

Abstract
Cyclic GMP analogs, 8-Br, 8-pCPT, and PET-cGMP, have been widely used for characterizing cellular functions of cGMP-dependent protein kinase (PKG) I and II isotypes. However, interpreting results obtained using these analogs has been difficult due to their low isotype specificity. Additionally, each isotype has two binding sites with different cGMP affinities and analog selectivities, making understanding the molecular basis for isotype specificity of these compounds even more challenging. To determine isotype specificity of cGMP analogs and their structural basis, we generated the full-length regulatory domains of PKG I and II isotypes with each binding site disabled, determined their affinities for these analogs, and obtained cocrystal structures of both isotypes bound with cGMP analogs. Our affinity and activation measurements show that PET-cGMP is most selective for PKG I, whereas 8-pCPT-cGMP is most selective for PKG II. Our structures of cyclic nucleotide binding (CNB) domains reveal that the B site of PKG I is more open and forms a unique π/π interaction through Arg285 at β4 with the PET moiety, whereas the A site of PKG II has a larger β5/β6 pocket that can better accommodate the bulky 8-pCPT moiety. Our structural and functional results explain the selectivity of these analogs for each PKG isotype and provide a starting point for the rational design of isotype selective activators.
Graphical abstract
As one of the main receptors of cGMP, cGMP dependent protein kinase (PKG) is a central mediator of the NO-cGMP signaling pathway that regulates a myriad of physiological responses.1,2 These include smooth muscle tone, bone growth, and memory formation. Mammalian cells have three types of PKGs, PKG Iα, Iβ, and II. PKG Iα and Iβ are splice variants. They differ only by the first ~100 amino acids sharing cyclic nucleotide binding (CNB) and catalytic domains. PKG isotypes show distinct subcellular localization, tissue expression, and substrates suggesting their nonredundant cellular functions, which are strongly supported by gene deletion studies of each isotype and distinct phenotypes.3,4
The regulatory (R) domains contain leucine zipper domains that dimerize PKG isotypes and target them to binding partners and substrates in an isotype-dependent manner.5–7 All human PKGs have tandem CNB domains (CNB-A and -B) with different binding profiles for cGMP and its analogs.1,2,8 The CNB domain consists of an eight-stranded β barrel and variable numbers of α helices.9,10 The β-barrel contains a highly conserved structural motif referred to as the phosphate binding cassette (PBC), which provides multiple contacts with the ribose cyclic-phosphate moiety.
Cyclic nucleotide selectivity of PKG isotypes and their structural basis were characterized recently.11–14 CNB-A domains of PKG I and II isotypes show little selectivity for cGMP despite their high affinity.11,13 In contrast, the CNB-B domains provide high cGMP selectivity required for cGMP-dependent activation, acting as gatekeeper domains for activation of each isotype.11,12,14 Consistent with these results, crystal structures of the CNB domains show that CNB-A domains lack cGMP specific contacts, whereas CNB-B domains show specific contacts with the guanine moiety that explain their high selectivity. Furthermore, while their overall structures are similar, only the CNB-B domains show isotype specific interactions with the guanine moiety and the distinct C-terminus, which differently shield the cGMP pocket. In PKG I, strand β5 provides critical contacts for cGMP binding, whereas the C-terminal region consisting of a short helix and loop only provides a limited and nonspecific capping interaction.12,14 Due to its limited contacts at the C-terminal region, its cGMP pocket is more accessible to solvent. Unlike PKG I, the C-terminus of the PKG II CNB-B domain is fully helical.11 This C-terminal helix (αC-helix) shields the base of the cGMP pockets and provides the majority of the guanine specific contacts. Because of these contacts, its pocket is more shielded from solvent compared to PKG I.
Numerous cGMP analogs have been developed and used for functional studies of PKG isotypes.15,16 Of these cGMP analogs, three have been widely used. These include 8-bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP), β-phenyl-1,N2-ethenoguanosine 3′,5′-monophosphate (PET-cGMP), and 8-(4-chlorophenylthio)guanosine-3′-5′-cyclic monophosphate (8-pCPT-cGMP). However, their usage is limited due to low specificity for each isoform and cross-activity with cAMP-dependent protein kinases (PKA) and other cGMP receptors.15,17,18 For example, cell permeable 8-Br-cGMP is commonly used in cellular studies, but it activates both isotypes. While PET-cGMP and 8-pCPT-cGMP are known to be selective for PKG I and II, respectively, they easily activate either isotypes at low micromolar concentrations. All three analogs activate PKA and interact with cyclic nucleotide gated channels and phosphodiesterases at high concentrations. Despite their wide usage in determining specific cellular functions of PKG isotypes, little is known about cGMP analog affinities and molecular details of their interactions with PKG isotypes. Here, we investigated how these three analogs interact specifically with CNB-A and -B domains by independently disabling either CNB domain in the dimeric full-length regulatory domain, measuring their EC50, and determining cocrystal structures. Consistent with the previous reports on activation, our affinity and activity measurements show that PET-cGMP is selective for PKG I, while 8-Br-cGMP and 8-pCPT-cGMP are more selective for PKG II.19–21 Our crystal structures of the CNB-B:analog complexes and previous structures of CNB-A domains reveal isotype specific features and contacts that explain their selectivity and provide key insights for designing new analogs with improved isotype specificity.
RESULTS
Binding and Activation Measurements
To investigate isotype selectivity of three cGMP analogs, we first measured their EC50 values of the wild type and mutant R domains for both isotypes using competitive surface plasmon resonance (SPR; Figure 1b and Table 1). PKG Iβ wild type measurements show that having a PET moiety significantly decreases its EC50 value (from 163 nM to 3.8 nM) compared to cGMP, while having either 8-Br or 8-pCPT modification has little effect on its EC50 values. This makes PET-cGMP the most selective analog for PKG Iβ (~40 fold selective). As for the PKG II wild type, both 8-Br and 8-pCPT show modest reductions (20 nM and 5 nM, respectively) compared to cGMP, while PET shows a slight increase (from 96 nM to 193 nM). Thus, 8-pCPT-cGMP is the most selective analog for PKG II (~19-fold compared to cGMP).
Figure 1.
