

Abstract
Background
Previously, we reported that high expression of nipped-B-like protein (NIPBL) was strongly correlated with poor prognosis, tumor differentiation, and lymph node metastasis. Survival analysis indicated that NIPBL expression was a potential prognostic factor for non-small cell lung cancer (NSCLC). Moreover, loss of NIPBL decreased lung cancer cells proliferation, migration, invasion and promoted apoptosis as well as sensitivity to chemotherapeutic agents. However, the deep mechanisms were not explored.
Purpose
The objective of this study was to identify the role of NIPBL in DNA damage response, as well as autophagy pathway, so as to interpret the mechanisms of how NIPBL knockdown enhances the chemosensitivity of lung cancer cell.
Methods
Cells (NCI-H1299 and NCI-H1650) were transfected by specific siRNAs before immunofluorescence and single-cell gel electrophoresis, which were mainly used to observe the differences of DNA damage in different groups. Additionally, protein were obtained and then analyzed by western blot and mass spectroscopy.
Results
In this study, we found that knockdown of NIPBL resulted in accumulation of phosphorylated H2AX (γ-H2AX) foci and higher levels of DNA damage, as revealed by comet assay. Western blot assay revealed that loss of NIPBL decreased expression of ATM/ATR, Rad3-related protein and Ku70/Ku80, but increased expression of LC3-B and depletion of p62. Using mass spectroscopy, we identified eight proteins that were significantly differentially expressed upon NIPBL knockdown. Gene Ontology analysis revealed that these proteins are mainly involved in DNA repair, mismatch repair, and binding to damaged DNA. The expression changes in two of the proteins, MSH2 and STAT1, were verified by Western blotting in NIPBL-knockdown cells.
Conclusions
In summary, these results reflected that loss of NIPBL impairs the DNA damage response and promotes autophagy. And NIPBL suppression may represent a novel strategy for preventing chemotherapy resistance in lung cancer.
Keywords: nipped-B-like protein, lung cancer, DNA damage response, double-strand break, autophagy
Introduction
In a previous study,1 our group confirmed that high expression of nipped-B-like (NIPBL) protein is associated with poor differentiation and prognosis in lung cancer patients. We also found that knockdown of NIPBL in non-small-cell lung cancer (NSCLC) cell lines (NCI-H1299 and NCI-H1650) significantly inhibited the proliferation, migration, and invasion abilities, and besides, also promoted apoptosis as well as sensitivity to chemotherapeutic agents. However, the underlying mechanisms remained to be elucidated.
Carcinogenic factors exert an influence on cells primarily by inducing DNA damage and mutations.2 When the DNA damage response (DDR) is defective, mutations gradually accumulate, exceed a certain threshold, and ultimately predispose cells to malignant transformation. To diminish the deleterious consequences of DNA damage, cells possess a series of signaling pathways that detect and repair lesions in DNA. Among all the types of DNA damage, DNA double-strand breaks (DSBs) are the most lethal.3 Phosphorylated H2AX (also called γ-H2AX or p-H2AX) is a hallmark of DSBs. γ-H2AX appears rapidly after damage (within a few minutes) and is thus the earliest DSB-induced chromatin modification. Once lesions are detected, cell cycle progression is temporarily blocked, and the repair machinery is activated. The main DNA repair systems are the homologous recombination (HR) and non-homologous end-joining (NHEJ) pathways;2,3 the relative prominence of these two pathways depends on specific cellular context.
NIPBL is the human homologue of Scc2 and functions as a loading factor to load cohesin onto chromosomes. The evolutionarily conserved cohesin complex, which plays a critical role in DSB repair, consists of the proteins Scc1, Scc3, and the heterodimer SMC1/SMC3.4–6 Several cohesin subunits, including SMC1/3, SMC5/6,7 and sororin,6,8 have been studied comprehensively and shown to be directly or indirectly involved in the DDR. In post-replicative cells, the Scc2/Scc4 protein complex is responsible for loading cohesin onto DSB sites, after which cohesin activates the ATM signal transduction pathway.6 Publications have reported that NIPBL is a multifunctional protein, which not only functions as a loading factor for cohesin but has also been implicated in gene expression.9,10 However, few studies have thoroughly explored the role of NIPBL in DNA repair.
