

Immunotherapy has transformed cancer treatment, but some patients do not benefit from these therapies. This article describes candidate biomarkers that have been shown to predict resistance to T‐cell‐mediated immunotherapy in the clinical setting. The practical application of these markers and potential targets for overcoming resistance are discussed.
Keywords: Immunotherapy, T cell, Resistance, Biomarker
Abstract
The emergence of immunotherapy has revolutionized cancer treatment in recent years. Inhibitors of immune checkpoints, including antibodies against cytotoxic T‐lymphocyte‐associated protein 4, programmed cell death protein 1, and programmed death ligand 1, have demonstrated notable efficacy in certain advanced cancers. Unfortunately, many patients do not benefit from these therapies and either exhibit primary resistance to treatment or develop acquired mechanisms of resistance after initially responding to therapy. Here, we review the genomic and immune traits that may promote resistance to T‐cell‐mediated immunotherapy, with a focus on identifying potential biomarkers that could eventually be used in the clinical setting to guide treatment selection. We summarize the clinical evidence for these markers and discuss how current understanding of resistance mechanisms can inform future studies and aid clinical decision‐making in order to derive maximum benefit from immunotherapy.
Implications for Practice.
Immunotherapy has rapidly progressed as a treatment modality for multiple cancers, but it is still unclear which patients are likely to benefit from these therapies. Studies of resistance mechanisms have only recently started to identify biomarkers that can help predict patient outcomes. This review summarizes the available clinical data in regard to immunotherapy resistance, with a focus on molecular biomarkers that may be useful in guiding clinical decision‐making. It discusses possible applications of these biomarkers and highlights opportunities for further clinical discovery.
Introduction
Cancer treatment has been transformed in recent years by advances in immunotherapy, in particular the advent of inhibitors targeting cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4), programmed cell death protein 1 (PD‐1), and programmed death ligand 1 (PD‐L1) [1], [2]. These agents block immune checkpoint pathways, thereby activating a tumor‐specific T‐cell immune response [3]. The efficacy of immunotherapy was initially demonstrated in patients with advanced melanoma who were treated with ipilimumab, an anti‐CTLA‐4 antibody [4], [5], [6]. Inhibition of PD‐1 with the antibodies nivolumab and pembrolizumab was subsequently found to be effective in a variety of malignancies, including melanoma [7], [8], [9], non‐small cell lung cancer (NSCLC) [10], [11], [12], Hodgkin's lymphoma [13], head and neck cancer [14], renal cell carcinoma [15], gastric cancer [16], and hepatocellular carcinoma [17]. More recently, antibodies against PD‐L1, which binds PD‐1, have shown efficacy in urothelial cancer [18], NSCLC [19], and Merkel cell carcinoma [20].
Despite these encouraging results, response rates vary widely across tumor types, and the majority of patients either do not respond to immunotherapy or subsequently exhibit disease progression [21]. These outcomes suggest several categories of resistance to immunotherapy (Fig. 1). In temporal terms, tumors can demonstrate primary resistance, in which patients do not exhibit a significant initial response to treatment, or develop acquired resistance, in which patients respond for some time but their disease subsequently progresses. This classification scheme is readily apparent from a clinical perspective, can be useful for algorithmic approaches to decision‐making, and thus will frame the ensuing discussion.
Figure 1.

Classifications for resistance biomarkers. Biomarkers of resistance to immunotherapy can be categorized via several relevant frameworks. A temporal division between primary and acquired resistance correlates with the real‐time observations of clinicians. The mechanism underlying each biomarker, in contrast, can be spatially sorted as being intrinsic to tumor cells or extrinsic in the microenvironment or systemic circulation. Finally, an immunological perspective describes resistance as mechanistically occurring because of failures in the cancer‐immunity cycle. An immune‐desert tumor is unable to elicit a strong immune response, an immune‐excluded tumor prevents immune cell infiltration despite sufficient immunogenicity, and an inflamed tumor suppresses immune actions despite ample immune cell intrusion.
Abbreviations: IFN, interferon; JAK, Janus kinase; MHC I, major histocompatibility complex class I; MMR, mismatch repair; PD‐L1, programmed cell death ligand 1; TIL, tumor‐infiltrating lymphocyte.
However, the underlying mechanisms of resistance can be fit into several other helpful frameworks. For instance, a resistance factor can be broadly categorized as being either intrinsic or extrinsic to tumor cells. The former encompasses internal characteristics, such as cancer‐specific genetic adaptations, whereas the latter acts to suppress immune function systemically or in the tumor microenvironment [22]. Another relevant schema links immunotherapy resistance to breakdowns in the cancer‐immunity cycle [23]. In this context, resistance occurs because tumors (a) fail to elicit an immune response (“immune desert”), (b) prevent infiltration of immune cells (“immune excluded”), or (c) suppress immune function despite adequate immune presence (“inflamed”).
Finally, resistance mechanisms can be discussed in relation to predictive biomarkers, which importantly have the potential ability to guide treatment decisions. Patients who are unlikely to respond based on these biomarkers might be directed toward alternative therapies and protected from avoidable toxicities. Identification of resistance biomarkers is thus crucial to the fully effective use of immunotherapy.
In this review, we describe candidate biomarkers that have been shown in the clinical setting to predict resistance to T‐cell‐mediated immunotherapy (Fig. 2). Furthermore, we discuss the practical application of these markers and survey potential targets for overcoming resistance.
Figure 2.

Biomarkers of resistance to immunotherapy. Tumor cells can evade T‐cell attack after immunotherapy via primary or acquired mechanisms of resistance. Potential biomarkers of primary resistance include the following: negative PD‐L1 expression (A); low neoantigen or mutation load (B); MMR proficiency leading to decreased neoantigens (C); low levels of TILs in the tumor parenchyma (D); increased frequency of circulating MDSCs (E); decreased levels of IDO (F); deleterious mutations in the IFN‐γ pathway, including in the IFNGR (G); loss of function mutations in JAK 1/2 (H); amplification of MDM2 (I); and increased VEGF signaling (J). Acquired resistance biomarkers include the following: loss of MHC I molecules, leading to inability of T cells to recognize neoantigens via the TCR (K); acquired mutations in JAK 1/2 (L); loss of immunodominant neoantigens (M); and upregulation of suppressors such as TIM‐3 or LAG‐3 (N). Biomarkers can be based on decreased or loss of function (red Xs) or may be upregulated during resistance (red upward arrows).