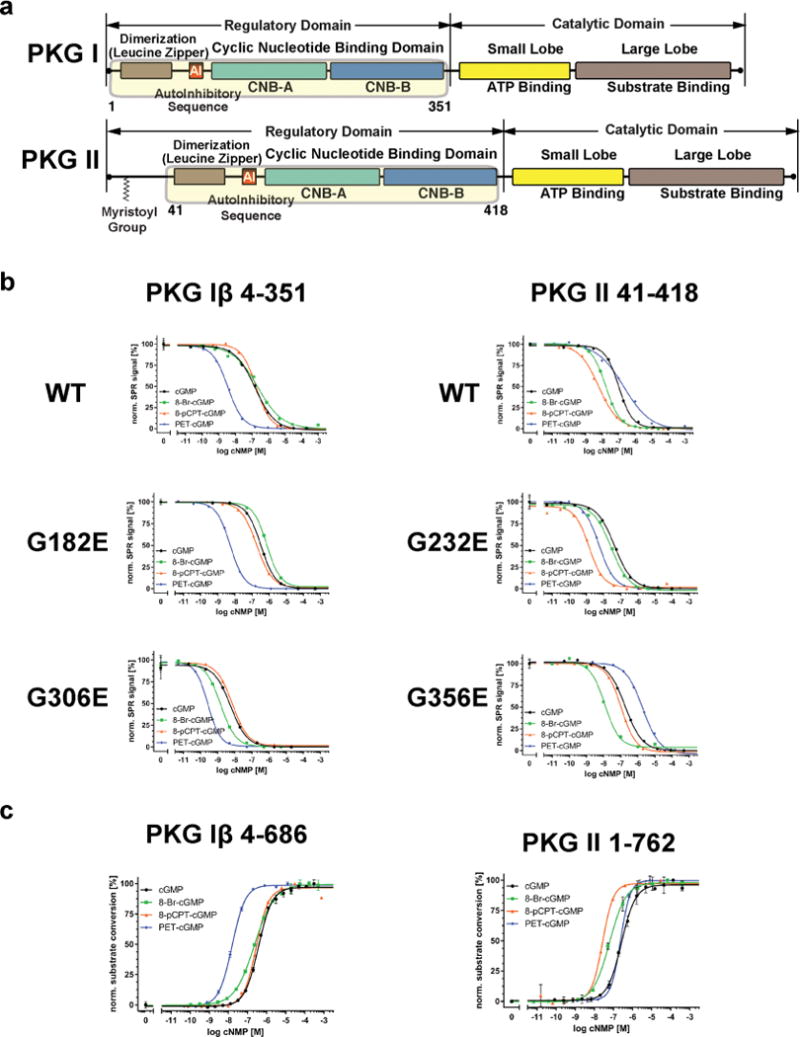
Domain organization and SPR data. (a) The domain organizations of PKG I and PKG II are shown. The regulatory domains used for SPR are highlighted with corresponding residue numbers. (b) SPR measurements of the PKG Iβ and II R domains for cGMP and analogs. Competition SPR curves are shown for wild type and mutant proteins. (c) Activation of PKG Iβ and II full-length by cGMP analogs.
Table 1.
Affinity Measurements of PKG Iβ and II R Domains for cGMP Analogsa
| PKG Iβ
| ||||
|---|---|---|---|---|
| hPKG Iβ 1–351 | cGMP | 8-Br-cGMP | 8-pCPT-cGMP | PET-cGMP |
| wt | 163 ± 6 nM (2) | 222 ± 4 nM (2) | 212 ± 20 nM (3) | 3.8 ± 0.4 nM (2) |
| G182E | 328 ± 6 nM (2) | 765 ± 25 nM (2) | 206 ± 18 nM (2) | 5.8 ± 0.4 nM (2) |
| G306E | 4.2 ± 0.1 nM (3) | <2 nM (2) | 7.7 ± 0.4 nM (2) | ≪ 2 nM (2) |
|
PKG II | ||||
| hPKG II 41–418 | cGMP | 8-Br-cGMP | 8-pCPT-cGMP | PET-cGMP |
|
| ||||
| wt | 96 ± 5 nM (3) | 20 ± 2 nM (3) | 5 ± 1 nM (3) | 193 ± 10 nM (3) |
| G232E | 184 ± 0.1 nM (2) | 11 ± 1 nM (2) | 116 ± 8 nM (2) | 1963 ± 12 nM (2) |
| G356E | 50 ± 3 nM (3) | 30 ± 2 nM (2) | ≤1 ± 0.3 nM (2) | 5.3 ± 0.3 nM (2) |
Determination of EC50 ± SEM in nanomolarity (nM). (n) = Number of measurements
Next, we investigated which CNB domain(s) of PKG Iβ and II is (are) responsible for the isotype selectivity by disabling either A or B sites within the full-length R domains and comparing their EC50 values with the wild types, again employing competitive SPR. To disable each site for cGMP binding, we replaced a conserved glycine within each PBC (G182 and G306 in PKG Iβ and G232 and G356 in PKG II, respectively) with a glutamate. These mutations within PBC are predicted to cause steric hindrance and charge repulsion with the negatively charged cyclic phosphate moiety. A similar strategy was taken in characterizing binding pockets of PKA.22,23 For the PKG Iβ mutants, our measurements show that disabling site A (G182E), thus having only intact site B, has no effect on its selectivity for PET, while slightly increasing its values for 8-Br-cGMP (from 328 nM to 765 nM). These values suggest that site B provides the selectivity for PET-cGMP. Disabling site B (G306E) reduces its EC50 values for all compounds, further suggesting site A plays little role in analog selectivity despite its high affinity.
As for the PKG II mutants, having the intact B site (G232E) decreases its EC50 for 8-Br and 8-pCPT-cGMP while increasing its value for PET (changed from 184 nM to 2.0 μM), suggesting that site B of PKG II is negatively selective for PET. Having the intact A site (G356E) reduces its EC50 values for both 8-pCPT and PET compared to cGMP (changed from 50 nM to <1 nM and 5.3 nM respectively), suggesting that site A is selective for both compounds.
Next, we investigated isotype selectivity in activation by measuring activation constants (Ka) of these analogs for each isotype using a microfluidic mobility-shift assay (Figure 1c and Table 2). Consistent with the binding data, our measurements show that PET-cGMP is most selective for PKG Iβ, showing a Ka value of 18 nM (compared to a Ka value of 370 nM with cGMP). 8-Br-cGMP and 8-pCPT provide little selectivity showing similar Ka values to cGMP. Also consistent, 8-pCPT is most selective for PKG II with a Ka value of 22 nM (compared to a Ka value of 257 nM with cGMP). 8-Br-cGMP is moderately selective for PKG II showing a Ka value of 60 nM. Somewhat inconsistent with the binding data, where PET-cGMP binds PKG II with a slightly higher EC50 value compared to cGMP (193 nM versus 96 nM), PET-cGMP shows a similar Ka value to cGMP, providing little selectivity against PKG II.
Table 2.
Activation Constants of PKG Iβ and II for cGMP Analogsa
| cGMP | 8-Br-cGMP | 8-pCPT-cGMP | PET-cGMP | |
|---|---|---|---|---|
| hPKG Iβ 5–676 | 370 ± 13 nM (4) | 206 ± 13 nM (3) | 249 ± 17 nM (3) | 18 ± 2 nM (3) |
| hPKG II 1–762 | 257 ± 11 nM (3) | 58 ± 5 nM (3) | 22 ± 3 nM (4) | 225 ± 13 nM (3) |
Ka values were measured using a microfluidic mobility shift assay, and errors denote SEM. (n) = Number of measurements.
Structure Solution and Overall Structure
To understand the molecular basis for isotype selectivity seen in EC50 values, we determined cocrystal structures between CNB-B domains and these analogs (Figure 2 and Table 3). We determined five structures in all at high resolutions (1.49–1.73 Å). Structures of PKG Iβ CNB-B were determined with 8-Br-cGMP, 8-pCPT-cGMP, and PET-cGMP bound (Figure 2a). The PKG II CNB-B domain was crystallized and solved with 8-Br-cGMP and 8-pCPT-cGMP (Figure 2b). All five structures show the same β-barrel fold without major conformational changes compared to the previous structures bound with cyclic nucleotides.11–14
Figure 2.
