

Abstract
While interest in the synthetic chemistry of radical cations continues to grow, controlling enantioselectivity in the reactions of these intermediates remains a challenge. Based on recent insights into the oxidation of tryptophan in enzymatic systems, we report a photocatalytic method for the generation of indole radical cations as hydrogen-bonded adducts with chiral phosphate anions. These non-covalent open-shell complexes can be intercepted by the stable nitroxyl radical TEMPO• to form alkoxyamine-substituted pyrroloindolines with high levels of enantioselectivity. Further elaboration of these optically-enriched adducts can be achieved via a catalytic single-electron oxidation/mesolytic cleavage sequence to furnish transient carbocation intermediates that may be intercepted by a wide range of nucleophiles. Taken together, this two-step sequence provides a simple catalytic method to access a wide range of substituted pyrroloindolines in enantioenriched form via a standard experimental protocol from a common synthetic intermediate. The design, development, mechanistic study, and scope of this process are presented, as are applications of this method to the synthesis of several dimeric pyrroloindoline natural products.
Graphical Abstract
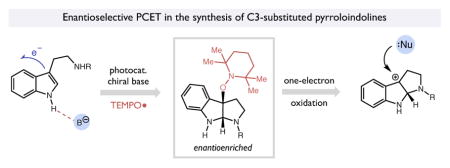
Introduction
The applications of radical cations in organic synthesis have expanded significantly in recent years, in parallel with the development of practical catalytic methods for their generation.1,2 However, strategies for controlling enantioselectivity in the reactions of these charged, open-shell intermediates are considerably less advanced.3 The high innate reactivity of radical cations presents a challenge to the design of such schemes, as the catalytic asymmetric pathway must be kinetically favored over a typically facile racemic background reaction. One strategy to address this issue is to couple the association of the substrate and chiral catalyst with the redox event, ensuring that the radical cations are only generated as catalyst-bound adducts. This approach has proven successful in a number of enantioselective organocatalytic transformations involving enamine radical cations that proceed via covalent association of a chiral amine.3 While powerful, such methods are inherently limited to systems where a reversible covalent equilibrium between substrate and catalyst is possible. If such processes could be generalized for non-covalent systems, the scope of potential applications could be expanded to include a broader range of asymmetric transformations.4
Along these lines, our group has recently reported an approach for generating neutral free radical intermediates as H-bonded adducts with chiral Brønsted bases via reductive proton-coupled electron transfer (PCET).5,6 In these reactions, a hydrogen-bonded complex between a ketone substrate and phosphoric acid can engage in an electron transfer reaction with a one-electron reductant. This electron exchange occurs in concert with proton transfer across the hydrogen bond interface to furnish a neutral ketyl radical intermediate. However, following PCET, the H-bond complex may persist to provide a discrete association between the nascent radical and the conjugate base of the proton donor, providing a basis for asymmetric induction when chiral phosphoric acids are employed.7
We recently questioned whether it would be possible to use a similar approach to control enantioselectivity in the reactions of indole radical cations generated via oxidative PCET. This proposal finds its basis in both advances in chiral counterion strategies in asymmetric catalysis8 and mechanistic work detailing the oxidation of redox-active tryptophans in enzymatic systems such as DNA photolyase and ribonucleotide reductase.9 In these processes, the N-H bond of a tryptophan residue forms a hydrogen bond to a proximal basic moiety within the protein interior, which decreases the potential required for one-electron oxidation of the indole (Figure 1a). In many cases, these participating H-bond acceptors are not sufficiently basic to fully deprotonate the resulting indole radical cations. Rather, they remain associated through a strong ionic hydrogen bond. We anticipated that this PCET framework might be adapted to non-enzymatic contexts as well, wherein a pre-equilibrium H-bonding association of a chiral phosphate anion (pKa = 12–13 for conjugate acid in MeCN)10 to an indole N-H bond will lower the potential required for indole oxidation without deprotonating the resultant radical cation (pKa ~ 14.5 in MeCN).11 This H-bond-coupled redox modulation ensures that the radical cation is only generated when the substrate is pre-associated with the phosphate, while the resulting H-bonded ion pair would remain strongly associated in solution. In turn, this complexation should provide a basis for controlling enantioselectivity in the subsequent bond forming steps when chiral proton donors are employed.12 To put this approach in context, Luo has recently reported a method for the intramolecular asymmetric hydroetherification of olefins that proceeds via chiral phosphate anions associated with alkene radical cations, albeit with modest enantioselectivities.13
Figure 1.

a) Tryptophan radical cation in enzymatic systems. b) proposal for an asymmetric reaction using indole radical cation. c) mesolytic bond cleavage enables a concise route to substituted pyrroloindolines and alkaloid natural products.
Here we report the successful realization of this goal in the context of a general two-step, photo-driven enantioselective synthesis of 3-substituted pyrroloindolines.14 In the first step, an oxidative PCET event as described above furnishes an ionic H-bonded complex between a tryptamine radical cation and a chiral phosphate base. Asymmetric capture of this intermediate by the persistent radical TEMPO• results in the formation of an optically enriched alkoxyamine-substituted pyrroloindoline (Figure 1b). Notably, Xia and coworkers have reported methods to access similar TEMPO-substituted pyrroloindoline adducts in both racemic and diastereoselective forms.15 In accord with our previous work, subsequent one-electron oxidation of this adduct in a second step directly forms a configurationally-biased carbocation and the stable nitroxyl radical TEMPO• via mesolytic cleavage of a transient pyrroloindoline radical cation.16,17 This tertiary carbocation can in turn react with a wide range of nucleophilic partners to furnish enantioenriched pyrroloindoline derivatives from a common synthetic precursor (Figure 1c). The design, mechanistic elucidation and synthetic outcomes of this study are presented below, including the development of concise enantioselective syntheses of the dimeric pyrroloindoline-containing natural products (−)-calycanthidine, (−)-chimonanthine, and (−)-psychotriasine.
Results and Discussion
Asymmetric synthesis of TEMPO-substituted pyrroloindolines
The first reaction in our proposed sequence was the asymmetric coupling of tryptamine and TEMPO• to furnish the key alkoxyamine-substituted pyrroloindoline adduct. We envisioned a prospective catalytic cycle wherein a chiral phosphate base would first form a hydrogen-bonded adduct with the indole N–H bond of a tryptamine substrate (Figure 2). Oxidation of this complex would occur via electron transfer with an excited state oxidant generated under visible light irradiation. The resultant indole radical cation – phosphate ion pair would then react with the stable nitroxyl TEMPO• to form a closed-shell intermediate with a new C–O bond in the C3 position. Capture of the resultant iminium ion by the pendant amine nucleophile would furnish the desired alkoxyamine-substituted pyrroloindoline product. Finally, the reduced state of the photocatalyst and the conjugate acid of the phosphate base would jointly reduce a second equivalent of TEMPO• in a PCET process to form TEMPO-H and complete the catalytic cycle. In this scheme, TEMPO• was expected to play the role of both reactant and stoichiometric oxidant.
Figure 2.
