

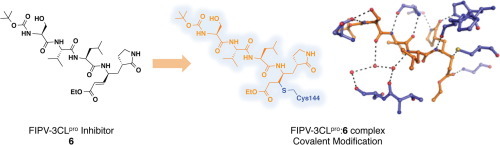
Graphical abstract
Keywords: Coronavirus, 3CLpro, Feline infectious peritonitis, Structure-based drug design, Peptidomimetics
Abstract
Feline infectious peritonitis (FIP) is a deadly disease that effects both domestic and wild cats and is caused by a mutation in feline coronavirus (FCoV) that allows the virus to replicate in macrophages. Currently, there are no treatments or vaccines available for the treatment of FIP even though it kills approximately 5% of cats in multi-cat households per year. In an effort to develop small molecule drugs targeting FIP for the treatment of cats, we screened a small set of designed peptidomimetic inhibitors for inhibition of FIPV-3CLpro, identifying two compounds with low to sub-micromolar inhibition, compound 6 (IC50 = 0.59 ± 0.06 μM) and compound 7 (IC50 = 1.3 ± 0.1 μM). We determined the first X-ray crystal structure of FIPV-3CLpro in complex with the best inhibitor identified, compound 6, to a resolution of 2.10 Å to better understand the structural basis for inhibitor specificity. Our study provides important insights into the structural requirements for the inhibition of FIPV-3CLpro by peptidomimetic inhibitors and expands the current structural knowledge of coronaviral 3CLpro architecture.
Feline infectious peritonitis (FIP) was first described in 1963 and was initially termed ‘chronic fibrinois peritonitis’.1 Today, FIP is a major killer of domestic cats, especially kittens—where over 50% of FIP infections occur in cats younger than 12 months.2 FIP is caused by feline coronavirus (FCoV), which belongs to the alphacoronavirus phylogeny. FIP affects domesticated and wild cats globally and is especially prevalent in multi-cat households because it is spread through the feces. Studies have shown that at least 50% of American cats have antibodies against FCoV.3 Typically, FCoV infection results in feline enteric coronavirus (FEC), which is highly prevalent in cats and causes asymptomatic to mild enteritis and diarrhea.4, 5 FEC is known to replicate in enterocytes; however, spontaneous mutation of a particular region in the FCoV genome allows for the phagocytosis of the FCoV virions into macrophages and their subsequent replication.6, 7, 8 The ability of mutated FCoV to replicate in macrophages allows the coronavirus to spread throughout the entire cat, ultimately resulting in FIP virus (FIPV).9 Approximately 5% of cats in multi-cat households develop fatal FIPV.10 Currently, there are no therapeutics on the market for the treatment of this deadly feline virus. Non-domestic cats are also affected by FIP; cheetahs are known to be highly susceptible to the disease, having a possible predisposition as a consequence of a genetic deficiency in their cellular immunity.11
Coronaviruses are positive-sense RNA viruses with some of the largest viral genomes reported to date, ranging from 27 to 32 kilobases in length. The coronaviral genome has two long open reading frames (ORFs), ORF1a and ORF1b, which are related by a −1 ribosomal frame-shift. Upon translation, ORF1a and ORF1b produce the coronaviral polyproteins pp1a and pp1ab.12, 13 The viral polyproteins pp1a and pp1ab are then processed by two cysteine proteases, the papain-like protease (PLpro) and the 3C-like protease (3CLpro), which is also known as the main protease (Mpro) or nsp5. Both PLpro and 3CLpro are essential for the coronaviral life cycle, making them appealing targets for the development of anti-coronaviral therapeutics.
