

Abstract
The structure-based design, synthesis, and biological evaluation of a series of nonpeptidic HIV-1 protease inhibitors with rationally designed P2′ ligands are described. The inhibitors are designed to enhance backbone binding interactions, particularly at the S2′ subsite. Synthesis of inhibitors was carried out efficiently. The stereochemistry of alcohol functionalities of the P2′ ligands was set by asymmetric reduction of the corresponding ketone using (R,R)- or (S,S)-Noyori catalysts. A number of inhibitors displayed very potent enzyme inhibitory and antiviral activity. Inhibitors 3g and 3h showed enzyme Ki values of 27.9 and 49.7 pM and antiviral activity of 6.2 and 3.9 nM, respectively. These inhibitors also remained quite potent against darunavir-resistant HIV-1 variants. An X-ray structure of inhibitor 3g in complex with HIV-1 protease revealed key interactions in the S2′ subsite.
Keywords: drug resistance, HIV-1 protease inhibitors, P2′ ligands, pharmacokinetics, structure-based design
Introduction
The arrival of HIV/AIDS in the early 1980s marked the onset of a global pandemic.[1,2] This led to intense worldwide research efforts in academic and pharmaceutical laboratories for treatment of HIV infection and AIDS.[3,4] Enormous research advances in the mid-1990s fundamentally changed the prognosis of HIV/AIDS treatment and management of the disease. The advent of combined antiretroviral therapy (cART), involving the use of HIV-1 protease inhibitors (PIs) and reverse transcriptase inhibitors drastically improved life expectancy and decreased HIV-related mortality of patients with access to this treatment regimen.[5,6] Recent improvement of first line cART and declines in the price of treatment have prompted further accessibility of cART to developing countries.[7,8] Despite these great strides, there are serious limitations to current treatment regimens including cost, toxicity, patient compliance, side effects, and resistance. Perhaps, the most imposing limitation is the rapid rate at which the HIV generates viable drug-resistant mutants under selection pressure.[9,10] PIs are an integral part of some of the most preferred cART treatment regimens.[11,12] In our continuing efforts to develop PIs to combat drug resistance, we designed several classes of novel compounds that display exceptional antiviral potencies across wild-type and multidrug-resistant HIV strains.[13,14] Inhibitor 1 (TMC-114 or darunavir, DRV, Figure 1) displayed picomolar enzyme inhibitory activity and potent antiviral activity.[15,16] It was also endowed with unprecedented activity against a range of known multidrug resistant HIV strains.[17,18] Darunavir received FDA approval in 2006 for use in adult patients harboring drug-resistant strains.[15,16] In 2008, it was approved for all HIV/AIDS patients and emerged as a front line anti-HIV therapy for use in both adult and pediatric patients.[19]
Figure 1.
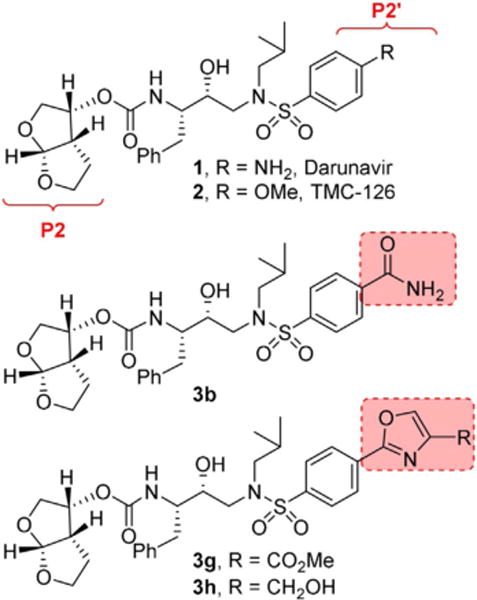
Structures of HIV-1 protease inhibitors 1 and 2 and of novel inhibitors 3b,g,h.
Our X-ray structural analysis of DRV-bound HIV-1 protease structure revealed an extensive network of hydrogen bonding interactions between DRV and backbone atoms of HIV-1 protease.[20] In particular, the bis-THF P2 ligand formed a pair of strong hydrogen bonds with Asp29 and Asp30 NHs in the S2 subsite. The P2′-aminosulfonamide forms a pair of hydrogen bonds with Asp30′ backbone amide NH as well as with the Asp30′ backbone carbonyl oxygen. These backbone interactions have been attributed to DRV′s wide range of activity against known PI-resistant mutants.[15,21] The bis-THF ligand is a privileged ligand for DRV and its extensive interactions are responsible for DRV′s unique activity. Over the years, we have investigated other structurally intriguing ligands that mimic the ligand-binding interactions of the bis-THF ligand of DRV in the S2 subsite. In an effort to promote further ligand-binding site interactions in the S2′ subsite, we have explored modifications of the P2′ ligand with hydrogen-bonding donor/acceptor groups to form enhanced backbone interactions. We are particularly interested in ligands and scaffolds that may improve metabolic stability. In a preliminary effort, based on the X-ray structure of DRV and HIV-1 complex, we incorporated carboxylic acid and carboxamide functionalities at the 4-position of the benzene ring of the P2′-sulfonamide ligand.[22,23] Both the carboxylic acid and the carboxamide ligand showed very potent picomolar enzyme inhibitory activity. The carboxamide ligand derived inhibitor 3b showed potent antiviral activity. To obtain ligand-binding site interactions of the carboxamide derivative, we determined an X-ray structure of 3b-bound HIV-1 protease.[23] The key inhibitor-binding site interactions are shown in Figure 2A. In particular, the P2′ moiety of 3b, similarly to DRV, shows hydrogen-bond interactions with the backbone of Asp30′. Interestingly, the P2′ benzamide moiety of 3b also showed a conserved water-mediated bridging hydrogen bond with Gly48′ (Figure 2B).[24] Based on this structure, we have now further modified the P2′ ligand to promote hydrogen bonding interactions and improve antiviral activity using a range of stereochemically defined functionalities and heterocyclic scaffolds. Herein we report the structure-based design, synthesis, X-ray structural studies and biological investigation of a series of HIV-1 PIs with novel P2′ ligands.
Figure 2.
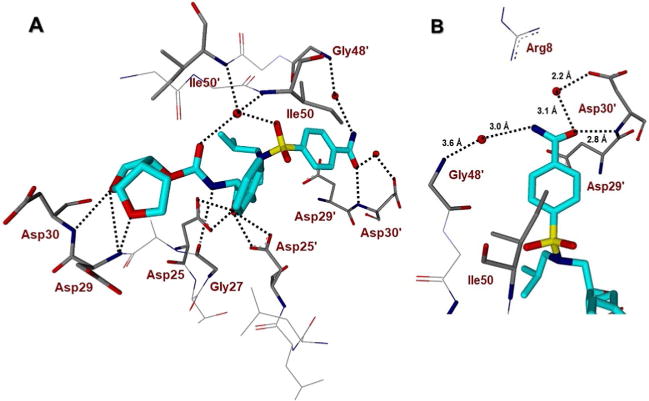
A) Inhibitor 3b-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in turquoise, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines. B) Specific hydrogen bonding interactions of carboxamide are shown. These interactions are indicated with respective distances.
Results and Discussion
Synthesis of ligands and inhibitors
All target PIs herein presented are accessible via diacetate intermediate 5, which has been prepared in two steps from commercially available chiral epoxide 4 as described previously.[25] The synthesis of compounds 3a–f is reported in Scheme 1. Deprotection of 5 to the corresponding aldehyde with potassium carbonate in methanol and subsequent Pinnick oxidation led to the key carboxylic acid intermediate 7 in excellent yield. Compound 7 was treated with trifluoroacetic acid (TFA) to remove the Boc group and the resulting TFA salt was treated with the known bis-THF activated carbonate 6[26] in a 10:1 mixture of acetonitrile/water in the presence of N,N-DIPEA to provide inhibitor 3a. Carboxamide inhibitor 3b was prepared via direct one-pot conversion of inhibitor 3a to a mixed anhydride with di-tert-butyldicarbonate in the presence of pyridine followed by condensation with ammonium carbonate in 76% yield.[27]
Scheme 1.