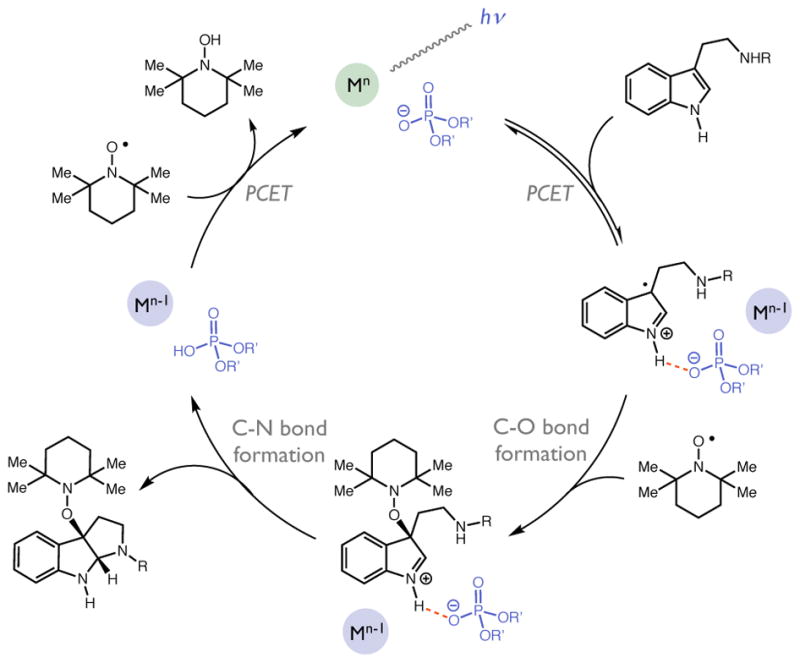
Prospective catalytic cycle for tryptamine PCET reaction.
Initial exploration of the reaction using N′-CO2Me-protected tryptamine, 2 mol% [Ru(bpy)3](BArF)2 with 20 mol% tetrabutylammonium (2,4,6-triisopropyl)phenyl (TRIP) BINOL phosphate, and 2 equivalents of TEMPO• in THF at room temperature irradiated by 7 W blue LEDs gave poor yields but promising enantioselectivities (21% yield, 52% ee) (Table 1, entry 1). Further exploration revealed that using an N′-Cbz protecting group on the tryptamine boosted the enantioselectivity to 86% ee, but unfortunately had little positive effect on the reaction yield (Table 1, entry 2). We hypothesized that the formation of TEMPO-H as a stoichiometric byproduct might be responsible for the poor reactivity profile. TEMPO-H is a well documented substrate in oxidative multi-site PCET reactions and exhibits a very weak O-H bond (O-H BDFE ~ 67 kcal/mol).18 As such, it could sequester the excited state oxidant and Brønsted base catalysts in a kinetically dominant but unproductive redox cycle. Indeed, we observed that addition of 20 mol% TEMPO-H to the model reaction under otherwise standard conditions led to no detectable formation of the desired product (Table 1, entry 3). Based on this finding, we explored the use of other stoichiometric oxidants that could serve the role of both terminal proton and electron acceptor. After considerable exploration, we found that use of iodonium oxidants with 0.5 mol% Ir(ppy)3 and 5 mol% TRIP BINOL phosphate greatly improved the overall yield (Table 1, entry 4). Further optimization revealed that TIPS-EBX provided optimal levels of reaction efficiency and maintained high enantioselectivity at phosphate loadings as low as 3 mol% (Table 1, entries 5). Further evaluation revealed that the H8-TRIP BINOL phosphate was an even more selective catalyst, providing the desired product in 93% ee (Table 1, entry 6). It is worth noting that TIPS-EBX is a poor one-electron oxidant (Ep/2 = −1.12 V vs. SCE), and it cannot directly engage in electron transfer reactions with the indole substrate. However, one electron reduction of this reagent by the excited state of the Ir(ppy)3 photocatalyst (*E1/2 = −1.73 V vs. SCE) is facile and liberates a carboxylate base that can serve as a stoichiometric acceptor for the proton liberated during each revolution of the catalytic cycle, negating the formation of TEMPO-H.
Table 1.
Optimization of asymmetric PCET reaction.a

| ||||||
|---|---|---|---|---|---|---|
| Entry |
|
photocatalyst | base (loading) | additive (equiv) | % yield | % ee |
| 1 | Me | [Ru(bpy)3](BArF)2 | TRIP (20 mol%) | — | 17 | 25 |
| 2 | CO2Me | [Ru(bpy)3](BArF)2 | TRIP (20 mol%) | — | 21 | 52 |
| 3 | Cbz | [Ru(bpy)3](BArF)2 | TRIP (20 mol%) | — | 14 | 86 |
| 4 | Cbz | [Ru(bpy)3](BArF)2 | TRIP (20 mol%) | TEMPO-H (0.2 equiv) | 0 | — |
| 5 | Cbz | Ir(ppy)3 | TRIP (5 mol%) | PhI(OAc)2 (1.0 equiv) | 71 | 58 |
| 6 | Cbz | Ir(ppy)3 | TRIP (5 mol%) | TIPS-EBX (1.0 equiv) | 73 | 89 |
| 7 | Cbz | Ir(ppy)3 | H8-TRIP (3 mol%) | TIPS-EBX (1.5 equiv) | 91 | 93 |
|
| ||||||
Optimization studies were performed on 0.05 mmol scale. Yields were determined by 1H-NMR analysis of the crude reaction mixtures relative to an internal standard. Enantiomeric excess was determined by HPLC analysis on a chiral stationary phase.
Following our optimization, we evaluated the scope of the reaction on preparative scale. Using the conditions from Entry 6 in Table 1, the model tryptamine substrate 1 was converted to 2 in 81% isolated yield and 93% ee after 12 h at room temperature under blue LED irradiation. Substitution of the indole core was also examined, with an emphasis on tryptamines that would grant opportunities for down-stream derivatization. To this end, we were able to achieve highly enantioselective couplings between TEMPO• and 4-Br, 5-Br, and 6-Br tryptamines (3, 4, and 5) in good yields. The chlorinated congener 6 was a similarly effective substrate. Methoxy- and alkyl-substitutions were also tolerated (7 and 8), as were methyl ester and borylated tryptamines (9 and 10). With respect to limitations, we found that tryptamines with substitution at the C2 and C7 positions were poor substrates in the reaction, which we attribute to disruption of key H-bonding interactions between the phosphate catalyst and radical cation. Lastly, to demonstrate the scalability of this protocol, we ran the model reaction in a simple illuminated flow reactor, performing the transformation of 1 to 2 on 10 mmol scale in 20 h with no loss of enantioselectivity or reaction efficiency.19,20
Mechanistic support for PCET
To better understand this process, we elected to study the mechanism of the charge transfer step involved in radical generation. Under the oxidizing conditions of the catalytic reaction, we expected that the excited-state of the Ir(ppy)3 photocatalyst (*E1/2 = −1.73 V vs SCE) would first be quenched by the TIPS-EBX iodonium reagent (Ep/2 = −1.13 V vs SCE) and that the resulting ground state IrIV complex (E1/2 = +0.80 V vs. SCE) would serve as the operative oxidant in the reaction. As such, the key charge transfer step with tryptamine 1 could not be studied directly by luminescence quenching. However, we reasoned that [IrIV(ppy)3]+ could be approximated in Stern–Volmer experiments by *[Ir(ppy)2(dtbbpy)]PF6, which is also an effective catalyst for the reaction (88% yield, 91% ee) and exhibits an excited state reduction potential that is similar to that of the IrIV/III couple of Ir(ppy)3. Using this system, we found that no quenching of *[Ir(ppy)2(dtbbpy)]PF6 is observed in the presence of tryptamine 1 (Ep/2 (R+•/R) = +1.16 V vs. SCE) alone. However, irradiation of [Ir(ppy)2(dtbbpy)]PF6 in the presence of a diphenyl phosphate base and 1 together results in strong phosphorescence quenching that exhibits a first-order kinetic dependence on the concentrations of both 1 and phosphate (Figure 3a). These results suggest that oxidation of the tryptamine-phosphate complex is kinetically more favorable than direct electron transfer with the tryptamine alone.21
Figure 3.

