

Abstract
Heterogeneous expression of neurotransmitter deficits results from onset and progression of Parkinson’s disease. Intervals, characterized by reappearance of motor and associated certain nonmotor symptoms, determine the end of good tolerability and efficacy of oral levodopa therapy. These “OFF” states result from levodopa pharmacokinetics and disease progression-related deterioration of the central buffering capacity for fluctuations of dopamine levels. This review discusses safinamide as an add-on therapeutic agent in orally levodopa-treated patients with “OFF” phenomena. Safinamide provided beneficial effects on “OFF” symptoms in pivotal trials with doses of 50 or 100 mg once daily. Safinamide reversibly inhibits mono-amine oxidase B and declines abnormal glutamate release by modulation of potassium- and sodium ion channels. An ideal candidate for combination with safinamide is opicapone. This inhibitor of peripheral catechol-O-methyltransferase supports continuous brain delivery of levodopa and, thus, the continuous dopaminergic stimulation concept. Both compounds with their once-daily application and good tolerability may complement each other by reduction of necessary oral levodopa intakes and “OFF” times. Thus, a promising, future option will be combination of safinamide and opicapone in one formulation. It will reduce adherence issues and may complement levodopa treatment. It will probably cause less nausea and edema than a dopamine agonist/levodopa regimen.
Keywords: safinamide, MAO-B inhibition, abnormal glutamate release inhibition, Parkinson’s disease, dopamine substitution, glutamate
Introduction
Incidence and prevalence
Parkinson’s disease (PD) is one of the most frequent chronic neurodegenerative diseases worldwide. Its incidence rate ranges from 8 to 18 per 100,000 human subjects each year. One estimates that prevalence of PD will increase in the future because of a further rise of life expectancy.1 Onset of PD is less frequent under the age of 50 and then considerably goes up after the age of 60.2,3
Symptoms
The term “idiopathic PD” describes a superordinate concept for a disease entity. PD onset presents as an individual different and pronounced combination of motor and nonmotor symptoms. The initially smouldering onset of at least one of the cardinal motor symptoms of rigidity, akinesia, and resting tremor mostly leads to the diagnosis in clinical practice. Response to levodopa or other dopamine substituting compounds is a further essential criterion.4 Well accepted is the differentiation of PD in akinetic-, rigid-, tremor-dominant-, and the equivalent forms.5 The tremor-dominant type mostly shows a slow progression;5 however, therapy, personality traits, and epigenetic and environmental components provide a substantial impact on the course of PD.6 Balance problems are rare in the beginning. They predominantly become manifest later in the further course of the disease. Functional imaging techniques for visualizing of the dopaminergic deficit are mostly only employed to confirm the clinical diagnosis or to monitor progression of PD. In clinical practice, mostly cheaper single-photon emission computed tomography is employed instead of more expensive positron emission tomography. The value of both techniques is nearly identical for the everyday maintenance of PD patients.7,8
Pitfalls of initial diagnosis and identification of “OFF” episodes in clinical trials
Various nonmotor symptoms often precede the initial temporary onset of disturbed motor behavior. Research initiatives now focus on a better awareness of these early nonmotor features to enable an earlier diagnosis of PD. They aim to describe a so-called “prodromal” or “premotor” interval of PD.9,10 Appearance and severity of motor symptoms is closely associated with manifestation of certain nonmotor features, for instance rigidity with pain syndromes. This is the case in early and in later stages of PD, particularly when patients fluctuate.11 Clinicians still predominantly diagnose and quantify the so-called “OFF” time in relation to motor behavior. Generally, “OFF” reflects a temporary dopamine deficit of the nigrostriatal system. Nonmotor features related to “OFF” episodes may at least result from an insufficient dopaminergic stimulation of mesolimbic structures. Certain nonmotor features, such as apathy and depression may sometimes lead the way to the slow or sudden manifestation of “wearing-OFF”, which is one form of the so-called “motor complications”. Generally, time without motor impairment is called “ON Time”. “OFF Time” describes the temporary reappearance of motor symptoms. Dopaminergic overstimulation causes onset of involuntary movements mostly of the upper and lower limbs. These so-called “dyskinesia” are more frequent during “ON Time”. They may be regarded as “troublesome” and limit quality of life. They may also be “nontroublesome”, when they show a slight severity. All these various types of motor complications are predominantly related to oral levodopa intake and occur after a variable period of time, but almost invariably in all patients. Patient diaries are mostly employed for the measurement of “OFF” intervals. This documentation is simple. It focuses on motor impairment. However, it disregards periods with “OFF”-related nonmotor features. These diaries have been used since the introduction of inhibitors of catechol-O-methyltransferase (COMT) as an adjunct to levodopa therapy in clinical trial protocols.12 They are well accepted as valuable assessment instruments by drug-approving authorities. As a result, drug development in PD aims to demonstrate a beneficial effect with the help of these diaries in fluctuating PD patients.
Drug therapy
Generally, dopamine substitution improves motor behavior in PD patients. Heterogeneity of motor and nonmotor symptoms and individual difference in drug tolerability inquires a patient-tailored drug combination.13 Mostly a personalized combination of PD drugs is performed, that is, combination of dopamine agonists with levodopa formulations. Dopamine-substituting agents often cause lowering of blood pressure and/ or nausea. It is advisable to add agents, such as midodrine and domperidon, respectively, if available. This treatment regimen may be supplemented by antidepressant or neuroleptic compounds, if necessary. Frequently, coexisting drug treatment of concomitant disorders, that is, high blood pressure, needs to be modified. Thus, in clinical practice, it is essential to perform a careful and slow drug titration with continuous and concomitant consideration of individually varying drug tolerability and safety adapted to the needs of the patients and their caregivers. These are the most important preconditions for a successful, tailored drug therapy of PD patients in the long term.13 Start of therapy is nowadays soon after diagnosis is made. In former times, initiation was delayed until quality of life was severely affected. Mostly, concomitant intensive physiotherapy, speech therapy, and so on are necessary to ameliorate PD patients in a considerable, satisfactory manner.13
The value of guidelines in PD
The heterogeneity of PD symptoms and their progression is well known. Therefore, a too strict adoption to standardized treatment approaches is sometimes not advantageous in clinical practice.14 Rough treatment guidelines exist for the balance of the dopaminergic deficit in PD. There are two essential, general approaches. One supports the delay of levodopa use as long as possible. This view considers findings on putative possible toxic or progression-accelerating effects of levodopa. An alternative approach suggests an earlier titration of levodopa. This drug is highly efficacious, well tolerated, but closely associated with motor complications. Subsequently, earlier interventions with drug-sparing neurosurgical procedures, that is, deep brain stimulation of the nucleus subthalamicus or the globus pallidus, may become necessary. An alternative is pump therapies, which enable infusion of levodopa or an dopamine agonist similar to an insulin pump in diabetes.15 All these therapies do not modify the slow or sometimes even relapse-like progression of PD.