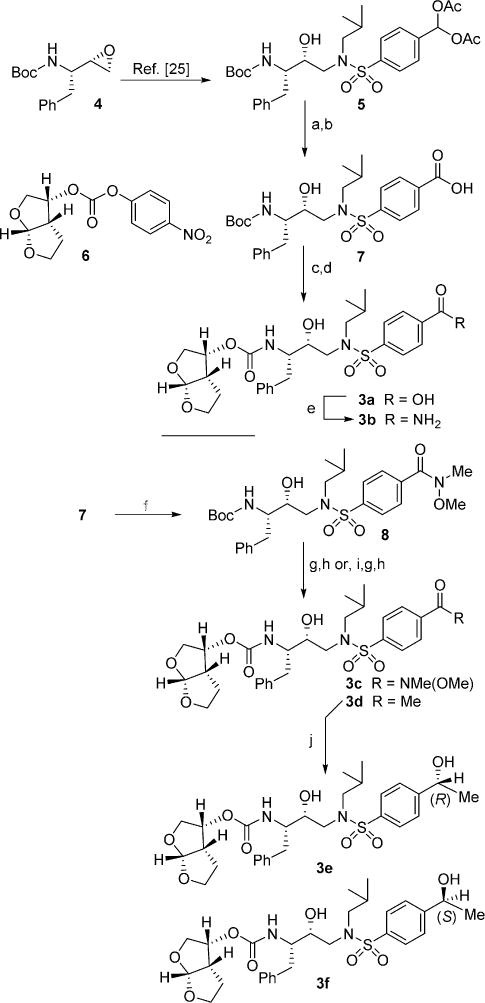
Reagents and conditions: a) K2CO3, MeOH, 23°C, 1 h, 98%; b) NaClO2, cis-2-methyl-2-butene, NaH2PO4. H2O, tBuOH-H2O, 23°C, 50 min, 95%; c) TFA, CH2Cl2, 23°C, 3 h; d) 6, N,N-DIPEA, MeCN/H2O, 23°C, 3 days 89% (over two steps); e) pyridine, Boc2O, (NH4)2CO3, THF, 23 °C, 14 h, 76%; f) iBuOCOCl, NMM then MeNHOMe·HCl, dry CH2Cl2, −15°C →23°C, 12 h, 76%; g) TFA, CH2Cl2, 23 °C, 2 h; h) 6, N,N-DIPEA, MeCN,23°C, 5 days, 73–78%; i) MeMgBr (3 M in Et2O), dry THF, −78°C, 4 h, 98%; j) RuCl[(R,R)-TsDPEN](mesitylene) for 3e or RuCl(p-cymene)[(S,S)-Ts-DPEN] for 3f, TEA, HCOOH, dry CH2Cl2, 0°C →23°C, 7 h, 94–98 %.
Treatment of acid 7 with isobutyl chloroformate in the presence of N-methylmorpholine, followed by N,O-dimethylhydrox-ylamine led to the corresponding Weinreb amide 8. Exposure to TFA followed by treatment with activated carbonate 6 and N,N-DIPEA in acetonitrile led to inhibitor 3c. Intermediate 8 was also converted into its methyl ketone derivative by exposure to methylmagnesium bromide in dry THF. Boc deprotection followed by reaction with bis-THF derivative 6 led to final compound 3d.
Inhibitors 3e,f were prepared starting from compound 3d. Exposure of 3d to a catalytic amount (0.5 mol%) of the (R,R)-or (S,S)-Noyori catalysts[28] in a 1:2 ratio of Et3N/HCO2H led to high yields (95–98%) of the corresponding (R) and (S) final alcohols 3e and 3f, respectively, with excellent enantioselectivity (see Experimental Section for details).
The synthesis of oxazole-containing inhibitors 3g,h is reported in Scheme 2. Key carboxylic acid intermediate 7 was submitted to coupling reaction with serine methyl ester hydrochloride in the presence of EDCI, HOBt and N,N-DIPEA in dry DMF affording the corresponding amide adduct in 91% yield.
Scheme 2.
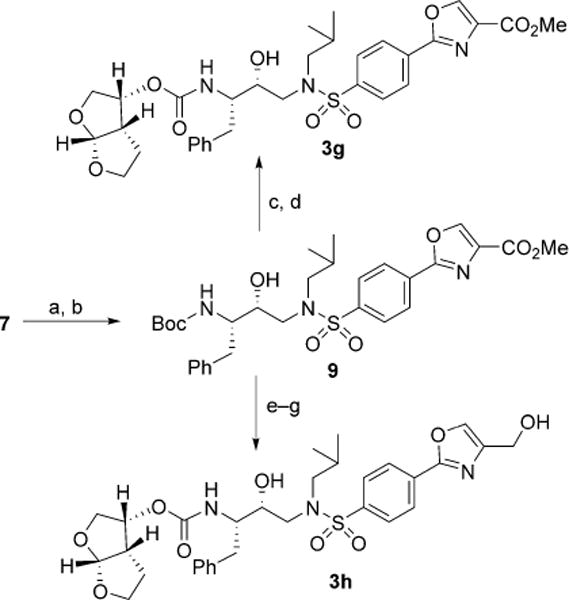
Reagents and conditions: a) D,L-Serine methyl ester hydrochloride, EDCI, HOBt, N,N-DIPEA, dry DMF, 0°C→23°C, 16 h, 91%; b) DAST, dry CH2Cl2, −78°C, 30 min, then BrCCl3, DBU, dry CH2Cl2, 0°C→23°C, 2.5 h, 64% (over two steps); c) TFA, CH2Cl2, 23°C, 4 h; d) 6, N,N-DIPEA, MeCN, 23°C, 6 days, 61% (over two steps); e) NaBH4, EtOH, 0°C→23°C, 24 h, 65%; f) TFA, CH2Cl2, 23°C, 4 h; g) 6, N,N-DIPEA, MeCN, 23°C, 5 days, 60% (over two steps).
Subsequent dehydrative cyclization conditions with DAST reagent at −78°C afforded the corresponding oxazoline intermediate immediately submitted to a radical bromination-reductive elimination protocol using DBU/BrCCl3 providing oxa-zole derivative 9 in 64% yield over two steps.[29] The reduction of the ester functionality of oxazole 9 with NaBH4 in absolute ethanol provided the corresponding hydroxymethyl intermediate in moderate yield. Both the oxazole ester 9 and its hydrox-ymethyl derivative were converted into the corresponding final inhibitors 3g,h via standard methods.
The second series of oxazole inhibitors 3i–l was prepared as shown in Scheme 3. Diacetate intermediate 5 was converted into the oxazole derivative 10 in high yield employing the procedure reported by van Leusen and co-workers with toluene-sulfonylmethyl isocyanide (TosMIC) in the presence of K2CO3 in methanol.[30] Exposure of compound 10 to TFA followed by treatment with activated bis-THF derivative 6 in the presence of N,N-DIPEA led to inhibitor 3i. Intermediate 10 was also treated with isopropylmagnesium chloride providing the 2-mag-nesiate derivative which was immediately reacted with the acetate-derived Weinreb amide to give the desired 2-acetyl ox- azole 11 in good yield.[31] Acetyl derivative 11 underwent Boc-deprotection with TFA and treatment with activated bis-THF derivative 6 in the presence of N,N-DIPEA providing inhibitor 3j. This latter also underwent Noyori asymmetric hydrogenation with both the (R,R)- or (S,S)-Noyori catalysts in a 1:2 ratio of Et3N/HCO2H leading to final compounds 3k,l in high yields and enantiomeric excess (see Experimental Section for details).
Scheme 3.
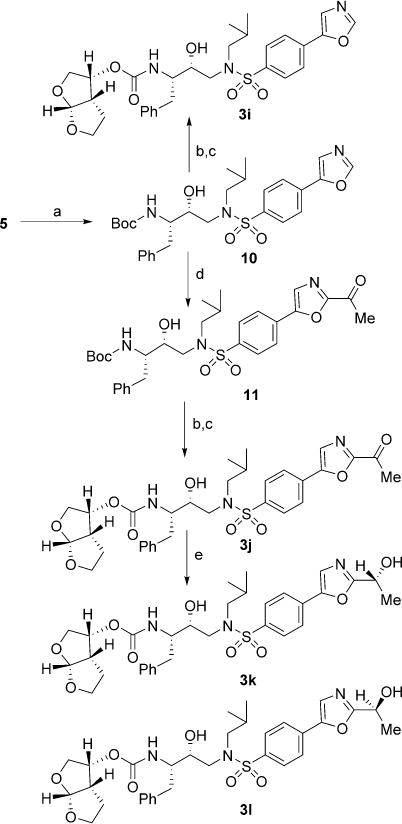
Reagents and conditions: a) TosMIC, K2CO3, MeOH, 55°C, 1.5 h, 96%; b) TFA, CH2Cl2, 23°C, 2.5 h; c) 6, N,N-DIPEA, MeCN, 23°C, 4–7 days, 47–80% (over two steps); d) iPrMgCl, dry THF, 15°C, 40 min, then AcN(-Me)OMe, −15°C→23°C, 5 h, 79%; e) RuCl[(−R,R)-TsDPEN](mesitylene) for 3k or RuCl(p-cymene)[(S,S)-Ts-DPEN] for 3l, TEA, HCOOH, dry CH2Cl2, 0°C→23°C, 7 h, 95–97%.
