

Abstract
Lessons Learned.
A phase I study of the pan‐class I phosphoinositide 3‐kinase inhibitor pilaralisib (in capsule formulation) in advanced solid tumors established the maximum tolerated dose as 600 mg once daily.
The current study investigated pilaralisib in tablet formulation.
Pilaralisib tablets were associated with a favorable safety profile and preliminary antitumor activity.
Based on pharmacokinetic data, the recommended phase II dose of pilaralisib tablets was established as 400 mg once daily.
Background.
A phase I trial of pilaralisib, an oral pan‐class I phosphoinositide 3‐kinase (PI3K) inhibitor, established the maximum tolerated dose (MTD) of the capsule formulation in patients with advanced solid tumors as 600 mg once daily. This phase I study investigated pilaralisib in tablet formulation.
Materials and Methods.
Patients with advanced solid tumors received pilaralisib tablets (100–600 mg once daily). Primary endpoints were MTD and safety; secondary and exploratory endpoints included pharmacokinetics (PK), pharmacodynamics, and efficacy.
Results.
Twenty‐two patients were enrolled. No dose‐limiting toxicities (DLTs) were reported. The most common treatment‐related adverse events were diarrhea (40.9%), fatigue (40.9%), decreased appetite (22.7%), and hyperglycemia (22.7%). Pilaralisib plasma exposure did not appear to increase dose‐proportionally. Steady‐state exposure was higher with pilaralisib tablet formulation at 400 mg than with pilaralisib capsule formulation at 400 or 600 mg (mean area under the curve [AUC0–24] 2,820,000 ng × h/mL vs. 2,653,000 and 1,930,000 ng × h/mL, respectively). Of 18 evaluable patients, 2 (11.1%) had a partial response (PR).
Conclusion.
Pilaralisib tablets were associated with a favorable safety profile and preliminary antitumor activity. MTD was not determined. The recommended phase II dose for pilaralisib tablets, based on PK data, was 400 mg once daily.
Abstract
经验总结
• 一项在晚期实体瘤中对泛I类磷脂酰肌醇‐3激酶抑制剂Pilaralisib(胶囊剂型)进行的I期研究确定了最大耐受剂量为600 mg每日一次。
• 当前研究对Pilaralisib片剂剂型进行了研究。
• Pilaralisib片剂与良好的安全性特征和初步抗肿瘤活性相关。
• 基于药代动力学数据, 将Pilaralisib片剂的推荐II期剂量确定为400 mg每日一次。
摘要
背景.Pilaralisib[一种口服泛I类磷脂酰肌醇‐3激酶(PI3K)抑制剂]的一项I期研究确定胶囊剂型在晚期实体瘤患者中的最大耐受剂量(MTD)为600 mg每日一次。本项I期研究对Pilaralisib片剂剂型进行了研究。
材料和方法.晚期实体瘤患者接受Pilaralisib片剂给药(100–600 mg每日一次)。主要终点是MTD和安全性;次要和探索性终点包括药代动力学(PK)、药效学和疗效。
结果.22例患者入组。未报告剂量限制性毒性(DLT)。最常见的治疗相关不良事件为腹泻(40.9%)、疲乏(40.9%)、食欲下降(22.7%)和高血糖症(22.7%)。Pilaralisib血浆暴露量未呈现与剂量成比例增加。400mg Pilaralisib片剂剂型的稳态暴露量高于400或600mg Pilaralisib胶囊剂型的稳态暴露量[平均曲线下面积(AUC0–24)分别为2 820 000 ng x h/mL、2 653 000和1 930 000 ng x h/mL]。在18例可评价患者中, 2例(11.1%)达到部分缓解(PR)。
结论.Pilaralisib片剂与良好的安全性特征和初步抗肿瘤活性相关。未确定MTD。基于PK数据, Pilaralisib片剂的推荐II期剂量为400 mg每日一次。
Discussion
The PI3K pathway has been implicated in the pathogenesis of solid tumors, and several PI3K inhibitors in development have shown evidence of clinical antitumor activity. Pilaralisib (SAR245408) is a highly selective and reversible pan‐class I PI3K inhibitor. A phase I study of pilaralisib capsules in 69 patients with advanced solid tumors (NCT00486135) established the MTD as 600 mg once daily. In addition, pharmacodynamic inhibition of the PI3K pathway and one PR were observed. The current study investigated pilaralisib in tablet formulation.