Mechanistic support for PCET oxidation of tryptamine a) luminescence quenching studies of [Ir(ppy)2(dtbbpy)]PF6 with varying concentrations of 1 b) cyclic voltammograms of 1 with varying amounts of phosphate c) cyclic voltammograms of Ir(ppy)3 upon addition of 1 and phosphate.a
aConditions: 1 mM of Ir(ppy)3, 3 mM of NBu4(PhO)2PO2, and 3 mM of 1 with 0.1 M tetrabutylammonium hexafluorophosphate. A glassy carbon working electrode, SCE reference electrode, and platinum mesh counter electrode were used. The experiment was conducted in THF at 23 °C with a scan rate of 0.1 V/s. Each voltammogram was obtained independently.
Further support for this conclusion was obtained from cyclic voltammetry studies. Scans of 1 in THF containing 0.1 M NBu4PF6 and varying concentrations of monobasic diphenyl phosphate (Figure 3b) produced a set of irreversible voltammograms wherein the onset potentials were shifted significantly more positive and the current response increased with increasing concentrations of phosphate. Control experiments revealed that scans of either tryptamine 1 or the phosphate base alone are not responsible for these current features. These results are consistent with a mechanism wherein a hydrogen bonded complex between the tryptamine substrate and the phosphate base is oxidized at a potential more positive than those required for oxidation of either its constituent parts. Such behavior is a characteristic feature of PCET processes, wherein specific hydrogen bonding interactions are known to modulate the redox potentials of the bound substrates.22 Increasing concentrations of the phosphate results in a higher equilibrium concentration of this key H-bonded complex, which in turn leads to increased current. Next, we sought to determine whether evidence for similar PCET chemistry could be obtained for the catalytic reactions mediated by Ir(ppy)3. Voltammograms of 1mM Ir(ppy)3 in a 0.1M THF solution of NBu4PF6 revealed a reversible current corresponding to the IrIV/III couple (Figure 3c). Addition of either the phosphate base or tryptamine 1 alone did not affect the reversibility or current response of this peak. However, addition of both the phosphate base and 1 together led to loss of reversibility and the observation of a new catalytic current. Together with the results above, this observation provides support for a mechanism wherein the putative IrIV species generated in the reaction is kinetically competent to react with the indole-phosphate complex, but not with either of its constituents individually. In the electrochemical experiment, the resulting IrIII species is then reoxidized to IrIV at the electrode and this cycle continues to produce the observed current response (Figure 3c).
The data above provide support for the one-electron oxidation of a tryptamine-phosphate complex, but they do not shed light on the nature of the post-PCET hydrogen bonded complex or the position of the proton within this interface. Seeking to address these questions, we turned to DFT calculations (UB3LYP 6-31G+(d,p) CPCM=THF), which indicated that the proton within the hydrogen bond between the indole nitrogen and the oxygen of a biphenyl phosphate is covalently associated with the indole nitrogen. This outcome is consistent with the solution pKa data in MeCN (vide supra), and supports the intermediacy of an indole radical cation rather than a neutral indolyl radical as the key intermediate in the reaction.
In addition to these spectroscopic, voltammetric, and computational results, a number of reactivity studies were also consistent with our mechanistic proposal. First, if the reaction proceeds through a phosphate-bound indole radical cation, we would expect that replacing the indole N–H bond with an N-alkyl group would disrupt both the H-bond donor-coupled electron transfer and the basis for asymmetric induction. To this end, we observed that a tryptamine substrate bearing an N-Me group on the indole nitrogen did not react at all under the standard reaction conditions and was recovered unchanged. This outcome is notable in the fact that the N-alkyl indole is easier to oxidize via direct outer sphere ET than the N–H compound by more than 70mV. This observation confirms the ability of an H-bond interface to modulate the potentials requirements in PCET reactions even in the absence of formal proton transfer. One could also imagine that TEMPO• is oxidized to its corresponding oxoammonium ion under the reported conditions. This oxoammonium cation could react with the indole as a polar electrophile that is associated with a chiral phosphate, in analogy to the recent asymmetric phase transfer work of Toste.23 However, control experiments wherein a pre-generated oxoammonium salt derived from TEMPO24 was added to the reaction provided only trace quantities of product as a racemic mixture, suggesting that this is not the operative pathway for product formation.
Lastly, we observed that while no reaction occurs in the absence of either the Ir photocatalyst or blue LED irradiation, this reaction could proceed in the absence of the chiral phosphate catalyst, demonstrating that a racemic background reaction is accessible under these conditions. We hypothesize that this background reactivity may be driven by the generation of a benzoate base in situ upon reduction of the iodonium oxidant. To evaluate the contribution of this background reaction in reactions containing the chiral phosphate, we examined the enantioselectivity of 2 as a function of phosphate loading. Remarkably, these experiments revealed that the observed level of enantioselectivity reaches a maximum at 3 mol% of added phosphate and does not increase above 93% ee with increased phosphate loading. Intriguingly, this outcome suggests that the racemic background reaction is effectively suppressed even when low concentrations of phosphate are present. Extending this line of reasoning, we found that the much more oxidizing photocatalyst [Ir(dF(CF3)ppy)2(bpy)](PF6) (*E1/2 = + 1.32 V vs SCE) can also be used in this protocol without any observable loss of enantioselectivity. Unlike [IrIV(ppy)3]+ (E1/2 = + 0.80 V vs SCE), this catalyst does not require H-bond pre-association of the phosphate to oxidize the tryptamine en route to radical cation formation. This observation suggests that ion pairing between the radical cation and the phosphate is faster than TEMPO• trapping and that the productive reaction proceeds exclusively through the phosphate-bound complex even if they are not complexed during the initial electron transfer event. If general, we anticipate that this surprising insight will prove useful in the design of future work on the asymmetric catalysis of radical cation reactions. Taken altogether, we believe that these experiments are consistent with the proposed mechanism of substrate activation and catalyst association, and represent a rare example of a highly enantioselective catalytic reaction of a radical cation intermediate mediated entirely by non-covalent interactions.