Biological assays
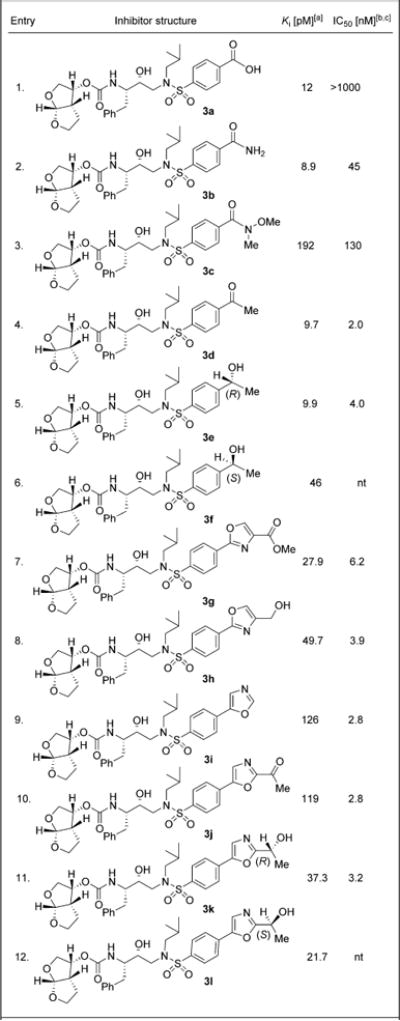
We evaluated all synthetic inhibitors in a HIV-1 protease inhibitory assay using the protocol developed by Toth and Mar-shall.[32] These results are shown in Table 1. Selected potent inhibitors were further evaluated in an antiviral assay following a reported protocol using MT-2 cells exposed to HIV-1LAI.[33] In an attempt to promote hydrogen bond formation with the back bone atoms at S2′ subsite, we investigated the feasibility of carboxylic acid, and various derivatives. As shown, incorporation of a para-carboxylic acid functionality provided inhibitor 3a which showed very potent HIV-1 inhibitory activity (Ki=12 pM). However, this carboxylic acid derivative showed an antiviral IC50 value greater than 1 μM. Interestingly, the corresponding carboxamide derivative 3b not only showed very potent HIV-1 protease inhibitory activity, but also displayed potent antiviral activity (IC50=45 nM). The reason for the poor antiviral activity for compound 3a may be due to the low cell penetration ability of carboxylic acids. Based on the X-ray structure of DRV-bound HIV-1 protease, we speculated that the carbonyl group of a carboxylic acid derivative or the heteroa-toms of the corresponding heterocyclic derivatives can interact with backbone atoms, particularly Asp29′ and Asp30′ backbone NHs. We therefore investigated other carbonyl derivatives such as the methoxymethyl amide in inhibitor 3c as well as the methyl ketone derivative in inhibitor 3d (entries 3, 4). Interestingly, methyl ketone derivative 3d displayed very potent enzyme Ki as well as antiviral activity. We have examined inhibitory properties of the corresponding reduced products, 3e and 3f (entries 5, 6). The stereochemical effect is apparent, as compound 3e with (R)-hydroxy configuration has shown a greater than fourfold improvement in enzyme Ki. Compound 3e has also shown potent antiviral IC50 of 4 nM.
Table 1.
Structures and activity of inhibitors 3a–l.
|
Ki values represent at least five data points; the standard error in all cases was <7%. Darunavir exhibited Ki=16 pM.
nt: Not tested.
Values are the mean of at least three experiments; standard error in all cases was <5%. Darunavir exhibited IC50=1.6 nM.
We then pursued carboxylic acid bioisosteres with a series of oxazole-derived P2′ ligands. As shown, compound 3g and 3h, bearing, respectively, a methyl ester and a hydroxymethyl functionality at the 4-position of the 2-phenyloxazole moiety, displayed a slight decrease of enzyme inhibitory potency with Ki=27.9 and Ki=49.7 pM, respectively (entries 7 and 8). The corresponding reduced product alcohol 3h displayed a slightly improved antiviral IC50 value of 3.9 nM. We have also synthe-sized a series of carboxylic acid derivatives with 5-phenyloxa-zole scaffolds (entries 9–12). These derivatives displayed decreased enzyme inhibitory potency; however, they maintained excellent antiviral activity similar to that of DRV. Monosubsti-tuted derivative 3i showed a Ki of 126 pM and an antiviral IC50 value of 2.8 nM (entry 9). Incorporation of methyl ketone at the 2-position of the oxazole ring provided 3j. This substitution did not improve potency and showed activity similar to 3i (entry 10). Reduction of the methyl ketone provided stereo-chemically defined alcohols with (R)- and (S)-configuration. Both derivatives showed potent antiviral activity, however, no improvement was observed over monosubstituted oxazole derivative 3i (entries 11 and 12).
Because compound 3g contains an interesting heterocyclic P2′ ligand, we planned to obtain molecular insight into its binding properties using an X-ray structure. To this end, we determined the X-ray structure of 3g-bound HIV-1 protease. To further correlate the structure with its antiviral properties, we examined both potent oxazole containing PIs (3g, 3h) against DRV-resistant HIV-1 variants. In these assays, MT-4 cells were exposed to wild-type HIV-1 and three drug-resistant variants HIV-1DRVR20, HIV-1DRVR30, and HIV-1DRVR40 and were treated with various concentrations of each inhibitor. These DRV-resistant HIV-1DRVRS variants are highly resistant to all current clinically used PIs including DRV and nucleoside/nucleotide reverse transcriptase inhibitors such as tenofovir. IC50 values were determined using p24 assay.[33,34] The results are shown in Table 2. Oxazole derivative 3g was less potent against HIVNL4-3 than DRV (3.4 nM vs. 22 nM). Its fold-change against HIVDRVRP20 is similar to that of DRV, however, fold-changes against HIVDRVRP30 and HIVDRVRP40 were more than 3- and 6-fold lower than DRV (fold differences for DRV 71- and >297-fold). Inhibitor 3h with a hydroxymethyl side chain on the oxazole ring showed similar IC50 values as DRV against HIVNL4-3. Its fold-change of antiviral activity against HIVDRVR20 was higher than compound 3g and DRV, although the fold-changes against HIVDRVRP30 and HIVDRVRP40 were similar to DRV. This data indicates an interesting profile for the novel inhibitor 3g toward DRV-resistant variants, and is to be considered a starting point for further optimization.
Table 2.
Antiviral activity of two novel compounds against highly DRV-resistant HIV-1 variants.
| HIV-1 | IC50 [nM] (fold change)a | ||
|---|---|---|---|
| DRV | 3g | 3h | |
| HIV-1NL4-3 | 3.4 | 22 | 3.0 |
| HIV-1DRVRP20 | 43 (13) | 320 (15) | 260 (88) |
| HIV-1DRVRP30 | 24 (71) | 430 (20) | 290 (99) |
| HIV-1DRVRP40 | >1 (>297) | >1 (>46) | >1 (>337) |
MT-4 cells (1×104) were exposed to 50 TCID50s of wild-type HIV-1NL4-3, HIV-1DRVRP20, HIV-1DRVRP30, or HIV-1DRVRP40 and cultured in the presence of various concentrations of each PI, and the IC50 values were determined using the p24 assay. The amino acid substitutions identified in protease of HIV-1DRVRP20, HIV-1DRVRP30, and HIV-1DRVRP40 HIV-1NL4-3 include L10I/I15V/K20R/L24I/V32I/M36I/M46L/L63P/V82A/L89M, L10I/I15V/K20R/L24I/V32I/M36I/M46L/L63P/K70R/V82A/I84V/L89M, and L10I/I15V/K20R/L24I/V32I/L33F/M36I/M46L/I54M/L63P/K70Q/V82I/I84V/L89M, respectively. All assays were conducted in triplicate, and the data are mean values derived from the results of three independent experiments; standard error in all cases <5%.
X-ray crystal structure description
To gain molecular insight into interactions of the oxazole inhibitors, we co-crystallized inhibitor 3g (GRL-5311) with wild-type HIV-1 protease and the X-ray structure was refined at 1.30 Å resolution to an Rwork/Rfree of 15.4/19.6 % (PDB ID: 6B4N). The structure contains HIV-1 protease dimer with inhibitor 3g bound in a single orientation. The HIV-1 protease dimer structure closely resembles our reported structure of protease-DRV complex with RMSD of 0.22 Å for all Cα atoms.[20] Larger differ ences between the corresponding Cα atoms are evident in one subunit of the dimer, where the differences of 0.75 Å occurred for residues 53′ to 54′ in the flap region and 0.77 Å for residues 80′ and 81′ in the 80s loop. These shifts are probably because the inhibitor 3g has a larger P2′ and P3′ substituted oxazole group relative to the P2′ ligand of DRV. The inhibitor binds in the active site showing similar interactions for P2-bis-THF, the urethane functionality, and the P1-phenylmethyl side chain to those observed for the HIV-1 protease-DRV X-ray structure.[20]
The major differences for inhibitor 3g interactions with HIV-1 protease are evident in the S2′ subsite where the 4-amino group of DRV was replaced by a substituted oxazole group. As shown in Figure 3, the oxazole nitrogen forms a strong hydro gen bond with the backbone NH of Asp30′. Interestingly, the carboxyl group of the ester side chain also forms a strong hydrogen bond with the Asp30′ backbone NH. The oxygen atom of the oxazole heterocycle forms a water-mediated interaction with Gly48′ amide NH. The carbonyl group of the oxazole side chain has two orientations and both stack against the carbox-ylic acid side chain of Asp30′. In one S-trans orientation of the ester, the methoxy oxygen is within hydrogen bonding distance of the Lys45′ amine side chain. Furthermore, the aromatic P2′ ring in 3g has shifted toward the flap region, as shown in the overlay of X-ray structures of DRV-bound HIV-1 protease and inhibitor 3g-bound HIV-1 protease (Figure 4). The side chain of Asp30′ has also shifted toward Asn88 to accommodate the oxazole ring. This heterocyclic core shows good van der Waals interactions with the side chains of Ile47′, Asp29′, and the main chain of Gly48′. These interactions may be responsible for the inhibitor′s high affinity for HIV-1 protease.