The utmost unmet need of therapy in PD
Cure from PD is still not available or on the horizon. There have been many attempts, such as transplantation trials with dopamine-synthesizing cells, gene therapy, infusion of growth factors or other disease course-modifying therapies. All of them were not successful, yet. Tested approaches worked in animal models of PD. They failed in the clinic. Heterogeneous forms of PD may hypothetically be one essential reason.16,17 Thus, to date, research and development of PD drugs still mainly aim to improve motor impairment and amelioration of motor complications, particularly “OFF” episodes. In these two indications, drug-approving authorities accept the correspondingly employed assessment and rating tools. Therefore, the development programme of safinamide primarily also focused on amelioration of motor complications.
Objectives
This narrative review aims to evaluate the efficacy of safinamide in the treatment of levodopa-treated PD patients based on the literature selected by the author.
Drug therapy in PD
Overview
Anticholinergics
They represent the oldest used compounds for the drug treatment of PD. These agents lower the intensity of tremor. Their chronic application may deteriorate short-time memory function. Therefore, they are nearly abandoned nowadays.18
Levodopa
The introduction of levodopa in the 1960s was the start of new era of drug therapy in PD.19 This amino acid may trespass the blood–brain barrier in contrast to its metabolic derivative dopamine. Following the conversion of levodopa to dopamine in presynaptic brain neurons and the release of dopamine to the synaptic cleft, this biogenic amine stimulates postsynaptic dopaminergic receptors.20 Thus, it counteracts the nigrostriatal dopamine deficiency. The well-accepted concept of “continuous dopaminergic stimulation” implicates that dopamine or dopamine-substituting compounds should activate postsynaptic nigrostriatal dopamine receptors as continuously as possible. In other words, only continuous but not pulsatile dopamine substitution induces normal movement behavior in PD patients. Out of the nowadays available dopamine-substituting compounds, levodopa is the most potent and efficacious agent for the amelioration of motor symptoms. Levodopa particularly improves akinesia and rigidity, and to a lesser extent tremor. However, there is a serious drawback of levodopa. This amino acid is rapidly metabolized, which limits it efficacy. Therefore, repeat oral levodopa administration supports a nonphysiologic, so-called “pulsatile” stimulation of the nigrostriatal system.20 Additionally, complex mechanisms of gastrointestinal absorption and transport further complicate levodopa therapy. However, therapeutic strategies with pharmacologic inhibition of the main levodopa-metabolizing enzymes, dopa decarboxylase (DDC) and COMT, eased oral levodopa application.21–23 Nevertheless, still today, all oral levodopa formulations suffer from a relatively short plasma half-life. Therefore, PD patients have to take currently available oral levodopa formulations several times per day. This oral intake pattern is a serious problem because of the well-known adherence problems of PD patients in general. Therefore, nowadays the main focus of research on novel levodopa formulations aims to deliver levodopa as continuously as possible to the brain over the blood–brain barrier. Currently, novel retarded-release levodopa formulations and novel subcutaneous infusion systems are under way in clinical trials.18 There is hope that they will reduce plasma fluctuations of levodopa. They are believed to be translated into ups and downs of dopamine concentrations in the synaptic cleft between the pre- and postsynaptic neurons. As a result, a temporary, too high stimulation with dopamine on the one hand and transient dopamine deficiency on the other hand occur. In the long run, this pulsatile pattern overexerts the existing buffering capacity within the dopaminergic system, which slowly diminishes in the further disease course. Finally, these dopamine fluctuations cause onset of quality-of-life-limiting, above-described fluctuations of movement behavior.
Enzyme blockers
There are further pharmacologic principles for drug treatment in PD. They focus on central inhibition of dopamine turnover by monoamine oxidase (MAO) and COMT in glial cells. They are well tolerated but only have limited efficacy, because they depend on endogenous and exogenous dopamine supply. Both approaches contribute to more continuous and stable dopamine levels in the synaptic cleft. Correspondingly, clinical trials showed an improvement of motor complications because of a longer stimulation of postsynaptic dopamine receptors.18
Dopamine agonists
An alternative to these aforementioned modes of action is the direct stimulation of postsynaptic, nigrostriatal receptors with dopamine agonists. Both affinity to the dopamine receptors and metabolic half-life, determine their efficacy. There are more older ergoline- and more newer non-ergot-dopamine agonists.18 All of them suffer from a limited tolerability and safety. Generally, titration and long-term dosing of dopamine agonists is complex. They may cause edema and nausea, and probably support impulsive control disorders via tonic long-term stimulation of ventral striatal structures in predisposed individuals.24 There is considerable evidence from clinical studies that dopamine agonists may delay onset and ameliorate all kinds of motor complications, probably due to sparing of oral levodopa.76
Receptor antagonism
Antagonizing of adenosine-2A receptors may be a further promising therapeutic principle. It modulates more downstream postsynaptic dopamine receptor functions. Tested compounds, such as tozedenant, enhance movement behavior in PD. Currently, istradefylline is the only approved and available agent, but its use is limited to Japan.25
Another approach is reduction of glutamatergic neurotransmission. The concept of antagonizing of N-methyl-D-aspartate (NMDA) receptor function is the mode of action of amantadine. This relatively old drug was initially employed as an antiviral compound. Clinicians discovered a moderate effect on motor symptoms. Antidyskinetic effects were then described in PD patients.26,27 These clinical findings initiated the research on the role of glutamate in PD.28,29 An alternative approach to NMDA antagonism is the decline of abnormal glutamate release. This pharmacologic principle represents one out of many modes of action of safinamide.
Safinamide
Chemistry
Safinamide, [(S)-(+)-2-[4-(3-fluorobenzyl) oxybenzyl) aminopropanamide methanesulfonate], is a small molecule, which is soluble in water but chemically and metabolically stable (Figure 1).
Figure 1.
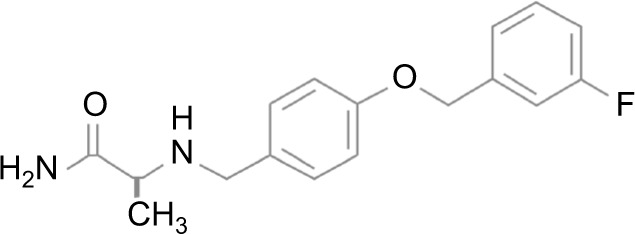
Chemical structure of safinamide.
