

Highlights
-
•
HTS and structure-based design produced naphthalene-based lead compounds with inhibition of SARS-CoV PLpro in the nM range.
-
•
These naphthalene-based lead compounds have antiviral potency against SARS-CoV in cell culture.
-
•
SARS-CoV PLpro naphthalene-based inhibitors are non-toxic and highly selective for SARS-CoV PLpro.
-
•
Designed SARS-CoV PLpro inhibitors act through a non-covalent, competitive mechanism of inhibition.
-
•
Lessons from design of SARS-CoV PLpro inhibitors have profound implications for other USPs implicated in disease processes.
Abbreviations: PLpro, papain-like protease; CoV, coronavirus; nsp, nonstructural protein; USP, ubiquitin-specific protease; DUB, de-ubiquitinating enzyme
Keywords: Papain-like protease, 3C-like protease, Nsp3, SARS-CoV, MERS-CoV, Ubiquitin
Abstract
Over 10 years have passed since the deadly human coronavirus that causes severe acute respiratory syndrome (SARS-CoV) emerged from the Guangdong Province of China. Despite the fact that the SARS-CoV pandemic infected over 8500 individuals, claimed over 800 lives and cost billions of dollars in economic loss worldwide, there still are no clinically approved antiviral drugs, vaccines or monoclonal antibody therapies to treat SARS-CoV infections. The recent emergence of the deadly human coronavirus that causes Middle East respiratory syndrome (MERS-CoV) is a sobering reminder that new and deadly coronaviruses can emerge at any time with the potential to become pandemics. Therefore, the continued development of therapeutic and prophylactic countermeasures to potentially deadly coronaviruses is warranted. The coronaviral proteases, papain-like protease (PLpro) and 3C-like protease (3CLpro), are attractive antiviral drug targets because they are essential for coronaviral replication. Although the primary function of PLpro and 3CLpro are to process the viral polyprotein in a coordinated manner, PLpro has the additional function of stripping ubiquitin and ISG15 from host-cell proteins to aid coronaviruses in their evasion of the host innate immune responses. Therefore, targeting PLpro with antiviral drugs may have an advantage in not only inhibiting viral replication but also inhibiting the dysregulation of signaling cascades in infected cells that may lead to cell death in surrounding, uninfected cells. This review provides an up-to-date discussion on the SARS-CoV papain-like protease including a brief overview of the SARS-CoV genome and replication followed by a more in-depth discussion on the structure and catalytic mechanism of SARS-CoV PLpro, the multiple cellular functions of SARS-CoV PLpro, the inhibition of SARS-CoV PLpro by small molecule inhibitors, and the prospect of inhibiting papain-like protease from other coronaviruses. This paper forms part of a series of invited articles in Antiviral Research on “From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses.”
1. Introduction
1.1. SARS-CoV genome and replication
Coronavirus (CoV) replication is a highly orchestrated process involving complex replication machineries to protect the virus genome, the largest known RNA genome, and the viral proteins from the host’s antiviral defense mechanisms (Brian and Baric, 2005). Before the zoonotic emergence of the first human coronavirus that caused the 2002/2003 epidemic of severe acute respiratory syndrome (SARS-CoV), there was a paucity of information pertaining to CoV genomes and mechanisms of replication. Since then, the numbers of CoVs sequenced and studied have increased dramatically and several potential drug targets have emerged. Many of the SARS-CoV drug targets are encoded in the 5′-terminal two thirds of the genome, within the two open reading frames (ORF) that encode for the replicase or non-structural proteins (nsps) (see Fig. 1 ) (Tong, 2009).
Fig. 1.
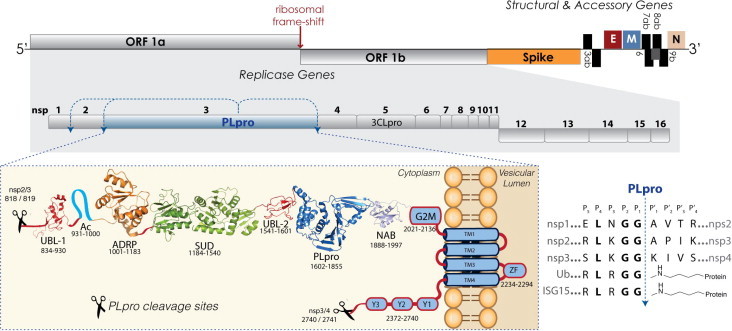
Genome and proteome organization of SARS-CoV non-structural proteins highlighting nsp3 domain organization and PLpro cleavage sites. The ∼30 kb genome of SARS-CoV and its associated replicase, structural and accessory proteins are indicated and the sizes of boxes representing each protein are to scale (top of figure). The replicase genes encoded by ORF1a and ORF1b are shaded in grey. The nsp3 multi-domain protein is shown with the amino acids defining the approximate boundaries of each domain indicated underneath. Downstream of the SARS-CoV PLpro cleavage site between nsp2/3 (818/819aa) is a ubiquitin-like domain (Ubl-1), PDB: 2GRI, a N-terminal Glu-rich acidic-domain (AC), ADP-ribose-1″-phosphatase (ADRP) domain (PDB: 2FAV) the SARS unique domain SUD (PDB: 2WCT, 2JZE, 2KAF,), a papain-like protease (PLpro) containing a second UBl-2 domain at the N-terminus (PDB: 2FE8) followed by the nucleic acid binding domain (NAB), PDB: 2K87, the marker domain G2M and four predicted transmembrane domains (TM1–TM4) forming an additional domain containing a metal-binding region (ZF). Finally, the remainder of nsp3 is composed of so-called Y domains (Y1–3), which precede the C-terminal PLpro cleavage sequence at nsp3/4 (2740/2741aa). An alignment of the SARS-CoV PLpro cleavage sequences (right bottom corner) shows a comparison of the P-sites and P′-sites from the nsps to the C-terminal sequences of the cellular proteins ubiquitin (Ub) and ISG15 shown with an isopeptide bond at the P′-sites.