Figure 3.
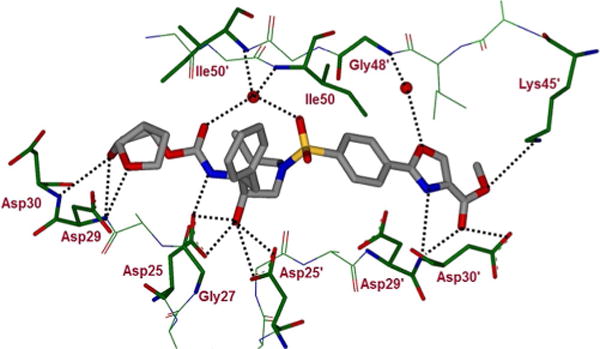
Inhibitor 3g-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in grey, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines (PDB ID: 6B4N).
Figure 4.
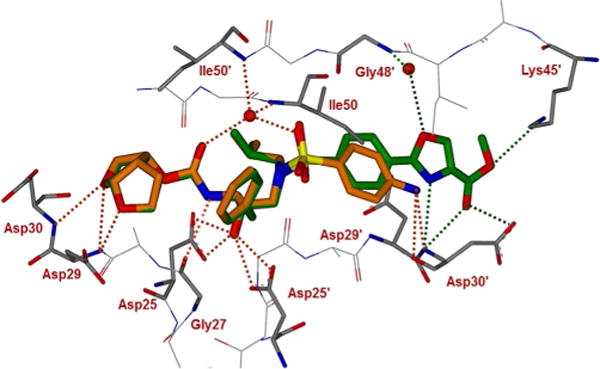
An overlay of the X-ray crystal structure of DRV-bound HIV-1 protease (orange) with the X-ray structure of inhibitor 3g (green)-bound HIV-1 protease.
Conclusions
In conclusion, we have designed, synthesized, and examined a number of HIV-1 PIs containing carboxylic acid derivatives and substituted oxazole derivatives as P2′ ligands. These ligands are designed to enhance interactions with backbone atoms and residue in the S2′ subsite. Inhibitor 3b with 4-carboxamide functionality showed enhanced potency over the carboxylic acid 3a. The X-ray structure of 3b-bound HIV-1 protease showed key hydrogen bonding interactions with Asp30′ backbone NH. Furthermore, the carboxamide also formed an interesting water-mediated hydrogen bond with the Gly48′ carbonyl group. Antiviral activity of 3b showed that it is highly active against HIVAO2 with EC50 value of 29 nM. Antiviral activity of 3b against HIV-2ROD or HIVDRVRP20 was 30 nM and 97 nM. There was only 2.6-fold increase in its EC50 value relative to its EC50 against HIVWT. Based on the X-ray structure of 3b-bound HIV-1 protease and its biological properties, we have examined a series of derivatives, including stereochemically defined alcohols, a ketone, a methoxymethylamide, 2-phenyl oxazole derivatives, and 5-phenyl oxazole derivatives. Both methyl ketone 3d and its reduced products provided very potent enzyme inhibitory and antiviral activity similar to DRV. Various oxazole derivatives also displayed very potent antiviral activity. In particular, inhibitors 3g and 3h with 2-phenyloxazoles as the P2′ ligands showed very potent antiviral activity. While inhibitor 3h has shown 10-fold better IC50 value than inhibitor 3g against HIVNL4-3, inhibitor 3h also showed similar fold-changes as DRV against HIVDRVRP30 and HIVDRVR40. However, its fold-changes are higher against HIVDRVRP20. While inhibitor 3g showed an IC50 value of 22 nM against HIVNL4-3, its fold-changes against DRV-resistant viruses are superior to inhibitor 3h or DRV. The X-ray crystal structure of 3g-bound HIV-1 protease revealed several new hydrogen bonds and water-mediated hydrogen bonding interactions in the S2′ subsite of HIV-1 protease. Further design and synthesis of new inhibitors using this molecular insight is in progress.
Experimental Section
General
All moisture-sensitive reactions were carried out in an oven dried flask under argon atmosphere. All chemicals and reagents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were obtained as follows: anhydrous tetrahydrofuran, diethyl ether, and benzene were distilled from sodium metal under argon. Anhydrous dichloromethane, toluene, methanol, and acetonitrile were dried via distillation from CaH2 under argon. All other solvents were HPLC grade. 1H NMR and 13C NMR spectra were recorded on Varian INOVA300-1, Bruker Avance ARX-400 and Bruker DRX-500 spectrometers. NMR data are reported as: δ value (chemical shift, J-value (Hz), integration, where s=singlet, d=doublet, t=triplet, q=quartet, br= broad). Low resolution mass analyses were performed on a Agilent 1290 Infinity II spectrometer. High-resolution mass spectrometric analyses were performed at the Purdue University Campus-wide Mass Spectrometry Center. TLC analysis was carried out with SiliCycle 60A-F254 plates. Flash chromatography was performed using SiliCycle 230–400 mesh, 60 Å pore diameter silica gel. HPLC analysis was performed on a Agilent 1260 Infinity instrument. All test inhibitors showed purity >95% by HPLC analysis.
4-(N-((2R,3S)-3-((tert-Butoxycarbonyl)amino)-2-hydroxy-4-phenyl-butyl)-N-isobutylsulfamoyl)benzoic acid (7)
To a solution of compound 5 (81 mg, 0.156 mmol) in MeOH (14 mL) was added K2CO3 (113 mg, 0.392 mmol) and the reaction was stirred at 23°C for 1 h. The reaction mixture was concentrated under reduced pressure and the residue partitioned between water and EtOAc. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (30% EtOAc in n-hexanes) providing tert-butyl ((2S,3R)-4-((4-formyl-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (98% yield). 1H NMR (400 MHz, CDCl3) δ=10.08 (s, 1H), 8.00 (d, J=8.4 Hz, 2H), 7.93 (d, J=8.3 Hz, 2H), 7.41–7.11 (m, 5H), 4.67 (d, J=8.2 Hz, 1H), 3.95–3.64 (m, 3H), 3.16 (d, J=5.8 Hz, 2H), 3.03–2.71 (m, 4H), 1.96–1.73 (m, 1H), 1.34 (s, 9H), 0.86 ppm (dd, J=8.8, 6.7 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ=190.9, 156.4, 144.2, 138.9, 137.8, 130.3, 129.6, 128.7, 128.1, 127.8, 126.7, 80.1, 72.7, 58.1, 55.1, 53.2, 35.7, 28.4, 27.2, 20.2, 20.0 ppm. LRMS-ESI (m/ z): 527.2 [M+Na]+. This aldehyde intermediate (41 mg, 0.081 mmol) was dissolved in tBuOH (5 mL) and 2-methyl-2-butene (2 mL) was added at 23°C; 2.6 mL of a stock solution obtained by dissolving NaH2PO4·H2O (600 mg) and NaClO2 (600 mg) in 6 mL of water was added and the reaction mixture was stirred at 23°C for 50 min. Water (3 mL) was added and the reaction mixture was extracted with EtOAc. The organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (5% to 10% MeOH in CH2Cl2) providing compound 7 as a white solid (95% yield). 1H NMR (400 MHz, CD3OD) δ=8.17 (d, J=8.3 Hz, 2H), 7.92 (d, J=8.3 Hz, 2H), 7.30–7.18 (m, 3H), 7.18–7.09 (m, 2H), 3.71 (t, J=7.1 Hz, 1H), 3.64–3.53 (m, 1H), 3.42 (dd, J=15.0, 1.9 Hz, 1H), 3.20–3.01 (m, 2H), 2.94 (dd, J=13.7, 6.7 Hz, 1H), 2.54 (dd, J=13.7, 10.8 Hz, 1H), 2.10–1.95 (m, 1H), 1.36–1.21 (m, 9H), 0.89 ppm (dd, J=15.4, 6.6 Hz, 6H). 13C NMR (100 MHz, DMSO) δ=168.3, 158.1, 144.9, 140.3, 135.8, 131.5, 130.6, 129.3, 128.7, 127.2, 80.2, 74.0, 58.3, 57.0, 53.6, 37.4, 28.8, 27.9, 20.5 ppm. LRMS-ESI (m/z): 521.2 [M+H]+, 543.1 [M+Na]+.