Mesolytic cleavage step
With a successful and scalable method to make the key enantioenriched alkoxyamine-substituted pyrroloindolines in hand, we sought to derivatize these products into a range of diverse pyrroloindoline structures. In accord with our previous work in catalytic carbocation generation,16 we envisioned that the nitrogen lone pair of this TEMPO-substituted pyrroloindoline adduct (Ep/2 = +0.92 V vs SCE) could be oxidized by an excited state photocatalyst to furnish a transient radical cation. The C–O bond of this intermediate would be significantly weakened relative to its neutral closed shell precursor, and undergo C–O mesolytic cleavage to generate TEMPO• and a configurationally biased tertiary pyrroloindoline carbocation. Based on prior art, we expect that this carbocation could react in turn with exogenous nucleophiles to furnish a wide variety of structurally diverse pyrroloindoline cores.25 Proton-coupled reduction of TEMPO• by the reduced form of the photocatalyst would form TEMPO-H as a stoichiometric byproduct and close the catalytic cycle. Notably, this protocol differs from prior work in cation generation in that it occurs under Brønsted neutral conditions, enabling the use of a range of acid-sensitive or Lewis basic nucleophiles that might prove incompatible with traditional methods involving acidic or electrophilic activators.
Preliminary experiments demonstrated that subjecting the C3-substituted pyrroloindoline alkoxyamine 2 to [Ir(dF,CF3-ppy)2(bpy)]PF6 in the presence of potassium trans-styrenyl trifluoroborate provided the desired vinylated product without loss of optical purity, but in low yield despite full conversion of starting material. Systematic evaluation of changes in temperature, solvent, and photocatalyst did little to improve the efficiency of the reaction. We hypothesized that the poor mass balance might be a consequence of the in situ generation of strongly Lewis acidic BF3 in the presence of the unprotected indoline in the substrate. Indeed, subjecting a N-Boc protected version of the pyrroloindoline alkoxyamine (11) (Ep/2 = +1.28 V vs SCE) to a more oxidizing photocatalyst [Ir(dF, CF3-ppy)2(dCF3-bpy)]PF6 (*E1/2 = +1.68 V vs SCE) in MeNO2, we could obtain the desired alkene product in 65% yield after 8 h at room temperature (Table 3, 12).
Table 3.
Scope of mesolytic bond cleavage induced nucleophilic substitution.a
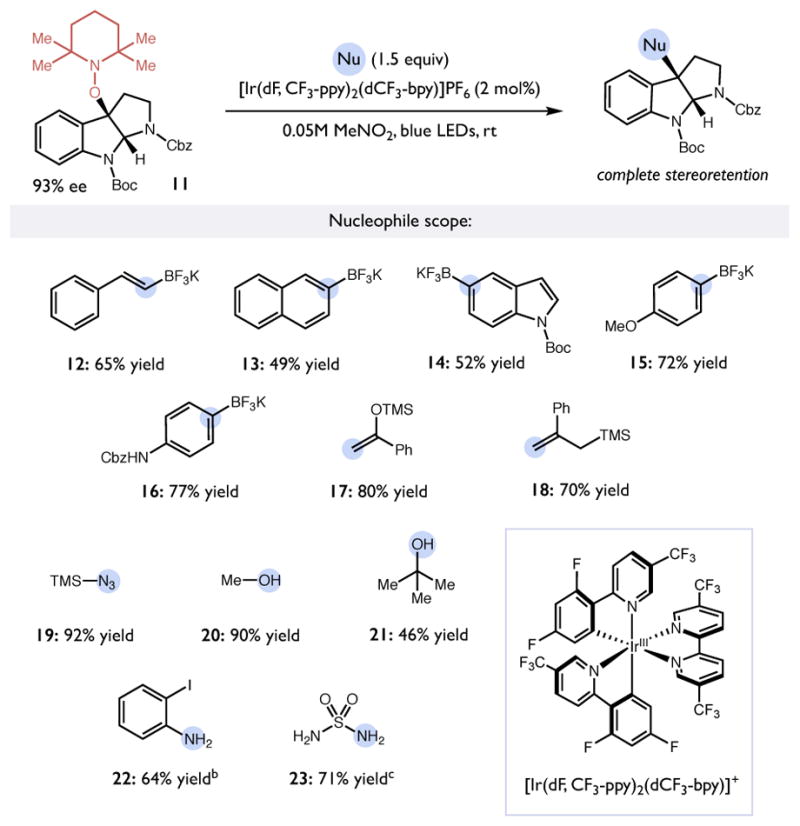
|
Reactions run on a 0.25 mmol scale. Reported yields are for isolated and purified materials. The optical purity of all products was confirmed by HPLC analysis.
Unprotected pyrroloindoline 2 as the substrate.
Conducted with 6 equiv. of sulfamide.
With these conditions, we next surveyed the scope of this nucleophilic substitution reaction. With the N-Boc-protected TEMPO-substituted pyrroloindoline (11) as the standard electrophile, we were able to employ a variety of C-, N-, and O- nucleophilic coupling partners (Table 3). In addition to the model vinyl trifluoroborate nucleophile, naphthyl, phenyl, and 5-indolyl BF3K salts could be employed to obtain the corresponding arylated pyrroloindoline adducts (Table 3, 13–16). Silyl nucleophiles were also competent in the reaction, with allyl silanes and silyl enol ethers producing alkylated products 17 and 18 and TMSN3 generating azide product 19 in excellent yield. Finally, a number of protic nucleophiles could be utilized. Aliphatic alcohols act as proficient nucleophiles in the reaction (20, 21). Of particular interest, tert-butanol was even a successful coupling partner with the tertiary pyrroloindoline carbocation to produce the sterically congested ether 21 in modest yield. Nitrogen nucleophiles were also amenable to the method, as evidenced by the generation of ortho-iodoaniline 22 and sulfamate 23. The latter functional group has been used to great effect by Movassaghi in recent work as precursor for the synthesis of diazene-linked pyrroloindoline dimers that can be coupled photochemically.26,27 In all the reactions above, we expect that the proton, boron, or silicon groups liberated in this reaction bind to and promote the single-electron reduction of TEMPO• to complete the catalytic cycle. Taken together, this approach represents a practical and highly adaptable method to generate a wide variety of C3-substituted pyrroloindoline structures in optically enriched form via a standard experimental protocol from a common intermediate. Our efforts to extend these reactions to accommodate more complex fragment couplings are detailed below.
Alkaloid natural product synthesis
The cyclotryptamine alkaloids are classic targets for chemical synthesis and have been the focus of sustained synthetic interest for decades.28,29 As such these natural products represent useful molecular benchmarks for assessing the value and applicability of new synthetic strategies and methods.30 In this vein, we questioned whether we could utilize the two catalytic radical cation chemistries described above to enable the development of concise asymmetric routes to the pyrroloindoline cores of these target structures. Specifically, we anticipated that the carbocation generated in the oxidatively-induced mesolytic cleavage step from alkoxyamine 2 could be intercepted by a tryptamine π-nucleophile to directly furnish the characteristic bis-pyrroloindoline core of the calycantheous alkaloids. To put this approach in context, Movassaghi has elegantly demonstrated that Ag-mediated ionization of related bromo-substituted pyrroloindolines can be trapped by aryl boron nucleophiles to furnish new C–C bonded products in his synthesis of (+)-naseseazines A and B.25h,31 The key question we hoped to address in this study was the ability to control the stereochemical outcome of the C–C bond-forming event with a prochiral tryptamine nucleophile to furnish the characteristic vicinal quaternary stereocenters in a diastereoselective fashion. With respect to the strategic advantages of this approach, we note that many of the dimeric cyclotryptamine natural products are unsymmetrical and differ in their substitution patterns on the four peripheral nitrogens. Consequently, dimerization-based strategies, wherein two identical fragments are coupled together, are not easily amenable to the construction of these targets. A clear advantage of the strategy outlined above is that the two pyrroloindoline units in the dimer arise from a coupling reaction between a distinct tryptamine nucleophile and a pyrroloindoline carbocation electrophile. As such, synthesizing the appropriately substituted reaction partners provides a common method to generate unsymmetrical targets directly.