Abbreviations: APC, antigen‐presenting cell; CTLA‐4, cytotoxic T‐lymphocyte‐associated protein 4; IDO, indoleamine 2,3‐dioxygenase; IFN‐γ, interferon‐γ; IFNGR, interferon‐γ receptor; JAK 1/2, Janus kinase 1 and 2; LAG‐3, lymphocyte‐activation gene‐3; MDM2, murine double minute 2; MDSC, myeloid‐derived suppressor cell; MHC I, major histocompatibility class I; MMR, mismatch repair; PD‐1, 2programmed cell death protein 1; PD‐L1, programmed cell death ligand 1; STING, stimulator of interferon genes; TCR, T‐cell receptor; TIL, tumor‐infiltrating lymphocyte; TIM‐3, T‐cell immunoglobulin mucin‐3; VEGF, vascular endothelial growth factor.
Primary Resistance to Immunotherapy
PD‐L1 Expression
Beginning with the earliest clinical trials of PD‐1 inhibitors, high tumor levels of PD‐L1 have been shown to be associated with improved clinical response [7], [24]. Subsequent trials have repeatedly linked PD‐L1 positivity with favorable outcomes, including in NSCLC [25] and melanoma [26], [27]. As a result, PD‐L1 immunohistochemistry (IHC) assays have obtained regulatory approval both as companion diagnostics that are mandatory for certain treatment indications and as complementary tests deemed likely to predict patient response [28].
Although the general relationship between PD‐L1 and clinical response has been consistent, the degree of association has been markedly variable. Of concern, as many as 20% of PD‐L1‐negative tumors exhibited response to anti‐PD‐1 treatment in certain cohorts.
The basis for this predictive quality was first explained by studies that correlated PD‐L1 positivity with increased numbers of tumor‐infiltrating lymphocytes (TILs) [29]. Notably, these TILs were often found in close proximity to PD‐L1‐expressing cells. Based on this data, Taube et al. proposed a mechanism termed “adaptive immune resistance,” in which tumor cells adapt to immune attack by suppressing the action of cytotoxic T cells, specifically, in this case, by upregulating PD‐L1 [29]. Increased PD‐L1 levels cause a dampening of the immune response via the regulatory effects of PD‐1 pathway activation on these TILs. In keeping with the adaptive immune resistance hypothesis, analysis of tumors in the Cancer Genome Atlas demonstrated that high cytolytic immune activity can lead to PD‐L1 amplification [30].
Although the general relationship between PD‐L1 and clinical response has been consistent, the degree of association has been markedly variable. Of concern, as many as 20% of PD‐L1‐negative tumors exhibited response to anti‐PD‐1 treatment in certain cohorts [31]. This inconsistency may represent true predictive uncertainty, or it may be related to issues with PD‐L1 assays themselves. A comparison of four PD‐L1 IHC assays found that changing the assay system led to a different PD‐L1 classification in 37% of cases [32]. One limitation of PD‐L1 staining is the arbitrary nature of cutoffs for “positive expression,” which are deceptive because PD‐L1 levels exist on a continuum [33]. Additionally, intratumor heterogeneity in PD‐L1 expression suggests that PD‐L1 assays are vulnerable to sampling variation [34]. This problem is particularly relevant to smaller‐sized biopsy samples, although one survey found over 90% concordance in PD‐L1 expression between needle biopsy and surgical resection specimens [35].
PD‐L1 assays have nevertheless been well validated as integral to patient evaluation prior to initiation of immunotherapy. Based on a phase II trial of patients with NSCLC [11], the U.S. Food and Drug Administration (FDA) has approved the PD‐L1 IHC 22C3 pharmDx as a companion diagnostic test for use with pembrolizumab [28]. Another assay using a different antibody, the PD‐L1 IHC 28‐8 pharmDx, has been approved as complementary to nivolumab in NSCLC [10] and melanoma [36]. In addition, the PD‐L1 SP142 assay has been approved for complementary use with the anti‐PD‐L1 antibody atezolizumab in urothelial carcinoma [18] and NSCLC [19], and the PD‐L1 SP263 assay has been approved as complementary to the anti‐PD‐L1 antibody durvalumab [37]. Although the utility of these assays remains somewhat controversial, they overall represent an important step forward in predicting patient outcomes using biomarkers.
Low Mutational Burden and Mismatch Repair Status
Specific targeting of tumors by T cells requires the presence of tumor‐specific antigens that are capable of eliciting an immune response [38]. Of particular interest are neoantigens, which are formed by somatic mutations and contain new epitopes that are recognized as foreign by the immune system [39]. Neoantigens have been postulated to serve as tumor‐ and even individual‐specific targets for T cells [40], whose antitumor activity can then be enhanced by immune checkpoint blockade [39], [41]. Consistent with this hypothesis, poor response to anti‐CTLA‐4 therapy has been correlated with both decreased tumor mutational burden and low neoantigen load [42], [43]. Similar associations between mutational burden and clinical benefit have also been reported with anti‐PD‐1 [44] and anti‐PD‐L1 [18] therapies.
Unfortunately, low mutational load remains a flawed biomarker. Although correlated with response, mutational load was unable to completely predict clinical benefit for anti‐CTLA‐4 therapy in melanoma [42]. Other studies have shown no difference in antigen density between tumors with and without evidence of tumor inflammation [45], [46]. This limitation may be explained in part by variable spatial patterns of neoantigen expression. Clonal neoantigens that are universally expressed within a tumor have been shown to confer greater treatment efficacy, whereas increased intratumor neoantigen heterogeneity is associated with poor response [47]. A more refined assessment of tumor neoantigen load may thus be necessary before it can be utilized clinically to predict immunotherapy response.
Tumor mutation rates are strongly influenced by DNA repair mechanisms [48], [49]. In colorectal cancer, defects in mismatch repair (MMR) can lead to an over 100‐fold increase in mutational burden [50]. MMR status can be assessed by measures of microsatellite instability (MSI), with high MSI indicating MMR deficiency [51]. The relevance of these findings was demonstrated by a phase II trial of pembrolizumab, which reported considerably decreased clinical benefit, including an objective response in 0 of 18 patients, for colorectal cancers with MMR proficiency (or low MSI) [52]. Studies in other solid tumors have similarly demonstrated links between MMR phenotype and clinical response to immune checkpoint inhibitors [52], [53]. A prospective cohort of 86 patients with MMR‐deficient tumors achieved objective radiographic response to PD‐1 blockade in an impressive 53% of cases [54]. Based on the above data, the FDA has now approved pembrolizumab for any MMR‐deficient solid tumors that have progressed on prior treatments. This landmark indication represents the unique development of a “pan‐tumor” biomarker applicable to all solid tumor types.