tert-Butyl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-(methoxy(methyl)-carbamoyl)phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (8)
Compound 7 (58 mg, 0.111 mmol) was dissolved in dry CH2Cl2 (6 mL) and N-methyl morpholine (27 μL, 0.244 mmol) was added. The solution was cooled to −15°C and iBuOCOCl (16 μL, 0.122 mmol) was added. The reaction mixture was stirred at −15°C for 15 min, then N,O-dimethylhydroxylamine hydrochloride (11 mg, 0.113 mmol) was added and the reaction was stirred at 23°C for 12 h. The mixture was then partitioned between water and CH2Cl2. Organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (5% MeOH in CH2Cl2) providing compound 8 as a white solid (76% yield). 1H NMR (400 MHz, CDCl3) δ=7.99–7.70 (m, 4H), 7.39–7.10 (m, 5H), 4.69 (d, J=7.6 Hz, 1H), 3.96–3.68 (m, 3H), 3.51 (s, 3H), 3.37 (s, 3H), 3.13 (d, J=5.7 Hz, 2H), 3.05–2.80 (m, 4H), 1.94–1.75 (m, 1H), 1.46–1.20 (m, 9H), 0.86 ppm (dd, J=11.1, 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ=168.3, 156.3, 140.5, 138.4, 138.0, 129.7, 129.0, 128.6, 127.2, 126.6, 79.9, 72.8, 61.5, 58.4, 55.1, 53.6, 35.6, 33.4, 28.4, 27.2, 20.2, 20.0 ppm. LRMS-ESI (m/z): 564.3 [M+H]+, 586.2 [M+Na]+.
Methyl 2-(4-(N-((2R,3S)-3-((tert-butoxycarbonyl)amino)-2-hy-droxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)oxazole-4-car-boxylate (9)
Compound 7 (32 mg, 0.0615 mmol) was dissolved in dry DMF (4 mL) and the solution was cooled to 0°C. EDCI (14 mg, 0.074 mmol), HOBt (10 mg, 0.074 mmol), N,N-DIPEA (20 mL, 0.153 mmol) and DL-serine methyl ester hydrochloride (12 mg, 0.074 mmol) were sequentially added to the solution and the reaction mixture was stirred at 23°C for 16 h. Water was added and the reaction mixture was extracted with EtOAc. Organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (EtOAc) providing methyl (4-(N-((2R,3S)-3-((tert-butoxycarbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)benzoyl)serinate as a colorless oil (91% yield). 1H NMR (400 MHz, CDCl3) δ=7.94 (d, J=8.4 Hz, 2H), 7.83 (d, J=8.3 Hz, 2H), 7.42–7.13 (m, 5H), 4.88 (dt, J=7.0, 3.3 Hz, 1H), 4.67 (d, J=8.1 Hz, 1H), 4.08–4.00 (m, 2H), 3.92– 3.86 (m, 1H), 3.86–3.68 (m, 5H), 3.13 (d, J=5.7 Hz, 2H), 3.04–2.82 (m, 4H), 2.63 (br, 1H), 1.92–1.78 (m, 1H), 1.67 (s, 1H), 1.65–1.51 (m, 1H), 1.34 (s, 9H), 0.87 ppm (dd, J=10.6, 6.6 Hz, 6H). The above intermediate (70 mg, 0.113 mmol) was dissolved in dry CH2Cl2 (5 mL) and the solution was cooled to −78°C. DAST (17 μL, 0.124 mmol) was added dropwise. The reaction mixture was stirred at −78°C for 30 min (TLC monitoring in 100% EtOAc) and then quenched with K2CO3 and passed quickly through a silica gel column pretreated with triethylamine and eluted with EtOAc. The crude material recovered from the column was dried under reduced pressure and dissolved in dry CH2Cl2 (5 mL) and the resulting solution was cooled to 0°C. BrCCl3 (13 μL, 0.124 mmol) and DBU (19 μL, 0.124 mmol) were sequentially added and the reaction mixture was then stirred at 23°C for 2.5 h. Solvents were evaporated under reduced pressure and the residue was purified by silica gel flash chromatography (50% EtOAc in n-hexane) providing compound 9 as a white solid (64% yield over two steps). 1H NMR (400 MHz, CDCl3) δ=8.35 (s, 1H), 8.24 (d, J=8.5 Hz, 2H), 7.87 (d, J=8.5 Hz, 2H), 7.40–7.16 (m, 5H), 4.67 (d, J=7.7 Hz, 1H), 3.97 (s, 3H), 3.94–3.86 (m, 1H), 3.86–3.70 (m, 2H), 3.16 (d, J=5.8 Hz, 2H), 3.08–2.83 (m, 4H), 1.94–1.80 (m, 1H), 1.34 (s, 9H), 0.86 ppm (dd, J=11.0, 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ=161.5, 160.9, 156.3, 144.7, 141.2, 137.9, 135.1, 130.2, 129.7, 128.7, 128.0, 127.6, 126.7, 108.9, 80.0, 72.8, 58.3, 55.1, 53.3, 52.6, 35.6, 28.4, 27.2, 20.2, 20.0 ppm. LRMS-ESI (m/z): 624.2 [M+Na]+, 502.1 [M−Boc+H]+.
tert-Butyl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-(oxazol-5-yl)phe-nyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (10)
Compound 5 (52 mg, 0.086 mmol) and TosMIC (40 mg, 0.206 mmol) were dissolved in MeOH (3 mL) in a sealed tube and K2CO3 (76 mg, 0.548 mmol) was added. The reaction mixture was stirred at 55°C for 1.5 h (TLC monitoring). The reaction mixture was then cooled to room temperature and filtered on a Celite pad. The crude residue was purified by silica gel flash chromatography (20% to 50% EtOAc in n-hexane) providing compound 10 as a white solid (96% yield). 1H NMR (500 MHz, CDCl3) δ=8.04 (s, 1H), 7.88 (d, J=8.5 Hz, 2H), 7.82 (d, J=8.4 Hz, 2H), 7.55 (s, 1H), 7.45–7.18 (m, 5H), 4.81 (d, J=8.4 Hz, 1H), 3.96–3.75 (m, 2H), 3.32–3.16 (m, 2H), 3.12–2.80 (m, 4H), 1.98–1.81 (m, 1H), 1.35 (s, 9H), 0.93 ppm (dd, J=11.2, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=156.3, 151.6, 150.0, 138.4, 137.9, 131.7, 129.6, 128.6, 128.2, 126.6, 124.9, 124.0, 79.9, 72.8, 58.4, 54.9, 53.5, 35.6, 28.4, 27.2, 20.2, 20.0 ppm.
tert-Butyl ((2S,3R)-4-((4-(2-acetyloxazol-5-yl)-N-isobutylphenyl)-sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (11)
Compound 10 (42 mg, 0.0775 mmol) was dissolved in dry THF (2 mL) and the resulting solution was cooled to −15°C. Then iPrMgCl (2.0M solution in THF, 116 μL, 0.232 mmol) was added dropwise and the mixture was stirred for 40 min at −15°C. N-Me-thoxy-N-methylacetamide (25 μL, 0.232 mmol) was added and the reaction mixture was allowed to slowly reach 23°C and stirred for 5 h. The reaction was quenched with a saturated solution of NH4Cl and extracted with EtOAc. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (20% to 50% EtOAc in n-hexane) providing compound 11 as an amorphous white solid (79% yield). 1H NMR (400 MHz, CDCl3) δ=7.65 (s, 1H), 7.40–7.14 (m, 5H), 4.62 (br, 1H), 3.97–3.66 (m, 3H), 3.16 (d, J=5.6 Hz, 2H), 3.06–2.83 (m, 4H), 2.72 (s, 3H), 1.96–1.79 (m, 1H), 1.35 (s, 9H), 0.88 (dd, J=11.1, 6.7 Hz, 6H), 8.01–7.79 ppm (m, 4H).