Pharmacodynamics
MAO-B inhibition
Interactions of safinamide with dopamine receptor subtypes and dopamine transporter systems were investigated in human recombinant receptors and a transfected cell line (CHO-K1 cells). Safinamide inhibited the dopamine transporter sites (50% at 10 μM). Safinamide did not show any specific affinities for the different isoforms of the D1, D2, D3, D4, and D5 receptor subtypes. Safinamide blocked the dopamine uptake (IC50=8.44 μM). In rat brain synaptosomes, safinamide diminished uptake of [3H]dopamine and [3H]serotonin at low concentrations (dopamine: IC50=12 μM; serotonin: IC50=21 μM). Safinamide directly influenced the function of the dopamine transporter (IC50=9 μM) and the serotonin transporter (IC50=6 μM). Safinamide reduced amphetamine-induced [3H] dopamine release (IC50=66 μM) and p-chloroamphetamine-induced [3H]serotonin release (IC50=5.9 μM). Safinamide and its major metabolites did not influence activity of the enzymes L-amino-acid decarboxylase and COMT. In rats, safinamide ~5000 times more selectively inhibited MAO-B (brain: IC50=0.098) compared to MAO-A (brain: IC50=485). In humans, safinamide 1000 times more selectively blocked MAO-B (brain: IC50=0.079) in comparison with MAO-A (brain: IC50=80; Table 1). No significant difference appeared between the IC50 obtained with and without preincubation between enzyme and safinamide in human platelet-rich plasma. In rat brain mitochondria, recovery of MAO-B activity was complete following the second washing of the enzyme– inhibitor complex. This is essential for the claim that MAO-B inhibition by safinamide is reversible and not time dependent. High-resolution X-ray analysis revealed that safinamide is noncovalently bound to the MAO-B protein. Safinamide blocked MAO-B in rat brain and in liver in a dose-dependent fashion according to ex vivo experiments, whereas MAO-A enzyme activity was not affected. The effective dose, for 50% of people receiving the drug (ED50) value for MAO-B in rat brain tissue was 1.1 mg/kg 1 hour after dosing. After administration of safinamide in a dose of 5 mg/kg, MAO-B inhibition in the brain was 79% 1 hour later and 13% 24 hours later. This outcome supports that safinamide acts like a reversible, short-acting MAO-B inhibitor.
Table 1.
Summary of pharmacologic properties
| Chemical structure | (S)-(+)-2-[4-(3-fluorobenzyl) oxybenzyl) aminopropanamide methanesulfonate (see Figure 1), water stable, small molecule |
|---|---|
| Drug name | Xadago®, safinamide (generic) |
| Phase | IV, approved in 2015 in the EU |
| Indication | Fluctuating levodopa-treated patients with Parkinson’s disease |
| Mechanism of action | Reversible inhibition of MAO-B, inhibition of abnormal glutamate release, modulation of NA+- and Ca++ channels |
| Administration route | Oral |
| Dosing | 50 or 100 mg once in 1 day |
| Pivotal trials in levodopa-treated PD patients | SETTLE Study:42; 016 Study,40 018 study extension41 |
| Absorption | Gastrointestinal |
| Inhibition of dopamine uptake (human; CHO-K1 cells) | IC50 =8.44 μM |
| Blocking of dopamine transporter sites (human; CHO-K1 cells) | 50% at 10 μM |
| Dopamine (rat brain) | IC50 =12 μM |
| Serotonine (rat brain) | IC50 =21 μM |
| Interaction with dopamine transporter (rat brain) | IC50 =9 μM |
| Interaction with serotonine transporter (rat brain) | IC50 =6 μM |
| MAO-B (rat brain) | IC50 =0.098 μM |
| MAO-A (rat brain) | IC50 =485 μM |
| MAO-B (human brain) | IC50 =0.079 μM |
| MAO-A (human brain) | IC50 =80 μM |
| Blocking of veratridine-induced glutamate release (rat) | IC50 =56 μM |
| Blocking of K+-induced glutamate release (rat) | IC50 =9 μM |
| Interval to maximum concentration | 2–4 hours |
| Bioavailability | 95% |
| Plasma protein binding | 88%–90% |
| Terminal half-life | 22 hours (range: 22–30) |
| Elimination | Hepatic |
Abbreviations: IC50, half-maximal inhibitory concentration; EU, European Union; MAO, monoamine oxidase.
Decline of abnormal glutamate release
Veratridine-induced glutamate release may be provoked by opening of voltage-dependent Na+ channels. High K+ ion concentrations trigger a Ca++-mediated glutamate release. Safinamide blocked this veratridine (IC50=56 μM)-induced effect on glutamate release. In rat hippocampal synaptosomes, safinamide blocked the glutamate release induced by K+ (IC50=9 μM). In conclusion, safinamide has two mechanisms – reversible inhibition of MAO-B and deterioration of abnormal glutamate release. Both of them are important for the treatment of the dopaminergic deficit in PD. Therefore, it was no surprise that safinamide improved impaired motor behavior in animal models of PD.30,31
Pharmacokinetic and metabolic properties of safinamide
Pharmacokinetic behavior
The gastrointestinal absorption of safinamide is good and fast. The maximum concentration was observed within 2–4 hours. The absolute bioavailability was 95%. Steady-state concentrations were reached within 1 week. Plasma protein binding was within a range between 88% and 90% with a volume distribution of ~165 L, which corresponds to 2.5-fold of the body volume. There was an extensive extravascular distribution of safinamide. The terminal half-life was about 22 hours (range 20–30 hours).32 The total clearance was 4.6 L/hour (Table 1).
Metabolic characteristics
Extensive degradation of safinamide is predominantly undertaken via amide hydrolytic oxidation with safinamide acid generation as main derivative (Table 2). There are further less important metabolic pathways. Ether bond oxidation synthesizes an O-debenzylated safinamide. Oxidative turnover of safinamide or safinamide acid generates the N-dealkylated acid. All these derivatives possess no pharmacologic activity. The beta-glucuronide of the N-dealkylated acid and the monohydroxy safinamide appeared in urine.33,34 The glycine conjugate of the N-dealkylated acid and 2-[4-hydroxybenzylamino] propanamide are minor urine derivatives of safinamide.34,35 Mild-to-moderate impairment of hepatic function may rise safinamide levels within a range between 30% and 80%.
Table 2.
Essential derivatives of safinamide, selegiline, and rasagiline
Dosing recommendations
Safinamide should be administered orally once daily (o.i.d.) in humans. Adjustment of oral safinamide application is not necessary in case of mild hepatic impairment. Safinamide 50 mg/day should only be given in case of moderate hepatic impairment. Severe hepatic dysfunction is a contraindication for safinamide intake. Renal dysfunction has no influence on safinamide exposure.36
Pharmacokinetic studies in humans
Four trials were performed to evaluate the effects of safinamide within a dose range between 25 and 10,000 μg/mL in healthy male volunteers (age range: 18–45 years). The first trial investigated single dosing of 2.5, 5, and 10 mg/kg safinamide vs. placebo. In a second study, four participants were exposed to 25 μg/kg o.i.d., four took 50 μg/kg o.i.d, four received 75 μg/kg o.i.d. and four were put on 150 μg/kg safinamide o.i.d. Then within a study period of 7 days, participants 1–8 were exposed to 2.5 mg/kg safinamide o.i.d. and the other ones took 5 mg/kg safinamide o.i.d on every day. The third trial investigated safinamide dosing 1.25 mg/kg o.i.d. over an interval of 7 days. Objective of the fourth trial was to exclude an impact of a high-fat-content breakfast on the absorption of 900 μg/kg safinamide in comparison with the fasting state in six men. All trial results described a linear pharmacokinetic behavior in relation to the applied dose of safinamide without any effects of accumulation or relevant interaction with food.33,37
Interactions of safinamide with other drugs
Pharmacokinetics
Systemic safinamide concentrations do not interfere with enzyme activities. The activity of the cytochrome P450 (CYP) systems was not altered by safinamide according to a screening for CYP2A6, 2B6, 2C9, 2C19, 2D6, 2E1, and 3 A3/5 interactions. CYP1A2 substrate caffeine and the CYP3A4 inhibitor ketoconazole did not change the pharmacokinetic behavior of safinamide. Chronic treatment with levodopa or with dopamine agonists did not influence the clearance of safinamide in PD patients.33,37
Pharmacodynamics
Generally, serious adverse reactions may occur during combination of pethidine or dextromethorphan with MAO inhibitors. Caution is also recommended for the use of MAO inhibitors in combination sympathomimetic drugs. A clear contraindication is combination of safinamide use with other MAO inhibitors, because this may elevate the risk for hypertensive crisis, triggered by the tyramine-induced, so-called “cheese” effect.36,38 As safinamide is a selective and reversible MAO-B inhibitor, one may combine safinamide with serotonin reuptake inhibitors, serotonin–norepinephrine reuptake inhibitors, and tricyclic and tetracyclic antidepressants in a cautious manner.38
Dosing routes
Safinamide is orally administered as a tablet containing 50 or 100 mg safinamide (Table 2).