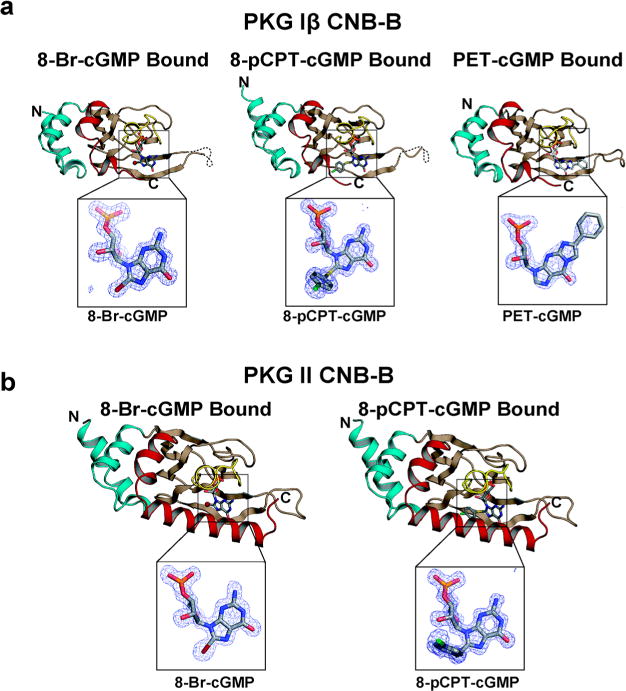
Overall structures of CNB-B:cGMP analog complexes. (a) Structures of PKG Iβ CNB-B:cGMP analog complexes. The N- and C-terminus are labeled. The structures are rendered as ribbon diagrams with the cGMP analogs shown as sticks. The phosphate-binding cassette (PBC) is colored in yellow, the αB/C helices in red, the N-terminal helices in teal, and the β barrel in light brown. cGMP analogs are shown as sticks, and the atoms are colored as follows: carbons in gray; oxygen, red; nitrogen, blue; sulfur, yellow; chlorine, green. Zoom-in views show |Fo−Fc| omit maps (contoured at 1.0 σ level) for the bound analogs. (b) Structures of PKG II CNB-B:cGMP analog complexes. All structure images were generated using PyMOL (Delano Scientific).
Table 3.
Data and Refinement Statistics
| PKG isoform
|
PKG Iβ CNB-B
|
PKG II CNB-B
|
|||
|---|---|---|---|---|---|
| analog | 8-Br-cGMP | 8-pCPT-cGMP | PET-cGMP | 8-Br-cGMP | 8-pCPT-cGMP |
| data collection | |||||
| wavelength (Å) | 0.99993 | 0.99993 | 0.97741 | 0.97741 | 0.97741 |
| space group | P4 212 | P1211 | P41212 | P212121 | P212121 |
| cell dimensions | |||||
| a, b, c (Å) | 47.01, 47.01, 102.6 | 35.51, 59.24, 67.32 | 46.29, 46.29, 102.5 | 41.62, 52.09, 68.64 | 42.21, 50.21, 65.51 |
| α, β, δ (deg) | 90.00, 90.00, 90.00 | 90.00, 100.2, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| resolution (Å) | 51.29–1.49 | 51.29–1.49 | 51.25–1.73 | 52.09–1.47 | 65.54–1.53 |
| Rsym or Rmergea | 0.105(1.721) | 0.105(0.429) | 0.184(2.248) | 0.093(1.788) | 0.082(0.521) |
| I/σI | 19.4(3.0) | 19.4(2.6) | 12.5(1.3) | 12.6(0.8) | 12.1(3.6) |
| CC b 1/2 | 0.999(0.929) | 0.999(0.852) | 0.999(0.994) | 0.999(0.372) | 0.999(0.606) |
| completeness (%) | 99.9(100) | 99(96) | 99.9(99.4) | 98.3(83.2) | 98.0(81.3) |
| redundancy | 26.3(19.6) | 4.2(3.8) | 21.4(13.3) | 7.9(5.6) | 4.0(3.3) |
| refinement | |||||
| resolution (Å) | 42.74–1.49 | 34.95–1.49 | 42.19–1.75 | 41.49–1.47 | 39.85–1.5 |
| no. reflections | 19698 | 44528 | 11850 | 25594 | 22476 |
| Rwork/Rfreec | 16.93/19.90 | 16.34/18.91 | 19.34/22.65 | 16.47/18.79 | 17.95/19.84 |
| no. atoms | |||||
| proteins | 1998 | 3955 | 1942 | 1942 | 2277 |
| ligand/ion | 37 | 175 | 47 | 154 | 100 |
| water | 127 | 280 | 80 | 119 | 180 |
| B-factors | |||||
| protein | 25.47 | 22.09 | 33.69 | 33.69 | 18.3 |
| ligand/ion | 35.42 | 18.78 | 32.68 | 33.45 | 25.5 |
| water | 30.70 | 35.58 | 34.68 | 34.17 | 28.93 |
| r.m.s.d deviations | |||||
| bond lengths (Å) | 0.009 | 0.012 | 0.012 | 0.009 | 0.013 |
| bond angles (deg) | 1.322 | 1.253 | 1.096 | 1.3 | 1.489 |
| PDB ID | 5JAX | 5J48 | 5JD7 | 5JIX | 5JIZ |
Highest resolution shell is shown in parentheses.
CC1/2, Pearson correlation coefficient.
5.0% of the observed intensities was excluded from refinement for cross validation purposes.
r.m.s., root-mean-square.
The PKG Iβ complexes with 8-Br-cGMP and PET-cGMP were crystallized in the P41212 space group with one molecule per asymmetric unit (Table 3). Both structures show clear electron density for the entire CNB-B domain (PKG Iβ 219–351) used for crystallization and the bound analogs excluding for the dynamic β4–β5 loop (residues 288–290). The PKG Iβ:8-pCPT-cGMP complex was crystallized in the P1211 space group and contains two molecules per asymmetric unit. While both molecules show clear electron density for the bound 8-pCPT-cGMP, only one molecule shows clear density for the C-terminal region that provides a capping interaction with the guanine moiety. This is due to each molecule having different crystal packing contacts. For the fully ordered molecule, the PBC of a neighboring symmetry mate packs onto its C-terminal region. No such packing interaction is seen for the other molecule of the asymmetric unit. We will focus on the fully ordered molecule for our structural analysis.
For all of the PKG Iβ: analog complexes, interactions with the ribose-cyclic phosphate and guanine moieties are nearly identical, as seen in the PKG Iβ:cGMP complex (Figure 2a).12,14 These include E307, R316, and T317 at the PBC, which bind the sugar-cyclic phosphate through hydrogen bonds. Also L296 and R297 at β5 interact selectively with the purine ring of the analogs through van der Waals contacts and hydrogen bonds at the bottom of the binding pocket (Figure 3a).
Figure 3.
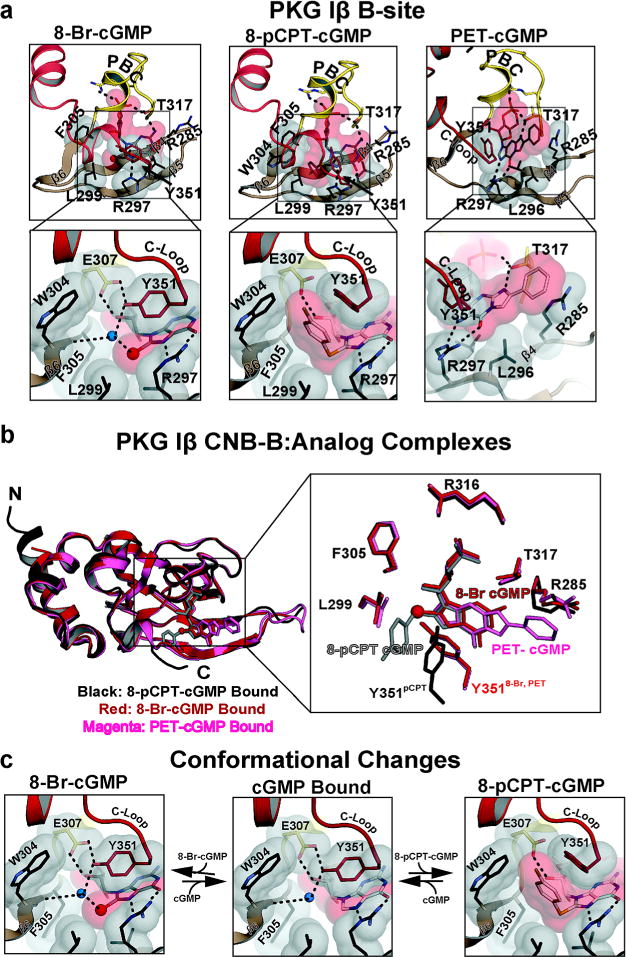
Interactions between PKG Iβ B site and cGMP analogs. (a) Detailed interactions between PKG Iβ B site and cGMP analogs. Only β4–6, the PBC, and the αB/C helices are shown. Residues contacting the cyclic nucleotides are shown as sticks, with transparent surfaces. Water molecules are shown as blue spheres. Hydrogen bonds are shown as dotted lines. Zoomed-in views highlight the interactions with the modified portion of cGMP. (b) Comparison between three analog bound structures. Three structures are aligned (left) and shown with the residues that directly interact with the analogs. The 8-pCPT, 8-Br, and PET-cGMP bound structures are colored black, red, and magenta, respectively. cGMP analogs are shown as sticks except for the bromine. The bromine is shown as a sphere. (c) Conformational changes associated with binding 8-Br and 8-pCPT. Regions near the cGMP pockets are shown. The middle panels show the cGMP bound conformation.