4-(N-((2R,3S)-3-(((((3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsul-famoyl)benzoic acid (3a)
Compound 7 (11.5 mg, 0.0221 mmol) was dissolved in dry CH2Cl2 (3 mL) and TFA (400 μL) was added. The resulting mixture was stirred at 23°C for 3 h. Solvents were evaporated under reduced pressure, then CH2Cl2 (5 mL) was added and evaporated twice to provide the corresponding Boc-deprotected compound, which was carried on without further purification. The compound was dissolved in a mixture of MeCN (4 mL) and water (0.4 mL), then N,N-DIPEA (39 μL, 0.221 mmol) and activated bis-THF carbonate 6 (7.8 mg, 0.0252 mmol) were sequentially added and the reaction mixture was stirred at 23°C for 3 days. Volatiles were evaporated under reduced pressure and the crude residue was purified by HPLC to provide pure 3a as a white solid (89% yield over two steps). 1H NMR (500 MHz, CD3OD) δ=8.22 (d, J=8.4 Hz, 2H), 7.97 (d, J=8.4 Hz, 2H), 7.35–7.24 (m, 4H), 7.24–7.13 (m, 1H), 5.62 (d, J=5.2 Hz, 1H), 3.97 (dd, J=9.7, 6.0 Hz, 1H), 3.85–3.74 (m, 4H), 3.74–3.63 (m, 1H), 3.55–3.47 (m, 1H), 3.27–3.17 (m, 2H), 3.08 (dd, J=15.0, 8.3 Hz, 1H), 2.98 (dd, J=13.7, 6.7 Hz, 1H), 2.94–2.86 (m, 1H), 2.55 (dd, J=13.8, 10.9 Hz, 1H), 2.14–2.00 (m, 1H), 1.59–1.46 (m, 1H), 1.41–1.28 (m, 1H), 0.94 ppm (dd, J=26.3, 6.6 Hz, 6H). 13C NMR (125 MHz, CD3OD) δ=167.2, 156.7, 143.7, 139.2, 130.5, 129.5, 128.3, 127.6, 126.3, 109.8, 73.6, 73.2, 71.1, 69.7, 57.4, 56.5, 52.5, 45.9, 36.1, 26.9, 26.0, 19.4 ppm.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-carba-moyl-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3b)
Compound 3a (3.4 mg, 0.0059 mmol) was dissolved in THF (4 mL) and pyridine (0.5 mL), Boc2O (2 mg, 0.0092 mmol) and (NH4)2CO3 (1 mg, 0.0104 mmol) were sequentially added. The reaction mixture was stirred at 23°C for 14 h and then concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (10% MeOH in CH2Cl2) providing 3b as a white solid (76% yield). 1H NMR (400 MHz, CDCl3) δ=7.95 (d, J=8.4 Hz, 2H), 7.84 (d, J=8.3 Hz, 2H), 7.36–7.12 (m, 5H), 6.34 (br, 1H), 5.82 (br, 1H), 5.64 (d, J=5.2 Hz, 1H), 5.03–4.87 (m, 2H), 3.98–3.79 (m, 4H), 3.76–3.58 (m, 2H), 3.51–3.41 (m, 1H), 3.21–3.02 (m, 2H), 3.00–2.84 (m, 3H), 2.78 (dd, J=13.9, 9.0 Hz, 1H), 1.85 (dt, J=13.6, 6.6 Hz, 1H), 1.74–1.56 (m, 2H), 1.55–1.42 (m, 1H), 0.90 ppm (dd, J=9.5, 6.6 Hz, 6H). LRMS-ESI (m/z): 576.3 [M+H]+, 598.2 [M+Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C28H37N3O8SNa 598.2199, found 598.2206.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-(methoxy(methyl)carbamoyl)phenyl)sulfonami-do)-1-phenylbutan-2-yl)carbamate (3c)
Compound 8 (6.5 mg, 0.0115 mmol) was dissolved in dry CH2Cl2 (3 mL) and TFA (200 μL) was added to the solution. The reaction mixture was stirred at 23°C for 2 h. Solvents were evaporated under reduced pressure, then CH2Cl2 (5 mL) was added and evaporated twice to provide the corresponding Boc-deprotected compound, which was carried on without further purification. The compound was dissolved in MeCN (4 mL), then N,N-DIPEA (20 μL, 0.115 mmol) and activated bis-THF carbonate 6 (3.4 mg, 0.0115 mmol) were sequentially added and the reaction mixture was stirred at 23°C for 5 days. Volatiles were evaporated under reduced pressure and the residue was purified by silica gel flash chromatography (50% EtOAc in n-hexane) to provide 3c as a white solid (73% yield over two steps). 1H NMR (400 MHz, CDCl3) δ=7.87–7.75 (m, 4H), 7.39–7.11 (m, 5H), 5.64 (d, J=5.2 Hz, 1H), 5.03 (dd, J=14.0, 6.2 Hz, 1H), 4.92 (d, J= 8.5 Hz, 1H), 4.00–3.80 (m, 4H), 3.75–3.62 (m, 2H), 3.59–3.47 (m, 4H), 3.39 (s, 3H), 3.21 (dd, J=15.0, 8.5 Hz, 1H), 3.14–2.96 (m, 3H), 2.95–2.71 (m, 3H), 1.93–1.78 (m, 1H), 1.50–1.38 (m, 2H), 0.90 ppm (dd, J=20.8, 6.6 Hz, 6H).
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-acetyl-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)-carbamate (3d)
Compound 8 (35 mg, 0.062 mmol) was dissolved in dry THF (2.5 mL) and the resulting solution was cooled to −78°C. MeMgBr (3M solution in Et2O, 105 μL, 0.310 mmol) was added dropwise and the reaction mixture was stirred at −78°C for 4 h and then quenched with a saturated solution of NH4Cl. The mixture was extracted with EtOAc and the organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (50% EtOAc in n-hexanes) providing tert-butyl ((2S,3R)-4-((4-acetyl-N-iso-butylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (98% yield). 1H NMR (400 MHz, CDCl3) δ=8.07 (d, J=8.3 Hz, 2H), 7.88 (d, J=8.3 Hz, 2H), 7.37–7.14 (m, 5H), 4.70 (d, J=7.5 Hz, 1H), 3.92 (br, 1H), 3.86–3.69 (m, 2H), 3.23–3.12 (m, 2H), 3.07–2.82 (m, 4H), 2.66 (s, 3H), 1.97–1.72 (m, 2H), 1.36 (s, 9H), 0.89 ppm (dd, J= 9.9, 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ=196.89, 156.37, 142.78, 140.14, 137.86, 129.65, 129.10, 128.69, 127.77, 126.68, 80.05, 72.71, 58.30, 55.08, 53.35, 35.68, 28.39, 27.21, 27.01, 20.20, 20.01 ppm. The above methyl ketone intermediate was then Boc-deprotected and submitted to coupling with the activated bis-THF derivative 6 as described for compound 3c, providing pure compound 3d as an amorphous white solid (78% yield over two steps). 1H NMR (400 MHz, CDCl3) δ=8.08 (d, J=8.4 Hz, 2H), 7.87 (d, J=8.4 Hz, 2H), 7.35–7.15 (m, 5H), 5.65 (d, J=5.2 Hz, 1H), 5.02 (dd, J=14.0, 6.2 Hz, 1H), 4.92 (d, J=8.3 Hz, 1H), 4.01–3.81 (m, 4H), 3.76–3.62 (m, 2H), 3.55 (br, 1H), 3.26–3.14 (m, 1H), 3.13–2.96 (m, 3H), 2.96–2.74 (m, 3H), 2.66 (s, 3H), 1.92–1.75 (m, 1H), 1.74–1.40 (m, 2H), 0.90 ppm (dd, J=19.9, 6.6 Hz, 6H). LRMS-ESI (m/z): 597.1 [M+Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C29H38N2O8SNa 597.2247, found 597.2243.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((R)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phe-nylbutan-2-yl)carbamate (3e)
Compound 3d (3 mg, 0.0052 mmol) was dissolved in dry CH2Cl2 (0.5 mL) and Et3N (280 μL) and HCO2H (140 μL) were added at 0°C. After stirring for 30 min, Noyori asymmetric transfer hydrogenation catalyst RuCl[(R,R)-TsDPEN](mesity-lene) (0.5 mol%) was added and the reaction mixture was stirred at 23°C for 7 h. The reaction mixture was then quenched saturated NaHCO3 and extracted with CH2Cl2. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (20% to 60% EtOAc in n-hexane) providing 3e as a white solid (94% yield). 1H NMR (500 MHz, CDCl3) δ=7.81 (d, J=8.3 Hz, 2H), 7.60 (d, J= 8.2 Hz, 2H), 7.41–7.21 (m, 5H), 5.70 (d, J=5.2 Hz, 1H), 5.11–4.93 (m, 3H), 4.04–3.97 (m, 1H), 3.96–3.84 (m, 3H), 3.80–3.66 (m, 2H), 3.27–3.11 (m, 2H), 3.11–3.00 (m, 2H), 3.00–2.79 (m, 3H), 1.96–1.82 (m, 1H), 1.77–1.65 (m, 1H), 1.58 (d, J=6.5 Hz, 3H), 1.55–1.46 (m, 1H), 0.98 ppm (dd, J=23.7, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ= 155.9, 151.7, 138.0, 137.2, 129.8, 129.0, 128.1, 127.1, 126.6, 109.7, 73.9, 73.3, 71.3, 70.0, 60.9, 59.4, 55.5, 54.3, 45.8, 36.1, 27.8, 26.3, 25.9, 20.6, 20.3 ppm. LRMS-ESI (m/z): 577.2 [M+H]+, 599.3 [M+ Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C29H40N2O8SNa 599.2404, found 599.2408. HPLC analysis: Chiralpak IA-3 column, 10% isopropanol in n-hexane, flow rate=1 mLmin−1, t=24°C, l=254 nm, tR=32.1 min, d.r. 98:2.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((S)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phe-nylbutan-2-yl)carbamate (3f)
Compound 3f was obtained from 3d following the same procedure described for compound 3e employing RuCl(p-cymene)[(S,S)-Ts-DPEN] as the catalyst. Compound 3f was obtained in 98% yield. 1H NMR (500 MHz, CDCl3) δ=7.81 (d, J=8.2 Hz, 2H), 7.60 (d, J=8.1 Hz, 2H), 7.41–7.18 (m, 5H), 5.70 (d, J=5.2 Hz, 1H), 5.10–4.90 (m, 3H), 4.04–3.97 (m, 1H), 3.97–3.87 (m, 3H), 3.79–3.64 (m, 3H), 3.23 (dd, J=15.1, 8.5 Hz, 1H), 3.14 (dd, J=14.2, 3.8 Hz, 1H), 3.11–3.02 (m, 2H), 3.00–2.93 (m, 1H), 2.92–2.80 (m, 2H), 2.20 (br, 1H), 1.96–1.85 (m, 1H), 1.76–1.64 (m, 1H), 1.58 (d, J=6.5 Hz, 3H), 1.55–1.46 (m, 1H), 0.98 ppm (dd, J=24.1,6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=155.9, 151.6, 138.0, 137.3, 129.8, 129.0, 128.0, 127.1, 126.6, 109.7, 73.9, 73.3, 71.3, 70.0, 59.4, 55.6, 54.3, 45.8, 36.1, 28.3, 27.8, 26.3, 25.9, 20.6, 20.3 ppm. LRMS-ESI (m/z): 577.2 [M+H]+, 599.2 [M+Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C29H40N2O8SNa 599.2404, found 599.2408. HPLC analysis: Chiralpak IA-3 column, 10% isopropanol in n-hexane, flow rate=1 mLmin−1, t=24°C, l=254 nm, tR=34.6 min, d.r. 98:2.