Apoptosis is a major cellular response to DNA damage, and recent reports show that autophagy also plays a role in determining cell fate. Autophagy, also known as macroautophagy, is a “self-eating” mechanism that helps to preserve cellular homeostasis.11,12 The main function of autophagy is to capture and degrade unfolded proteins and organelles, enabling the recycling of their components. Autophagy participates in multiple physiologic and pathologic processes, including cancer,13 but the role of autophagy is context-dependent.13–15 On one hand, it suppresses the accumulation of toxic materials to prevent tumorigenesis and tumor progression, but on the other hand, it enables cancer cells to survive in diverse stress conditions. The role of NIPBL in autophagy remains unclear.
Based on the observations described earlier, we speculated that NIPBL may be involved in the DDR and autophagy pathway, and manipulation of this protein could promote apoptosis and chemosensitivity. To test this conjecture, we carried out the experiments described in the following section. The results revealed that NIPBL plays an important role in chemoresistance, and that the previously observed sensitization of NIPBL-knockdown cells most likely results from effects on both the DDR and autophagy pathways.
Materials and methods
Cell culture and transfection
The human NSCLC cell line NCI-H1299 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). NCI-H1650 cell line was obtained from Cell Bank at the Chinese Academy of Sciences (Shanghai, China). Cells were grown and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
siRNAs were constructed by GenePharma (Shanghai, China). The sequences of siRNAs (siNIPBL-N2 and siNIPBL-N3) were reported previously.1 For transfections, cells were plated on six-well plates at 3×105 cells/well and cultured overnight to 40%–50% confluence. Transfections were performed using Lipofectamine 3000 according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA).
Immunofluorescence staining
Lung cancer cells were grown on cover slips in six-well plates. After transfection for 48 h, cells were washed once in cold phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 30 min at room temperature, followed by three washes with PBS. The fixed cells were permeabilized for 10 min with 0.1% Triton X-100, followed by three washes with PBS. Cover slips were blocked for 30 min in blocking buffer (PBS containing 1% bovine serum albumin [BSA]), incubated with anti-γ-H2AX antibody in PBS (Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature, washed three times in PBS, incubated in the dark with Alexa Fluor-conjugated secondary antibody (ProteinTech, Manchester, UK) in PBS for 50 min, and then washed again three times in PBS. Next, a drop of DAPI solution was applied to the cover slip and incubated for 10 min. Images were acquired on a fluorescence microscope (Olympus, Tokyo, Japan).
Single-cell gel electrophoresis (comet assay)
The alkaline comet assay was performed as described by Wu et al.16 Briefly, after transfection for 48 h, cells were embedded in 0.65% low-melting point agarose at a suitable concentration. Seventy-five microliters of cellular suspension was spread onto a frosted slide previously covered with 100 μL of 1% normal-melting point agarose (the first layer). The slides were immersed in freshly prepared lysis solution at 4°C for 1 h, and then placed in a horizontal electrophoresis unit covered with fresh buffer for 20 min. Electrophoresis was performed for 20 min at 1.5 V/cm and 300 mA. Subsequently, the slides were gently washed twice in neutralization buffer. Each slide was stained with 40 μL of Gel Red. All the abovementioned steps were conducted in a darkroom to avoid additional DNA damage.
Images were acquired on a fluorescence microscope (Olympus). For each sample, more than 40 cells were counted in each independent experiment, and the CASP software was used to analyze the comets. The tail DNA percentage was taken as a quantified index of DNA damage.
Mass spectroscopy
Proteomic analysis was carried out in biological duplicates. Briefly, after transfection for 48 h, cells were lysed with 0.1 M Tris in 4% sodium deoxycholate solution (pH adjusted to 8.0). Equal amounts of proteins from each sample (50 μg) were mixed with 100 mM dithiothreitol for 20 min at 60°C, and then alkylated with 50 mM iodoacetamide for 30 min in the dark. Proteins were digested with mass spectrometry-grade trypsin overnight at 37°C. The digestion was stopped by addition of 10% trifluoroacetic acid, after which peptides were desalted using C18 tips. Extracts were then centrifuged in a SpeedVac to reduce the volume. Next, the peptides were redissolved in 30 μL of 0.1% formic acid. The labeled samples were analyzed on an Orbitrap Mass Spectrometer system (Thermo Fisher Scientific), which was directly connected to a Q Exactive mass spectrometer (Thermo Fisher Scientific).17
Statistical analysis
For mass spectroscopy, responsive proteins were defined as those whose abundance changed by ≥1.5-fold. Data were analyzed in DAVID Bioinformatics Resources version 6.8. Venn diagrams were drawn using the Venny 2.1.0 online software.