Safety and tolerability
Safinamide (50-100 mg/day) was well tolerated and safe in PD patients in the pivotal trials.32,39
Clinical efficacy
Adding of safinamide to chronic levodopa/DDC therapy in PD patients
The 016 study was the first big trial that investigated the combination of safinamide with levodopa in PD patients. It lasted 6 months. It was a randomized, double-blind, placebo-controlled trial.40 Participants were 669 mid- to late-stage-treated idiopathic PD patients. Inclusion criteria were a disease duration of at least 3 years, a preexisting stable levodopa regimen and occurrence of motor fluctuations with an “OFF” time duration for at least 1.5 hours daily. Levodopa therapy was optimized for 4 weeks after inclusion. Then the optimum titrated participants took either safinamide 50 mg o.i.d. (N=223) or 100 mg o.i.d. (N=224) or placebo (N=222) as an adjunct to their existing drug regimen. Increase of mean daily “ON Time” without dyskinesia plus “ON Time” with minor or “nontroublesome” dyskinesia was the primary endpoint of this trial. Monitoring of motor behavior over an interval of 18 hours was performed with a patient diary. Compared with baseline, safinamide 50 or 100 mg o.i.d. increased daily total “ON Time” for 1.3 hours per day (mean), whereas placebo elevated “ON Time” for 0.7 hours (mean). Therapy with safinamide was superior to placebo (safinamide 50 mg o.i.d. vs. placebo: p=0.022; safinamide 100 mg o.i.d. vs. placebo: p=0.013). Scores of Unified Parkinson’s Disease Rating Scale (UPDRS) part complications of therapy also improved; however, this effect turned out significant only in the safinamide 100 mg o.i.d. arm. Eighty nine patients on safinamide completed the study (50 mg: 91%; 100 mg: 87%; placebo: 89%).40 More than 90% of patients, who completed the initial study interval lasting 24 weeks, were included in a placebo-controlled double-blind extension trial, called 018, which lasted 78 weeks. That study investigated an effect on dyskinesia as primary objective with the Dyskinesia Rating Scale (DRS). This trial was negative concerning the DRS outcome, but safinamide improved motor fluctuations without worsening of dyskinesia. Safinamide also ameliorated motor behavior, activities of daily living and maintained this efficacy up to 2 years in total.41
SafinamidE Treatment as add-on To LEvodopa in idiopathic Parkinson’s disease with motor fluctuations (SETTLE) was a Phase III trial. Participants received either 50 or 100 mg of safinamide or placebo for 24 weeks. Five hundred and forty nine patients participated. Prior to randomization, the dopamine-substituting drug regime was optimized. All of participants suffered from motor fluctuations. Four hundred eighty four PD patients finished the study. Safinamide lowered (p<0.001) “OFF” time (1.03±0.21 [mean±SEM] hours), elevated “ON Time” (0.96±0.19 hours), and improved UPDRS scores (−1.82±0.61) compared with placebo.42
Regulatory affairs
Safinamide was approved by the European Medicines Agency in the European Union for the therapy of fluctuating, levodopa-treated PD patients in 2015. Safinamide was launched in Germany in 2015. To date, safinamide is available in 11 European countries, such as Sweden, UK, Switzerland and Italy. It was also approved in the US in 2017.
Conclusion
Outcomes of clinical trials in chronic levodopa/DDC-treated patients provided convincing evidence that safinamide particularly ameliorated motor impairment and “OFF” intervals in PD. Generally, levodopa therapy is efficacious but complex in clinical practice. One reason is the variability of peripheral duodenal absorption with its further dependency from gastric emptying. Degradation by the levodopa-metabolizing enzymes differs in the periphery and in the brain. Further important components are the short half-life of levodopa, impact of body weight on levodopa pharmacokinetics, and competition between amino acids and levodopa concerning the transport over gastrointestinal membranes and the blood–brain barrier.43
Pharmacokinetics of safinamide in relation to other PD drugs
However, the pharmacokinetics of levodopa have a considerable impact on its effects in PD patients.23 On the contrary, plasma behavior of safinamide may be described as a linear one-compartment model with first-order absorption and first-order elimination. Age, gender, renal function, and exposure to levodopa have no impact on the pharmacokinetic behavior of safinamide. The beneficial response to safinamide, as shown by an increase of “ON Time” over weeks, may also result from a certain long-time effect.44 Generally, there is a certain interindividual variability of drug response in humans. Age-related decline of renal function or overall metabolism capacity may impact pharmacokinetic and pharmacodynamic behavior of a drug. Safinamide predominantly provides its beneficial effects in the central nervous system for PD. Safinamide is easier to use as an adjunct to a levodopa/DDC regimen compared with COMT inhibitors. Entacapone and tolcapone provide a fluctuating inhibition of COMT activity, which may also impact the ups and downs of levodopa in plasma in relation to its dosing frequency over the day. Here, opicapone provides a more stable COMT inhibition over the day. A possible impact of the well-known COMT polymorphism on levodopa pharmacokinetics has not adequately been investigated in PD patients yet.12 Meta-analyses or comparisons with other compounds in terms of change of “ON”- or “OFF”-time intervals may be initial steps for the scientific evaluation of these phenomena in PD patients. To date, all of them suffer from the still not sufficient pharmacogenetic characterization of the response to PD drugs in each individual PD patient. In terms of drug effects, enzyme activities of levodopa-and dopamine-degrading enzymes may play an eminent role. In terms of use of safinamide, rasagiline or selegiline, a still rather underestimated component is the variability of MAO-A and -B enzyme activity. This is still present even after drug withdrawal over night.45,46,72,73
Safinamide and its competitors
Safinamide combines two well-known and proven pharmacologic principles in the treatment of PD: inhibition of MAO-B and decline of abnormal glutamate release. One may postulate that safinamide resembles rasagiline or selegiline. However, safinamide does not act in an irreversible fashion on MAO-B enzyme activity like rasagiline or selegiline. Therefore, it is not just another MAO-B inhibitor. Safinamide does not block NMDA receptor function. NMDA receptor antagonism is the mode of action of amantadine. This agent has also some anticholinergic features in addition to its dopamine-mimicking properties.47 In contrast, safinamide modulates sodium- and potassium-ion channels in a way that finally induces a declined abnormal glutamate release. Thus, safinamide is different from NMDA antagonists.48,49