Methyl 2-(4-(N-((2R,3S)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)oxazole-4-carboxylate (3g)
Compound 3g was obtained from compound 9 and activated bis-THF derivative 6 following the same procedure described for compound 3c (61% yield over two steps). 1H NMR (500 MHz, CDCl3) δ=8.42 (s, 1H), 8.33 (d, J=8.5 Hz, 2H), 7.95 (d, J=8.5 Hz, 2H), 7.43–7.17 (m, 5H), 5.71 (d, J=5.2 Hz, 1H), 5.09 (dd, J=14.2, 6.3 Hz, 1H), 5.01 (d, J=8.4 Hz, 1H), 4.13–3.98 (m, 4H), 3.98–3.84 (m, 3H), 3.85–3.69 (m, 2H), 3.64 (s, 1H), 3.29 (dd, J=15.1, 8.3 Hz, 1H), 3.22–3.05 (m, 3H), 3.03–2.93 (m, 2H), 2.87 (dd, J=14.0, 9.1 Hz, 1H), 1.96–1.81 (m, 1H), 1.78–1.61 (m, 1H), 1.59–1.45 (m, 1H), 0.96 ppm (dd, J=25.2, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=161.6, 160.9, 155.7, 144.8, 140.8, 137.7, 135.1, 130.4, 129.5, 128.8, 128.1, 127.7, 126.9, 109.5, 73.7, 72.9, 71.0, 69.8, 58.7, 55.4, 53.7, 52.7, 45.6, 35.8, 27.4, 26.0, 20.3, 20.0 ppm. LRMS-ESI (m/z): 658.2 [M+H]+, 680.3 [M+Na]+.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-(4-(hydroxymethyl)oxazol-2-yl)-N-isobutylphenyl)sulfona-mido)-1-phenylbutan-2-yl)carbamate (3h)
Compound 9 (8.5 mg, 0.014 mmol) was dissolved in absolute EtOH (3 mL) and the resulting solution was cooled to 0°C. NaBH4 (1.6 mg, 0.042 mmol) was added and the reaction mixture was stirred at 23°C for 24 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated. The crude was purified by silica gel flash chromatography (60% EtOAc in n-hexanes) providing the corresponding alcohol derivative (65% yield). This intermediate underwent Boc-deprotection and coupling with the bis-THF derivative 6 as described for compound 3c (60% yield over two steps). 1H NMR (500 MHz, CDCl3) δ=8.17 (d, J=8.5 Hz, 2H), 7.86 (d, J=8.6 Hz, 2H), 7.73 (s, 1H), 7.36–7.11 (m, 5H), 5.64 (d, J=5.2 Hz, 1H), 5.01 (dd, J=14.2, 6.3 Hz, 1H), 4.94 (d, J=8.5 Hz, 1H), 4.70 (s, 2H), 3.96 (dd, J=9.6, 6.3 Hz, 1H), 3.94–3.77 (m, 3H), 3.78–3.61 (m, 3H), 3.56 (s, 1H), 3.20 (dd, J=15.2, 8.4 Hz, 1H), 3.14–2.98 (m, 3H), 2.96–2.84 (m, 2H), 2.84–2.74 (m, 1H), 1.91–1.79 (m, 1H), 1.71–1.53 (m, 1H), 1.52–1.41 (m, 1H), 0.98–0.84 ppm (m, 6H). 13C NMR (125 MHz, CDCl3) δ=160.5, 155.7, 142.4, 139.9, 137.7, 136.4, 131.4, 129.5, 128.8, 128.1, 127.2, 126.9, 109.5, 73.7, 72.9, 70.9, 69.8, 58.8, 57.3, 55.4, 53.7, 45.5, 35.8, 27.4, 26.0, 20.3, 20.1 ppm. LRMS-ESI (m/z): 630.3 [M+H]+, 652.2 [M+Na]+.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-(oxazol-5-yl)phenyl)sulfonamido)-1-phenylbu-tan-2-yl)carbamate (3i)
Compound 3i was obtained from compound 10 and activated bis-THF derivative 6 following the same procedure described for compound 3c (47% over two steps). 1H NMR (500 MHz, CDCl3) δ=8.00 (s, 1H), 7.84 (d, J=8.7 Hz, 2H), 7.80 (d, J=8.6 Hz, 2H), 7.52 (s, 1H), 7.33–7.26 (m, 2H), 7.25–7.17 (m, 3H), 5.64 (d, J=5.2 Hz, 1H), 5.09–4.95 (m, 2H), 4.01–3.79 (m, 4H), 3.79–3.54 (m, 3H), 3.20 (dd, J=15.1, 8.4 Hz, 1H), 3.14–2.96 (m, 3H), 2.96–2.87 (m, 2H), 2.86–2.70 (m, 1H), 1.93–1.78 (m, 1H), 1.74–1.56 (m, 1H), 1.50–1.39 (m, 1H), 0.90 ppm (dd, J=22.2, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=155.4, 151.4, 149.6, 137.7, 137.4, 131.7, 129.2, 128.5, 128.0, 126.5, 124.7, 123.9, 109.2, 73.4, 72.7, 70.7, 69.5, 58.5, 55.1, 53.5, 45.3, 35.5, 27.1, 25.7, 20.0, 19.7 ppm. LRMS-ESI (m/z): 600.3 [M+H]+, 622.2 [M+Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C30H37N3O8SNa 622.2199, found 622.2205.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-(2-ace-tyloxazol-5-yl)-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phe-nylbutan-2-yl)carbamate (3j)
Compound 3j was obtained from compound 11 and activated bis-THF derivative 6 following the same procedure described for compound 3c (80% over two steps). 1H NMR (500 MHz, CDCl3) δ=7.92 (d, J=8.4 Hz, 2H), 7.86 (d, J=8.5 Hz, 2H), 7.66 (s, 1H), 7.35–7.26 (m, 3H), 7.26–7.17 (m, 2H), 5.64 (d, J=5.2 Hz, 1H), 5.01 (dd, J=14.0, 6.1 Hz, 1H), 5.00–4.92 (m, 1H), 4.00–3.80 (m, 4H), 3.75–3.62 (m, 2H), 3.59 (s, 1H), 3.21 (dd, J=15.2, 8.4 Hz, 1H), 3.15–2.97 (m, 3H), 2.95–2.85 (m, 2H), 2.80 (dd, J=13.8, 9.2 Hz, 1H), 2.72 (s, 3H), 1.85 (dt, J=13.3, 6.8 Hz, 1H), 1.73–1.56 (m, 1H), 1.51–1.39 (m, 1H), 0.90 ppm (dd, J=22.9, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=185.6, 158.0, 155.8, 152.4, 139.5, 137.6, 130.9, 129.5, 128.8, 128.4, 126.9, 126.1, 126.0, 109.5, 73.8, 72.9, 71.0, 69.8, 58.7, 55.4, 53.7, 45.6, 35.8, 27.4, 26.9, 26.0, 20.3, 20.0 ppm. LRMS-ESI (m/z): 664.2 [M+Na]+; HRMS-ESI (m/z) [M+Na]+ calculated for C32H39N3O9SNa 664.2305, found 664.2314.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-(2-((R)-1-hydroxyethyl)oxazol-5-yl)-N-isobutylphenyl)sulfo-namido)-1-phenylbutan-2-yl)carbamate (3k)
Compound 3k was obtained from compound 3j following the same procedure described for compound 3e (97% yield). 1H NMR (400 MHz, CDCl3) δ=7.93–7.63 (m, 4H), 7.43 (s, 1H), 7.35–7.04 (m, 5H), 5.64 (d, J= 5.2 Hz, 1H), 5.13–4.87 (m, 3H), 4.02–3.75 (m, 4H), 3.76–3.35 (m, 4H), 3.29–2.95 (m, 4H), 2.96–2.58 (m, 4H), 1.93–1.81 (m, 1H), 1.69 (d, J=6.7 Hz, 3H), 0.90 ppm (dd, J=15.4, 6.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=166.7, 157.4, 155.4, 137.7, 135.9, 131.8, 129.2, 128.8, 128.5, 128.0, 127.9, 126.5, 124.5, 124.0, 109.2, 78.5, 73.4, 70.9, 69.8, 69.5, 63.7, 58.5, 55.8, 53.4, 46.4, 35.8, 29.6, 27.1, 25.7, 21.3, 20.0, 19.9 ppm. LRMS-ESI (m/z): 644.4 [M+H]+, 666.4 [M+Na]+. HPLC analysis: Chiralpak IC column, 75% isopropanol in n-hexane, flow rate=0.8 mLmin−1, t=24°C, l=254 nm, tR=13.8 min, d.r. >99:1.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-(2-((S)-1-hydroxyethyl)oxazol-5-yl)-N-isobutylphenyl)sulfon-amido)-1-phenylbutan-2-yl)carbamate (3l)
Compound 3l was obtained from compound 3j following the same procedure described for compound 3f (95% yield). 1H NMR (400 MHz, CDCl3) δ=7.90–7.71 (m, 4H), 7.44 (s, 1H), 7.31–7.10 (m, 5H), 5.64 (d, J=5.2 Hz, 1H), 5.07–4.98 (m, 2H), 4.88–4.83 (m, 1H), 3.98–3.80 (m, 4H), 3.71–3.52 (m, 4H), 3.10–2.88 (m, 4H), 2.85–2.72 (m, 4H), 1.87– 1.79 (m, 1H), 1.68 (d, J=6.7 Hz, 3H), 0.91 ppm (dd, J=15.3, 6.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ=166.7, 157.6, 155.4, 137.4, 135.8, 131.7, 129.2, 128.8, 128.5, 128.0, 127.4, 126.5, 124.5, 109.3, 78.5, 73.0, 70.8, 69.8, 69.5, 63.7, 58.4, 53.4, 46.5, 35.8, 29.5, 27.1, 24.8, 21.2, 20.0, 19.8 ppm. LRMS-ESI (m/z): 644.4 [M+H]+, 666.3 [M+ Na]+. HPLC analysis: Chiralpak IC column, 75% isopropanol in n-hexane, flow rate=0.8 mLmin−1, t=24°C, l=254 nm, tR= 11.2 min, d.r. >99:1.
Determination of X-ray structure of HIV-1 protease and inhibitor 3g complex