PKG Iβ CNB-B Bound with 8-Br-cGMP
The PKG Iβ CNB-B:8-Br-cGMP complex reveals that the 8-Br moiety docks to a hydrophobic pocket adjacent to the guanine binding site (Figure 3a, left panel). The pocket consists of V283, R297, L299, F305, and Y351. V283 at β4 and neighboring R297 at β5 form the base of the pocket. A sharp turn connects β5 and β6, which places L299 and F305 within VDW distance; these residues form the left wall of the pocket. The capping residue, Y351, interacts with the 8-Br moiety through a water molecule and shields the guanine at the top of the pocket. Even with these contacts, the 8-Br moiety is partially exposed to solvent due to its limited contacts with the C-terminal loop. The complex shows little structural changes compared to the cGMP bound structure (Figure 3b and c).
PKG Iβ CNB-B Bound with 8-pCPT-cGMP
8-pCPT-cGMP has a para-chlorophenyl moiety added to the C8 position of guanine through a thiol linkage (Figure 3a, middle panel). The structure shows that, while the thiol moiety docks to the same hydrophobic pocket at β5/β6 as the 8-Br moiety, the para-chlorophenyl moiety interacts with an additional β6 residue, W304, and the capping residue, Y351, at the C-terminal loop. In particular, the para-chlorophenyl group forms a π-stacking interaction with Y351. This is due to the bond angle of the aryl-thioether substitution, which is ~90°. Therefore, the para-chlorophenyl moiety is roughly perpendicular to the plane of the guanine ring and protruding toward the C-terminal loop. This places the phenyl ring near the side chains of W304 and Y351. The side chain of Y351 rotates about 90°, providing the capping interaction for the para-chlorophenylthio ring instead of the guanine ring (Figure 3a). This change in the side chain orientation of Y351 disrupts its hydrogen bonds with the neighboring residues and slightly reduces the VDW contact surface with the guanine moiety compared to the cGMP bound structure (Figure 3b and c). Superpositioning with the cGMP bound structure reveals that the C-loop moves approximately 2 Å away from the rest of the pocket to accommodate the bulky chlorophenylthio moiety (Figure 3b).
PKG Iβ CNB-B Bound with PET-cGMP
PET-cGMP is modified from cGMP by adding a two-carbon bridge between the ring N1 and exocyclic C2 nitrogen, resulting in a 1,N2 etheno adduct. This is followed by the addition of a phenyl ring at the β position of the etheno group. The CNB-B:PET–cGMP complex shows two interactions specific for PET-cGMP (Figure 3a, right panel). Formation of the ethenoguanosine moiety creates a secondary amine at N2, which allows for a more stable hydrogen bonding interaction with T317 compared to the primary amine at N2 in cGMP. The second interaction is a π/π interaction between the phenyl ring of PET-cGMP and R285, which is not seen with the other analogs. The structure reveals that the phenyl ring reaches out to the side chain of R285 at the end of the β4 strand providing this unique π/π interaction.
Overall Structures of PKG II CNB-B Bound with Analogs
We attempted to cocrystallize the PKG II CNB-B domain with the same set of cGMP analogs, but only obtained crystals in the presence of 8-Br-cGMP and 8-pCPT-cGMP. Both complexes are crystallized in the P212121 space group with similar unit cell dimensions showing only one molecule per asymmetric unit. Each complex showed clear density for the entire CNB-B domain (residues 269–418) and the bound analog (Figure 2b). Both structures show few changes in their overall conformation and interactions with the shared cGMP moiety compared to our previous cocrystal structures of the PKG II CNB domain with cGMP.11 These include the hydrogen-bonding network that recognizes the guanine moiety (S367, D412, and R415) and the several hydrophobic interactions (V333, I346, K358, and A368) that form the purine pocket.
PKG II CNB-B Bound with 8-Br-cGMP
The interactions between the PKG II CNB-B domain and 8-Br cGMP are significantly different compared to the interactions seen with PKG I due to the αC-helix that shields the rest of the cGMP binding pocket (Figure 4, left panel). Similarly to PKG Iβ, the PKG II CNB-B domain also has a β5/β6 hydrophobic pocket next to the cGMP binding pocket, where the 8-Br moiety docks. The structure shows that K347, L349, and F355 form an analogous hydrophobic pocket in PKG II. Additionally, the structure reveals that two key cGMP binding residues, K347 and E357, and an αC-helix residue, Y404, form strong hydrogen bonds with the 8-Br moiety indirectly through ordered water molecules. As shown in Figure 4a, the hydroxyl group of Y404 at the cGMP pocket facing surface of the αC-helix points perpendicular to the 8-Br-guanine moiety forming strong hydrogen bonds through two water molecules. Due to the guanine induced polarization of bromine, the 8-Br moiety has a ring of negative electrostatic potential perpendicular the 8-Br-gunaine bond. This electronegative annulus captures these water molecules and bridges Y404, E357, K347, and 8-Br-cGMP through hydrogen bonds.24
Figure 4.
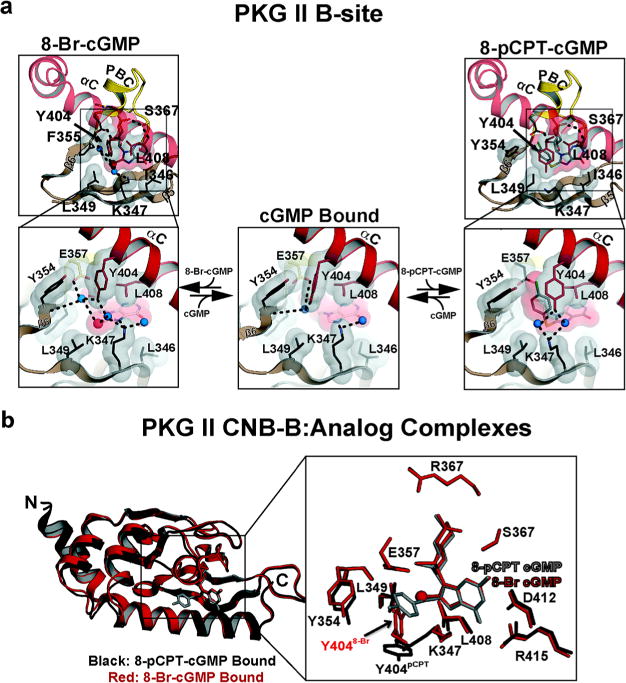
Interactions between PKG II B site and cGMP analogs. (a) Detailed interactions between PKG II B site and cGMP analogs. Only β4–6, the PBC, and the αB/C helices are shown. Residues contacting the cyclic nucleotides are shown as sticks, with transparent surfaces. Water molecules are shown as blue spheres. Hydrogen bonds are shown as dotted lines. Zoomed-in views highlight the interactions with the modified portion of cGMP. The left and right panels show 8-Br-cGMP and 8-pCPT-cGMP bound, respectively. The middle panel shows the cGMP bound conformation. (b) Comparison between 8-Br-cGMP and 8-pCPT-cGMP bound structures. The 8-Br-cGMP and 8-pCPT-cGMP bound structures are colored red and black, respectively. cGMP analogs are shown as sticks except for the bromine. The bromine is shown as a red sphere.
