibitor for DLP1).
8. DRP1-MEDIATED MITOCHONDRIAL ABNORMALITIES IN DIABETES
Mitochondria are dynamic organelles that undergo continuous fission and fusion. Fission events are regulated by dynamin-related protein (Drp1), while fusion events are regulated by the large dynamin-related GTPases known as Mfn1 and Mfn2 as well as optic atrophy 1 (OPA1).131 Alterations in mitochondrial dynamics affect mitochondrial numbers and shape, respiratory enzyme activity, and ATP production. Imbalance between mitochondrial fission and fusion in diabetes results predominantly from upregulation of Drp1, which induces mitochondrial dysfunction (impaired respiration and ATP production) in a variety of cell types, including dorsal root ganglion neurons and β cells.41 Mitochondrial dysfunction has been implicated in the development of insulin resistance in skeletal muscle cells and hyperglycemia.132
A novel and pivotal role of mitochondrial dysfunction in diabetes-induced synaptic impairment involves a GSK3b/Drp1-dependent connection between mitochondrial dysfunction in diabetic neurons and synaptic dysfunction including decline in long-term potentiation. These findings are consistent with diabetic neuropathy as shown by increased Drp1 expression and mitochondrial fission in dorsal root ganglion neurons of 6-month-old type II diabetes (db/db) mice.2 In contrast to the greater numbers of mitochondria in dorsal root ganglion neurons, hippocampal neurons in 5- to 6-month-old db/db mice displayed smaller numbers of mitochondria, such a decrease was not seen in mice younger than 3 months. Between 3 and 6 months of age, complex I enzyme activity significantly declined by 15%–35% and ATP content was significantly altered. Pharmacologic or genetic inactivation of Drp1 prevented changes in mitochondrial morphology and function in db/db mouse hippocampus or human neuronal cells under hyperglycemic conditions, indicating the role of Drp1 in diabetes-induced mitochondrial dysfunction.2 Furthermore, genetic activation of GSK3β without high glucose treatment can also promote mitochondrial fragmentation, while inactivation of GSK3β prevents high glucose-induced mitochondrial dysfunction. Taken together, these data suggest that GSK3β likely acts as an upstream signaling mechanism for Drp1 upregulation in diabetes-induced mitochondrial dysfunction.2
9. CONCLUSION
Several lines of evidence suggest that age-related AD and diabetes are predominantly associated with mitochondrial dysfunction. Mitochondrial defects result in increased ROS generation, abnormal protein–protein interactions, and decreased mitochondrial ATP production. Overproduction of ROS and mPTP formation with attendant compromised mitochondrial function contribute importantly to neuronal perturbation. Several other factors including intracellular Ca2+, Aβ, and CypD also play an important role in mPTP formation, leading to mitochondrial dysfunction. In addition, disruption of mitochondrial dynamics by altered mitochondrial fusion and fission events contributes to mitochondrial and synaptic injury and cognitive decline relevant to the pathogenesis of AD and diabetes (Fig. 3). Thus, inhibition of mPTP opening by blocking CypD and regulation of mitochondrial dynamics are rational targets for potential therapeutic strategies for AD and diabetes.
Fig. 3.
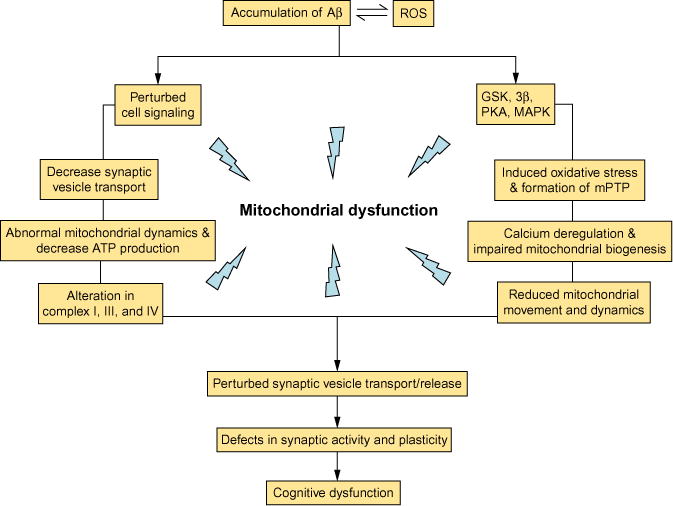
The cellular factors and related pathways contribute to Aβ-mediated mitochondrial defects and synaptic damage. Aβ accumulation perturbs mitochondrial transport and dynamics, cell signaling, synaptic mitochondrial structure and function, leading to decreased energy metabolism/ATP production, deregulation of calcium homeostasis, perturbed cell signaling cascades, altered key enzymes associated with mitochondrial respiratory chain, induced oxidative stress, and, eventually, synaptic injury and cognitive decline.