Toward the goal of developing a therapeutic to target FIPV, we developed an over-expression system for FIPV-3CLpro and purified the enzyme to homogeneity and to high-yield using hydrophobic-interaction and anion-exchange chromatography (see Supplementary material). We then tested a series of eleven peptidomimetic compounds (1–11, Table 1 ) using well established methods in our lab,14 that we recently designed and synthesized for the inhibition of Middle East Respiratory Syndrome (MERS) 3CLpro, for FIPV-3CLpro inhibition.15 This set of peptidomimetic inhibitors contains a Michael acceptor capable of reacting with the catalytic cysteine (Cys144) of FIPV-3CLpro, and they are similar to other previously reported peptidyl inhibitors of various 3CLpro’s.16, 17, 18, 19 The synthesis of the eleven tested peptidomimetics in Table 1 has been previously reported.15, 20
Table 1.
Peptidomimetic inhibitors of FIPV-3CLpro
| Compd | Structure | %I (50 μM) | %I (10 μM) | %I (5 μM) |
|---|---|---|---|---|
| 1 | 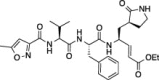 |
54.8 ± 5.7 | 36.4 ± 0.4 | 30.1 ± 1.9 |
| 2 | 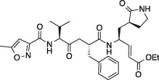 |
39.2 ± 0.5 | 12.8 ± 2.2 | 15.6 ± 0.8 |
| 3 | 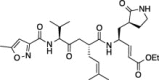 |
52.3 ± 1.4 | 36.0 ± 3.9 | 31.3 ± 1.1 |
| 4 | 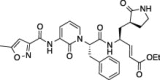 |
55.6 ± 1.9 | 34.4 ± 2.9 | 32.7 ± 1.3 |
| 5 | 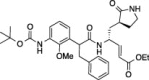 |
17.6 ± 1.2 | 11.2 ± 3.4 | 2.9 ± 2.7 |
| 6 | 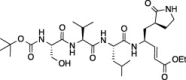 |
99.7 ± 0.2 | 92.1 ± 4.3 | 89.8 ± 2.1 |
| 7 | 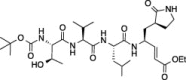 |
99.4 ± 0.02 | 92.2 ± 1.4 | 85.8 ± 4.9 |
| 8 | 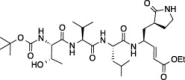 |
94.7 ± 1.3 | 69.1 ± 1.7 | 53.7 ± 4.0 |
| 9 | 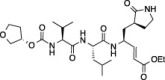 |
82.5 ± 1.8 | 33.3 ± 0.2 | 26.9 ± 0.5 |
| 10 | 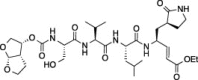 |
97.0 ± 0.9 | 58.6 ± 3.9 | 37.0 ± 2.4 |
| 11 | 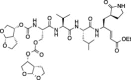 |
66.3 ± 0.2 | 20.5 ± 1.9 | 13.4 ± 3.7 |
Compounds (1–11) were initially tested at three concentrations (5 μM, 10 μM, and 50 μM) to identify inhibitors of FIPV-3CLpro and the results are reported in Table 1. A trend is immediately observed in the six compounds with the highest inhibition of FIPV-3CLpro (6–11), that is, having greater than 60% inhibition at a concentration of 50 μM, where a leucine residue is favored in the P 2 position of the inhibitor over phenyl or isopentenyl groups (1–3). Analysis of the eleven cleavage sites within the FIPV replicase polyprotein 1ab shows that nine of the eleven cleavage sites have a leucine residue at the P 2 position and none have a phenylalanine residue (Fig.1 A), which is the P 2 residue that is present in compounds 1, 2 and 4, 5.21 Furthermore, the two best peptidomimetic inhibitors, compounds 6 and 7, have either a serine or threonine residue at the P 4 position. A P 4 serine is present in two of the eleven FIPV-3CLpro cleavage sites, while a P 4 threonine is present in four of the eleven cleavage sites (Fig.1A).
Figure 1.