Data were analyzed using the Statistical Package for the Social Sciences (SPSS), version 21 (IBM Corporation, Armonk, NY, USA). For continuous variables, Student’s t-test was applied for paired samples. All experiments were performed at least in duplicate. Data are shown as mean ± standard deviation (SD) from independent experiments. The two-tailed p-value test was used for all analyses, and differences were considered to be statistically significant at values of p<0.05.
Results
NIPBL-silenced lung cancer cells exhibit distinct DNA damage traits
To determine whether NIPBL is involved in the DDR, we performed the alkaline comet assay, which allowed us to assess the degree of damage directly and quantify single-or double-strand breaks by measuring the percentage of DNA in the comet tail. A higher tail DNA percentage represents more severe DNA damage, ie, a larger number of DNA strand breaks. The tail DNA percentage was higher in NIPBL-silenced H1299 cells than in controls (Figure 1A and B, p=0.000 and 0.000, respectively). Similar results were obtained in H1650 cells (p=0.000 and 0.000, respectively). Based on these results, we concluded that NIPBL is involved in the DDR; specifically, downregulation of NIPBL resulted in more extensive DNA damage.
Figure 1.

Knockdown of NIPBL aggravates DNA damage and increases the level of DSBs in lung cancer cells.
Notes: (A) Knockdown of NIPBL in H1299 and H1650 cells markedly aggravated DNA damage, as revealed by comet assay. (B) Quantitative analysis of tail DNA percentage in comet assay. Magnification ×200. (C) Knockdown of NIPBL induced more γ-H2AX foci in H1299 and H1650 cells. Magnification ×400. (D) Average number of γ-H2AX foci per cell in the indicated treatment groups. (E) Elevated γ-H2AX expression in NIPBL-knockdown cells, as revealed by Western blot. **p<0.01; ***p<0.001.
Abbreviations: DSB, double-strand break; NC, negative control.
NIPBL-silenced lung cancer cells contain more DNA DSBs
In the light of the difference between the control and treated groups in the comet assay, we further explored the potential relationship between NIPBL and the DDR. Given that γ-H2AX foci are markers of DSBs, we asked whether NIPBL was associated with γ-H2AX. To this end, we performed immunofluorescence staining and Western blotting for γ-H2AX. The results revealed that both NIPBL-silenced groups had significantly higher levels of γ-H2AX foci than controls (Figure 1C and D, H1299: p=0.000 and 0.001, H1650: p=0.004 and 0.003, respectively). The elevated γ-H2AX level in the NIPBL-knockdown cells was confirmed by Western blot analysis (Figure 1E). The stable increase in γ-H2AX expression in NIPBL-silenced cells suggested that NIPBL is involved in DDR via the γ-H2AX-mediated damage response pathway.
Knockdown of NIPBL influences key molecules in the DNA repair pathway
We hypothesized that NIPBL acts as positive regulator of the DDR via its interaction with γ-H2AX. To elucidate the role of NIPBL in the DDR, we carried out Western blot assays to detect several key damage-related molecules in the DDR pathway. Upon NIPBL knockdown, ATM and ATR were also downregulated (Figure 2A) in both cell lines; these molecules act as sensors for DNA repair. Activated ATM and ATR localize at damaged DNA marked by γ-H2AX foci, where they regulate numerous downstream mediators that coordinate the DDR. In addition, the levels of the core NHEJ proteins Ku70 and 80 were also reduced (Figure 2A). We suspected that NIPBL is involved in DNA repair by recruiting ATM and ATR to damaged sites, where they initiate the downstream repair reactions. Furthermore, we concluded that NIPBL also plays a role in the NHEJ pathway to promote DSB repair.
Figure 2.

Knockdown of NIPBL influences key molecules in the DNA repair and autophagy pathway.
Notes: (A) Knockdown of NIPBL in H1299 and H1650 cell lines obviously decreased DNA repair-related molecules. (B) Knockdown of NIPBL in lung cancer cells influenced autophagy pathway molecules. H1299 cell line is p53 null.
Abbreviation: NC, negative control.