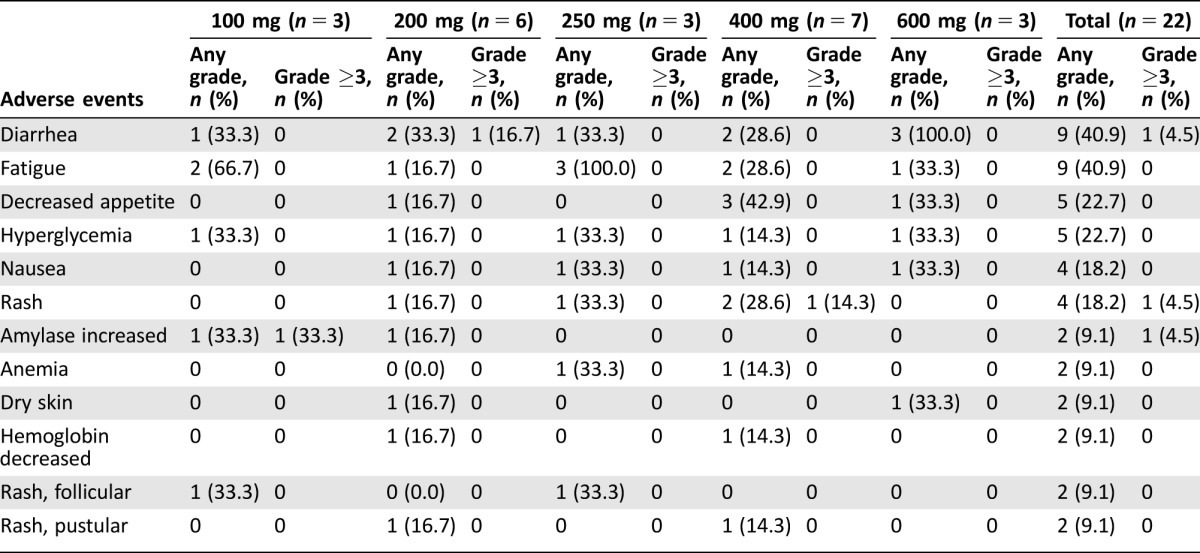
Twenty‐two patients were enrolled and received at least one dose of pilaralisib tablets. The median duration of exposure was 56.0 days. No DLTs were observed, and the MTD was not established. Safety findings with pilaralisib tablets were generally consistent with previous findings in patients with solid tumors who received the capsule formulation. The most common treatment‐related toxicities included gastrointestinal toxicities, fatigue, and hyperglycemia (Table 1), consistent with other PI3K inhibitors in clinical development.
Table 1. Treatment‐related adverse events occurring in >5% of patients treated with pilaralisib tablets once daily (graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0).
In PK analyses, pilaralisib plasma exposure did not appear to increase in a dose‐proportional manner and steady‐state plasma exposure appeared to be slightly higher with pilaralisib 400 mg tablets compared with capsules at 400 mg and 600 mg once daily (Fig. 1); however, small patient numbers prevented firm conclusions. Pharmacodynamic impact on glucose homeostasis was evaluated in plasma samples from patients receiving pilaralisib tablets at 250, 400, and 600 mg (n = 2, n = 3, and n = 2, respectively); no impact was observed on C‐peptide or glucose levels in the limited sample set analyzed. Similarly, no impact was observed on C‐peptide or glucose levels in plasma samples from five patients with lymphoma receiving 600 mg pilaralisib capsules.
Figure 1.