Lack of Tumor‐Infiltrating Lymphocytes
A long‐observed element of an effective antitumor immune response is the accumulation of TILs in the tumor parenchyma [55]. TILs represent a complex set of immune cells and include the tumor‐specific CD8+ T cells that are potentiated by immune checkpoint inhibitors [56], [57]. In patients with melanoma treated with anti‐CTLA‐4 therapy, tumor regression was associated with corresponding increases in CD8+ T‐cell infiltration [58]. A phase II trial of ipilimumab in advanced melanoma again observed a significant relationship between TILs and clinical activity [59].
The predictive value of TILs in immunotherapy was demonstrated in a cohort of patients with metastatic melanoma treated with anti‐PD‐1 inhibitors. Decreased expression of PD‐1, PD‐L1, and CD8 within the tumor margins were all correlated with poor response [60]. Similar conclusions were obtained in a study of anti‐PD‐L1 therapy against multiple cancer types [27]. These studies suggested that the effects of immunotherapy were mediated by T cells that had already infiltrated tumors but were then negatively regulated by PD‐L1. In mouse models, anti‐PD‐1 therapy was effective even after blocking T‐cell exit from lymphatic tissue, further supporting the idea that pre‐existing TILs are responsible for the clinical response seen with immunotherapy [61].
Tumors can thus be described as “inflamed” or “noninflamed” based on the presence of TILs and related proinflammatory cytokines [56]. A non‐T‐cell‐inflamed tumor can become immunologically “cold” either because of lack of immune activation (as with neoantigen‐poor cancers) or as a result of elements in the microenvironment that exclude T cells from the tumor interior [23], [62]. These factors potentially represent additional biomarkers of resistance. For example, gain‐of‐function mutations in beta‐catenin have been reported to cause decreased T‐cell infiltration by downregulating chemokines such as CCL4 [63]. Of note, these mutations accounted for less than half of non‐T‐cell‐inflamed tumors in this study, indicating the likely presence of multiple complex pathways that promote T‐cell exclusion.
Activation of Immune Regulatory Pathways
Even with adequate TIL presence (i.e., an inflamed tumor state), antitumor activity can be blunted by regulatory elements that suppress immune cells. These inhibitory molecules include of course CTLA‐4 and PD‐1 themselves, which can be expressed not only in effector T cells but also in other immune cells [64]. CTLA‐4 is constitutively expressed on regulatory T cells (Tregs), which diminish the immune response [64]. Depletion of these Tregs has been correlated with greater clinical benefit in patients treated with ipilimumab [65], [66], and therefore changes in Treg levels can serve as an on‐treatment predictor of response. Factors that influence Treg activity, meanwhile, might represent markers of resistance. For example, soluble CD25, an interleukin‐2 receptor whose binding has been hypothesized to stimulate Treg proliferation, was reported as a negative correlate to overall survival with CTLA‐4 blockade [67].
Several other cell types have also been described as exerting an immunosuppressive effect [68]. Myeloid‐derived suppressor cells (MDSCs) negatively regulate immune activity in cancer, and increased circulating MDSCs have been identified as a poor prognostic factor [69], [70]. Tumors resistant to immune checkpoint inhibitors in mouse models became susceptible to therapy when treated with drugs that reduced MDSCs [71]. Among patients treated with ipilimumab for melanoma, higher frequencies of MDSCs were associated with poor outcomes [72].
Thus, multiple regulatory pathways act independently from PD‐1 and CTLA‐4 to abet tumor resistance. One biomarker that has raised considerable interest from a therapeutic standpoint is indoleamine 2,3‐dioxygenase (IDO) [61]. This enzyme suppresses T‐cell function by catabolizing the essential amino acid tryptophan [73] and is notably activated in dendritic cells after engagement with CTLA‐4 [74]. Histological studies from a phase II trial of ipilimumab in melanoma noted decreased baseline expression of IDO in poorly responding tumors [59]. Because IDO is upregulated by CTLA‐4 during adaptive immune resistance, a low IDO level might signify a lack of suppressed TILs available to be reactivated by immunotherapy.
Mutations in Janus Kinase 1 and 2 and Interferon‐γ Pathway
Tumor cells can also exploit genetic alterations in order to resist antiproliferative signaling by immune cells. The interferon‐γ (IFN‐γ) signaling pathway has been recognized as a critical component of immunotherapy. During adaptive immune resistance, tumors will upregulate PD‐L1 expression in direct response to IFN‐γ production by TILs [29]. IFN‐γ signaling also mediates many of the antitumor actions of immune cells. Increased expression of IFN‐γ‐inducible genes, including chemokines that promote T‐cell infiltration and activation, has been observed after anti‐CTLA‐4 therapy [75]. Meanwhile, defective mutations in the IFN‐γ pathway were found in 9 of 12 melanoma tumors resistant to ipilimumab, and increased copy‐number alterations in IFN‐γ was associated with poor response to ipilimumab [76]. These data together imply that reduced IFN‐γ signaling can lead to primary resistance in tumors.
The IFN‐γ signaling pathway contains the enzymes Janus kinase (JAK) 1 and 2, which act downstream of IFN‐γ [29]. In melanoma cell lines, lack of PD‐L1 upregulation in response to IFN‐γ was traced to mutations in either IFN‐γ receptor 1 or JAK 1/2 [77]. Genetic analysis of nonresponders to anti‐PD‐1 therapy with high mutational loads revealed inactivating JAK 1/2 mutations in one case of melanoma and one case of MMR‐deficient colon cancer [78]. These mutations led to decreased signaling via IFN‐γ and resulted in negative PD‐L1 tumor expression. JAK inactivation thus may be useful as a resistance biomarker, and mutations in other immune signaling effectors should be explored as potential contributors to primary resistance.
Other Biomarkers of Primary Resistance
A plethora of other mechanisms have been implicated in immunotherapy resistance, although these all require further study. The growing acknowledgment of the influence of gut microbiota on the immune system has opened up new avenues of study. Resistance to ipilimumab was recently linked to the enrichment of Bacteriodes species in a cohort of 26 patients with metastatic melanoma [79], and oral administration of Bifidobacterium was synergistic with PD‐1 inhibition in mouse models [80]. Despite these encouraging results, the optimum microbiota composition remains in question. In contrast to the above study, an analysis of 25 patients treated with CTLA‐4 blockade for melanoma showed increased outgrowth of Bacteriodes species, and fecal transfer of these bacteria was actually associated with positive outcomes in mice [81]. Evidence is mounting in favor of the strong influence of the intestinal microbiome on immune function, but more clinical data are clearly needed in order to direct therapeutic options.