Downregulation of NIPBLinduces autophagy in lung cancer cells
DNA-damaging agents that cause DSBs initiate cell cycle arrest, as well as autophagy.2 Hence, we asked whether NIPBL also participated in cellular autophagy. To this end, we analyzed p-mTOR, p53, p62, and LC3-B proteins after siRNA treatment. The upregulation of LC3-B and downregulation of p62 indicate promotion of autophagy.13 The results of this study revealed that NIPBL-silenced cells had a promoted autophagy (Figure 2B). To further elucidate the mechanistic role of NIPBL in autophagy, we investigated the mTOR signaling pathway in H1299 and H1650 cells. As shown in Figure 2B, the levels of phosphorylated mTOR and p53, two principal regulators of both the DDR and autophagy, were significantly reduced after siRNA treatment. The mTOR signaling pathway negatively regulates autophagy in response to DNA damage, whereas p53 can regulate autophagy in either direction, depending on the location of the molecule within the cell: nuclear p53 facilitates autophagy, whereas cytoplasmic p53 functions as a repressor of autophagy.
The results described in this section show that NIPBL-silenced lung cancer cells can induce autophagy via suppressing the mTOR signaling pathway and p53 (mainly cytoplasmic p53). These results were consistent with our previous observations in breast cancer cell lines.18
DNA damage-related proteins are altered in siRNA-treated cells, as determined by mass spectroscopy
To more comprehensively elucidate the role of NIPBL in lung cancer cells, we performed mass spectrometry to identify proteins whose levels were altered in NIPBL siRNA-treated cells. All proteins identified were subjected to Gene Ontology (GO) functional classification analysis in DAVID Bioinformatics Resources. To interpret separately, we easily acquired the fact that the altered proteins were not exactly the same after the treatment of different siRNAs in biological processes, cellular components and molecular functions (Figure 3A–C). This may be ascribed to the fact that different siRNAs act on different loci. On the other hand, in terms of regulation of gene expression, NIPBL knockdown primarily affected biological regulation, protein and nucleic acid processing, and DNA binding.
Figure 3.

Mass spectrum analysis of NCI-H1299 and -H1650 cell lines following siRNA treatment.
Notes: (A–C) GO functional classification analysis, performed in DAVID Bioinformatics Resources. (D) Venn diagram of 19 proteins whose levels were changed in both cell lines after siRNA treatment. (E) MSH2 and STAT1 were downregulated upon NIPBL knockdown.
Abbreviations: GO, Gene Ontology; NC, negative control.
On combining the mass spectrographic data of the two cell lines, we identified 19 proteins whose abundance was changed following treatment with both siRNAs in both cell lines (Figure 3D). Afterward, we eliminated the proteins who were inconsistent in different types of siRNAs or cells. Ultimately, eight of these proteins were shown to be simultaneously upregulated or downregulated after siRNA treatment. To characterize the role of NIPBL in the DDR, we selected the MSH2 and STAT1 proteins, both of which are implicated in damage repair, for validation. The Western blotting results confirmed the mass spectrographic data the other way round (Figure 3E).
Discussion
Cancer has become a major public health concern in China, among which lung cancer is the most common and the leading cause of cancer-related death.19 Among the pathological types of lung cancer, NSCLC is predominant, representing 85% of cases. Chemotherapy is one of the most effective solutions, but with chemotherapy regimens frequently changing chemotherapy resistance is a major problem in clinical practice.
In our previous study, we found that knockdown of NIPBL in NSCLC lines (NCI-H1299 and NCI-H1650) significantly sensitized the cells to chemotherapeutic agents such as cisplatin, paclitaxel, and gemcitabine hydrochloride.1 Mechanistically, these agents function by generating DNA damages. Therefore, inhibition of the DDR pathway by siRNAs or small molecules represents a promising approach to improving the efficacy of chemotherapy. However, DDR inhibition is controversial because it could also trigger normal cells to undergo malignant transformation. Several independent studies have described the function of NIPBL in the DDR. Kong et al reported that NIPBL is localized to DSB sites,20,21 and Bot et al also showed that the NIPBL–MAU2 heterodimer is recruited to damaged DNA sites.5 These observations implied that NIPBL is involved in the DDR, but no previous study had systematically analyzed the mechanisms of NIPBL in DNA damage and repair.
In this study, we discovered that NIPBL-silenced cells had a greater degree of DNA damage. Furthermore, we confirmed that part of the damage was caused by DSBs, the most hazardous form of DNA damage, as reflected by the accumulation of γ-H2AX in NIPBL-silenced cells. NIPBL might initiate the NHEJ system to take part in DSB repair, but it remains unclear whether it is also involved in the HR system.