Calycanthidine
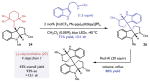
To this end, our initial efforts focused on the construction of the unsymmetrical dimer, calycanthidine (27).26,32,33 This alkaloid exhibits local C2 symmetry at the bisquaternary centers, but possesses a methyl group on one of the two isomeric aniline nitrogens that breaks the overall symmetry of the structure. Enantioselective syntheses of calycanthidine reported in the literature are comparatively lengthy relative to the synthesis of its symmetrical congener, chimonanthine. We proposed that the sequence of mesolytic cleavage and nucleophilic capture could be readily adapted to enable a concise enantioselective synthesis of this target in just four steps from tryptamine 1. As described above, one-electron oxidation of the unprotected pyrroloindoline 2 (Ep/2 = + 0.92 V vs SCE) by the excited state of [Ir(dCF3Me-ppy)2(dtbbpy)]PF6 (*E1/2 = + 1.22 V vs SCE) at room temperature in THF results in mesolytic bond cleavage to furnish a tertiary carbocation electrophile. This cation was then intercepted by N-Me-N′-Cbz tryptamine 25 in a Friedel-Crafts process to form the unsymmetrical dimer 26 in moderate yield as a 1:1 mixture of diastereomers at the bisquaternary centers (Table 4, entry 1). Further evaluation of the solvent and temperature led to little improvement in the stereoselectivity (Table 4, entry 2), prompting us to explore an alternative strategy involving N-methylation of the pyrroloindoline electrophile, which was readily accomplished upon treatment of 2 with MeI and NaHMDS in THF to furnish pyrroloindoline 24 in 93% yield (Table 4). We observed that reactions of this cation precursor (Ep/2 = + 0.81 V vs SCE) with N′-Cbz tryptamine in the presence of [Ir(dCF3Me-ppy)2(dtbbpy)]PF6 at room temperature gave rise to higher diastereoselectivities (5:1), albeit with lower overall yields of 26 (Table 4, entry 3). Lowering the temperature of the reaction improved the diastereoselectivity of the reaction but did not benefit the reaction efficiency (Table 4, entry 4). More extensive optimization revealed that the addition of 1 equivalent of trifluoroacetic acid enhanced the yield of the reaction while maintaining the higher levels of diastereoselectivity (Table 4, entry 5). With these conditions in hand, we were able to perform the heterodimerization on a 2 mmol scale to afford 71% isolated yield (849mg) of the desired product 26 as a 13:1 mixture of diastereomers. Reduction of the two benzyl carbamate groups in the resulting adduct with Red-Al proceeds smoothly (80% yield) to furnish (−)-calycanthidine (27). This asymmetric synthesis proceeds in four chemical steps from tryptamine 1 in 43% overall yield.
Table 4.
Optimization of heterodimerizationa and synthesis of (−)-calycanthidine.
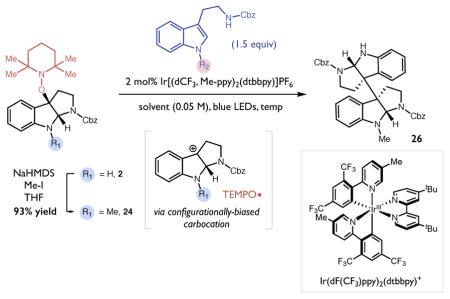
| |||||||
|---|---|---|---|---|---|---|---|
| Entry |
|
|
solvent | additive | temp | % yield | dr (C2:meso) |
| 1 | H | Me | THF | — | rt | 21 | 1:1 |
| 2 | H | Me | THF | — | −40 ºC | 10 | 2:1 |
| 3 | Me | H | CH2Cl2 | — | rt | 48 | 5:1 |
| 4 | Me | H | CH2Cl2 | — | −40 ºC | 30 | 14:1 |
| 5 | Me | H | CH2Cl2 | TFA (1 equiv) | −40 ºC | 71 | 14:1 |
| Diastereoselective synthesis of (−)-calycanthidine | |||||||
| |||||||
Optimization reactions performed on a 0.025 mmol scale. Yields and diastereomeric ratios were determined by 1HNMR analysis of the crude reaction mixtures. Optimized heterodimerization reaction performed on 2 mmol scale.
Chimonanthine
We next turned our attention to the synthesis of the C2-symmetrical dimer (−)-chimonanthine.34,35 In analogy to the results described above, oxidation of the unprotected pyrroloindoline 2 (Ep/2 = + 0.92 V vs SCE) using the excited state of [Ir(dCF3Meppy) 2(dtbbpy)]PF6, generated a carbocation intermediate that could be efficiently captured by tryptamine 1 to form symmetrical dimer 28 in moderate yield (26%) with diastereoselectivities of up to 3:1 C2:meso. As in the calycanthidine system, we observed that addition of trifluoroacetic acid enhanced the efficiency of the reaction (54% yield). However the diastereomeric ratio of isolated products under these conditions proved to be highly irreproducible on small scale, ranging from 3:1 to >20:1.
Additional experiments revealed that the when an isolated 3:1 mixture (C2:meso) of the dimeric material was re-subjected to the reaction conditions under an air atmosphere, the isomeric ratio enriched over time to furnish the C2 isomer in >20:1 dr in 69% yield after 24 hr at −40 °C. Notably, the enantiopurity of the C2 isomer (93% ee) in this reaction was unchanged during the enrichment process. Also, when this enrichment reaction was carried out in the presence of an exogenous N′-Boc tryptamine nucleophile, no crossover products were observed. Together these results suggest that reversible cleavage of the central C–C bond was not the operative mechanism for isomeric enrichment. Rather, when these enrichment reactions were monitored by NMR, the meso diastereomer appeared to be selectively consumed over time, while the C2 isomer was not degraded (Figure 4). Control experiments revealed that this process requires both the photocatalyst and exposure to oxygen, suggesting an oxidative pathway for selective decomposition of the meso isomer. However, despite extensive efforts, none of the complex mixture of degradation byproducts could be identified. This enrichment process was performed on a 3:1 mixture of isolated dimer diastereomers in batch under blue light irradiation at −40 °C in THF to yield the desired C2 isomer in 69% yield on >20:1 dr with 93% ee. This adduct was subsequently reduced with Red-Al in 79% yield to provide (−)-chimonanthine (29). Altogether, this stereoselective synthesis proceeds in 4 steps from tryptamine 1 in 24% overall yield (Figure 4).
Figure 4.

Synthesis of (−)-chimonanthine and origin of diastereoselectivity. 500MHz 1H NMR spectra obtained in CD3CN at rt. aDiastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixture.