Mean (+ standard deviation [SD]) plasma concentration of pilaralisib administered once daily in patients with solid tumors on Cycle 1, Day 1 (A), and Cycle 1 Day 28 (B). SD was not calculable for 100 mg and 250 mg tablets at Cycle 1, Day 1, for the 24‐hour time point or for 600 mg tablets at Cycle 1, Day 28 (n = 2 for each).
Of 18 evaluable patients, two patients (11.1%) had a PR (overall response rate 11.1%) and six patients (33.3%) had stable disease as best response. Median progression‐free survival was 1.9 months (90% confidence interval 1.7–5.5); three patients (16.7%) were alive and progression free at 6 months.
Based on PK data, the recommended phase II dose of pilaralisib tablets was established as 400 mg once daily.
Trial Information
- Disease
Advanced cancer/Solid tumor only
- Stage of Disease/Treatment
Metastatic/Advanced
- Prior Therapy
No designated number of regimens
- Type of Study – 1
Phase I
- Type of Study – 2
Null
- Primary Endpoint
Maximum tolerated dose
- Primary Endpoint
Toxicity
- Secondary Endpoint
Pharmacokinetics
- Secondary Endpoint
Pharmacodynamic
- Secondary Endpoint
Efficacy
- Additional Details of Endpoints or Study Design
- This was a phase I, multicenter, open‐label, single‐arm study. The primary endpoints were MTD and safety. Secondary or exploratory endpoints included pharmacokinetics, pharmacodynamics, and efficacy. Eligible patients were aged ≥18 years, with an Eastern Cooperative Oncology Group (ECOG) performance status ≤2 and histologically confirmed metastatic or unresectable solid tumors. Patients were required to have adequate organ and bone marrow function, fasting plasma glucose <160 mg/dL, and hemoglobin A1c <8%. Patients who had previously received treatment with a PI3K inhibitor were excluded. All patients provided written informed consent.
- Panel for Copy Number Alterations in Squamous Non‐Small Cell Lung Cancer
- DNA was isolated from macrodissected, 5‐micron, formalin‐fixed, paraffin‐embedded tumor tissue sections enriched for 50% tumor cell content, using standard protocols. DNA was analyzed by droplet digital polymerase chain reaction (ddPCR; BioRad, Mountain View, CA) for copy number amplifications at the following 12 genic loci: BCL2, CCND1, CDK4, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, FGFR4, MET, PDGFRA, and PIK3CA. Two locus‐specific TaqMan minor groove binder probes (ThermoFisher Scientific, Waltham, MA) were used to measure the concentration of target‐specific gene sequences relative to the concentration of reference loci. Quantification of these concentrations provided a ratio of the target to reference loci and was expressed as an absolute copy number. To minimize aneusomy at the reference loci, the reference locus (RPP30, c.16_c.77, chr10q23) was interrogated against two additional reference loci (AP3B1, c.2578–23605_c.2578_23543, chr5q14; NFAT5, c.327_c.395, chr16q22) such that each target locus was normalized to the average concentration of the three reference loci.
- Whole‐Exome Sequencing
- Whole‐exome sequencing was performed as previously described [Nat Med 2014;20:682–688].
- PTEN Immunohistochemistry
- Immunohistochemistry for PTEN was performed using clone 138G6 (Cell Signaling Technology, Danvers, MA) within a Clinical Laboratory Improvement Amendments‐certified immunohistochemistry lab. PTEN loss was defined as <10% tumor cells staining at any intensity [PLoS One 2012; 7:e30427].
- Investigator's Analysis
Drug tolerable, hints of efficacy
Drug Information for Phase I Pilaralisib
- Drug 1
- Generic/Working Name
Pilaralisib
- Drug Type
Small molecule
- Drug Class
PI3 kinase
- Dose
100–600 mg (tablets) per day
- Route
Oral (p.o.)