PKG II CNB-B Bound with 8-pCPT-cGMP
The structure shows that the para-chlorophenylthio moiety docks to the same hydrophobic pocket at β5/6 as well as a new pocket created at the αC-helix as a result of conformational change. The para-chlorophenyl moiety perpendicular to the guanine points toward the αC-helix and docks to an additional hydrophobic pocket consisting of residues from β6, the PBC, and αC-helix. The structure reveals that Y354 (β6), E347 (PBC), Y404 (αC-helix), and L408 (αC-helix) surround the para-chlorophenyl providing VDW contacts (Figure 4a, right panel). In the cGMP bound structure, these residues interact with one another and form a hydrophobic patch. The PKG II:8-pCPT complex structure shows that the para-chlorophenyl group displaces Y404 away from the rest. Additionally, the bulky chlorophenylthio moiety pushes the side chain of K347 away from the guanine pocket. These conformational changes rearrange neighboring solvent molecules as well as solvent mediated hydrogen bond networks (Figure 4a and b). As previously mentioned, both cGMP and 8-Br-cGMP bound structures show a water molecule captured between Y354, E357, and Y404. However, the 8-pCPT-cGMP complex lacks this water molecule because the 8-pCPT moiety occupies the same pocket. Thus, Y404 no longer interacts with E357 and the ribose but interacts only with K347 through newly positioned solvent molecules (Figure 4a).
DISCUSSION
While understanding cellular functions of PKG isotypes requires inhibitors and activators with high selectivity and potency, no such chemical tools are currently available. Improving selectivity and potency of known compounds and identifying new effector molecules would serve as a good starting point to overcome this challenge. However, improving these properties requires molecular-level understanding of how these compounds interact with PKG I and II. This has been difficult due to the following reasons. First, PKG isotypes have multiple cGMP binding sites with different affinities, making their individual measurements impossible. Second, bacterially expressed proteins were often contaminated with cAMP due to the high level of cAMP in the media during expression and unexpectedly high affinity for cAMP, making subsequent characterization difficult. We resolved these issues by disabling each cGMP binding pocket and by expressing in E. coli Δcya TP2000, which lacks adenylyl cyclase activity.25 This allowed us to obtain reproducible binding constants and diffraction quality crystals.
Our binding and activation data suggest that PET-cGMP and 8-pCPT-cGMP are most selective in binding and activating PKG Iβ and II, respectively, while 8-Br-cGMP is moderately selective for PKG II, which is consistent with the previously reported activation data.15,19–21
Previous activation data show that
PKG Iβ: PET-cGMP (20 nM20) ≫ cGMP (250 nM20)/8-Br-cGMP (210 nM20)/8-pCPT-cGMP (440 nM20)
PKG II: 8-pCPT-cGMP (3.5 nM-80 nM19,21) > 8-Br-cGMP (25 nM19) > cGMP (40–360 nM19,21) ≫PET-cGMP (60 nM ~ 4.2 μM19,21).
Consistent with these trends, our affinity measurements show that
PKG Iβ: PET-cGMP ≫ cGMP > 8-Br-cGMP ≥ 8pCPT-cGMP
PKG II: 8-pCPT-cGMP > 8-Br-cGMP > cGMP > PET-cGMP.
Our activation data show that
PKG Iβ: PET-cGMP ≫ 8-Br-cGMP ≥ 8pCPT-cGMP ≥ cGMP
PKG II: 8-pCPT-cGMP > 8-Br-cGMP > PET-cGMP ≥ cGMP
Our results indicate that PET-cGMP and 8-pCPT-cGMP are ~40–50-fold selective in binding and ~10-fold in activating each isotype, whereas 8-Br-cGMP is only ~10-fold selective in binding and activating PKG II and ~4-fold in activating. 8-Br-cGMP and 8-pCPT-cGMP similarly bind and activate PKG Iβ compared to cGMP. In contrast, 8-Br-cGMP and 8-pCPT-cGMP show gradual increases in both EC50 and Ka values for PKG II. Unexpectedly, PET-cGMP binds and activates PKG II with similar potencies as cGMP, providing little selectivity against PKG II.
The trends seen in the interactions between mutants of PKG Iβ and cGMP analogs are consistent with cGMP analog specificity reported in a dissociation study.26 In this study, PKG I is preincubated with [3H]cGMP and the ability of cGMP analogs to compete with [3H]cGMP was measured. The authors first identified two sites with different exchange profiles, slowly exchanging (high affinity, A site) and rapidly exchanging (low affinity, B site) with unlabeled cGMP. They also showed that 8-Br-cGMP binds to the slowly exchanging site (A site) 3.4-fold stronger than cGMP, whereas PET-cGMP binds to the B site 10-fold stronger. Our affinity data show that 8-Br-cGMP binds to the A site over 2-fold stronger than cGMP. Somewhat consistent with the dissociation studies, PET-cGMP binds to the B site over 50-fold stronger.
Comparing EC50 values of wild types and site-specific mutants suggests that the B site plays a dominant role in determining EC50 values for cGMP analogs in PKG Iβ, whereas both A and B sites are critical for PKG II. Our data show that EC50 values of the B site closely resemble those of the wild type in PKG Iβ, not those of the A site. Unlike PKG Iβ, the higher EC50 values of the B site are compensated by the lower values of the A site in PKG II. These trends are particularly true for PET-cGMP and 8-pCPT-cGMP, where their respective EC50 values of the B or combined values of A/B sites are reflected in wild types of PKG Iβ and II. These differences are maybe due to different accessibilities of these sites when they are bound to their respective C-domain (inactive state), which is supported by the different assembly of holoenzymes seen in PKA isoforms and variable linkers that connect the R and C domains in PKG isotypes.27–30 Since no structural information is currently available for the inactive state of PKG isotypes, structures of PKA holoenzyme complexes serve as models for PKG holoenzymes. Several holoenzyme structures of PKA isotypes show different R–C assemblies depending on the isotype, resulting in very different R–C and interdomain contacts. For example, the RIα holoenzyme shows a compact tetramer assembly where its C subunits surround CNB-A of the R subunits, making the A site relatively inaccessible.27 In contrast, RIβ and RIIβ holoenzymes display an extended assembly with the R subunits more exposed to solvent; therefore both A and B sites are accessible.28,30 Furthermore, sequence alignment of PKG Iβ and II shows that the linker regions are highly variable with PKG II having an over 20 amino acid long insert, which may cause a different R–C assembly and contacts compared to the PKG Iβ isotype. Thus, we hypothesize that PKG holoenzymes have different assembly and dimeric contacts, which makes the A site less accessible in PKG I and more or equally accessible compared to the B site in PKG II, and this may explain different roles of two distinct sites in binding cGMP analogs.