Psychotriasine
Lastly, we also adapted this method to develop a concise enantioselective synthesis of psychotriasine, another dimeric pyrroloindoline natural product that features an unusual C-N linkage.36,37 Using the mesolytic cleavage conditions described above with ortho-iodoaniline as the nucleophile resulted in formation of expected N-aryl pyrroloindoline 22 in 64% yield. Adopting a strategy originally developed by Baran in his synthesis of psychotriasine, the iodoaniline was subjected to a Larock annulation with alkyne 30 to furnish the core of the natural product.38 Subsequent reduction of the carbamate protecting groups with Red-Al resulted in the synthesis of (−)-psychotriasine 31 in 93% ee in four chemical steps and 38% overall yield from tryptamine 1 (Figure 5). Taken together, these syntheses highlight the versatility of this method and suggest that its use in even more complex contexts might prove feasible. Efforts to evaluate this hypothesis are the subject of ongoing efforts.
Figure 5.
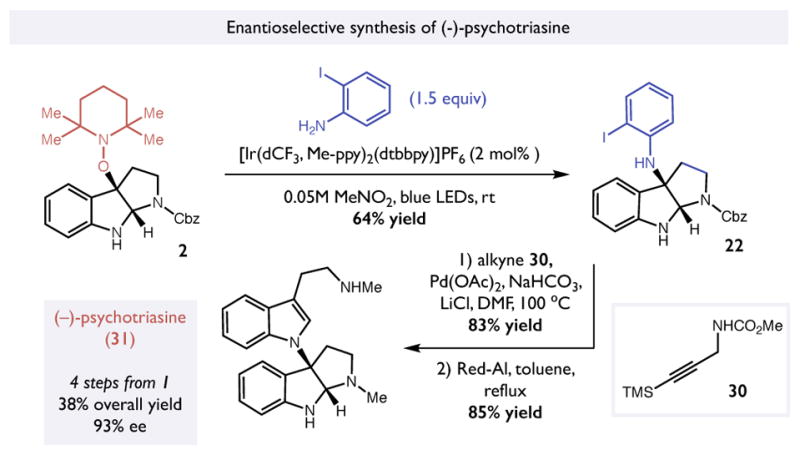
Synthesis of (−)-psychotriasine.
Conclusions
In conclusion, we have developed a new protocol for the asymmetric synthesis of a range of substituted pyrroloindolines that proceeds via sequential reactions of catalytically generated radical cation intermediates. In the first step, single electron oxidation with an excited state redox catalyst produces a tryptamine radical cation that exists as a hydrogen-bonded complex with a chiral phosphate base, enabling an enantioselective coupling with the stable nitroxyl radical TEMPO•. This transformation represents a rare example of a highly enantioselective reaction of a radical cation intermediate that is mediated entirely by non-covalent interactions. In the second step, the resulting pyrroloindoline adduct undergoes single-electron oxidation and the resulting radical cation fragments mesolytically to reveal a carbocation intermediate that can be trapped by a range of different nucleophiles. This sequence is notable in the scope of nucleophilic partners that it accommodates, and for its ability to enable stereoselective couplings with prochiral tryptamine nucleophiles to furnish asymmetric access to a range of dimeric pyrroloindoline natural products via short synthetic sequences. We are optimistic that these reports will facilitate future advances in both the asymmetric reactions of radical cation intermediates as well as in the synthesis of more complex alkaloid natural products and related structures.
Supplementary Material
Table 2.
Substrate scope of enantioselective PCET reaction.

|
Reactions run on a 0.5 mmol scale. Reported yields are for isolated and purified materials. Enantiomeric excess determined by HPLC analysis.
Acknowledgments
Financial support was provided by NIH R01 GM120530.
Footnotes
NOTES
The authors declare no competing financial interest.
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org. Experimental details; characterization data and NMR spectra; voltammetric, computational and luminescence quenching data.
References
- 1.(a) Schmittel M, Burghart A. Angew Chem Int Ed Engl. 1997;36:2550–2589. [Google Scholar]; (b) Bauld N, Bellville DJ, Harirchian B, Lorenz KT, Pabon RA, Jr, Reynolds DW, Wirth DD, Chiou HS, Marsh BK. Acc Chem Res. 1987;20:371–378. [Google Scholar]
- 2.For representative examples: Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y.Lin S, Ischay MA, Fry CG, Yoon TP. J Am Chem Soc. 2011;133:19350–19353. doi: 10.1021/ja2093579.Margrey KA, Nicewicz DA. Acc Chem Res. 2016;49:1997–2006. doi: 10.1021/acs.accounts.6b00304.Morris SA, Wang J, Zheng N. Acc Chem Res. 2016;49:1957–1968. doi: 10.1021/acs.accounts.6b00263.Musacchio AJ, Nguyen LQ, Beard GH, Knowles RR. J Am Chem Soc. 2014;136:12217–12220. doi: 10.1021/ja5056774.
- 3.(a) Beeson TD, Mastracchio A, Hong JB, Ashton A, MacMillan DWC. Science. 2007;316:582–585. [PubMed] [Google Scholar]; (b) Jang HY, Hong JB, MacMillan DWC. J Am Chem Soc. 2007;129:7004–7005. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]; (c) Kim H, MacMillan DWC. J Am Chem Soc. 2008;130:398–399. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]; (d) Rendler S, MacMillan DWC. J Am Chem Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected reports on role of non-covalent interactions in electron transfer reactions, see: Turek AK, Hardee DJ, Ullman AM, Nocera DG, Jacobsen EN. Angew Chem Int Ed. 2015;55:549–554. doi: 10.1002/anie.201508060.Okamoto K, Ohkubo K, Kadish KM, Fukuzumi S. J Phys Chem A. 2004;108:10405–10413.Yuasa J, Yamada S, Fukuzumi S. J Am Chem Soc. 2008;130:5808–5820. doi: 10.1021/ja8001452.Fukuzumi S, Kitaguchi H, Suenobu T, Ogo S. Chem Commun. 2002:1984–1985. doi: 10.1039/b205517a.Ge Y, Lilienthal RR, Smith DK. J Am Chem Soc. 1996;118:3976–3977.Clare LA, Pham AT, Magdaleno F, Acosta J, Woods JE, Cooksy AL, Smith DK. J Am Chem Soc. 2013;135:18930–18941. doi: 10.1021/ja410061x.Neumann M, Kirsten Zeitler K. Chem Eur J. 2013;19:6950–6955. doi: 10.1002/chem.201204573.Greaves MD, Niemz A, Rotello VM. J Am Chem Soc. 1999;121:266–267.Breinlinger EC, Rotello VM. J Am Chem Soc. 1997;119:1165–1166.
- 5.(a) Gentry EC, Knowles RR. Acc Chem Res. 2016;49:1546–1556. doi: 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi GJ, Knowles RR. J Am Chem Soc. 2015;137:9226–9229. doi: 10.1021/jacs.5b05377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yayla HG, Wang H, Orbe HS, Knowles RR. J Am Chem Soc. 2016;138:10794–10797. doi: 10.1021/jacs.6b06517. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tarantino KT, Liu P, Knowles RR. J Am Chem Soc. 2013;135:10022–10015. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 6.For general references on PCET, see: Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Phil Trans R Soc, B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j.Hammes-Schiffer S. Acc Chem Res. 2009;42:1881–1889. doi: 10.1021/ar9001284.Mayer JM. Ann Rev Phys Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446.Miller DC, Tarantino KT, Knowles RR. Top Curr Chem. 2016;374:145–203. doi: 10.1007/s41061-016-0030-6.