- Schedule of Administration
Once daily, continuous 28‐day cycles
Patient Characteristics for Phase I Pilaralisib
- Number of Patients, Male
8
- Number of Patients, Female
14
- Stage
-
Stage II — 1
Stage III — 2
Stage IIIA — 2
Stave IV — 9
Unknown stage — 8
- Age
Median (range), years: 63.0 (27–86)
- Number of Prior Systemic Therapies
-
Median (range): 3.0 (0–12)
0–1 prior systemic therapies — 2 patients
2–4 prior systemic therapies — 14 patients
≥5 prior systemic therapies — 6 patients
- Performance Status: ECOG
-
0 — 10
1 — 12
2 —
3 —
Unknown —
- Other
- Median Time from Initial Diagnosis
Years (range): 2.63 (0.8–14.2)
- Patients with Prior Anticancer Treatment
-
Prior systemic anticancer therapy alone — 11
Prior radiation therapy alone — 1
Prior systemic anticancer therapy or radiation — 22
Both prior systemic anticancer therapy and radiation — 10
Cancer Types or Histologic Subtypes
- Adenocarcinoma
6
- Bladder cancer
1
- Carcinosarcoma
1
- Chordoma (pelvis)
1
- Colorectal adenocarcinoma
1
- Ductal with lobular component (breast)
1
- Endometrial cancer
3
- Leiomyosarcoma (uterus)
1
- Mesothelioma
2
- Non-small cell lung cancer
2
- Ovarian carcinoma
1
- Prostate cancer
1
- Sarcoma (synovial)
1
Primary Assessment Method for Phase I Pilaralisib
- Number of Patients Enrolled
22
- Number of Patients Evaluable for Toxicity
22
- Number of Patients Evaluated for Efficacy
18
- Evaluation Method
RECIST 1.0
- Response Assessment CR
0%
- Response Assessment PR
11.1%
- Response Assessment SD
33.3%
- Response Assessment PD
55.6%
- (Median) Duration Assessments PFS
1.9 months; CI, 1.7–5.5
- (Median) Duration Assessments Duration of Treatment
56.0 days
Assessment, Analysis, and Discussion
- Completion
Study completed
- Investigator's Assessment
Drug tolerable, hints of efficacy
The phosphoinositide 3‐kinase (PI3K) pathway is pivotal in the growth and survival of normal cells, and dysregulation of this pathway has been implicated in the pathogenesis of various solid tumors [1], [2], [3]. Several PI3K inhibitors in development have shown antitumor activity in patients with advanced solid tumors [4], [5], [6].
Pilaralisib is a highly selective and reversible pan‐class I PI3K inhibitor. This single‐arm study was part of a multicenter, open‐label, phase I study (NCT00486135), investigating single‐agent pilaralisib capsules in patients with advanced solid tumors, which established the maximum tolerated dose (MTD) for the capsule formulation at 600 mg once daily (QD) [5]. Pharmacodynamic analyses demonstrated that pilaralisib treatment inhibited the PI3K pathway, and one patient with advanced non‐small cell lung cancer had a partial response (PR). The cohort described in this report investigated a tablet formulation of pilaralisib. Primary endpoints were MTD and safety. Secondary and exploratory endpoints included pharmacokinetics (PK), pharmacodynamics and efficacy.
In total, 22 patients with solid tumors received pilaralisib tablets QD at 100 mg (n = 3), 200 mg (n = 6), 250 mg (n = 3), 400 mg (n = 7), or 600 mg (n = 3). Median age was 63.0 years (range 27–86), and median number of prior systemic therapies was 3 (range 0–12). All 22 patients discontinued study treatment. The most common reasons for study discontinuation were disease progression as defined by RECIST version 1.0 (15 patients, 68.2%), disease progression based on clinical deterioration (three patients, 13.6%), and adverse event (AE; two patients, 9.1%). One patient was enrolled into an extension study (TED12414).