References
- 1.Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid Redox Signal. 2012;16(12):1467–1475. doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang S, Wang Y, Gan X, et al. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015;64(5):1728–1742. doi: 10.2337/db14-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du H, ShiDu Yan S. Unlocking the door to neuronal woes in Alzheimer’s disease: Abeta and mitochondrial permeability transition pore. Pharmaceuticals (Basel) 2010;3(6):1936–1948. doi: 10.3390/ph3061936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu Q, Fang D, Swerdlow RH, et al. Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J Alzheimers Dis. 2016;54(2):679–690. doi: 10.3233/JAD-160532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du H, Guo L, Yan S, Sosunov AA. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA. 2010;107(43):18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheng B, Gong K, Niu Y, Liu L. Inhibition of gamma-secretase activity reduces Abeta production, reduces oxidative stress, increases mitochondrial activity and leads to reduced vulnerability to apoptosis: implications for the treatment of Alzheimer’s disease. Free Radic Biol Med. 2009;46(10):1362–1375. doi: 10.1016/j.freeradbiomed.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 7.Yao J, Irwin RW, Zhao L, et al. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caspersen C, Wang N, Yao J, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19(14):2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 9.Manczak M, Anekonda TS, Henson E, et al. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15(9):1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 10.Takuma K, Yao J, Huang J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19(6):597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 11.Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 12.Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer’s disease. J Neurochem. 2006;96(1):1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Su B, Siedlak SL, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008;105(49):19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shukkur EA, Shimohata A, Akagi T, et al. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum Mol Genet. 2006;15(18):2752–2762. doi: 10.1093/hmg/ddl211. [DOI] [PubMed] [Google Scholar]
- 15.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crouch PJ, Blake R, Duce JA, et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25(3):672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devi L, Prabhu BM, Galati DF, et al. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26(35):9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillardon F, Rist W, Kussmaul L, et al. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7(4):605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- 19.Mungarro-Menchaca X, Ferrera P, Morán J, Arias C. beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res. 2002;68(1):89–96. doi: 10.1002/jnr.10193. [DOI] [PubMed] [Google Scholar]
- 20.Casley CS, Canevari L, Land JM, et al. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80(1):91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 21.Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nat Med. 2006;12(11):1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 22.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14(2):45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14(10):1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89(6):1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 25.Cardoso SM, et al. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15(8):1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 26.Mao P, Manczak M, Calkins MJ, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21(13):2973–2990. doi: 10.1093/hmg/dds128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy PH, Tripathi R, Troung Q, et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta. 2012;1822(5):639–649. doi: 10.1016/j.bbadis.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang D, Wang Y, Zhang Z, et al. Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum Mol Genet. 2015;24(18):5198–5210. doi: 10.1093/hmg/ddv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt RE, Parvin CA, Green KG. Synaptic ultrastructural alterations anticipate the development of neuroaxonal dystrophy in sympathetic ganglia of aged and diabetic mice. J Neuropathol Exp Neurol. 2008;67(12):1166–1186. doi: 10.1097/NEN.0b013e318190d6db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tay SS, Wong WC. Ultrastructural changes in the gracile nucleus of alloxan-induced diabetic rats. Acta Anat (Basel) 1990;139(4):367–373. doi: 10.1159/000147025. [DOI] [PubMed] [Google Scholar]
- 31.Greenwood CE, Winocur G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol Aging. 2005;26(1):42–45. doi: 10.1016/j.neurobiolaging.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Desrocher M, Rovet J. Neurocognitive correlates of type 1 diabetes mellitus in childhood. Child Neuropsychol. 2004;10(1):36–52. doi: 10.1076/chin.10.1.36.26241. [DOI] [PubMed] [Google Scholar]
- 33.Biessels GJ, Kamal A, Ramakers GM, et al. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes. 1996;45(9):1259–1266. doi: 10.2337/diab.45.9.1259. [DOI] [PubMed] [Google Scholar]