Safinamide was approved because of its demonstrated reduction of “OFF” time in levodopa/DDC-treated PD patients according to pivotal trial outcomes. Study participants received a prior optimum titration with dopamine-substituting compounds over an interval lasting at least 4 weeks. Then they received safinamide additionally. This study design is different to trials with dopamine agonists, MAO-B inhibitors or COMT inhibitors in cohorts with chronic levodopa/DDI-treated PD patients, who suffered from “OFF” phenomena. They were not optimized before they received the study drug. From this point of view, one must scrutinize discussions, whether safinamide provides a clinical relevant benefit on “OFF” periods at all or will provide a minimal clinically important benefit.50 Safinamide was investigated under different, more demanding conditions and still outcomes were positive. The long-term data of the pivotal trials additionally showed that safinamide provides a sustained effect on motor impairment in PD. Clinical handling of safinamide is easier than therapy with dopamine agonists because of the better safety and tolerability of safinamide. Dopamine agonists cause edema and nausea in the long term. They increase sleepiness during higher dosing. Safinamide reduces presynaptic abnormal glutamate release and thus glutamatergic stimulation. NMDA antagonists, such as amantadine or memantine, also reduce glutamatergic stimulation but by NMDA receptor antagonism. It is well known that memantine or amantadine also exerts positive effects on vigilance.51,52 It warrants an investigation, whether safinamide also improves vigilance, attention, alertness, apathy, cognitive slowing, and depression, because this symptom complex of nonmotor features also responds to MAO-B inhibitors.53 These compounds elevate biogenic amines, such as serotonin or norepinenephrine, in the synaptic cleft.53 The glial MAO-B inhibition improves neurotransmission of bio-genic amines. This mode of action resembles the impact on biogenic amine metabolism provided by glial COMT inhibition.54–56 Central COMT inhibition has beneficial effects on cognition and cortical information processing. Increase of biogenic amine concentrations in the prefrontal cortex and in the mesolimbic system may relatively selectively improve slowness of thinking, apathy, and motivation. All these symptoms are closely related to “OFF” states.57–59 To date, there are no clinical data available for these still hypothetical considerations. One may suppose that perhaps the real world of patient maintenance may realize that safinamide may be superior to selegiline or rasagiline in terms of patients’ coping with nonmotor features of “OFF” periods. To date, one may classify selegiline, rasagiline, and safinamide as similar because of their MAO-B-inhibiting properties. However, their derivatives and modes of action differ (Tables 1 and 2). This may impact pain thresholds60 or occurrence of nonmotor features, such as sleep and depression, as shown by a switch study from selegiline with its amphetamine-like derivatives to rasagiline with its pharmacologic inactive metabolites.61 Selegiline is degraded to desmethylselegiline and amphetamine derivatives, rasagiline to aminoindane, and safinamide to inactivated dealkylated derivatives (Table 2). Mean elimination half-life of selegiline is 1.5 hours, the one of rasagiline is 1.0 hour in controls, and 1.3 hours in patients,62 whereas the one of safinamide is 22 hours. As selegiline and rasagiline inhibit MAO-B in an irreversible fashion, generation of new MAO-B enzyme is necessary after stopping selegiline and rasagiline. In contrast, safinamide is a reversible MAO-B inhibitor. It only blocks the enzyme activity, which may more easily and quickly recover following the stopping of safinamide intake.53 One may hypothesize based on these pharmacologic considerations that combination with serotonin reuptake inhibitors may be easier and less risky compared with selegiline or rasagi-line.63 However, it is emphasized here that combination of rasagiline with a serotonin reuptake inhibitor was generally safe according to a postmarketing evaluation and a trial in healthy volunteers.64,65 To date, no information from clinical study outcomes is available for safinamide regarding this topic. This is relevant for the maintenance of PD patients, who recurrently suffer from depressive episodes in the course of the disease.53 Therefore, they need concomitant antidepressant drug therapy, particularly when they experience “OFF” periods. Taken together, these pharmacological differences underline that safinamide is a unique compound.
Place in therapy and the future
It will be interesting, whether safinamide use supports long-term sparing of levodopa similar to selegiline, as demonstrated in the selegiline plus levodopa (SELEDO) trial.67 A further future role of safinamide in the real world of maintenance of PD patients will be that this compound may allow lower dosing of dopamine agonist- and levodopa/DDI formulations. A proven strategy for the treatment of “OFF” periods in levodopa/DDI-treated patients is to initiate a dopamine agonist or to increase its dosage. As a result, the risk for development of impulsive control disorders may emerge because of a more continuous, tonic stimulation of dopamine receptors in the ventral striatum.24 Onset of peripheral side effects, particularly edema and nausea, became also more likely. They also limit the dosing of the dopamine agonist. In view of the good side effect profile of safinamide and the worldwide future increasing availability of opicapone, a peripherally acting COMT inhibitor with one-time daily intake only, an alternative is on the horizon to this “dopamine agonist”-mediated approach for the reduction of “OFF” times.12 A “triple concept”, consisting of levodopa/DDI, safinamide, and opicapone, may provide a better quality of life for patients because of its superior safety and tolerability profile. Due to the reversibility of MAO-B inhibition the risk for onset of a serotonergic syndrome less severe with safinamide compared with selegiline or rasagi-line. Manifestation of diarrhea is lower in case of opicapone compared with entacapone or tolcapone.12 Both safinamide and opicapone have to be taken only once daily. Opicapone provides a more stable and continuous COMT inhibition compared to entacapone or the nowadays more rarely used tolcapone.12 As a result, a more continuous levodopa brain delivery may occur. It is well known that less fluctuations of levodopa in plasma occur during COMT inhibition.12 Less ups and downs of levodopa in the periphery contribute to more central stable dopamine concentrations in the synaptic cleft, particularly during additional MAO-B inhibition. Thus, these pharmacologic principles complement each other and contribute to a more continuous dopaminergic stimulation of postsynaptic dopaminergic receptors. Accordingly, one may expect less frequent and intense “OFF” episodes with a combination of safinamide and opicapone. As both agents require once-daily intake only, they may also complement each other in terms of reduction of the number of oral levodopa intakes in the long term. It would be worth while to consider the combination of both compounds in one formulation (the SAFinamide plus OPIcapone “Safopi”® capsule). This approach may also reduce the considerable compliance problems of PD patients.67 This combination may become an ideal adjunct therapy for prevention, delay, and treatment of the frequency and severity of levodopa-related “OFF” phenomena.66 Particularly, observational data from the real world may support a future long-term evaluation of this concept.