Other biomarkers that have evidence in the clinical setting include vascular endothelial growth factor (VEGF). Elevated levels of VEGF were associated with decreased overall survival in patients with melanoma treated with ipilimumab [82], and VEGF has been linked to both decreased T‐cell infiltration and immunosuppressive effects [83]. Loss of phosphatase and tensin homolog (PTEN) has emerged as another potential biomarker of resistance and was correlated with greater tumor reduction in patients with melanoma treated with anti‐PD‐1 therapy [84]. PTEN has been associated with greater PD‐L1 positivity on tumor cells [85], as have alterations in a number of oncogenic drivers [86], [87], [88], [89]. Mutations in another family of tumorigenesis drivers, the serpins (including the genes SERPINB3 and SERPINB4), were associated with increased survival after anti‐CTLA‐4 therapy for melanoma, although the exact mechanism remains unclear [90]. Tumor hypoxia has also raised interest as a resistance factor, and signals activated by hypoxic environments were associated with worse clinical outcomes in mice treated with PD‐1 blockade [91]. Metformin has been proposed as a method of reducing oxygen consumption and improving susceptibility to anti‐PD‐1 treatment and is being studied in clinical trials [92]. Lastly, one particularly concerning category of resistance involves patients who exhibit “hyper‐progression” after immunotherapy [93]. Amplifications of the E3 ubiquitin‐ligase protein murine double minute 2, which inhibits the p53 tumor suppressor, have been associated with these hyper‐progressor phenotypes [94], although the mechanism of such an effect remains unknown.
Acquired Resistance to Immunotherapy
Even with an excellent initial response to immunotherapy, patients are at risk of subsequently relapsing because of acquired resistance. This issue is drawing increased attention as clinicians gain longitudinal experience with immune checkpoint inhibitors.
Loss of Major Histocompatibility Complex Class I Expression
Some of the earliest examples of immunotherapy failure were attributed to acquired defects in major histocompatibility complex (MHC) class I molecules [95]. These patients were found to have tumor cells that specifically lacked functional beta‐2 microglobulin (B2M), a protein necessary for CD8‐mediated T‐cell recognition. Biopsies obtained prior to immunotherapy exhibited normal B2M levels, pointing toward an acquired escape mutation that allowed tumor cells to evade immune recognition. Other mechanisms of MHC class I loss have since been reported, including the downregulation of transporter associated with antigen processing 2 and low‐molecular‐weight protein 7 in MSI‐negative colorectal tumors [96]. Direct loss of the gene encoding the human leukocyte antigen (HLA)‐C*08:02 class I molecule was recently described in a colorectal tumor that progressed after initial response to T‐cell transfer therapy [97]. These T cells were HLA‐C*08:02‐restricted TILs, and thus this mutation directly allowed immune evasion by tumor cells.
There is evidence to suggest that even with intact immune‐antigen recognition, tumors can avoid detection by reducing expression of these antigens. In one study, NSCLC tumors that progressed on immune checkpoint blockade were shown to have developed a loss of 6–18 neoantigens via chromosomal deletions [98]. These neoantigens were able to elicit T‐cell responses during in vitro assays, implying an integral role in facilitating the effects of immunotherapy. Similarly, another report described dedifferentiation and resulting antigen loss in melanoma cells as a method of acquiring resistance to adoptive T‐cell transfer [99]. These examples together reinforce a theme of tumors developing resistance by limiting the antigens available for recognition by immune cells.
IFN‐γ/JAK Pathway Mutations
As with primary resistance, tumor cells may attain genetic or molecular alterations that inhibit downstream effects of immune signaling. An important clinical report analyzed the molecular characteristics of four patients who responded to pembrolizumab and subsequently relapsed [100]. One patient had tumor cells that exhibited a B2M mutation leading to loss of MHC I expression, as described above. Notably, one patient had homozygous mutations in JAK 1 (Q503* nonsense mutation) and another had homozygous mutations in JAK 2 (F547 splice‐site mutation), both of which were predicted to lead to nonsense‐mediated decay or truncation of the protein prior to the active kinase domain. Tumors of both patients exhibited decreased downstream signaling in response to IFN‐γ. In conjunction with the previously described data on JAK 1/2 inactivation in primary resistance, these data establish IFN‐γ/JAK pathway aberrations as a route that tumors can exploit in order to avoid the effects of immunotherapy. Of concern, these mutations may exist at high frequency in tumors, with pretreatment melanoma biopsies demonstrating IFN‐γ pathway mutations in as many as 19% of samples [101].
Activation of Alternative Immunosuppressive Pathways
Tumors have also been shown to utilize alternative immune checkpoints that can take over immunosuppressive functions when CTLA‐4‐ or PD‐1‐dependent pathways are blocked. For example, T‐cell immunoglobulin mucin 3 (TIM‐3), which has been previously described as a marker of T‐cell exhaustion [102], was found to be upregulated in two patients who developed resistance to anti‐PD‐1 therapy [103]. In a mouse model of TIM‐3‐mediated resistance to anti‐PD‐1 therapy, administration of anti‐TIM‐3 antibodies resulted in enhanced therapeutic efficacy [103]. In addition, the combination anti‐PD‐1 and anti‐TIM‐3 therapy has shown synergistic effects in preclinical studies [104]. Other immune checkpoints might prove similarly useful in identifying resistance. For example, lymphocyte‐activation gene 3 (LAG‐3) has been shown to coexpress with PD‐1 as an immune suppressor [105], and anti‐LAG‐3 plus anti‐PD‐1 therapy resulted in enhanced tumor regression in mouse models [106].
Overall, mechanisms of acquired resistance remain poorly understood, given the paucity of clinical data on the subject. It can, however, be hypothesized that many of the mechanisms involved in primary resistance are able to develop later in the treatment course. This is especially true of genetic mutations, such as those in JAK 1/2. In another example, biallelic loss of PTEN was recently found in a patient with uterine leiomyosarcoma who acquired resistance to PD‐1 blockade [107]. As trials continue to come forth on the efficacy of immunotherapies, additional markers of acquired resistance should be identified. It is telling that in the above‐mentioned study by Zaretsky and colleagues, the authors were unable to identify an explanatory mutation in one of the four relapsed patients [100], exemplifying the shortcomings in our current understanding.
Discussion and Practical Application
T‐cell‐based immunotherapy is an exciting treatment modality that holds great promise but remains limited in part by our incomplete understanding of resistance. Given the vast genetic and molecular variability among and within tumors, it is not surprising that identifying definitive resistance biomarkers has been elusive. However, several candidates discussed in this review have shown promise and accumulated some clinical evidence as predicting primary (Table 1) and acquired (Table 2) resistance. Certainly, FDA‐approved biomarkers for PD‐L1 status and MMR proficiency should be taken into account in all applicable cases, despite lingering concerns over issues such as assay consistency. These flaws instead highlight the fallibility of single tests and underscore the need for comprehensive tumor analysis and patient assessment in order to optimize treatment decisions.
Table 1. Clinical evidence for biomarkers of primary resistance to immunotherapy.

Ranges in parentheses indicate 95% confidence interval.