The mean (± standard deviation) duration of exposure was 158.0 days (±258.97 days) and ranged from 6 to 1,009 days. Median duration of exposure was 56.0 days. Median number of cycles received was 2 (range 1–36). The majority of patients (90.9%) received >90% of the planned doses of pilaralisib.
No dose‐limiting toxicities were reported. Treatment‐related grade ≥3 AEs reported were increased alanine aminotransferase, increased amylase, increased aspartate aminotransferase, diarrhea, increased gamma‐glutamyl transferase, increased lipase, and rash (one patient each, all grade 3). Hepatic toxicity was reported in six patients (27.3%) and was grade ≥3 in four patients (18.2%). There were no events of bilirubin increased and no cases of Hy's law. There was no evidence of cardiovascular toxicity as determined in ECG assessments. Seven patients (31.8%) reported one or more serious AEs (SAEs), of which only disease progression (three patients, 13.6%) was reported in more than one patient. All SAEs were assessed to be either not related or unlikely to be related to treatment. Six patients (27.3%) had one or more AEs leading to a dose delay or interruption. Two patients (9.1%) permanently discontinued treatment because of an AE (grade 5 encephalopathy and grade 4 ischemic stroke); both AEs were not treatment related. No deaths occurred during the study; there were four deaths within 30 days after the last dose of study drug, all related to disease progression.
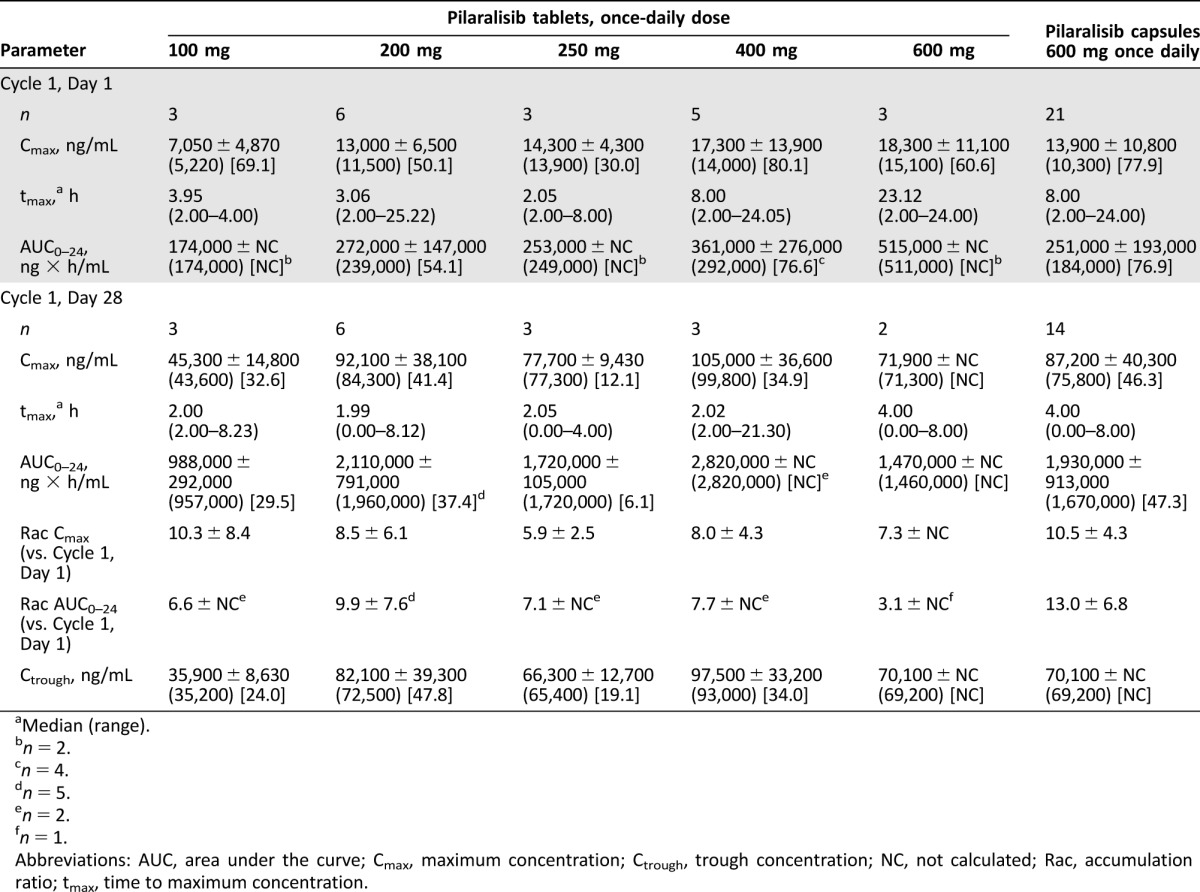
Figure 1 shows the mean pilaralisib plasma concentration‐time profile after the first single dose on Cycle 1, Day 1, and repeated daily dosing at Cycle 1, Day 28. After repeated daily administration of pilaralisib tablets (100–600 mg), steady state was reached by Cycle 1, Day 28, and median time to maximum concentration ranged from 1.99 to 23.1 hours (Table 2). The mean accumulation ratio for maximum concentration (Cmax) and area under the curve (AUC0–24) for Cycle 1, Day 28, compared with Cycle 1, Day 1, ranged from 5.9‐fold to 10.3‐fold and 3.1‐fold to 9.9‐fold, respectively. Exposure (Cmax and AUC0–24) did not appear to increase dose proportionally for the 100–600 mg dose levels, but conclusions were limited by the small patient numbers. On Cycle 1, Day 28, exposure was higher with pilaralisib 400 mg tablets administered QD compared with 600 mg tablets QD and the capsule formulation administered at 400 mg and 600 mg QD (mean AUC0–24 2,820,000 [n = 2] vs. AUC0–24 1,470,000 [n = 2], 2,653,000 [n = 2] and 1,930,000 ng × h/mL [n = 14], respectively).