There are also alternative future drug developments on the horizon, which may provide a negative impact on the above-mentioned triple combination with its focus on continuous dopaminergic stimulation. These approaches aim to improve the delivery mode of well-known compounds. These new formulations, such as inhaled levodopa/DDC or apomorphine strips with buccal absorption, intend to get PD patients out of “OFF” episodes as quickly as possible.68,69 However, one may also express some concerns. Recurrent and frequent use of these short-acting compounds aggravates pulsatile stimulation of the dopaminergic nigrostriatal system. As a result, buffering capacity of pulsatile stimulations may decline faster and manifestations of “OFF” episodes may become more frequent and intense in the long term. A misuse is likely in PD patients with an addictive behavior to dopamine substitution.70,71 Another future relevant issue may be that one will again consider heterogeneity of MAO enzyme activity in conjunction with titration of an MAO-B inhibitor. To date, this is possible with selegiline and safinamide, but not with rasagiline. There are individuals, who experience a pronounced MAO inhibition following drug intake, whereas others did not.72,73 They may have a more elevated risk to develop tyramine-induced hypertension during MAO inhibition. Therefore, future assessment of MAO-A and -B activity during MAO inhibition may hypothetically help to identify these “cheese effect at risk” individuals and allow them to be recommended preventive dietary restrictions. Generally, such assessments of enzyme activity may be helpful to identify individuals with a certain risk for onset of a serotonergic syndrome. Future necessary research will give deeper insight in the complexity of drug-induced regulation of enzyme activity. To date, certain long-term effects, such as induction of enzyme generation, during chronic levodopa/DDC supplementation are not well investigated. A consequence of this future determination of these heterogeneous enzyme activities will be that fixed dosing of enzyme inhibitors will not be recommended any more in clinical practice for chronic treated PD patients. It will be better to titrate and apply MAO-B- and COMT-inhibitors in various dosages to achieve the optimum motor response in relation to levodopa dosing.
Footnotes
Disclosure
The author participated in the trials on safinamide as a principal investigator and gave advice to safinamide-developing pharmaceutical companies Newron, Merck Serono, and Zambon. The author reports no other conflicts of interest in this work.
References
- 1.Bach JP, Riedel O, Klotsche J, Spottke A, Dodel R, Wittchen HU. Impact of complications and comorbidities on treatment costs and health-related quality of life of patients with Parkinson’s disease. J Neurol Sci. 2012;314(1–2):41–47. doi: 10.1016/j.jns.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 2.de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54(11 Suppl 5):S21–S23. [PubMed] [Google Scholar]
- 3.Rajput AH, Birdi S. Epidemiology of Parkinson’s disease. Parkinsonism Relat Disord. 1997;3(4):175–86. doi: 10.1016/s1353-8020(97)00029-1. [DOI] [PubMed] [Google Scholar]
- 4.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 5.Poewe W, Gerstenbrand F. Clinical subtypes of Parkinson disease. Wien Med Wochenschr. 1986;136(15–16):384–387. [PubMed] [Google Scholar]
- 6.Weiner WJ. There is no Parkinson disease. Arch Neurol. 2008;65(6):705–708. doi: 10.1001/archneur.65.6.705. [DOI] [PubMed] [Google Scholar]
- 7.Apostolova I, Taleb DS, Lipp A, et al. Utility of follow-up dopamine transporter SPECT With 123I-FP-CIT in the diagnostic workup of patients with clinically uncertain Parkinsonian syndrome. Clin Nucl Med. 2017;42(8):589–594. doi: 10.1097/RLU.0000000000001696. [DOI] [PubMed] [Google Scholar]
- 8.Müller T, Farahati J, Kuhn W, et al. [123I]beta-CIT SPECT visualizes dopamine transporter loss in de novo parkinsonian patients. Eur Neurol. 1998;39(1):44–48. doi: 10.1159/000007896. [DOI] [PubMed] [Google Scholar]
- 9.Berg D, Godau J, Seppi K, et al. The PRIPS study: screening battery for subjects at risk for Parkinson’s disease. Eur J Neurol. 2013;20(1):102–108. doi: 10.1111/j.1468-1331.2012.03798.x. [DOI] [PubMed] [Google Scholar]
- 10.Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm. 2004;111(2):201–216. doi: 10.1007/s00702-003-0102-y. [DOI] [PubMed] [Google Scholar]
- 11.Müller T, Öhm G, Eilert K, et al. Benefit on motor and non-motor behavior in a specialized unit for Parkinson’s disease. J Neural Transm (Vienna) 2017;124(6):715–720. doi: 10.1007/s00702-017-1701-3. [DOI] [PubMed] [Google Scholar]
- 12.Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75(2):157–174. doi: 10.1007/s40265-014-0343-0. [DOI] [PubMed] [Google Scholar]
- 13.Müller T. Nondopaminergic therapy of motor and nonmotor symptoms in Parkinson’s disease: a clinician’s perspective. Neurodegener Dis Manag. 2016;6(5):385–398. doi: 10.2217/nmt-2016-0025. [DOI] [PubMed] [Google Scholar]
- 14.Weiner WJ. What do clinical trials tell us about treating patients. Par-kinsonism Relat Disord. 2009;15(Suppl 3):S34–S37. doi: 10.1016/S1353-8020(09)70776-X. [DOI] [PubMed] [Google Scholar]
- 15.Müller T. Drug therapy in patients with Parkinson’s disease. Transl Neurodegener. 2012;1(1):1–10. doi: 10.1186/2047-9158-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 17.Olanow CW, Goetz CG, Kordower JH, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54(3):403–414. doi: 10.1002/ana.10720. [DOI] [PubMed] [Google Scholar]
- 18.Riederer P, Gerlach M, Müller T, Reichmann H. Relating mode of action to clinical practice: dopaminergic agents in Parkinson’s disease. Parkinsonism Relat Disord. 2007;13(8):466–479. doi: 10.1016/j.parkreldis.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Birkmayer W, Hornykiewicz O. The effect of l-3,4-dihydroxyphe-nylalanine (= DOPA) on akinesia in parkinsonism. 1961. Wien Klin Wochenschr. 2001;113(22):851–854. [PubMed] [Google Scholar]
- 20.Müller T, Russ H. Levodopa, motor fluctuations and dyskinesia in Par-kinson’s disease. Expert Opin Pharmacother. 2006;7(13):1715–1730. doi: 10.1517/14656566.7.13.1715. [DOI] [PubMed] [Google Scholar]
- 21.Müller T, Erdmann C, Muhlack S, Bremen D, Przuntek H, Woitalla D. Inhibition of catechol-O-methyltransferase contributes to more stable levodopa plasma levels. Mov Disord. 2006;21(3):332–336. doi: 10.1002/mds.20717. [DOI] [PubMed] [Google Scholar]