The optimized HIV-1 protease was expressed and purified as described.[35] The protease–inhibitor complex was crystal-lized by the hanging drop vapor diffusion method with well solutions of 1.3M NaCl, 0.1M sodium acetate buffer (pH 5.5). Diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, IL, USA) with X-ray wavelength of 0.8 Å, and processed by HKL-2000[36] with an Rmerge of 6.2%. Using the isomorphous structure[37] the crystal structure was solved by PHASER[38] in the CCP4i Suite[39–41] and refined in SHELX-97[42–44] with 1.3 Å resolution data. COOT[43,45] was used for manual modification of the atomic structure. PRODRG-2[46] was used to construct the inhibitor and the restraints for refinement. Alternative conformations were modeled, anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules, and hydrogen atoms were added in the final round of refinement. The final refined solvent structure comprised two Na+ ions, four Cl− ions, and 234 water molecules. The crystallographic statistics are listed in a Table in the supporting information. The coordinates and structure factors of the PR with GRL-05311A (3g) structure have been deposited in the RCSB Protein Data Bank[47] with PDB ID: 6B4N.
Supplementary Material
Acknowledgments
This research was supported by the US National Institutes of Health (Grant GM53386 to A.K.G. and Grant GM62920 to I.T.W.). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu-Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. The authors thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/cmdc.201700614.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.UNAIDS/WHO. AIDS Epidemic Update. 2006 Dec; http://data.u-naids.org/pub/report/2006/2006_gr_en.pdf.
- 2.Greene WC. Eur J Immunol. 2007;37:S94–S102. doi: 10.1002/eji.200737441. [DOI] [PubMed] [Google Scholar]
- 3.Sepkowitz KA. N Engl J Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 4.Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Nap-ravnik S, Cole SR, Van Rie A, Behets F. PLoS Med. 2011:8e1001044. doi: 10.1371/journal.pmed.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen MS, Chen YQ, McCauley M. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieffenbach CW, Fauci AS. Ann Intern Med. 2011;154:766–771. doi: 10.7326/0003-4819-154-11-201106070-00345. [DOI] [PubMed] [Google Scholar]
- 7.Maartens G, Celum C, Lewin S. Lancet. 2014;384:258–271. doi: 10.1016/S0140-6736(14)60164-1. [DOI] [PubMed] [Google Scholar]
- 8.Cihlar T, Fordyce M. Curr Opin Virol. 2016;18:50–56. doi: 10.1016/j.coviro.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Patel K, Hernμn MA, Williams PL, Seeger JD, McIntosh K, Van Dyke RB, Seage GR., III Clin Infect Dis. 2008;46:507–515. doi: 10.1086/526524. [DOI] [PubMed] [Google Scholar]
- 10.Gupta R, Hill A, Sawyer AW, Pillay D. Clin Infect Dis. 2008;47:712–722. doi: 10.1086/590943. [DOI] [PubMed] [Google Scholar]
- 11.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Osswald HL, Prato G. J Med Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.“Darunavir, a New PI with Dual Mechanism: From a Novel Drug Design Concept to New Hope against Drug-Resistant HIV”; Ghosh AK, Chapsal B, Mitsuya H. Aspartic Acid Proteases as Therapeutic Targets. Wiley-VCH; Weinheim: 2010. pp. 205–243. [Google Scholar]
- 14.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 15.“Design of the Anti-HIV Protease Inhibitor Darunavir”; Ghosh AK, Chapsal BD. In: Introduction to Biological and Small-Molecule Drug Research and Development. Ganellin CR, Roberts SM, Jefferis R, editors. Elsevier; Amsterdam: 2013. pp. 355–384. [Google Scholar]
- 16.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. J Med Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 19.On June 23, 2006, FDA approves Darunavir for HIV treatment-experienced patients, and on December 13, 2008 for treatment-naïve patients and pediatrics. Available at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm, (accessed September 2017).
- 20.Tie Y, Boross PI, Wang YF, Gaddis L, Hussian AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:1812–1838. [Google Scholar]
- 22.Yedidi RS, Maeda K, Fyvie WS, Steffey M, Davis DA, Palmer I, Aoki M, Kaufman JD, Stahl SJ, Garimella H, Das D, Wingfield PT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2013;57:4920–4927. doi: 10.1128/AAC.00868-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. J Med Chem. 2006;49:1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yedidi RS, Garimella H, Aoki M, Aoki-Ogata H, Desai DV, Chang SB, Davis DA, Fyvie WS, Kaufman JD, Smith DW, Das D, Wingfield PT, Maeda K, Ghosh AK, Mitsuya H. Antimicrob Agents Chemo-ther. 2014;58:3679–3688. doi: 10.1128/AAC.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kova-levsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 27.Couty F, David O, Larmanjat B, Marrot J. J Org Chem. 2007;72:1058–1061. doi: 10.1021/jo062221e. [DOI] [PubMed] [Google Scholar]
- 28.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J Am Chem Soc. 1996;118:2521–2522. [Google Scholar]
- 29.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 30.Van Leusen AM, Hoogenboom BE, Siderius H. Tetrahedron Lett. 1972:2369–2372. [Google Scholar]
- 31.Pippel DJ, Mapes CM, Mani NS. J Org Chem. 2007;72:5828–5831. doi: 10.1021/jo070646a. [DOI] [PubMed] [Google Scholar]
- 32.Toth MV, Marshall GR. Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 33.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. J Virol. 2010;84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh Y, Das D, Leschenko S, Nakata H, Ogata-Aoki H, Amano M, Nakayama M, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2009;53:997–1006. doi: 10.1128/AAC.00689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins Struct Funct Bioinf. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. In: Processing of X-ray Diffraction Data Collected in Oscillation Mode Methods in Enzymology, 276: Macromolecular Crystallography, Part A. Carter CW Jr, Sweet RM, editors. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.Shen CH, Wang YF, Kovalevsky AY, Harrison RW, Weber IT. FEBS J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storo-ni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A. Acta Crys-tallogr Sect D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project Number 4. Acta Crystallogr Sect D. 1994;50:760–763. [Google Scholar]
- 41.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr Sect D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 42.Sheldrick GM. Acta Crystallogr Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr Sect D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Schüttelkopf AW, van Aalten DMF. Acta Crystallogr Sect D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 47.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.