- 34.Biessels GJ, Kamal A, Urban IJ, et al. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800(1):125–135. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- 35.Li XL, Aou S, Oomura Y, et al. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113(3):607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 36.Stranahan AM, Arumugam TV, Cutler RG, et al. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11(3):309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41(10):1846–1854. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial dynamics in diabetes. Antioxid Redox Signal. 2011;14(3):439–457. doi: 10.1089/ars.2010.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaffe K, Lindquist K, Schwartz AV, et al. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology. 2011;77(14):1351–1356. doi: 10.1212/WNL.0b013e3182315a56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79(2):341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards JL, Quattrini A, Lentz SI, et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia. 2010;53(1):160–169. doi: 10.1007/s00125-009-1553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clodfelder-Miller BJ, Zmijewska AA, Johnson GV, et al. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes. 2006;55(12):3320–3325. doi: 10.2337/db06-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Planel E, Tatebayashi Y, Miyasaka T, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27(50):13635–13648. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ke YD, Delerue F, Gladbach A, et al. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4(11):e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56(7):1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 46.Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta. 2009;1792(5):482–496. doi: 10.1016/j.bbadis.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Palmeira CM, Rolo AP, Berthiaume J, et al. Hyperglycemia decreases mitochondrial function: the regulatory role of mitochondrial biogenesis. Toxicol Appl Pharmacol. 2007;225(2):214–220. doi: 10.1016/j.taap.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53(8):1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103(8):2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galloway CA, Lee H, Nejjar S, et al. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. 2012;61(8):2093–2104. doi: 10.2337/db11-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosca MG, Mustata TG, Kinter MT, et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. 2005;289(2):420–430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 52.Toth C, Schmidt AM, Tuor UI, et al. Diabetes, leukoencephalopathy and rage. Neurobiol Dis. 2006;23(2):445–461. doi: 10.1016/j.nbd.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 53.Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA. 2010;107(15):7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jolivalt CG, Hurford R, Lee CA, et al. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol. 2010;223(2):422–431. doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burdo JR, Chen Q, Calcutt NA, et al. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol Aging. 2009;30(12):1910–1917. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Wu L, Li J, et al. Synergistic exacerbation of mitochondrial and synaptic dysfunction and resultant learning and memory deficit in a mouse model of diabetic Alzheimer’s disease. J Alzheimers Dis. 2015;43(2):451–463. doi: 10.3233/JAD-140972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alonso-Nanclares L, Gonzalez-Soriano J, Rodriguez JR, et al. Gender differences in human cortical synaptic density. Proc Natl Acad Sci USA. 2008;105(38):14615–14619. doi: 10.1073/pnas.0803652105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- 59.Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18(20):8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548(Pt 2):425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinez M, Hernández AI, Martínez N, et al. Age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1996;731(1–2):246–248. doi: 10.1016/0006-8993(96)00708-1. [DOI] [PubMed] [Google Scholar]
- 62.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 63.Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281(17):11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Guo L, Lu L, et al. Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS One. 2016;11(3):e0150441. doi: 10.1371/journal.pone.0150441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan S, Du F, Wu L, et al. F1F0 ATP synthase-cyclophilin D interaction contributes to diabetes-induced synaptic dysfunction and cognitive decline. Diabetes. 2016;65(11):3482–3494. doi: 10.2337/db16-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernández-Vizarra P, Fernández AP, Castro-Blanco S, et al. Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol Histopathol. 2004;19(3):823–844. doi: 10.14670/HH-19.823. [DOI] [PubMed] [Google Scholar]
- 67.Hansson Petersen CA, Alikhani N, Behbahani H, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA. 2008;105(35):13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abramowski D, Rabe S, Upadhaya AR, et al. Transgenic expression of intraneuronal Abeta42 but not Abeta40 leads to cellular Abeta lesions, degeneration, and functional impairment without typical Alzheimer’s disease pathology. J Neurosci. 2012;32(4):1273–1283. doi: 10.1523/JNEUROSCI.4586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takuma K, Fang F, Zhang W, et al. RAGE-mediated signaling contributes to intra-neuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA. 2009;106(47):20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teng FY, Tang BL. Widespread gamma-secretase activity in the cell, but do we need it at the mitochondria? Biochem Biophys Res Commun. 2005;328(1):1–5. doi: 10.1016/j.bbrc.2004.12.131. [DOI] [PubMed] [Google Scholar]