Abbreviations: CNA, copy number alterations; EIA, enzyme immunoassay; IDO, indoleamine 2,3‐dioxygenase; IFN‐γ, interferon‐γ; IHC, immunohistochemistry; JAK 1/2, Janus kinase 1 and 2; MB, ; MDM2, murine double minute 2; MDSC, myeloid‐derived suppressor cell; MMR, mismatch repair; MSI, microsatellite instability; NSCLC, non‐small cell lung cancer; ORR, objective response rate; PBMC, peripheral blood mononuclear cell; PCR, polymerase chain reaction; PD‐1, programmed cell death protein 1; PD‐L1, programmed cell death ligand 1; PFS, progression‐free survival; Ref., reference; TIL, tumor‐infiltrating lymphocyte; TTF, time to treatment failure; VEGF, vascular endothelial growth factor; WES, whole exome sequencing.
Table 2. Clinical evidence for biomarkers of acquired resistance to immunotherapy.
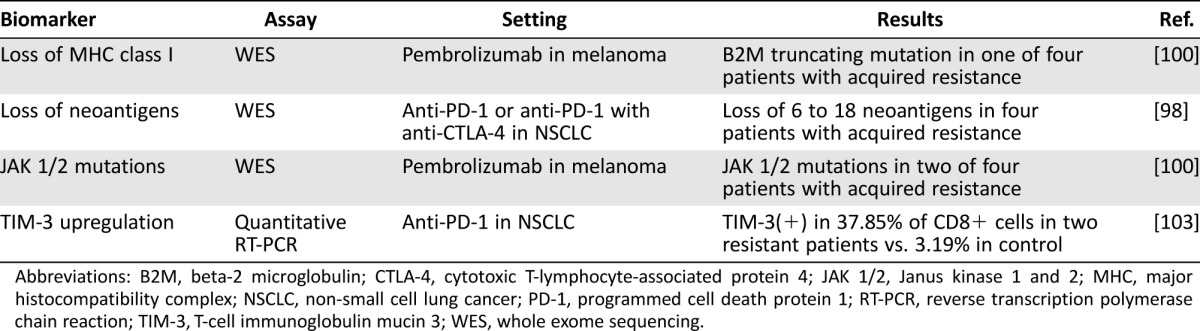
Abbreviations: B2M, beta‐2 microglobulin; CTLA‐4, cytotoxic T‐lymphocyte‐associated protein 4; JAK 1/2, Janus kinase 1 and 2; MHC, major histocompatibility complex; NSCLC, non‐small cell lung cancer; PD‐1, programmed cell death protein 1; RT‐PCR, reverse transcription polymerase chain reaction; TIM‐3, T‐cell immunoglobulin mucin 3; WES, whole exome sequencing.
Multifactorial Resistance Biomarkers
Several investigators have proposed combining multiple biomarkers to arrive at a more accurate predictor of treatment resistance [108], [109]. For example, both inadequate immunogenicity and a noninflamed tumor milieu have accumulated substantial evidence as contributing to negative prognostics with immunotherapy. Neither of these measures is perfectly predictive of response [110], an issue that may be ameliorated by aggregate measures. In one analysis of melanoma samples, an immune signature termed innate anti‐PD‐1 resistance, or “IPRES”—consisting of resistance‐associated genes involved in mesenchymal transition [111], matrix remodeling [112], and angiogenesis [83]—was associated with improved response to PD‐1 inhibitors [113]. Another study found that increased copy number alterations and low mutation load were nonredundant predictors of poor response, again suggesting the benefit of a combinatorial biomarker [114].
Multifactorial assays have also been shown to be useful for longitudinal monitoring [115]. An immune signature utilized as an early on‐treatment marker was highly predictive of response to immunotherapy [115]. Ultimately, effective biomarker analysis may entail the monitoring of multiple genetic and molecular factors in a longitudinal manner, thereby providing comprehensive and dynamic information regarding response to treatment. This would unfortunately require sequential biopsies, a logistical challenge that could be circumvented by using markers in peripheral blood. For instance, a score using on‐treatment levels of exhausted‐phenotype T cells that had been “reinvigorated” was found to correlate with patient response to PD‐1 blockade [116]. A variety of other blood‐based immune cell markers can be measured to assess patient response [117], but MDSC level represents one of very few such biomarkers that can be predictive prior to treatment. Identification of peripheral blood biomarkers thus represents an area greatly in need of further investigation.
Loss of MHC class I expression represents a particularly problematic resistance mechanism, as it can negate the ability of the immune system to act on tumor cells.
Treatment Strategies in Response to Resistance
The search for therapies that circumvent immunotherapy resistance has been progressing in parallel to mechanistic inquiries. Multiple targets described in this review have shown promise both as biomarkers of resistance and as targets of combination therapy. The most visible example of combination therapy in this field involves the dual targeting of PD‐1 and CTLA‐4. A clinical trial of patients who progressed on anti‐CTLA‐4 therapy showed improved response to nivolumab as compared with chemotherapy [118], suggesting that primary resistance can be overcome by targeting related pathways. Subsequent studies in melanoma have in fact demonstrated a synergistic effect of anti‐PD‐1 and anti‐CTLA‐4 treatment [36], [119], [120]. Ongoing clinical trials of combination therapies include those targeting immunosuppressive factors, such as IDO [121]. Alternative checkpoint molecules like TIM‐3 have shown efficacy in preclinical combination studies and are pending clinical evaluation [122]. Emerging data on oncogenic driver mutations in immunotherapy have additionally justified several exploratory trials for dual targeting [22]. Forthcoming results could confirm these combination treatments as prime methods for combating resistance.
Mechanisms of acquired resistance have only recently begun to be elucidated and will become increasingly relevant to clinical practice. Loss of MHC class I expression represents a particularly problematic resistance mechanism, as it can negate the ability of the immune system to act on tumor cells. In more fortunate cases, the emergence of acquired resistance can be localized to a single metastasis or recurrent tumor. In a case series of 36 patients who developed acquired resistance, 15 patients continued to respond well to immunotherapy after treating a solitary resistant lesion with surgery and/or radiation [123]. In situations of more widespread resistance, chemotherapy may be indicated unless novel methods of reactivating MHC class I expression are discovered.
Genetic mutations in signaling cascades, such as those in the IFN‐γ/JAK pathway, at least offer the ability to intervene at other steps. Inactivating mutations in JAK 1 or JAK 2, for example, might be overcome by stimulating downstream or parallel pathways. The stimulator of interferon genes (STING) pathway, for example, has been shown to upregulate type I IFN production [124] and may additionally play a role downstream of JAK 1/2 [125]. Activation of STING has also been shown in mouse models to induce an inflammatory microenvironment and cause tumor regression [126]. Such methods of immune enhancement may be helpful in converting non‐T‐cell‐inflamed tumors into inflamed ones that are more conducive to immunotherapy [22]. A variety of other techniques have been proposed to promote this transition to an inflamed tumor, including epigenetic modifications that reduce T‐cell exhaustion [127] and the use of BRAF inhibition to improve tumor antigen recognition [128] and promote a more favorable microenvironment [129].