- 7.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. J Am Chem Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- 8.(a) Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. J Am Chem Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. [DOI] [PubMed] [Google Scholar]; (b) Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496–499. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]; (d) Phipps RJ, Hamilton GL, Toste FD. Nat Chem. 2012;4:603–614. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]; (e) García-García P, Lay F, García-García P, Rabalakos C, List B. Angew Chem, Int Ed. 2009;48:4363–4366. doi: 10.1002/anie.200901768. [DOI] [PubMed] [Google Scholar]; (f) Brak K, Jacobsen EN. Angew Chem, Int Ed. 2013;52:534–561. doi: 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Migliore A, Polizzi NF, Therien MJ, Beratan DN. Chem Rev. 2014;114:3381–3465. doi: 10.1021/cr4006654. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Z, Tan C, Guo X, Li J, Wang L, Sancar A, Zhong D. Proc Nat Acad Sci. 2013;110:12966–12971. doi: 10.1073/pnas.1311073110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aubert C, Vos MH, Mathis P, Eker APM, Brettel K. Nature. 2000;405:586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]; (d) Byrdin M, Villette S, Espagne A, Eker APM, Brettel K. J Phys Chem B. 2008;112:6866–6871. doi: 10.1021/jp711435y. [DOI] [PubMed] [Google Scholar]; (e) Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]
- 10.Kaupmees K, Tolstoluzhsky N, Raja S, Rueping M, Leito I. Angew Chem, Int Ed. 2013;52:11569–11572. doi: 10.1002/anie.201303605. [DOI] [PubMed] [Google Scholar]
- 11.Calculated from N-H BDFE = 93 kcal/mol and Ep/2 = +0.78 V vs. Fc+/Fc in MeCN for indole.
- 12.(a) Crich D, Huang W. J Am Chem Soc. 2001;123:9239–9245. doi: 10.1021/ja010841l. [DOI] [PubMed] [Google Scholar]; (b) Adamo MFA, Aggarwal VK, Sage MA. J Am Chem Soc. 2000;122:8317–8318. [Google Scholar]; (c) Crich D, Shirai M, Brebion F, Rumthao S. Tetrahedron. 2006;62:6501–6518. [Google Scholar]; (d) Turek AK, Hardee DJ, Ullman AM, Nocera DG, Jacobsen EN. Angew Chem, Int Ed. 2016;55:539–544. doi: 10.1002/anie.201508060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fukuzumi S, Ohkubo K, D’Souza F, Sessler JL. Chem Comm. 2012;48:9801–9815. doi: 10.1039/c2cc32848h. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Li H, Li S, Zhang MT, Luo S. Org Chem Front. 2017;4:1037–1041. [Google Scholar]
- 14.For selected methods for the synthesis of pyrroloindolines, see: Taniguchi M, Hino T. Tetrahedron. 1981;37:1487–1494.Crich D, Huang X. J Org Chem. 1999;64:7218–7223. doi: 10.1021/jo982385y.Marsden SP, Depew KM, Danishefsky SJ. J Am Chem Soc. 1994;116:11143–11144.Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702–7703.Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc Natl Acad Sci USA. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101.Trost BM, Quancard J. J Am Chem Soc. 2006;128:6314–6315. doi: 10.1021/ja0608139.Newhouse T, Baran PS. J Am Chem Soc. 2008;130:10886–10887. doi: 10.1021/ja8042307.Espejo VR, Li XB, Rainier JD. J Am Chem Soc. 2010;132:8282–8284. doi: 10.1021/ja103428y.Repka LM, Ni J, Reisman SE. J Am Chem Soc. 2010;132:14418–14420. doi: 10.1021/ja107328g.Kieffer ME, Chuang KV, Reisman SE. Chem Sci. 2012;3:3170–3174. doi: 10.1039/C2SC20914D.Zhu S, MacMillan DWC. J Am Chem Soc. 2012;134:10815–10818. doi: 10.1021/ja305100g.
- 15.Deng X, Liang K, Tong X, Ding M, Li D, Xia C. Org Lett. 2014;16:3276–3279. doi: 10.1021/ol501287x. [DOI] [PubMed] [Google Scholar]
- 16.(a) Zhu Q, Gentry EC, Knowles RR. Angew Chem, Int Ed. 2016;55:9969–9973. doi: 10.1002/anie.201604619. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O’Bryan G, Braslau R. Macromolecules. 2006;39:9010–9017. [Google Scholar]
- 17.For pioneering studies of radical ion fragmentation: Kumar VS, Floreancig PE. J Am Chem Soc. 2001;123:3842–3843. doi: 10.1021/ja015526d.Floreancig PE. Synlett. 2007;2:191–203.Wang L, Seiders JR, Floreancig PE. J Am Chem Soc. 2004;126:12596–12603. doi: 10.1021/ja046125b.Cho DW, Yoon UC, Mariano PS. Acc Chem Res. 2011;44:204–215. doi: 10.1021/ar100125j.Fagnoni M, Albini A. Acc Chem Res. 2005;38:713–721. doi: 10.1021/ar0402356.Popielarz R, Arnold DR. J Am Chem Soc. 1990;112:3068–3082.
- 18.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hook BDA, Dohle W, Hirst PR, Pickworth M, Berry MB, Booker-Milburn KI. J Org Chem. 2005;70:7558–7564. doi: 10.1021/jo050705p. [DOI] [PubMed] [Google Scholar]
- 20.For details of flow experiments and apparatus, see Supporting Information.
- 21.(a) Zhang M-TL, Hammarstrom L. J Am Chem Soc. 2011;133:8806–8809. doi: 10.1021/ja201536b. [DOI] [PubMed] [Google Scholar]; (b) Reece S, Stubbe J, Nocera D. BBA – Bioenergetics. 2005;1706:232–238. doi: 10.1016/j.bbabio.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Costentin C. Chem Rev. 2008;108:2145–2175. doi: 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- 23.Neel AJ, Hehn JP, Tripet PF, Toste FD. J Am Chem Soc. 2013;135:14044–14047. doi: 10.1021/ja407410b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercadante MA, Kelly CB, Bobbitt JM, Tilley LJ, Leadbeater NE. Nat Protoc. 2013;8:666–676. doi: 10.1038/nprot.2013.028. [DOI] [PubMed] [Google Scholar]
- 25.Selected examples of cation generation strategies for pyrroloindoline synthesis: Marsden SP, Depew KM, Danishefsky SJ. J Am Chem Soc. 1994;116:11143–11144.Kim J, Movassaghi M. J Am Chem Soc. 2011;133:14940–14943. doi: 10.1021/ja206743v.Schiavi BM, Richard DJ, Joullie MM. J Org Chem. 2002;67:620–624. doi: 10.1021/jo0106593.Ley SV, Cleator E, Hewitt PR. Org Biomol Chem. 2003;1:3492–3294. doi: 10.1039/b308288a.Hewitt PR, Cleator E, Ley SV. Org Biomol Chem. 2004;2:2415–2417. doi: 10.1039/B410180D.Adhikari AA, Chisholm JD. Org Lett. 2016;18:4100–4103. doi: 10.1021/acs.orglett.6b02024.Wang Y, Kong C, Du Y, Song H, Zhang D, Qin Y. Org Biomol Chem. 2012;10:2793–2797. doi: 10.1039/c2ob00014h.Xie W, Jiang G, Liu H, Hu J, Pan X, Zhang H, Wan X, Lai Y, Ma D. Angew Chem, Int Ed. 2013;52:12924–12927. doi: 10.1002/anie.201306774.