Table 2. Pilaralisib pharmacokinetic parameters in patients with solid tumors. Mean ± standard deviation (geometric mean) [% coefficient of variation].
Median (range).
n = 2.
n = 4.
n = 5.
n = 2.
n = 1.
Abbreviations: AUC, area under the curve; Cmax, maximum concentration; Ctrough, trough concentration; NC, not calculated; Rac, accumulation ratio; tmax, time to maximum concentration.
The impact of pilaralisib tablets on glucose homeostasis was evaluated in post‐dose plasma samples after overnight fasting (up to 2 hours after dosing) on Days 1, 8, and 28. Pilaralisib had no effect on glucose and C‐peptide levels. No differences were observed between the capsule and tablet formulation.
Of 18 evaluable patients, two patients (11.1%) had a PR (overall response rate 11.1%): a female patient aged 53 years with poorly differentiated vaginal adenocarcinoma who had progression‐free survival (PFS) of 33.0 months (200 mg cohort; Fig. 2), and a female patient aged 41 years with squamous cell lung carcinoma who had PFS of 27.6 months (250 mg cohort). A squamous lung cancer copy number panel performed on a prior pneumonectomy specimen demonstrated PIK3CA amplification (copy number alteration 4.6; >4 considered amplified), with weak focal staining for PTEN in tumor cells. Mutational analysis on whole exome sequencing demonstrated TP53 P151H, KRAS T20M, and PIK3C2B R296Q. A supraclavicular lymph node biopsy performed after progression was inadequate for genomic analyses, but tumor cells identified were negative for PTEN staining. PIK3CA amplification, coupled with PTEN loss and PIK3C2B mutation [7], possibly caused PI3K dependence that resulted in the clinical benefit observed. The patient had PR at 11.6 months, maintained for 16 months (maximum reduction in tumor burden 50.3%). Six patients (33.3%) had stable disease as best response. Median PFS was 1.9 months (90% confidence interval 1.7–5.5); three patients (16.7%) were alive and progression free at 6 months.
Figure 2.