- 22.Müller T, Erdmann C, Bremen D, et al. Impact of gastric emptying on levodopa pharmacokinetics in Parkinson disease patients. Clin Neuropharmacol. 2006;29(2):61–67. doi: 10.1097/00002826-200603000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Müller T. The impact of COMT-inhibition on gastrointestinal levodopa absorption in patients with Parkinson’s disease. Clin Med Insights Ther. 2010;2:155–168. [Google Scholar]
- 24.Voon V, Napier TC, Frank MJ, et al. Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: an update. Lancet Neurol. 2017;16(3):238–250. doi: 10.1016/S1474-4422(17)30004-2. [DOI] [PubMed] [Google Scholar]
- 25.LeWitt PA, Guttman M, Tetrud JW, et al. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off ” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005) Ann Neurol. 2008;63(3):295–302. doi: 10.1002/ana.21315. [DOI] [PubMed] [Google Scholar]
- 26.Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78(14):1096–1099. doi: 10.1212/WNL.0b013e31824e8f0d. [DOI] [PubMed] [Google Scholar]
- 27.Verhagen ML, Del DP, van den MP, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50(5):1323–1326. doi: 10.1212/wnl.50.5.1323. [DOI] [PubMed] [Google Scholar]
- 28.Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56(11):1383–1386. doi: 10.1001/archneur.56.11.1383. [DOI] [PubMed] [Google Scholar]
- 29.Pahwa R, Tanner CM, Hauser RA, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study) Mov Disord. 2015;30(6):788–795. doi: 10.1002/mds.26159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregoire L, Jourdain VA, Townsend M, Roach A, Di PT. Safinamide reduces dyskinesias and prolongs l-DOPA antiparkinsonian effect in parkinsonian monkeys. Parkinsonism Relat Disord. 2013;19(5):508–514. doi: 10.1016/j.parkreldis.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Podurgiel S, Collins-Praino LE, Yohn S, et al. Tremorolytic effects of safinamide in animal models of drug-induced Parkinsonian tremor. Pharmacol Biochem Behav. 2013;105:105–111. doi: 10.1016/j.pbb.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Marzo A, Dal Bo L, Monti NC, et al. Pharmacokinetics and pharmaco-dynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol Res. 2004;50(1):77–85. doi: 10.1016/j.phrs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Krosser S, Marquet A, Gallemann D, et al. Effects of ketoconazole treatment on the pharmacokinetics of safinamide and its plasma metabolites in healthy adult subjects. Biopharm Drug Dispos. 2012;33(9):550–559. doi: 10.1002/bdd.1822. [DOI] [PubMed] [Google Scholar]
- 34.Leuratti C, Sardina M, Ventura P, Assandri A, Müller M, Brunner M. Disposition and metabolism of safinamide, a novel drug for Parkinson’s disease, in healthy male volunteers. Pharmacology. 2013;92(3–4):207–216. doi: 10.1159/000354805. [DOI] [PubMed] [Google Scholar]
- 35.Seithel-Keuth A, Johne A, Freisleben A, Kupas K, Lissy M, Krosser S. Absolute bioavailability and effect of food on the disposition of safinamide immediate release tablets in healthy adult subjects. Clin Pharmacol Drug Dev. 2013;2(1):79–89. doi: 10.1002/cpdd.2. [DOI] [PubMed] [Google Scholar]
- 36.Müller T, Foley P. Clinical pharmacokinetics and pharmacodynamics of safinamide. Clin Pharmacokinet. 2017;56(3):251–261. doi: 10.1007/s40262-016-0449-5. [DOI] [PubMed] [Google Scholar]
- 37.Fariello RG, McArthur RA, Bonsignori A, et al. Preclinical evaluation of PNU-151774E as a novel anticonvulsant. J Pharmacol Exp Ther. 1998;285(2):397–403. [PubMed] [Google Scholar]
- 38.Cattaneo C, Caccia C, Marzo A, Maj R, Fariello RG. Pressor response to intravenous tyramine in healthy subjects after safinamide, a novel neuroprotectant with selective, reversible monoamine oxidase B inhibition. Clin Neuropharmacol. 2003;26(4):213–217. doi: 10.1097/00002826-200307000-00012. [DOI] [PubMed] [Google Scholar]
- 39.Di Stefano AF, Rusca A. Pressor response to oral tyramine during co-administration with safinamide in healthy volunteers. Naunyn Schmie-debergs Arch Pharmacol. 2011;384(6):505–515. doi: 10.1007/s00210-011-0674-2. [DOI] [PubMed] [Google Scholar]
- 40.Borgohain R, Szasz J, Stanzione P, et al. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov Disord. 2014;29(2):229–237. doi: 10.1002/mds.25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borgohain R, Szasz J, Stanzione P, et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord. 2014;29(10):1273–1280. doi: 10.1002/mds.25961. [DOI] [PubMed] [Google Scholar]
- 42.Schapira AH, Fox SH, Hauser RA, et al. Assessment of safety and efficacy of safinamide as a levodopa adjunct in patients with Parkin-son disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2016;74(2):216–224. doi: 10.1001/jamaneurol.2016.4467. [DOI] [PubMed] [Google Scholar]
- 43.Müller T, Woitalla D, Saft C, Kuhn W. Levodopa in plasma correlates with body weight of parkinsonian patients. Parkinsonism Relat Disord. 2000;6(3):171–173. doi: 10.1016/s1353-8020(00)00005-5. [DOI] [PubMed] [Google Scholar]
- 44.Loprete L, Leuratti C, Cattaneo C, Thapar MM, Farrell C, Sardina M. Population pharmacokinetic and pharmacodynamic analyses of safinamide in subjects with Parkinson’s disease. Pharmacol Res Perspect. 2016;4(5):e00251. doi: 10.1002/prp2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jimenez-Jimenez FJ, onso-Navarro H, Garcia-Martin E, Agundez JA. Advances in understanding genomic markers and pharmacogenetics of Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2016;12(4):433–448. doi: 10.1517/17425255.2016.1158250. [DOI] [PubMed] [Google Scholar]
- 46.Schnitker J, Müller T. Meta-analysis of placebo-controlled clinical trials of safinamide and entacapone as add-on therapy to levodopa in the treatment of Parkinson’s disease. Eur Neurol Rev. 2015;10(1):15–22. [Google Scholar]
- 47.Muhlack S, Musch P, Konietzka S, Woitalla D, Przuntek H, Müller T. Impact of oral fast release amantadine on movement performance in patients with Parkinson’s disease. Pharmaceutics. 2010;2(3):313–320. doi: 10.3390/pharmaceutics2030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pahwa R, Tanner CM, Hauser RA, et al. ADS-5102 (Amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–949. doi: 10.1001/jamaneurol.2017.0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varanese S, Howard J, Di RA. NMDA antagonist memantine improves levodopa-induced dyskinesias and “on-off ” phenomena in Parkinson’s disease. Mov Disord. 2010;25(4):508–510. doi: 10.1002/mds.22917. [DOI] [PubMed] [Google Scholar]