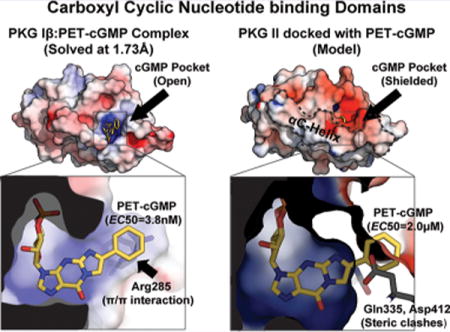
The site-specific mutant data suggest that the B sites provide a high preference of PET-cGMP in PKG Iβ, and the crystal structures of the CNB-B domains explain why. The B site of PKG Iβ binds PET-cGMP with 5.8 nM, whereas the B site of PKG II binds with 2 μM. Our structure of PKG Iβ CNB-B bound to PET-cGMP reveals that this is due to the B site providing a unique π/π interaction through Arg285 (β4) as well as having a more open pocket, which accommodates the bulky PET without any clashes. Consistent with our affinity measurements and structure, PET-cGMP was reported to have a higher affinity for the B site of PKG I than cGMP and only a modest difference in the A site compared to cGMP.26 Furthermore, our structures of PKG II CNB-B explain its low affinity for PET-cGMP.11 The structures show a glutamine (Gln335) at the analogous position to Arg285 of PKG Iβ, unlikely to form a stable interaction. The PKG II CNB-B domain also has a more shielded cGMP pocket, which may cause steric hindrance with the PET moiety reducing its affinity for the B site.11 Indeed, when we dock PET-cGMP into PKG II CNB-B, the PET moiety clashes with the key guanine-binding residue at the αC-helix (D412; Supporting Information Figure 1), which explains its higher EC values for PKG II R domains and our difficulty in obtaining its cocrystal structure.
The site-specific mutant data suggest that the A site is responsible for the high preference of 8-pCPT-cGMP in PKG II, which may be explained by our previous structures of CNB-A domains.11,13 The A site of PKG II binds 8-pCPT-cGMP with high affinity (≤1.1 nM), which represents more than a 50-fold reduction compared to cGMP, while the affinity of PKG Iβ for 8-pCPT-cGMP shows little change (Table 1). Our previous structures of the CNB-A domains reveal that PKG II has a larger β5/β6 hydrophobic pocket that can better accommodate a bulky functional group compared to PKG Iβ (Supporting Information Figure 2), which may explain its lower EC50 value.11,13 Our models of CNB-A domains bound with 8-pCPT-cGMP show that the β5/β6 pocket of PKG II has residues with less bulky side chains (I225 and T229), which may provide more favorable interactions with the 8-pCPT moiety. These residues are replaced with more bulky residues (M175 and K179) in PKG Iβ, which makes its interactions with the 8-pCPT moiety less favorable. For the interactions between the B sites and 8-pCPT-cGMP, both PKG isotypes show similar EC50 values, suggesting that the B sites contribute little to PKG II’s high preference for 8-pCPT-cGMP. Consistent with this, our structures show that the B sites of PKG Iβ and II bind the 8-pCPT moieties mainly through hydrophobic interactions.
The SPR data show that 8-Br-cGMP is more selective for PKG II because of its tighter interactions with the B site, and this is consistent with our structures. The wild type measurements show that adding Br at the 8 position reduces its EC50 value only for PKG II (from 96 nM to 20 nM) without affecting PKG Iβ (from 163 nM to 188 nM; Table 1). The site-specific mutant data further show that site B of PKG II is responsible for this reduction (184 nM to 11 nM). Our structures of CNB-B domains bound with 8-Br-cGMP reveal that, while both CNB-B domains have a hydrophobic β5/β6 pocket adjacent to the guanine-binding site where 8-Br docks without forming any clashes, only PKG II has an extended hydrogen bonding network which provides strong interactions with the 8-Br moiety. No such interactions are seen in PKG Iβ, consistent with higher EC50 values seen in the wild type and G182E of the PKG Iβ R domain.
Our results combined with previous studies suggest new strategies in designing isotype specific analogs. For designing PKG I specific agonists, the data suggest that derivatizing the C1 or C2 position to extend the guanine ring and substituting the β-phenyl of PET-cGMP with more negatively charged functional groups may improve its specificity for PKG I. We envision that making a bulkier compound by expanding these positions would still allow its interaction with the open pocket of PKG I while causing steric clashes with the more shielded pocket in PKG II. Alternatively, substituting the β-phenyl for a negative charged group may allow for a favorable electrostatic interaction with Arg285 because its flexible side chain can adopt multiple conformations. For PKG II, further improving site B’s affinity while preserving affinity for site A may improve its selectivity. Our structures suggest that placing hydrogen bond accepting groups at the 8-position of guanine moiety may increase its specificity for the B site. While both PKG I and II have a hydrophobic pocket adjacent to the guanine pocket, only PKG II shows a conserved tyrosine residue at the αC-helix with its hydroxyl group proximal to the 8-position. Thus, we envision that placing a hydrogen bond accepting group may allow direct hydrogen bonding interaction with Y404 and increase overall specificity for PKG II.
MATERIALS AND METHODS
Expression and Purification of Recombinant Proteins
Mutated regulatory domains were generated using mutagenesis PCR with pQTEV vectors containing either PKG Iβ 1–351 or PKG II 41–418 as a template. The site directed mutagenesis was performed with the following primers: 5′-GGAAAAGTGTTTGAGGAATTGGCT-ATTC-3′ and 5′-GAATAGCCAATTCCTCAAACACTTTTCC-3′ (G182E), 5′-GGAGACTGGTTTGAAGAGAAAGCC-3′ and 5′-GGCTTTCTCTTCAAACCAGTCTCC-3′ (G306E), 5′-GTGGACCACATTTGAGGAGCTTG-3′ and 5′-CAAGCTCC-TCAAATGTGGTCCAC-3′ (G232E), and 5′-GGAGAATA-CTTTGAAGAAAAAGCTCTTATC-3′ and 5′-GATAAGAG-CTTTTTCTTCAAAGTATTCTCC-3′ (G356E). All His-tagged regulatory domain constructs were expressed in Δcya TP2000 E. coli.25 The R-domains were purified using Ni-NTA agarose (Sigma-Aldrich) in a batch approach or a HisTrap excel affinity column (GE Healthcare). Impurities were further removed by a Hiload 16/60 Superdex 75 column (GE Healthcare). The sequence encoding wild-type PKG II was cloned into a pcDNA 3.0 SF-TAP vector31 from a template containing full-length human PKG II (Addgene plasmid # 23435)32 using the following primers: 5′-GGGGCTAGCATGGGAA-ATGGTTCAGTGAAACCT-3′ and 5′-GGGCTCGAGTCAGAAG-TCTTTATCCCAGCCTGA-3′.
FLAG-tagged PKG Iβ (5–686) and FLAG-Strep-Strep-tagged PKG II (1–762) were expressed in HEK293T cells as previously described.11,12,14 The cells were grown to 80% confluency in complete medium and transfected with the constructs using polyethylenimine. After harvesting, the cells were lysed with 20 mM MOPS (pH 7.4), 300 mM NaCl, 1% Tween20, 1 mM DTT, and protease inhibitors. The purification was performed using Anti-FLAG-M2 Agarose beads (Sigma-Aldrich) and eluted with 1 × MOPS buffer containing 5 mg mL−1 3×FLAG peptide (Sigma-Aldrich).