Final Recommendations
Although biomarker analysis remains in its infancy, it would still be appropriate to assess any patients being considered for treatment with immunotherapeutic agents (Fig. 3). Candidate patients should be evaluated for PD‐L1 status and MMR status; if sufficient resources exist, they can also be assessed for mutation load, TIL levels, IFN‐γ/JAK pathway mutations, and biomarkers relevant to any indications or trials for combination therapies. Re‐evaluation should occur periodically and with any evidence of acquired resistance. Ultimately, a tumor profile that meets criteria for multiple resistance biomarkers should be deemed unlikely to respond to simple immune checkpoint blockade, and alternative options should be discussed. Treatment regimens in such a situation should depend on whether resistance involves (a) deficits in tumor immunogenicity and inflammation requiring general immune enhancement, (b) coexisting molecular markers that can be targeted with combination therapy, or (c) compromise of immune pathways to an extent that would preclude immunotherapy as an appropriate treatment modality. If possible, patients who do not respond to treatment should be evaluated for causative mechanisms in order to inform future decisions and to further develop our understanding of immunotherapy resistance.
Figure 3.

Proposed evaluation of prospective candidates for immunotherapy. Candidate patients should undergo an initial evaluation for resistance biomarkers (A). A clinical profile consistent with a noninflamed tumor microenvironment may warrant enhancement of immune activity. Biomarkers that can be targeted would suggest benefit from combination therapies. Primary resistance might also be suggested by negative prognostic indicators during early on‐treatment (B). Patients whose disease progresses after initial response should receive workup for acquired resistance (C). These resistance mechanisms can similarly be addressed by enhancing the immune response or targeting immunosuppressive pathways with combination therapy. Finally, loss of MHC‐I may prohibit further use of immunotherapy and indicate alternative modalities.
Abbreviations: B2M, beta‐2 microglobulin; HLA, human leukocyte antigen; IDO, indoleamine 2,3‐dioxygenase; JAK, Janus kinase; LAG‐3, lymphocyte‐activation gene‐3; MDSCs, myeloid‐derived suppressor cells; MHC‐I, major histocompatibility class I; MSI, microsatellite instability; PD‐L1, programmed cell death ligand 1; STING, stimulator of interferon genes; TAP2, transporter associated with antigen processing 2; TIL, tumor‐infiltrating lymphocytes; TIM‐3, T‐cell immunoglobulin mucin‐3; Treg, regulatory T cells; VEGF, vascular endothelial growth factor.
Conclusion
Cancer immunotherapy, although only recently implemented as a foundation of treatment, has already yielded exciting results and holds significant promise for the future. As mechanisms of resistance are more comprehensively revealed, clinicians may be able to more effectively treat patients based on their specific genetic and molecular characteristics. Finding biomarkers that can predict patient response to immunotherapy remains a primary objective for investigators, although clinicians should approach any single marker with caution, given the complexity of tumor biology. Nevertheless, the identification and proper use of resistance biomarkers should remain a focus of ongoing study in order to extract the greatest benefit from these treatments.
Contributed equally
Footnotes
For Further Reading: Jonathan L. Messerschmidt, George C. Prendergast, Gerald L. Messerschmidt. How Cancers Escape Immune Destruction and Mechanisms of Action for the New Significantly Active Immune Therapies: Helping Nonimmunologists Decipher Recent Advances. The Oncologist 2016;21:233‐243; first published on February 1, 2016.
Implications for Practice: Oncologists have tremendous experience with therapies that target the cancer cells. New biologic agents have been rapidly introduced recently that target not cancer cells, but the patient's immune cells. The mechanisms of action of these immune‐based biologic agents are within the host immune system. To understand these new biologic therapies, basic knowledge of normal and abnormal immune function is essential. The present report explains the up‐to‐date basic immune normal and abnormal function and prepares the oncologist to understand how the new drugs work, why they work, and why there are associated adverse events.
Author Contributions
Conception/design: Young Kwang Chae
Manuscript writing: Michael S. Oh
Final approval of manuscript: Young Kwang Chae, Michael S. Oh, Francis J. Giles
Disclosures
The authors indicated no financial relationships.
References
- 1. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015;27:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56–61. [DOI] [PubMed] [Google Scholar]
- 3. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hodi FS, O'Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robert C, Thomas L, Bondarenko I et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–2526. [DOI] [PubMed] [Google Scholar]
- 6. Schadendorf D, Hodi FS, Robert C et al. Pooled analysis of long‐term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robert C, Long GV, Brady B et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–330. [DOI] [PubMed] [Google Scholar]
- 9. Robert C, Schachter J, Long GV et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–2532. [DOI] [PubMed] [Google Scholar]
- 10. Borghaei H, Paz‐Ares L, Horn L et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 2015;373:1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garon EB, Rizvi NA, Hui R et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 2015;372:2018–2028. [DOI] [PubMed] [Google Scholar]
- 12. Herbst RS, Baas P, Kim DW et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): A randomised controlled trial. Lancet 2016;387:1540–1550. [DOI] [PubMed] [Google Scholar]
- 13. Ansell SM, Lesokhin AM, Borrello I et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferris RL, Blumenschein G Jr, Fayette J et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med 2016;375:1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Motzer RJ, Escudier B, McDermott DF et al. Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015;373:1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuchs CS, Doi T, Jang RWJ et al. KEYNOTE‐059 cohort 1: Efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. J Clin Oncol 2017;35(suppl 15):4003a. 29040031 [Google Scholar]
- 17. El‐Khoueiry AB, Sangro B, Yau T et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenberg JE, Hoffman‐Censits J, Powles T et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: A single‐arm, multicentre, phase 2 trial. Lancet 2016;387:1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fehrenbacher L, Spira A, Ballinger M et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): A multicentre, open‐label, phase 2 randomised controlled trial. Lancet 2016;387:1837–1846. [DOI] [PubMed] [Google Scholar]
- 20. Kaufman HL, Russell J, Hamid O et al. Avelumab in patients with chemotherapy‐refractory metastatic Merkel cell carcinoma: A multicentre, single‐group, open‐label, phase 2 trial. Lancet Oncol 2016;17:1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol 2014;8:1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma P, Hu‐Lieskovan S, Wargo JA et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature 2017;541:321–330. [DOI] [PubMed] [Google Scholar]
- 24. Brahmer JR, Drake CG, Wollner I et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Velcheti V, Schalper KA, Carvajal DA et al. Programmed death ligand‐1 expression in non‐small cell lung cancer. Lab Invest 2014;94:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taube JM, Klein A, Brahmer JR et al. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti‐PD‐1 therapy. Clin Cancer Res 2014;20:5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herbst RS, Soria JC, Kowanetz M et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hansen AR, Siu LL. PD‐L1 testing in cancer: Challenges in companion diagnostic development. JAMA Oncol 2016;2:15–16. [DOI] [PubMed] [Google Scholar]