- 71.Pavlov PF, Wiehager B, Sakai J, et al. Mitochondrial gamma-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 2011;25(1):78–88. doi: 10.1096/fj.10-157230. [DOI] [PubMed] [Google Scholar]
- 72.Behbahani H, Pavlov PF, Wiehager B, et al. Association of Omi/HtrA2 with gamma-secretase in mitochondria. Neurochem Int. 2010;7(6):668–675. doi: 10.1016/j.neuint.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 73.Chui DH, Dobo E, Makifuchi T, et al. Apoptotic neurons in Alzheimer’s disease frequently show intracellular Abeta42 labeling. J Alzheimers Dis. 2001;3(2):231–239. doi: 10.3233/jad-2001-3208. [DOI] [PubMed] [Google Scholar]
- 74.Chui DH, Tanahashi H, Ozawa K, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5(5):560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 75.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 76.Hsia AY, Masliah E, McConlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96(6):3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li QX, Maynard C, Cappai R, et al. Intracellular accumulation of detergent-soluble amyloidogenic A beta fragment of Alzheimer’s disease precursor protein in the hippocampus of aged transgenic mice. J Neurochem. 1999;72(6):2479–2487. doi: 10.1046/j.1471-4159.1999.0722479.x. [DOI] [PubMed] [Google Scholar]
- 78.Takahashi RH, Milner TA, Li F, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161(5):1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reddy PH, McWeeney S, Park BS, et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13(12):1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 80.Reddy PH, Mani G, Park BS, et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7(2):103–117. doi: 10.3233/jad-2005-7203. [DOI] [PubMed] [Google Scholar]
- 81.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 82.Billings LM, Oddo S, Green KN, et al. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 83.Leissring MA, Farris W, Wu X, et al. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem J. 2004;383(Pt. 3):439–446. doi: 10.1042/BJ20041081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yan SD, Fu J, Soto C, et al. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer’s disease. Nature. 1997;389(6652):689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]
- 85.Du H, Guo L, Zhang W, et al. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2009;32(3):398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2009;1802(1):198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Du H, Guo L, Zhang W, et al. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32(3):398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yao J, Du H, Yan S, et al. Inhibition of Amyloid-{beta} (A{beta}) peptide-binding alcohol dehydrogenase-A{beta} interaction reduces A{beta} accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31(6):2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manczak M, Mao P, Calkins MJ, et al. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20(2):609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yin X, Manczak M, Reddy PH. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum Mol Genet. 2016;25(9):1739–1753. doi: 10.1093/hmg/ddw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fang D, Zhang Z, Li H, et al. Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J Alzheimers Dis. 2016;51(2):571–580. doi: 10.3233/JAD-150917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802(1):198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin LJ, Semenkow S, Hanaford A, et al. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol Aging. 2014;35(5):1132–1152. doi: 10.1016/j.neurobiolaging.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martin LJ, Gertz B, Pan Y, et al. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218(2):333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martin LJ. The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim Biophys Acta. 2010;1802(1):186–197. doi: 10.1016/j.bbadis.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dougherty SE, Hollimon JJ, McMeekin LJ, et al. Hyperactivity and cortical disinhibition in mice with restricted expression of mutant huntingtin to parvalbumin-positive cells. Neurobiol Dis. 2014;62:160–171. doi: 10.1016/j.nbd.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brustovetsky N, Brustovetsky T, Purl KJ, et al. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23(12):4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Y, Wu L, Li J, et al. Synergistic exacerbation of mitochondrial and synaptic dysfunction and resultant learning and memory deficit in a mouse model of diabetic Alzheimer’s disease. J Alzheimers Dis. 2014;43(2):451–463. doi: 10.3233/JAD-140972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Z, Wang Y, Yan S, et al. NR2B-dependent cyclophilin D translocation suppresses the recovery of synaptic transmission after oxygen-glucose deprivation. Biochim Biophys Acta. 2015;1852(10):2225–2234. doi: 10.1016/j.bbadis.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Du H, Guo L, Wu X, et al. Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta. 2013;1842(12):2517–2527. doi: 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo L, Du H, Yan S, et al. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One. 2013;8(1):e54914. doi: 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Du H, Guo L, Wu X, et al. Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta. 2014;1842(12):2517–2527. doi: 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valasani KR, Vangavaragu JR, Day VW, et al. Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J Chem Inf Model. 2014;54(3):902–912. doi: 10.1021/ci5000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valasani KR, Sun Q, Fang D, et al. Identification of a small molecule cyclophilin D inhibitor for rescuing abeta-mediated mitochondrial dysfunction. ACS Med Chem Lett. 2016;7(3):294–299. doi: 10.1021/acsmedchemlett.5b00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Falkevall A, Alikhani N, Bhushan S, et al. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281(39):29096–29104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 106.Alikhani N, Guo L, Yan S, et al. Decreased proteolytic activity of the mitochondrial amyloid-beta degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J Alzheimers Dis. 2011;27(1):75–87. doi: 10.3233/JAD-2011-101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fang F, Lue LF, Yan S, et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010;24(4):1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang H, Wang Y, Yan S, et al. Genetic deficiency of neuronal RAGE protects against AGE-induced synaptic injury. Cell Death Dis. 2014;5:e1288. doi: 10.1038/cddis.2014.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Origlia N, Bonadonna C, Rosellini A, et al. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. 2010;30(34):11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Origlia N, Criscuolo C, Arancio O, et al. RAGE inhibition in microglia prevents ischemia-dependent synaptic dysfunction in an amyloid-enriched environment. J Neurosci. 2014;34(26):8749–8760. doi: 10.1523/JNEUROSCI.0141-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Origlia N, Capsoni S, Cattaneo A, et al. Abeta-dependent inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. J Alzheimers Dis. 2009;17(1):59–68. doi: 10.3233/JAD-2009-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Origlia N, Righi M, Capsoni S, et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;28(13):3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yan SD, Chen X, Fu J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 114.Näslund J, Schierhorn A, Hellman U, et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA. 1994;91(18):8378–8382. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moskovitz J, Du F, Bowman CF, Yan SS. Methionine sulfoxide reductase A affects beta-amyloid solubility and mitochondrial function in a mouse model of Alzheimer’s disease. Am J Physiol Endocrinol Metab. 2016;310(6):388–393. doi: 10.1152/ajpendo.00453.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frazier AE, Kiu C, Stojanovski D, et al. Mitochondrial morphology and distribution in mammalian cells. Biol Chem. 2006;387(12):1551–1558. doi: 10.1515/BC.2006.193. [DOI] [PubMed] [Google Scholar]
- 117.Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 119.Fang D, Yan S, Yu Q, et al. Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci Rep. 2016;6:31462. doi: 10.1038/srep31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pham AH, Meng S, Chu QN, et al. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21(22):4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Karakaya T, Fußer F, Schröder J, Pantel J. Pharmacological treatment of mild cognitive impairment as a prodromal syndrome of Alzheimer s disease. Curr Neuropharmacol. 2013;11(1):102–108. doi: 10.2174/157015913804999487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gan X, Wu L, Huang S, et al. Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic Biol Med. 2014;75:230–240. doi: 10.1016/j.freeradbiomed.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gan X, Huang S, Wu L, et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta. 2014;1842(2):220–231. doi: 10.1016/j.bbadis.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21(11):2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chou CH, Lin CC, Yang MC, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One. 2012;7(11):e49112. doi: 10.1371/journal.pone.0049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hong YR, Chen CH, Cheng DS, et al. Human dynamin-like protein interacts with the glycogen synthase kinase 3beta. Biochem Biophys Res Commun. 1998;249(3):697–703. doi: 10.1006/bbrc.1998.9253. [DOI] [PubMed] [Google Scholar]
- 127.Chen CH, Hwang SL, Howng SL, et al. Three rat brain alternative splicing dynamin-like protein variants: interaction with the glycogen synthase kinase 3beta and action as a substrate. Biochem Biophys Res Commun. 2000;268(3):893–898. doi: 10.1006/bbrc.2000.2197. [DOI] [PubMed] [Google Scholar]
- 128.Park J, Choi H, Min JS, et al. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem. 2013;127(2):221–232. doi: 10.1111/jnc.12361. [DOI] [PubMed] [Google Scholar]
- 129.Motori E, Puyal J, Toni N, et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013;18(6):844–859. doi: 10.1016/j.cmet.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 130.Dong XB, Yang CT, Zheng DD, et al. Inhibition of ROS-activated ERK1/2 pathway contributes to the protection of H2S against chemical hypoxia-induced injury in H9c2 cells. Mol Cell Biochem. 2012;362(1–2):149–157. doi: 10.1007/s11010-011-1137-2. [DOI] [PubMed] [Google Scholar]
- 131.Palmer CS, Osellame LD, Stojanovski D, et al. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011;23(10):1534–1545. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 132.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]