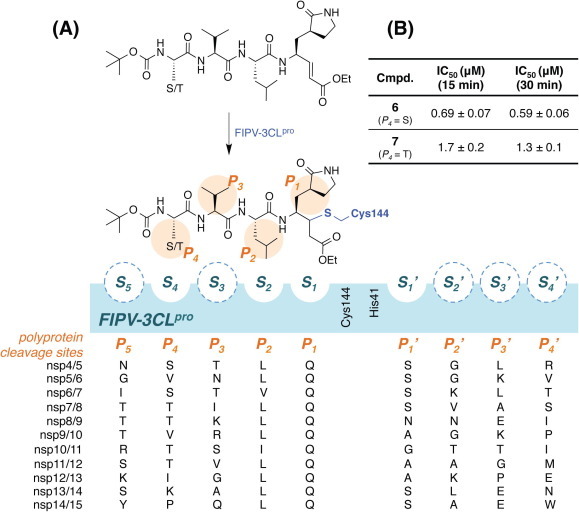
FIPV-3CLpro cleavage sites in polyprotein 1ab and inhibition by compounds 6 and 7. (A) The eleven polyprotein 1ab recognition sequences for FIPV-3CLpro are shown from P5 to P4′ under the blue shaded box in their respective binding locations. FIPV-3CLpro is represented by the blue shaded box where the subsites are represented as pockets and labeled accordingly. Subsites with no clear residue preferences are outlined by a dashed line, indicating they may not exist. Peptidomimetic inhibitor 6 (P4 = S) or 7 (P4 = T) binds to FIPV-3CLpro via nucleophilic attack of Cys144 at the β-carbon of the α,β-unsaturated ethyl ester, where the pyrrolidinonyl methyl acts as the P1 residue, Leu as the P2 residue, Val as the P3 residue, and either Thr or Ser as the P4 residue. (B) IC50 values for 6 and 7 against FIPV-3CLpro after both 15 and 30 min incubation periods.
Comparison of compounds 6 and 10, which are only different at the N-termini where 6 has a tert-butyl carbamate and 10 has a bis-tetrahydrofuranyl carbamate, shows that the increase in steric bulk associated with the N-terminus of 10 decreases FIPV-3CLpro inhibition at every concentration of 10. Furthermore, comparison of 10 to 11, which are only different in that 10 has a free P 4 serine while 11 has a P 4 bis-tetrahydrofuranyl carbonate, shows that the increase in steric bulk at P 4 of 11 decreases FIPV-3CLpro inhibition at every concentration of 11. Comparison of 7 to 8, which are only different in that 7 has a P 4 threonine and 8 has a P 4 l-allo-threonine, shows that the stereochemistry of the P 4 residue is important for good FIPV-3CLpro inhibition, where 7 has increased inhibition of FIPV-3CLpro at all inhibitor concentrations tested.
We then determined the IC50 values for the two best peptidomimetic inhibitors of FIPV-3CLpro, 6 and 7. These compounds were chosen because they are the only compounds in the series that have greater than 80% inhibition of FIPV-3CLpro at a concentration of 5 μM. The IC50 values of 6 and 7 were determined after pre-incubation of FIPV-3CLpro with the respective peptidomimetic inhibitor at both 15 and 30 min. We found compound 6, which showed slightly better inhibition of FIPV-3CLpro than compound 7, to have an IC50 of 0.69 ± 0.07 μM after 15 min of pre-incubation and an IC50 of 0.59 ± 0.06 μM after 30 min of pre-incubation. We found 7 to have an IC50 of 1.7 ± 0.2 μM after 15 min of pre-incubation and 1.3 ± 0.1 μM after 30 min of pre-incubation (Fig.1B).
To gain a more complete structural understanding of how the peptidomimetic compounds bind to and inhibit FIPV-3CLpro, we crystallized and determined the X-ray structure of FIPV-3CLpro bound to compound 6 via a covalent bond with the active site cysteine (Cys144). The statistics for X-ray data collection, processing, and refinement are given in the Supplementary material.22, 23, 24 The X-ray structure of the FIPV-3CLpro:6 complex was determined to 2.1 Å resolution (Rpim = 4.0%, Rwork = 17.6% Rfree = 23.0%) with four monomers in the asymmetric unit (Fig.2 A and B). The four monomers in the asymmetric unit represent two complete, biologically active dimers of the FIPV-3CLpro:6 complex. Electron density associated with compound 6 is clearly visible in final, FO − FC electron density omit maps contoured to +3.0σ (Fig.2C).