- 26.Lathrop SP, Movassaghi M. Chem Sci. 2014;5:333–340. doi: 10.1039/C3SC52451E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindovska P, Movassaghi M. J Am Chem Soc. 2017;139:17590–17596. doi: 10.1021/jacs.7b09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.For cyclotryptamine natural products, see: Anthoni U, Christophersen C, Nielsen PH. Naturally Occurring Cyclotryptophans and Cyclotryptamines. In: Pelletier SW, editor. Alkaloids: Chemical & Biological Perspectives. Vol. 13. Pergamon; Oxford: 1999. pp. 163–236.
- 29.For reviews on of pyrroloindoline natural product synthesis, see: Crich D, Banerjee A. Acc Chem Res. 2007;40:151–161. doi: 10.1021/ar050175j.Steven A, Overman LE. Angew Chem, Int Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612.Kim J, Movassaghi M. Chem Soc Rev. 2009;38:3035–3050. doi: 10.1039/b819925f.Ruiz-Sanchis P, Savina SA, Albericio F, Alvarez M. Chem-Eur J. 2011;17:1388–1408. doi: 10.1002/chem.201001451.
- 30.For selected examples of pyrroloindoline natural product synthesis, see: Movassaghi M, Schmidt MA. Angew Chem Int Ed. 2007;46:3725–3728. doi: 10.1002/anie.200700705.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777.Boyer N, Movassaghi M. Chem Sci. 2012;3:1798–1803. doi: 10.1039/C2SC20270K.Furst L, Narayanam JMR, Stephenson CRJ. Angew Chem, Int Ed. 2011;50:9655–9659. doi: 10.1002/anie.201103145.Kodanko JJ, Overman LE. Angew Chem, Int Ed. 2003;42:2528–2531. doi: 10.1002/anie.200351261.Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702–7703.Overman LE, Peterson EA. Angew Chem, Int Ed. 2003;42:2525–2528. doi: 10.1002/anie.200351260.Lebsack AD, Link JT, Overman LE, Stearns BA. J Am Chem Soc. 2002;124:9008–9009. doi: 10.1021/ja0267425.DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v.Snell RH, Woodward RL, Willis MC. Angew Chem, Int Ed. 2011;50:9116–9119. doi: 10.1002/anie.201103864.Newhouse T, Lews CA, Baran PS. J Am Chem Soc. 2009;131:6360–6361. doi: 10.1021/ja901573x.Horning BD, MacMillan DWC. J Am Chem Soc. 2013;135:6442–6445. doi: 10.1021/ja402933s.Jamison CR, Badillo JJ, Lipshultz JL, Comito RJ, MacMillan DWC. Nat Chem. 2017;9:1165–1169. doi: 10.1038/nchem.2825.Kieffer ME, Chuang KV, Reisman SE. J Am Chem Soc. 2013;135:5557–5560. doi: 10.1021/ja4023557.
- 31.For additional examples of Ag-mediated ionizations in natural product synthesis: Kim J, Movassaghi M. Acc Chem Res. 2015;48:1159–1171. doi: 10.1021/ar500454v.Loach RP, Fenton OS, Movassaghi M. J Am Chem Soc. 2016;138:1057–1064. doi: 10.1021/jacs.5b12392.
- 32.For isolation and characterization of (−)-calycanthidine: Barger G, Jacob A, Madinaveitia J. Trav Chim. 1938;57:548–554.Hodson HF, Robinson B, Smith GF. Proc Chem Soc. 1961:465–466.Saxton JE, Barsley WG, Smith GF. Proc Chem Soc. 1962:148.Grant IJ, Hamor TA, Robertson JM, Sim GA. Proc Chem Soc. 1962:148–149.Grant IJ, Hamor TA, Robertson JM, Sim GA. J Am Chem Soc. 1965:5678–5696.
- 33.Movassaghi M, Ahmad OK, Lathrop SP. J Am Chem Soc. 2011;133:13002–13005. doi: 10.1021/ja2057852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.For isolation, characterization, and biosynthetic hypothesis of chimonanthine: Woodward RB, Yang NC, Katz TJ, Clark VM, Harley-Mason J, Ingleby RFJ, Sheppard N. Proc Chem Soc. 1960:76–78.Robinson R, Teuber HJ. Chem Ind. 1954:783–784.Scott AI, McCapra F, Hall ES. J Am Chem Soc. 1964;86:302–303.Hendrickson JB, Rees R, Goschke R. Proc Chem Soc. 1962:383–384.Hino T, Yamada S. Tet Lett. 1963;4:1757–1760.
- 35.For previous asymmetric syntheses of chimonanthine: Link JT, Overman LE. J Am Chem Soc. 1996;118:8166–8167.Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702–7703.Overman LE, Larrow JF, Stearns BA, Vance JM. Angew Chem, Int Ed. 2000;39:213–215. doi: 10.1002/(sici)1521-3773(20000103)39:1<213::aid-anie213>3.0.co;2-z.Movassaghi M, Schmidt MA. Angew Chem, Int Ed. 2007;46:3725–3728. doi: 10.1002/anie.200700705.Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S. Angew Chem, Int Ed. 2012;51:5217–5221. doi: 10.1002/anie.201201132.Ding M, Liang K, Pan R, Zhang H, Xia C. J Org Chem. 2015;80:10309–10316. doi: 10.1021/acs.joc.5b01907.
- 36.Zhao H, He H-P, Wang Y-H, Hao X-J. Helv Chem Acta. 2010;93:1650–1652. [Google Scholar]
- 37.(a) Li Q, Xia T, Yao L, Deng H, Liao X. Chem Sci. 2015;6:3599–3609. doi: 10.1039/c5sc00338e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gallego S, Lorenzo P, Alvarez R, de Lera AR. Tet Lett. 2017;58:210–212. [Google Scholar]; (c) Liu C, Yi JC, Zheng ZB, Tang Y, Dai LX, You SL. Angew Chem, Int Ed. 2016;55:751–754. doi: 10.1002/anie.201508570. [DOI] [PubMed] [Google Scholar]; (d) Tayu M, Ishizaki T, Higuchi K, Kawasaki T. Org Biomol Chem. 2015;13:3863–3865. doi: 10.1039/c5ob00190k. [DOI] [PubMed] [Google Scholar]
- 38.(a) Newhouse T, Baran PS. J Am Chem Soc. 2008;130:10886–10887. doi: 10.1021/ja8042307. [DOI] [PubMed] [Google Scholar]; (b) Chuang KV, Kieffer ME, Reisman SE. Org Lett. 2016;18:4750–4753. doi: 10.1021/acs.orglett.6b02477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.