Figure 4 depicts a hypothetical model of NIPBL function. Once DNA damage (mainly DSBs) occurs, NIPBL rapidly recruits ATM/ATR, the sensors and key regulators of DNA DSB repair,2 to the damaged sites. Subsequently, the Ku70/80 proteins assemble the full DNA-dependent protein kinase (DNA-PK) complex.3 ATM/ATR then cooperates with DNA-PK to initiate downstream processes, such as phosphorylation of effector molecules (such as γ-H2AX), and ultimately launch the repair systems.
Figure 4.
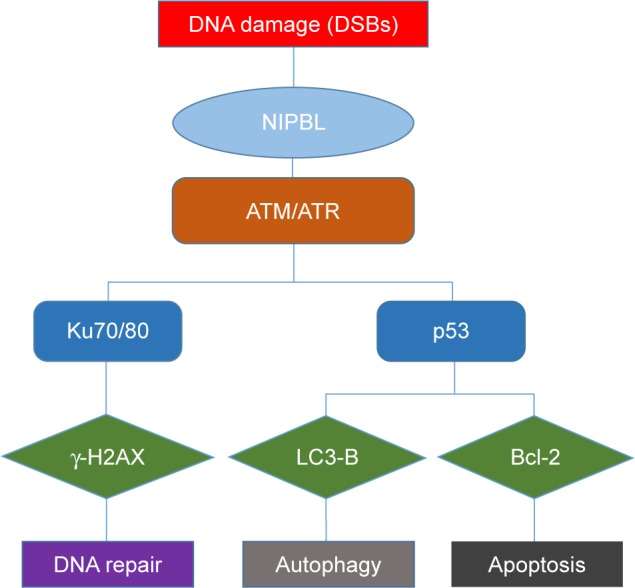
Potential processes when cells suffer DNA damage.
Notes: Cells suffering from DNA damage can have different fates, which mainly depends on the ability of repair systems.
Abbreviation: DSB, double-stranded break.
Apoptosis and autophagy are both cellular outcomes of DNA damage, and cells choose between the two fates in part as a function of DNA repair capacity. If the damage is irreparable, cells will initiate the apoptosis and/or autophagy pathway to prevent deterioration. In the former case, ATM/ATR activates p53, followed by activation of Bcl-2 and other apoptosis-related proteins (c-Myc, Mcl-1, and STAT3 in our last article), to initiate apoptosis. In the latter case, p53 can also induce autophagy by inhibiting mTOR, a negative regulator of autophagy.3 In contrast to nuclear p53, cytoplasmic p53 represses autophagy,22,23 which could explain why autophagy was promoted but p53 levels were reduced in the cells we examined. In our previous study,1 we showed that NIPBL knockdown can induce apoptosis, and here we showed that it can also induce autophagy.
Our previous study1 showed that about one-third of lung adenocarcinoma samples express high levels of NIPBL, that the level of NIPBL is inversely correlated with overall survival, and that loss of NIPBL sensitizes human lung cancer cells to chemotherapeutic agents. In this study, we performed a deeper analysis based on our previous results.
When cells undergo DNA damage, especially DSBs, firstly they made NIPBL to recruit ATM/ATR, leading to one of three cellular fates: repair, autophagy, or apoptosis. Knockdown of NIPBL blocks initiation of the DDR, preventing activation of downstream molecules such as Ku70/80 and increasing the accumulation of DSBs (reflected by γ-H2AX foci). In addition, we found that NIPBL knockdown also inhibits the mTOR cascade, a negative regulator of autophagy. The elevated expression of LC3-B and depletion of p62 in the knockdown cells indicated the promotion of autophagy. These results are consistent with the findings of Sandra et al that autophagy can induce autophagic cell death, thereby increasing sensitivity to chemotherapy and radiotherapy.24
Chemotherapeutic agents act on DNA strands to generate damage that normal cancer cells will repair or eliminate effectively. However, loss of NIPBL would make cells more susceptible, leading directly to death. Our results reveal that loss of NIPBL impairs the DDR while activating the autophagy and apoptosis pathways. This explains, at least in part, our previous observation that NIPBL-silenced cells are more sensitive to chemotherapeutic agents.
The finding that NIPBL is involved in DDR and autophagy represents a significant step forward in our understanding of the highly dynamic role of NIPBL in chemotherapy resistance. More detailed and comprehensive studies are necessary to fully elucidate the roles of NIPBL. Targeting NIPBL represents a promising novel approach to treating NSCLC, and would be in accordance with the increasing drive to translate laboratory-based findings into clinical applications.