Figure 2.
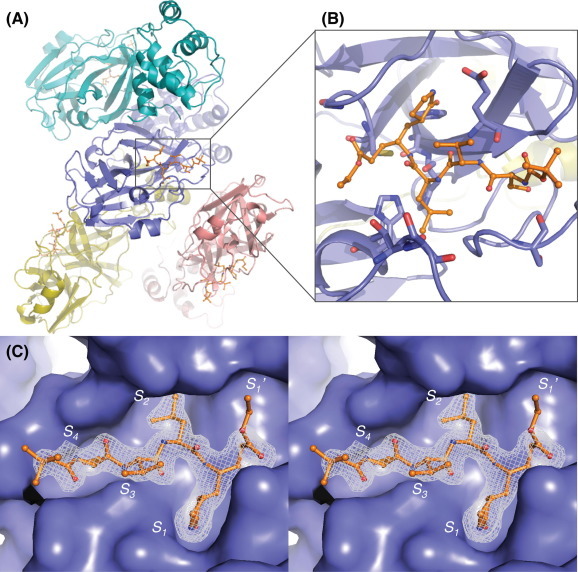
X-ray crystal structure of the FIPV-3CLpro:6 complex (PDB ID = 4ZRO). (A) The four monomers of FIPV-3CLpro are shown as ribbons and are colored teal, slate blue, yellow and pink. One of the biologically relevant FIPV-3CLpro dimers is shown with chain A colored in slate blue and chain B colored in teal. The two other monomers, which also form biologically relevant dimers though not depicted here in this representation of the asymmetric unit, are shown as chain C (pink) and D (yellow). 6 is represented as ball and stick with its carbon atoms colored orange, nitrogens colored blue and oxygens colored red. (B) Zoomed image of 6 within the active site of chain A. The residues of chain A that interact with 6 are colored according to atom and shown as sticks: His41, Thr47, Ser48, Gly142, Cys144, His162, His163, Glu165, Ser189. (C) Wall-eye stereoview of 6 bound in the active site of FIPV-3CLpro chain A, which is represented as a solvent accessible surface colored in slate blue. An electron density omit map (FO − FC) surrounding compound 6 is shown in grey mesh and it is contoured to +3.0σ.
Compound 6 binds in the active site of FIPV-3CLpro, mimicking the native polyprotein substrate (Figure 2, Figure 3 ). The pyrrolidinonyl methyl acts as the P 1 residue imitating glutamine in the polyprotein, while the β-carbon of the α,β-unsaturated ethyl ester is covalently bonded (distance of 1.9 Å) to the sulfur atom of the catalytic cysteine, Cys144 (Figs.2B and 3). The leucine residue of 6 acts as the P 2 residue and occupies the S 2 subsite pocket, whereas the valine and serine residues act as the P 3 and P 4 residues. The terminal tert-butyl carbamate group extends to the edge of the FIPV-3CLpro active site cleft (Figs.2C and 3).
Figure 3.
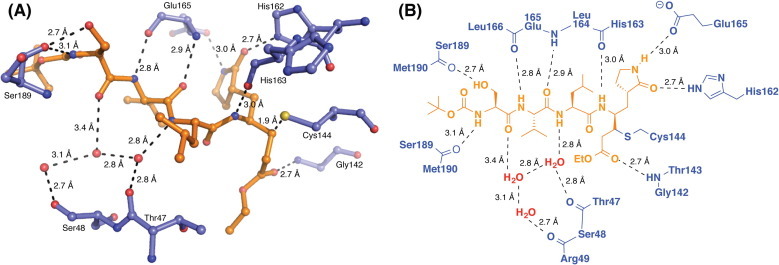
Hydrogen bond interactions between FIPV-3CLpro and compound 6 (PDB ID = 4ZRO). (A) 3D-representation of the hydrogen-bonding interactions of 6 with residues of FIPV-3CLpro residing in the substrate binding cavity. (B) 2D-representation of the same hydrogen-bonding interactions shown in A. Distances are shown between heteroatoms.