Surface Plasmon Resonance
All surface plasmon resonance (SPR) measurements were performed at 25 °C with 20 mM MOPS, at pH 7.0, 300 mM sodium chloride, and 0.05% (v/v) surfactant polysorbate P20 as a running buffer. The interaction analyses were monitored on a Biacore T100/T200 system (GE Healthcare) and a Biacore 3000 (GE Healthcare).
Cyclic nucleotide analogs were coupled to sensor chip surfaces of CM5 chips (GE Healthcare). All coupling steps were performed at a flow rate of 10 μL/min. Sensor chip surfaces were activated with N-hydroxysuccinimide/N-ethyl-N′-(dimethylaminopropyl)-carbodii-mide for 10 min. Then 3 mM of the respective analog in 100 mM borate, at pH 8.5, was coupled for 15 min. Subsequently, the surface was deactivated by injecting 1 mM ethanolamine-HCl, at pH 8.5 for 10 min. As a reference, the first flow cell of each chip was only activated and deactivated without any cyclic nucleotide coupled. cGMP-analog chips were produced by coupling N2-(6-aminohexyl)-cGMP, 8-(2-aminoethylthio)-cGMP, and 8-(6-aminohexylthio)-cGMP (Biolog life science) to the respective flow cells.
Solution competition assays were performed at a flow rate of 30 μL min−1 as previously described.33 Protein was preincubated with various concentrations of cGMP, 8-Br-cGMP, 8-pCPT-cGMP, and PET-cGMP (Biolog life science) before injection. Association and dissociation phases were monitored for at least 90 and 30 s, respectively. The surfaces were regenerated by two injections of 0.5% (w/v) SDS and a single injection of 1 M sodium chloride for 30 and 60 s, respectively.
The resonance signal (RU) 10 s before the end of the respective association phase was plotted against the cyclic nucleotide concentration, and EC50 values were calculated from sigmoidal dose response curves using GraphPad Prism 6.0.1.
Microfluidic Mobility-Shift Assay
Kinase activity was determined employing a microfluidic mobility-shift assay on a Caliper DeskTop Profiler (Caliper Life Sciences, PerkinElmer). The protein was incubated for 2 h at ambient temperature in a 384-well assay plate (Corning LV, nonbinding surface) in 20 μL of buffer (20 mM 3-(N-morpholino)propanesulfonic acid, at pH 7.0, 300 mM sodium chloride, 1 mM dithiothreitol, 0.05% L-31, 10 μM fluorescein isothiocyanate-kemptide, 990 μM kemptide, 1 mM ATP, 10 mM magnesium chloride) and various concentrations of cGMP, 8-Br-cGMP, 8-pCPT-cGMP, and PET-cGMP (Biolog life science), respectively. Reaction mixtures without cyclic nucleotide were used as controls. A ProfilerPro LabChip (4-sipper mode; PerkinElmer) was used for electrophoretic separation of the substrate and product. The following conditions were applied: downstream voltage −150 V, upstream voltage −1800 V with a screening pressure of −1.7 psi. Substrate conversion was plotted against the logarithmic cyclic nucleotide concentration, and activation constants (Ka) were calculated from sigmoidal dose–response curves employing GraphPad Prism 6.0.1.
Crystallization and Structure Determination
The CNB-B domains of PKG Iβ and II were expressed and purified as described previously.11,12 To cocrystallize CNB-B domains of PKG Iβ with the cGMP analogs, a protein sample (60 mg mL−1) was mixed with an equal volume of 10 mM 8-Br-cGMP, 8-pCPT-cGMP, and PET-cGMP (Biolog life science) and incubated for 1 h at 4 °C. The mixture was concentrated to reach the original concentration. This procedure was repeated twice using fresh analog solution each time. The sample was then crystallized using the hanging drop method at RT in 30% PEG 400, 200 mM calcium acetate, and 0.1 M sodium acetate, at pH 4.5–4.8. The crystals containing PET-cGMP and 8-pCPT were cryoprotected using the same crystallization buffer supplemented with 25% ethylene glycol. The crystals containing 8-Br-cGMP were cryoprotected using perfluoropolyether. Diffraction data were processed using iMosflm.34 To crystallize PKG II CNB-B bound to 8-Br and 8-pCPT-cGMP, we followed the same procedure as described above using a 50 mg mL−1 protein sample. Co-crystals with 8-Br-cGMP and 8-pCPT-cGMP were obtained in 30% 2-methyl-2,4-pentanediol, 20 mM calcium chloride, and 100 mM sodium acetate, at pH 4.6 and 4 °C, using the hanging drop method. Diffraction data were processed using iMosflm.34 The structures of the PKG Iβ and PKG II complexes were determined by Phaser-MR35 using the CNB-B domain structures of PKG Iβ and PKG II (PDB codes 4KU7 and 5BV6, respectively) as MR probes. Diffraction experiments were performed at the Advanced Light Source at Berkeley, California. All models were manually built using Coot36 and refined using Phenix.Refine.37
Supplementary Material
Acknowledgments
We thank D. Casteel (University of California, San Diego), G. Melacini (McMaster University), and Kim lab members for critical reading of the manuscript. C.K. was funded by the NIH grant R01 GM090161. J.C.C is supported by the Training Program in Pharmacological Sciences fellowship, National Institute of General Medical Science grant no. 32GM089657-04. F.W.H. and P.H. were funded by the Deutsche Forschungsgemeinschaft (DFG HE 1818/10) and the funding line “Future” project Phosmorg, University of Kassel. E.F. is supported by a Ph.D. fellowship of the University of Kassel as a member of the graduate program Functionomics. We acknowledge the Center for Interdisciplinary Nanostructure Science and Technology (CINSaT) of the University of Kassel for support of this work. The Berkeley Center for Structural Biology is supported in part by the NIH, the National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.7b00369.
(Figure S1) Structural model of PET-cGMP bound to the PKG II CNB-B domain and (Figure S2) structural models of PKG Iβ and II CNB-A domains bound to 8-pCPT-cGMP (PDF)
Accession Codes
Coordinates and structural factors for the structure for PKG Iβ CNB-B bound with 8-Br-cGMP, 8-pCPT-cGMP, and PET-cGMP have been deposited at the RCSB Protein Data Bank with an accession codes 5JAX, 5J48, and 5JD7, respectively. Coordinates and structural factors for PKG II CNB-B bound with 8-Br-cGMP and 8-pCPT-cGMP have been deposited with an accession codes 5JIX and 5JIZ.