Partial response in a patient aged 54 years with poorly differentiated vaginal adenocarcinoma treated with pilaralisib tablets 200 mg once daily. Sequential computed tomography scans of the abdomen show a 32% interval reduction of a left para‐aortic soft tissue mass in Cycle 8, compared with baseline. Overall tumor reduction was 21% in Cycle 8.
In summary, pilaralisib tablets were associated with a favorable safety profile, similar to that observed in patients who received the capsule formulation [5]. The most common treatment‐related toxicities included gastrointestinal toxicities, fatigue, and hyperglycemia (Table 1), consistent with other PI3K inhibitors in clinical development [4], [6], [8], [9], [10], [11]. Steady‐state plasma exposure was higher with pilaralisib 400 mg tablets than with 400 and 600 mg capsules [5]. Neither tablets nor capsules affected glucose homeostasis [5]. Pilaralisib tablets showed preliminary antitumor activity, including two PRs. The MTD for pilaralisib tablets was not determined and the recommended phase II dose, based on PK data, is 400 mg QD.
Figures and Tables
Acknowledgments
This study was funded by Sanofi and Exelixis. The authors received editorial support from Simone Blagg of MediTech Media, funded by Sanofi.
Footnotes
ClinicalTrials.gov Identifier: NCT00486135
Sponsor(s): Sanofi, Exelixis
Principal Investigator: Geoffrey I. Shapiro
IRB Approved: Yes
Disclosures
Jordi Rodon: Peptomyc, Novartis, Eli Lilly, Orion, Servier, (C/A), Bayer, Novartis (RF); Joanne Lager: Sanofi (E, OI); Jason Jiang: Sanofi (E, OI); Eliezer M. Van Allen: Invitae, Genome Medical, Tango Therapeutics (C/A), Novartis, Bristol‐Myers Squibb (RF),Genome Medical, Tango Therapeutics (OI); Nikhil Wagle: Novartis (C/A, RF), Foundation Medicine (OI); Lynette M. Sholl: GfK (C/A); Geoffrey I. Shapiro: Eli Lilly, Pfizer, G1 Therapeutics, Merck/EMD Serono, Roche (C/A); Lilly, Pfizer, Merck/EMD Serono (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010;28:1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Samuels Y, Wang Z, Bardelli A et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 3. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer 2011;11:289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papadopoulos KP, Tabernero J, Markman B et al. Phase I safety, pharmacokinetic and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res 2014;20:2445–2456. [DOI] [PubMed] [Google Scholar]
- 5. Shapiro GI, Rodon J, Bedell C et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan‐class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2014;20:233–245. [DOI] [PubMed] [Google Scholar]
- 6. Bendell C, Rodon J, Burris HA et al. Phase I, dose‐escalation study of BKM120, an oral pan‐Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282–290. [DOI] [PubMed] [Google Scholar]
- 7. Wheeler M, Domin J. Recruitment of the class II phosphoinositide 3‐kinase C2beta to the epidermal growth factor receptor: Role of Grb2. Mol Cell Biol 2001;21:6660–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Markman B, Tabernero J, Krop I et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol‐3‐kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol 2012;23:2399–2408. [DOI] [PubMed] [Google Scholar]
- 9. Arkenau HT, Fields Jones S, Kurkjian C et al. The PI3K/mTOR inhibitor BEZ235 given twice daily for the treatment of patients (pts) with advanced solid tumors. J Clin Oncol 2012;30(suppl 15):3097A. [Google Scholar]
- 10. Wagner AJ, Bendell JC, Dolly S et al. A first‐in‐human phase I study to evaluate GDC‐0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors. J Clin Oncol 2011;29(suppl 15):3020A. [Google Scholar]
- 11. Hong DS, Bowles DW, Falchook GS et al. A multicenter phase I trial of PX‐866, an oral irreversible phosphatidylinositol 3‐kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2012;18:4173–4182. [DOI] [PubMed] [Google Scholar]