Compound 6 forms eight direct hydrogen bonds to five active site residues of FIPV-3CLpro. In addition, two water-mediated hydrogen bonds are formed between compound 6 and backbone carbonyl oxygens of Thr47 and Ser48 (Fig. 3). The backbone amide –NH of Gly142 forms a 2.7 Å hydrogen bond to the carbonyl of the ethyl ester, which sits in the S 1′ pocket of FIPV-3CLpro. The pyrrolidinonyl methyl of 6 is anchored into the S 1 subsite by two hydrogen bonds, a 2.7 Å hydrogen bond from the side-chain tele-nitrogen of His162 to the lactam carbonyl, and a 3.0 Å hydrogen bond from the lactam –NH to the side chain carboxylate of Glu165. Each of the four backbone NHs of 6 forms a hydrogen-bond to an FIPV-3CLpro active site residue—three direct hydrogen bonds and one water-mediated hydrogen bond. Two of the four backbone carbonyls of 6 participate in hydrogen-bonding to FIPV-3CLpro active site residues—a direct hydrogen bond from the backbone –NH of Glu165 and a water-mediated hydrogen-bond to the backbone carbonyls of Thr47 and Ser48 (Fig. 3).
Examination of the FIPV-3CLpro S 2 pocket shows that it is large enough to accommodate a P 2 leucine residue, but may not be sufficiently large enough to allow optimal binding of peptidomimetic inhibitors with larger P 2 groups, such as phenylalanine or larger alkyl groups, which introduce more steric bulk (i.e. inhibitors 1–5, Table 1). Interestingly, Kim and coworkers have shown that peptidomimetic compounds, similar to our compounds 1–3, with sterically bulky P 2 substituents (leucine, benzyl or cyclohexylmethyl) are effective against FCoV in cell-based antiviral assays.18 This suggests that despite the reduced inhibition of FIPV-3CLpro by compounds 1–5 relative to 6–11, these compounds may be effective in cell-based antiviral assays. Kim et al. did not test their compounds against purified FIPV 3CLpro enzyme suggesting that our compounds 6–11 may be even more potent in FCoV antiviral assays. Furthermore, Kim et al. have demonstrated that peptidomimetics akin to 1–3 and 5–11 are capable of reducing viral titers and pathological lesions in the livers of mice infected with murine hepatitis virus (MHV), a coronavirus similar to FCoV.18 Together, our studies and those of Kim et al. suggest that peptidomimetic compounds like 1–3 and 6–11 may have significant potential for being developed into effective therapeutics against FIPV infection.
In conclusion, we have over-expressed and purified FIPV-3CLpro and determined its inhibition by a set of eleven peptidomimetic inhibitors. We found that the six best inhibitors of FIPV-3CLpro (6–11) had a leucine residue at the P 2 position and a valine residue at the P 3 position, which is consistent with these residues being preferred at the P 2 and P 3 positions of the polyprotein substrate (Fig.1A). We determined the X-ray structure of FIPV-3CLpro in complex with the best peptidomimetic inhibitor identified, compound 6, which is the first reported structure of FIPV-3CLpro. The molecular details elucidated from the X-ray structure of FIPV-3CLpro in complex with 6 provide insights into the key interactions between the inhibitor and the enzyme that can be targeted in the design of more potent FIPV-3CLpro inhibitors.
Acknowledgements
We wish to thank Sakshi Tomar for designing the FIPV-3CLpro expression plasmid. This work was supported in part by Grants (AI085089 and AI26603) to A.D.M. from the National Institutes of Health via the National Institute of Allergy and Infectious Diseases and Purdue University. A.D.M. also wishes to also acknowledge partial support from the Walther Cancer Foundation. Crystallization and DNA sequencing were partially supported by the Purdue Center for Cancer Research Macromolecular Crystallography and DNA Sequencing Shared Resources which are partially supported by an NIH Grant (P30 CA023168). Finally, the authors would like to acknowledge the LS-CAT beamline staff for their help in acquiring X-ray data. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Footnotes
Supplementary data (materials and methods and X-ray data-collection and refinement statistics for FIPV-3CLpro:6 complex (PDB ID = 4ZRO)) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.10.023.