- 50.Hauser RA, Auinger P. Determination of minimal clinically important change in early and advanced Parkinson’s disease. Mov Disord. 2011;26(5):813–818. doi: 10.1002/mds.23638. [DOI] [PubMed] [Google Scholar]
- 51.Ambrozi L, Danielczyk W. Treatment of impaired cerebral function in psychogeriatric patients with memantine–results of a phase II double-blind study. Pharmacopsychiatry. 1988;21(3):144–146. doi: 10.1055/s-2007-1014666. [DOI] [PubMed] [Google Scholar]
- 52.Comi G, Leocani L. Assessment, pathophysiology and treatment of fatigue in multiple sclerosis. Expert Rev Neurother. 2002;2(6):867–876. doi: 10.1586/14737175.2.6.867. [DOI] [PubMed] [Google Scholar]
- 53.Riederer P, Müller T. Use of monoamine oxidase inhibitors in chronic neurodegeneration. Expert Opin Drug Metab Toxicol. 2017;13(2):233–240. doi: 10.1080/17425255.2017.1273901. [DOI] [PubMed] [Google Scholar]
- 54.Apud JA, Mattay V, Chen J, et al. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsy-chopharmacology. 2007;32(5):1011–1020. doi: 10.1038/sj.npp.1301227. [DOI] [PubMed] [Google Scholar]
- 55.Fava M, Rosenbaum JF, Kolsky AR, et al. Open study of the catechol-O-methyltransferase inhibitor tolcapone in major depressive disorder. J Clin Psychopharmacol. 1999;19(4):329–335. doi: 10.1097/00004714-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 56.Moreau JL, Borgulya J, Jenck F, Martin JR. Tolcapone: a potential new antidepressant detected in a novel animal model of depression. Behav Pharmacol. 1994;5(3):344–350. [PubMed] [Google Scholar]
- 57.Gasparini M, Fabrizio E, Bonifati V, Meco G. Cognitive improvement during Tolcapone treatment in Parkinson’s disease. J Neural Transm. 1997;104(8–9):887–894. doi: 10.1007/BF01285556. [DOI] [PubMed] [Google Scholar]
- 58.Kayser AS, Allen DC, Navarro-Cebrian A, Mitchell JM, Fields HL. Dopamine, corticostriatal connectivity, and intertemporal choice. J Neurosci. 2012;32(27):9402–9409. doi: 10.1523/JNEUROSCI.1180-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roussos P, Giakoumaki SG, Bitsios P. Tolcapone effects on gating, working memory, and mood interact with the synonymous catechol-O-meth-yltransferase rs4818c/g polymorphism. Biol Psychiatry. 2009;66(11):997–1004. doi: 10.1016/j.biopsych.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 60.Cattaneo C, Barone P, Bonizzoni E, Sardina M. Effects of safinamide on pain in fluctuating Parkinson’s disease patients: a post-hoc analysis. J Parkinsons Dis. 2016;7:95–101. doi: 10.3233/JPD-160911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller T, Hoffmann JA, Dimpfel W, Oehlwein C. Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J Neural Transm. 2013;120(5):761–765. doi: 10.1007/s00702-012-0927-3. [DOI] [PubMed] [Google Scholar]
- 62.Hoy SM, Keating GM. Rasagiline: a review of its use in the treatment of idiopathic Parkinson’s disease. Drugs. 2012;72(5):643–669. doi: 10.2165/11207560-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 63.Bartl J, Müller T, Grunblatt E, Gerlach M, Riederer P. Chronic mono-amine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J Neural Transm. 2014;121(4):379–383. doi: 10.1007/s00702-013-1120-z. [DOI] [PubMed] [Google Scholar]
- 64.Hilli J, Korhonen T, Laine K. Lack of clinically significant interactions between concomitantly administered rasagiline and escitalopram. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(8):1526–1532. doi: 10.1016/j.pnpbp.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 65.Panisset M, Chen JJ, Rhyee SH, Conner J, Mathena J. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO) Pharmacotherapy. 2014;34(12):1250–1258. doi: 10.1002/phar.1500. [DOI] [PubMed] [Google Scholar]
- 66.Przuntek H, Conrad B, Dichgans J, et al. SELEDO: a 5-year long-term trial on the effect of selegiline in early parkinsonian patients treated with levodopa. Eur J Neurol. 1999;6(2):141–150. doi: 10.1111/j.1468-1331.1999.tb00007.x. [DOI] [PubMed] [Google Scholar]
- 67.Malek N, Grosset DG. Medication adherence in patients with Parkin-son’s disease. CNS Drugs. 2015;29(1):47–53. doi: 10.1007/s40263-014-0220-0. [DOI] [PubMed] [Google Scholar]
- 68.Hauser RA, Olanow CW, Dzyngel B, et al. Sublingual apomorphine (APL-130277) for the acute conversion of OFF to ON in Parkinson’s disease. Mov Disord. 2016;31(9):1366–1372. doi: 10.1002/mds.26697. [DOI] [PubMed] [Google Scholar]
- 69.Lewitt PA, Hauser RA, Grosset DG, et al. A randomized trial of inhaled levodopa (CVT-301) for motor fluctuations in Parkinson’s disease. Mov Disord. 2016;31(9):1356–1365. doi: 10.1002/mds.26611. [DOI] [PubMed] [Google Scholar]
- 70.Ambermoon P, Carter A, Hall WD, Dissanayaka NN, O’Sullivan JD. Impulse control disorders in patients with Parkinson’s disease receiving dopamine replacement therapy: evidence and implications for the addictions field. Addiction. 2011;106(2):283–293. doi: 10.1111/j.1360-0443.2010.03218.x. [DOI] [PubMed] [Google Scholar]
- 71.Castrioto A, Kistner A, Klinger H, et al. Psychostimulant effect of levodopa: reversing sensitisation is possible. J Neurol Neurosurg Psychiatry. 2013;84(1):18–22. doi: 10.1136/jnnp-2012-302444. [DOI] [PubMed] [Google Scholar]
- 72.Müller T, Riederer P, Grunblatt E. Simultaneous determination of MAO-A and -B activity following first time intake of an irreversible MAO-B inhibitor in patients with Parkinson’s disease. J Neural Transm (Vienna) 2017;124(6):745–748. doi: 10.1007/s00702-017-1705-z. [DOI] [PubMed] [Google Scholar]
- 73.Müller T, Riederer P, Grunblatt E. Determination of MAO-A and –B activity in long-term treated patients with Parkinson’s disease. Clin Neuropharmacol. 2017 Jul;:4. doi: 10.1097/WNF.0000000000000233. Epub. [DOI] [PubMed] [Google Scholar]
- 74.Magyar K. The pharmacology of selegiline. Int Rev Neurobiol. 2011;100:65–84. doi: 10.1016/B978-0-12-386467-3.00004-2. [DOI] [PubMed] [Google Scholar]
- 75.Müller T. Pharmacokinetic/pharmacodynamic evaluation of rasagiline mesylate for Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2014;10(10):1423–1432. doi: 10.1517/17425255.2014.943182. [DOI] [PubMed] [Google Scholar]
- 76.Baker WL, Silver D, White CM, et al. Dopamine agonists in the treatment of early Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord. 2009;15(4):287–294. doi: 10.1016/j.parkreldis.2008.07.004. [DOI] [PubMed] [Google Scholar]