- 29. Taube JM, Anders RA, Young GD et al. Colocalization of inflammatory response with B7‐h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rooney MS, Shukla SA, Wu CJ et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lipson EJ, Forde PM, Hammers HJ et al. Antagonists of PD‐1 and PD‐L1 in cancer treatment. Semin Oncol 2015;42:587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirsch FR, McElhinny A, Stanforth D et al. PD‐L1 immunohistochemistry assays for lung cancer: Results from phase 1 of the blueprint PD‐L1 IHC assay comparison project. J Thorac Oncol 2017;12:208–222. [DOI] [PubMed] [Google Scholar]
- 33. Kerr KM, Nicolson MC. Non‐small cell lung cancer, PD‐L1, and the pathologist. Arch Pathol Lab Med 2016;140:249–254. [DOI] [PubMed] [Google Scholar]
- 34. McLaughlin J, Han G, Schalper KA et al. Quantitative assessment of the heterogeneity of PD‐L1 expression in non‐small‐cell lung cancer. JAMA Oncol 2016;2:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kitazono S, Fujiwara Y, Tsuta K et al. Reliability of small biopsy samples compared with resected specimens for the determination of programmed death‐ligand 1 expression in non‐small‐cell lung cancer. Clin Lung Cancer 2015;16:385–390. [DOI] [PubMed] [Google Scholar]
- 36. Larkin J, Chiarion‐Sileni V, Gonzalez R et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Powles T, O'Donnell PH, Massard C et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open‐label study JAMA Oncol 2017;3:e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kvistborg P, van Buuren MM, Schumacher TN. Human cancer regression antigens. Curr Opin Immunol 2013;25:284–290. [DOI] [PubMed] [Google Scholar]
- 39. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 40. Yadav M, Jhunjhunwala S, Phung QT et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572–576. [DOI] [PubMed] [Google Scholar]
- 41. Gubin MM, Zhang X, Schuster H et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature 2014;515:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Snyder A, Makarov V, Merghoub T et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014;371:2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van Allen EM, Miao D, Schilling B et al. Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 2015;350:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spranger S, Luke JJ, Bao R et al. Density of immunogenic antigens does not explain the presence or absence of the T‐cell‐inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA 2016;113:E7759–E7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tran E, Ahmadzadeh M, Lu YC et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McGranahan N, Furness AJ, Rosenthal R et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci USA. 1996;93:14800–14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Howitt BE, Shukla SA, Sholl LM et al. Association of polymerase e‐mutated and microsatellite‐instable endometrial cancers with neoantigen load, number of tumor‐infiltrating lymphocytes, and expression of PD‐1 and PD‐L1. JAMA Oncol 2015;1:1319–1323. [DOI] [PubMed] [Google Scholar]
- 50. Timmermann B, Kerick M, Roehr C et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 2010;5:e15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–2087.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Le DT, Uram JN, Wang H et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Colle R, Cohen R, Cochereau D et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull Cancer 2017;104:42–51. [DOI] [PubMed] [Google Scholar]
- 54. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galon J, Costes A, Sanchez‐Cabo F et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960–1964. [DOI] [PubMed] [Google Scholar]
- 56. Gajewski TF. The next hurdle in cancer immunotherapy: Overcoming the non‐T‐cell‐inflamed tumor microenvironment. Semin Oncol 2015;42:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Im SJ, Hashimoto M, Gerner MY et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature 2016;537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ribas A, Comin‐Anduix B, Economou JS et al. Intratumoral immune cell infiltrates, FoxP3, and indoleamine 2,3‐dioxygenase in patients with melanoma undergoing CTLA4 blockade. Clin Cancer Res 2009;15:390–399. [DOI] [PubMed] [Google Scholar]
- 59. Hamid O, Schmidt H, Nissan A et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med 2011;9:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tumeh PC, Harview CL, Yearley JH et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spranger S, Koblish HK, Horton B et al. Mechanism of tumor rejection with doublets of CTLA‐4, PD‐1/PD‐L1, or IDO blockade involves restored IL‐2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2014;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Salmon H, Franciszkiewicz K, Damotte D et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 2012;122:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature 2015;523:231–235. [DOI] [PubMed] [Google Scholar]
- 64. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res 2017;27:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hodi FS, Butler M, Oble DA et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte‐associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA 2008;105:3005–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liakou CI, Kamat A, Tang DN et al. CTLA‐4 blockade increases IFNgamma‐producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA 2008;105:14987–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hannani D, Vétizou M, Enot D et al. Anticancer immunotherapy by CTLA‐4 blockade: Obligatory contribution of IL‐2 receptors and negative prognostic impact of soluble CD25. Cell Res 2015;25:208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lindau D, Gielen P, Kroesen M et al. The immunosuppressive tumour network: Myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013;138:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Talmadge JE, Gabrilovich DI. History of myeloid‐derived suppressor cells. Nat Rev Cancer 2013;13:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ostrand‐Rosenberg S, Sinha P. Myeloid‐derived suppressor cells: Linking inflammation and cancer. J Immunol 2009;182:4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim K, Skora AD, Li Z et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid‐derived cells. Proc Natl Acad Sci USA 2014;111:11774–11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Meyer C, Cagnon L, Costa‐Nunes CM et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014;63:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Platten M, von Knebel Doeberitz N, Oezen I et al. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol 2014;5:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Munn DH, Sharma MD, Mellor AL. Ligation of B7‐1/B7‐2 by human CD4+ T cells triggers indoleamine 2,3‐dioxygenase activity in dendritic cells. J Immunol 2004;172:4100–4110. [DOI] [PubMed] [Google Scholar]
- 75. Ji RR, Chasalow SD, Wang L al. An immune‐active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012;61:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gao J, Shi LZ, Zhao H et al. Loss of IFN‐γ pathway genes in tumor cells as a mechanism of resistance to anti‐CTLA‐4 therapy. Cell 2016;167:397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shin D, Garcia‐Diaz A, Zaretsky J et al. Innate resistance of PD‐1 blockade through loss of function mutations in JAK resulting in inability to express PD‐L1 upon interferon exposure. J Immunother Cancer 2015;2(suppl 2):P311. [Google Scholar]