Conclusion
The molecular findings of our study highlight NIPBL as a promising biomarker that sensitizes the chemosensitivity for NSCLC patients. Moreover, this research represents a further step to reveal the role of NIPBL in DDR and autophagy pathway. It is our firm conviction that our findings of NIPBL in chemotherapy resistance are still a corner of the iceberg. More detailed and comprehensive studies are still required.
Acknowledgments
The authors thank Professor Jianguo Feng, Wei Chen, and Zhiguo Zheng for their help to complete this work. The authors also thank the Zhejiang Academy of Medical Sciences for providing experimental platform. This work was supported by the grants from the Natural Science Foundation of Zhejiang Province (LY16H160039) and the National Nature Science Foundation of China (81672315).
Footnotes
Author contributions
Lei Zheng contributed to the acquisition, analysis and interpretation of data, and drafting of the manuscript. Huanhuan Zhou contributed to the analysis and interpretation of data, revising manuscript critically for important intellectual content. Liwei Guo contributed to drafting the article and revising it critically. Xiaoling Xu and Shengjie Zhang contributed to the final approval of the version to be published. Weizhen Xu and Weimin Mao contributed to the conception and design, analysis and interpretation of data. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Xu W, Ying Y, Shan L, et al. Enhanced expression of cohesin loading factor NIPBL confers poor prognosis and chemotherapy resistance in non-small cell lung cancer. J Transl Med. 2015;13:153. doi: 10.1186/s12967-015-0503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R. DNA damage and autophagy. Mutat Res. 2011;711(1–2):158–166. doi: 10.1016/j.mrfmmm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang D, Tang B, Xie X, Xiao YF, Yang SM, Zhang JW. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol Ther. 2015;16(7):1005–1013. doi: 10.1080/15384047.2015.1046022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jahnke P, Xu W, Wulling M, et al. The Cohesin loading factor NIPBL recruits histone deacetylases to mediate local chromatin modifications. Nucleic Acids Res. 2008;36(20):6450–6458. doi: 10.1093/nar/gkn688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bot C, Pfeiffer A, Giordano F, Manjeera DE, Dantuma NP, Strom L. Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J Cell Sci. 2017;130(6):1134–1146. doi: 10.1242/jcs.197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooker AS, Berkowitz KM. The roles of cohesins in mitosis, meiosis, and human health and disease. Methods Mol Biol. 2014;1170:229–266. doi: 10.1007/978-1-4939-0888-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potts PR, Porteus MH, Yu H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006;25(14):3377–3388. doi: 10.1038/sj.emboj.7601218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitz J, Watrin E, Lenart P, Mechtler K, Peters JM. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol. 2007;17(7):630–636. doi: 10.1016/j.cub.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Yuen KC, Xu B, Krantz ID, Gerton JL. NIPBL Controls RNA biogenesis to prevent activation of the stress kinase PKR. Cell Rep. 2016;14(1):93–102. doi: 10.1016/j.celrep.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorsett D, Merkenschlager M. Cohesin at active genes: a unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol. 2013;25(3):327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 13.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. doi: 10.1158/1078-0432.CCR-15-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W, Jiang H, Guo X, et al. The protective role of hyaluronic acid in Cr(VI)-induced oxidative damage in corneal epithelial cells. J Ophthalmol. 2017;2017:3678586. doi: 10.1155/2017/3678586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H, Chen X, Xu X, et al. Lysine acetylation and succinylation in HeLa cells and their essential roles in response to UV-induced stress. Sci Rep. 2016;6:30212. doi: 10.1038/srep30212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Zheng L, Lu K, et al. Downregulation of cohesin loading factor nipped-B-like protein (NIPBL) induces cell cycle arrest, apoptosis, and autophagy of breast cancer cell lines. Med Sci Monit. 2017;23:4817–4825. doi: 10.12659/MSM.906583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 20.Kong X, Ball AR, Jr, Pham HX, et al. Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol Cell Biol. 2014;34(4):685–698. doi: 10.1128/MCB.01503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oka Y, Suzuki K, Yamauchi M, Mitsutake N, Yamashita S. Recruitment of the cohesin loading factor NIPBL to DNA double-strand breaks depends on MDC1, RNF168 and HP1gamma in human cells. Biochem Biophys Res Commun. 2011;411(4):762–767. doi: 10.1016/j.bbrc.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22(2):181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14(1):90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]