Supplementary data
Materials and methods and X-ray data-collection and refinement statistics for FIPV-3CLpro:6 complex (PDB ID = 4ZRO).
References and notes
- 1.Holzworth J. Cornell Vet. 1963;53:157. [PubMed] [Google Scholar]
- 2.Hartmann K. Vet. Clin. N. Am-Small. 2005;35:39. doi: 10.1016/j.cvsm.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horzinek M.C., Osterhaus A.D.M.E. Am. J. Vet. Res. 1979;40:1487. [PubMed] [Google Scholar]
- 4.Pedersen N.C., Allen C.E., Lyons L.A. J. Feline Med. Surg. 2008;10:529. doi: 10.1016/j.jfms.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogel L., Van der Lubben M., Lintelo E.G.T., Bekker C.P.J., Geerts T., Schuijff L.S., Grinwis G.C.M., Egberink H.F., Rottier P.J.M. Vet. Res. 2010;41:71. doi: 10.1051/vetres/2010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedersen N.C. Feline Pract. 1995;23:7. [Google Scholar]
- 7.Vennema H., Poland A., Foley J., Pedersen N.C. Virology. 1998;243:150. doi: 10.1006/viro.1998.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vennema H., Poland A., Hawkins K.F., Pedersen N.C. Feline Pract. 1995;23:40. [Google Scholar]
- 9.Ward J.M. Virology. 1970;41:191. doi: 10.1016/0042-6822(70)90070-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Addie D.D., Jarrett O. Vet. Rec. 1992;131:202. doi: 10.1136/vr.131.9.202-a. [DOI] [PubMed] [Google Scholar]
- 11.Brown E.W., Olmsted R.A., Martenson J.S., Obrien S.J. Zoo Biol. 1993;12:135. [Google Scholar]
- 12.Kocherhans R., Bridgen A., Ackermann M., Tobler K. Virus Genes. 2001;23:137. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dye C., Siddell S.G. J. Gen. Virol. 2005;86:2249. doi: 10.1099/vir.0.80985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomar S., Johnston M.L., St. John S.E., Osswald H.L., Nyalapatla P.R., Paul L.N., Ghosh A.K., Denison M.R., Mesecar A.D. J. Biol. Chem. 2015 doi: 10.1074/jbc.M115.651463. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim Y., Lovell S., Tiew K.C., Mandadapu S.R., Alliston K.R., Battaile K.P., Groutas W.C., Chang K.O. J. Virol. 2012;86:11754. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y., Mandadapu S.R., Groutas W.C., Chang K.O. Antiviral Res. 2013;97:161. doi: 10.1016/j.antiviral.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y., Shivanna V., Narayanan S., Prior A.M., Weerasekara S., Hua D.H., Kankanamalage A.C.G., Groutas W.C., Chang K.O. J. Virol. 2015;89:4942. doi: 10.1128/JVI.03688-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prior A.M., Kim Y.J., Weerasekara S., Moroze M., Alliston K.R., Uy R.A.Z., Groutas W.C., Chang K.O., Hua D.H. Bioorg. Med. Chem. Lett. 2013;23:6317. doi: 10.1016/j.bmcl.2013.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 21.UniProt-Consortium UniProthttp://www.uniprot.org/uniprot/Q98VG9, 2002–2015.
- 22.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. Acta Crystallogr., Sect. D. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P., Cowtan K. Acta Crystallogr., Sect. D. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Acta Crystallogr., Sect. D. 2010;66:486. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods and X-ray data-collection and refinement statistics for FIPV-3CLpro:6 complex (PDB ID = 4ZRO).