ORCID
Choel Kim: 0000-0002-3152-0020
Author Contributions
J.C.C. and C.K. conceived and designed the project. J.C.C. purified the proteins used for crystallization, obtained the cocrystals, and solved their cocrystal structures. B.S. performed the diffraction experiments and data collection. P.H., E.F., and F.W.H. generated and purified the R domains and the full-length PKG I and II for affinity and activation measurements. P.H. and E.F. measured binding and activation constants. J.C.C. and C.K. wrote the bulk of the manuscript and created the figures. All authors commented on the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hofmann F, Wegener JW. cGMP-Dependent Protein Kinases (cGK) In: Krieg T, Lukowski R, editors. Guanylate Cyclase and Cyclic GMP: Methods and Protocols. Humana Press; Totowa, NJ: 2013. pp. 17–50. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-Dependent Protein Kinases as Revealed by Gene Deletion. Physiol Rev. 2006 doi: 10.1152/physrev.00015.2005. 86.110.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 4.Schlossmann J, Desch M. cGK Substrates. In: Schmidt HHHW, Hofmann F, Stasch J-P, editors. cGMP: Generators, Effectors and Therapeutic Implications. Springer; Berlin: 2009. pp. 163–193. [Google Scholar]
- 5.Casteel DE, Smith-Nguyen EV, Sankaran B, Roh SH, Pilz RB, Kim C. A Crystal Structure of the Cyclic GMP-dependent Protein Kinase Iβ Dimerization/Docking Domain Reveals Molecular Details of Isoform-specific Anchoring. J Biol Chem. 2010;285:32684–32688. doi: 10.1074/jbc.C110.161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin L, Reger AS, Guo E, Yang MP, Zwart P, Casteel DE, Kim C. Structures of cGMP-Dependent Protein Kinase (PKG) Ialpha Leucine Zippers Reveal an Interchain Disulfide Bond Important for Dimer Stability. Biochemistry. 2015;54:4419–22. doi: 10.1021/acs.biochem.5b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reger AS, Yang MP, Koide-Yoshida S, Guo E, Mehta S, Yuasa K, Liu A, Casteel DE, Kim C. Crystal Structure of the cGMP-dependent Protein Kinase II Leucine Zipper and Rab11b Protein Complex Reveals Molecular Details of G-kinase-specific Interactions. J Biol Chem. 2014;289:25393–25403. doi: 10.1074/jbc.M114.575894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlossmann J, Hofmann F. cGMP-dependent protein kinases in drug discovery. Drug Discovery Today. 2005;10:627–634. doi: 10.1016/S1359-6446(05)03406-9. [DOI] [PubMed] [Google Scholar]
- 9.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rehmann H, Wittinghofer A, Bos JL. Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat Rev Mol Cell Biol. 2007;8:63–73. doi: 10.1038/nrm2082. [DOI] [PubMed] [Google Scholar]
- 11.Campbell JC, Kim JJ, Li KY, Huang GY, Reger AS, Matsuda S, Sankaran B, Link TM, Yuasa K, Ladbury JE, Casteel DE, Kim C. Structural Basis of Cyclic Nucleotide Selectivity in cGMP-Dependent Protein Kinase II. J Biol Chem. 2016;291:5623–33. doi: 10.1074/jbc.M115.691303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang GY, Kim JJ, Reger AS, Lorenz R, Moon EW, Zhao C, Casteel DE, Bertinetti D, Vanschouwen B, Selvaratnam R, Pflugrath JW, Sankaran B, Melacini G, Herberg FW, Kim C. Structural basis for cyclic-nucleotide selectivity and cGMP-selective activation of PKG I. Structure. 2014;22:116–124. doi: 10.1016/j.str.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JJ, Casteel DE, Huang G, Kwon TH, Ren RK, Zwart P, Headd JJ, Brown NG, Chow DC, Palzkill T, Kim C. Co-crystal structures of PKG Iβ (92–227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PLoS One. 2011;6:e18413. doi: 10.1371/journal.pone.0018413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JJ, Lorenz R, Arold ST, Reger AS, Sankaran B, Casteel DE, Herberg FW, Kim C. Crystal Structure of PKG I:cGMP Complex Reveals a cGMP-Mediated Dimeric Interface that Facilitates cGMP-Induced Activation. Structure. 2016;24:710–720. doi: 10.1016/j.str.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 16.Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 17.Ji HL, Nie HG, Chang Y, Lian Q, Liu SL. CPT-cGMP Is A New Ligand of Epithelial Sodium Channels. Int J Biol Sci. 2016;12:359–366. doi: 10.7150/ijbs.13764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valtcheva N, Nestorov P, Beck A, Russwurm M, Hillenbrand M, Weinmeister P, Feil R. The Commonly Used cGMP-dependent Protein Kinase Type I (cGKI) Inhibitor Rp-8-Br-PET-cGMPS Can Activate cGKI in Vitro and in Intact Cells. J Biol Chem. 2009;284:556–562. doi: 10.1074/jbc.M806161200. [DOI] [PubMed] [Google Scholar]
- 19.Pohler D, Butt E, Meissner J, Muller S, Lohse M, Walter U, Lohmann SM, Jarchau T. Expression, purification, and characterization of the cGMP-dependent protein kinases Iβ and II using the baculovirus system. FEBS Lett. 1995;374:419–425. doi: 10.1016/0014-5793(95)01168-e. [DOI] [PubMed] [Google Scholar]
- 20.Sekhar KR, Hatchett RJ, Shabb JB, Wolfe L, Francis SH, Wells JN, Jastorff B, Butt E, Chakinala MM, Corbin JD. Relaxation of pig coronary arteries by new and potent cGMP analogs that selectively activate type I alpha, compared with type Iβ, cGMP-dependent protein kinase. Mol Pharmacol. 1992;42:103–108. [PubMed] [Google Scholar]
- 21.Vaandrager AB, Edixhoven M, Bot AGM, Kroos MA, Jarchau T, Lohmann S, Genieser HG, de Jonge HR. Endogenous Type II cGMP-dependent Protein Kinase Exists as a Dimer in Membranes and Can Be Functionally Distinguished from the Type I Isoforms. J Biol Chem. 1997;272:11816–11823. doi: 10.1074/jbc.272.18.11816. [DOI] [PubMed] [Google Scholar]
- 22.Ogreid D, Doskeland SO, Gorman KB, Steinberg RA. Mutations that prevent cyclic nucleotide binding to binding sites A or B of type I cyclic AMP-dependent protein kinase. J Biol Chem. 1988;263:17397–17404. [PubMed] [Google Scholar]
- 23.Steinberg RA, Gorman KB, Ogreid D, Doskeland SO, Weber IT. Mutations that alter the charge of type I regulatory subunit and modify activation properties of cyclic AMP-dependent protein kinase from S49 mouse lymphoma cells. J Biol Chem. 1991;266:3547–3553. [PubMed] [Google Scholar]
- 24.Scholfield MR, Ford MC, Vander Zanden CM, Billman MM, Ho PS, Rappe AK. Force Field Model of Periodic Trends in Biomolecular Halogen Bonds. J Phys Chem B. 2015;119:9140–9149. doi: 10.1021/jp509003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JJ, Huang GY, Rieger R, Koller A, Chow D, Kim C. A Protocol for Expression and Purification of Cyclic Nucleotide–Free Protein in Escherichia coli. In: Cheng X, editor. Cyclic Nucleotide Signaling. CRC Press; Boca Raton, FL: 2015. pp. 191–201. [Google Scholar]
- 26.Corbin JD, Ogreid D, Miller JP, Suva RH, Jastorff B, Doskeland SO. Studies of cGMP analog specificity and function of the two intrasubunit binding sites of cGMP-dependent protein kinase. J Biol Chem. 1986;261:1208–1214. [PubMed] [Google Scholar]
- 27.Boettcher AJ, Wu J, Kim C, Yang J, Bruystens J, Cheung N, Pennypacker JK, Blumenthal DA, Kornev AP, Taylor SS. Realizing the allosteric potential of the tetrameric protein kinase A RIalpha holoenzyme. Structure. 2011;19:265–276. doi: 10.1016/j.str.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, Taylor SS. Localization and quaternary structure of the PKA RIbeta holoenzyme. Proc Natl Acad Sci U S A. 2012;109:12443–12448. doi: 10.1073/pnas.1209538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, Taylor SS. Structure and allostery of the PKA RIIbeta tetrameric holoenzyme. Science. 2012;335:712–716. doi: 10.1126/science.1213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- 32.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moll D, Prinz A, Gesellchen F, Drewianka S, Zimmermann B, Herberg FW. Biomolecular interaction analysis in functional proteomics. J Neural Transm. 2006;113:1015–1032. doi: 10.1007/s00702-006-0515-5. [DOI] [PubMed] [Google Scholar]
- 34.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.