- 78. Shin DS, Zaretsky JM, Escuin‐Ordinas H et al. Primary resistance to PD‐1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chaput N, Lepage P, Coutzac C et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 2017;28:1368–1379. [DOI] [PubMed] [Google Scholar]
- 80. Sivan A, Corrales L, Hubert N et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science 2015;350:1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vétizou M, Pitt JM, Daillère R et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science 2015;350:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yuan J, Zhou J, Dong Z et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res 2014;2:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: An overview of rationale, preclinical evidence, and initial clinical data. Front Oncol 2015;5:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Peng W, Chen JQ, Liu C et al. Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov 2016;6:202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parsa AT, Waldron JS, Panner A et al. Loss of tumor suppressor PTEN function increases B7‐H1 expression and immunoresistance in glioma. Nat Med 2007;13:84–88. [DOI] [PubMed] [Google Scholar]
- 86. Taube JM, Young GD, McMiller TL et al. Differential expression of immune‐regulatory genes associated with PD‐L1 display in melanoma: Implications for PD‐1 pathway blockade. Clin Cancer Res 2015;21:3969–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Akbay EA, Koyama S, Carretero J et al. Activation of the PD‐1 pathway contributes to immune escape in EGFR‐driven lung tumors. Cancer Discov 2013;3:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Johnson DB, Lovly CM, Flavin M et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Marzec M, Zhang Q, Goradia A et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD‐L1, B7‐H1). Proc Natl Acad Sci USA 2008;105:20852–20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Riaz N, Havel JJ, Kendall SM et al. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti‐CTLA4 immunotherapy. Nat Genet 2016;48:1327–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Scharping NE, Menk AV, Whetstone RD et al. Efficacy of PD‐1 blockade is potentiated by metformin‐induced reduction of tumor hypoxia. Cancer Immunol Res 2017;5:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chae YK, Arya A, Malecek MK et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016;7:40767–40780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Champiat S, Dercle L, Ammari S et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti‐PD‐1/PD‐L1. Clin Cancer Res 2017;23:1920–1928. [DOI] [PubMed] [Google Scholar]
- 94. Kato S, Goodman A, Walavalkar V et al. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Restifo NP, Marincola FM, Kawakami Y et al. Loss of functional beta 2‐microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst 1996;88:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cabrera CM, Jiménez P, Cabrera T et al. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: Beta2‐microglobulin inactivation in MSI‐positive tumors and LMP7/TAP2 downregulation in MSI‐negative tumors. Tissue Antigens 2003;61:211–219. [DOI] [PubMed] [Google Scholar]
- 97. Tran E, Robbins PF, Lu YC et al. T‐cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med 2016;375:2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Anagnostou V, Smith KN, Forde PM et al. Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov 2017;7:264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Landsberg J, Kohlmeyer J, Renn M et al. Melanomas resist T‐cell therapy through inflammation‐induced reversible dedifferentiation. Nature 2012;490:412–416. [DOI] [PubMed] [Google Scholar]
- 100. Zaretsky JM, Garcia‐Diaz A, Shin DS et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016;375:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sucker A et al. Acquired IFNγ resistance impairs anti‐tumor immunity and gives rise to T‐cell‐resistant melanoma lesions. Nat Commun 2017;8:15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fourcade J, Sun Z, Benallaoua M et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010;207:2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Koyama S, Akbay EA, Li YY et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sakuishi K, Apetoh L, Sullivan JM et al. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med 2010;207:2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Huang CT, Workman CJ, Flies D et al. Role of LAG‐3 in regulatory T cells. Immunity 2004;21:503–513. [DOI] [PubMed] [Google Scholar]
- 106. Woo SR, Turnis ME, Goldberg MV et al. Immune inhibitory molecules LAG‐3 and PD‐1 synergistically regulate T‐cell function to promote tumoral immune escape. Cancer Res 2012;72:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. George S, Miao D, Demetri GD et al. Loss of PTEN is associated with resistance to anti‐PD‐1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017;46:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J 2010;16:399–403. [DOI] [PubMed] [Google Scholar]
- 109. Blank CU, Haanen JB, Ribas A et al. Cancer immunology. The “cancer immunogram.” Science 2016;352:658–660. [DOI] [PubMed] [Google Scholar]
- 110. Linnemann C, van Buuren MM, Bies L et al. High‐throughput epitope discovery reveals frequent recognition of neo‐antigens by CD4+ T cells in human melanoma. Nat Med 2015;21:81–85. [DOI] [PubMed] [Google Scholar]
- 111. Lou Y, Diao L, Cuentas ER et al. Epithelial‐mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res 2016;22:3630–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol 2012;196:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hugo W, Zaretsky JM, Sun L et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Roh W, Chen PL, Reuben A et al. Integrated molecular analysis of tumor biopsies on sequential CTLA‐4 and PD‐1 blockade reveals markers of response and resistance. Sci Transl Med 2017;9:eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chen PL, Roh W, Reuben A et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 2016;6:827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Huang AC, Postow MA, Orlowski RJ et al. T‐cell invigoration to tumour burden ratio associated with anti‐PD‐1 response. Nature 2017;545:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res 2016;22:1865–1874. [DOI] [PubMed] [Google Scholar]
- 118. Weber JS, D'Angelo SP, Minor D et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‐CTLA‐4 treatment (CheckMate 037): A randomised, controlled, open‐label, phase 3 trial. Lancet Oncol 2015;16:375–384. [DOI] [PubMed] [Google Scholar]
- 119. Postow MA, Chesney J, Pavlick AC et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wolchok JD, Kluger H, Callahan MK et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Vacchelli E, Aranda F, Eggermont A et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2014;3:e957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015;14:561–584. [DOI] [PubMed] [Google Scholar]
- 123. Wang DY, ERoglu Z, Ozgun A et al. Clinical features of acquired resistance to anti‐PD‐1 therapy in advanced melanoma. Cancer Immunol Res 2017;5:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell‐inflamed tumor microenvironment. Trends Immunol 2015;36:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen H, Sun H, You F et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011;147:436–446. [DOI] [PubMed] [Google Scholar]
- 126. Corrales L, Glickman LH, McWhirter SM et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015;11:1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ghoneim HE, Fan Y, Moustaki A et al. De novo epigenetic programs inhibit PD‐1 blockade‐mediated T cell rejuvenation . Cell 2017;170:142–157.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Boni A, Cogdill AP, Dang P et al. Selective BRAFV600E inhibition enhances T‐cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010;70:5213–5219. [DOI] [PubMed] [Google Scholar]
- 129. Frederick DT, Piris A, Cogdill AP et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]