Production of the replicase proteins is initiated by the translation of ORF1a and ORF1ab via a –1 ribosomal frame-shifting mechanism (Bredenbeek et al., 1990). This mechanism produces two large viral polyproteins, pp1a and pp1ab, that are further processed by two virally encoded cysteine proteases (Fig. 1), the papain-like protease (PLpro) and a 3C-like protease (3CLpro), which is sometimes referred to as main protease (Mpro) (Thiel et al., 2003, Ziebuhr, 2004, Ziebuhr et al., 2000, Ziebuhr et al., 2001). Processing of the viral polyprotein is required for the release and maturation of 16 viral proteins (non-structural proteins or nsps) involved in the formation of a membrane-associated, cytoplasmic enzyme complex, the replicase complex, which is responsible for directing the replication and transcription of the viral genome. It is thought that the establishment of viral replication sites is initiated by the recruitment of replicase proteins to host membranes, a process mediated by several viral transmembrane domain-containing proteins such as the nsp3, nsp4 and nsp6 (Kanjanahaluethai et al., 2007, Oostra et al., 2008, Oostra et al., 2007, van Hemert et al., 2008).
Electron tomography and three-dimensional reconstruction imaging studies have revealed that the replicase complex utilizes a reticulovesicular network of double-membrane vesicles (DMVs) with interconnected outer membranes originating from the endoplasmic reticulum (ER) (Angelini et al., 2013, Gosert et al., 2002, Knoops et al., 2008). As a result, the viral replication mechanism is localized, increasing the concentration and cooperation of viral macromolecules and forming a framework of RNA synthesis. Most importantly, this mechanism provides a microenvironment for the protection of viral RNA from host nucleases, and of double-stranded RNA intermediates from the host cell’s innate immune surveillance. Among the components of the replicase complex, nsp3 is of special interest since it is believed to be part of the central scaffolding protein of the replicase complex due to the large number of interactions with other nsp’s (Angelini et al., 2013, Imbert et al., 2008, Snijder et al., 2003).
1.2. The multi-domain protein nsp3
The SARS-CoV nsp3 multi-domain protein is the largest replicase subunit at 1,922 amino acids (Snijder et al., 2003, Thiel et al., 2003). Nsp3 is thought to play an essential role during the formation of virus replication complexes via its insertion into host membranes and its numerous interactions with other nsps (Imbert et al., 2008) especially nsp4 and nsp6 (Angelini et al., 2013). Numerous domains have now been identified in nsp3 (Fig. 1) and many are predicted to be conserved in all CoV (Neuman et al., 2008). Due to the large size of the nsp3 multi-domain protein, in vitro and in cellular studies have mainly utilized truncated nsp3 constructs that represent the predicted domains boundaries (Fig. 1). Using this approach, the three dimensional structures of most of the domains from the nsp3 of SARS-CoV have been determined by X-ray crystallography or NMR spectroscopy (Chatterjee et al., 2009, Egloff et al., 2006, Johnson et al., 2010, Ratia et al., 2006, Saikatendu et al., 2005, Serrano et al., 2007, Serrano et al., 2009, Tan et al., 2009).
The N-terminal region of the nsp3 (181–1000) is highly conserved among CoVs, containing a ubiquitin-like (Ubl) globular fold followed by a flexible, extended acidic-domain (AC domain) rich in glutamic acid (38%) (Serrano et al., 2007). Next to the AC domain is a catalytically active ADP-ribose-1″-phosphatase (ADRP, app-1″-pase) domain (also called macro domain or X domain) thought to play a role during synthesis of viral subgenomic RNAs (Egloff et al., 2006, Saikatendu et al., 2005). SARS Unique Domain (SUD), a domain not yet identified in other coronaviruses from alphacoronavirus and betacoronavirus, follows next (Tan et al., 2007). The SUD domain binds oligonucleotides known to form G-quadruplexes (Tan et al., 2009). Downstream of the SUD domain is a second Ubl domain and the catalytically active PLpro domain (Barretto et al., 2006) that proteolytically processes the nsp1/2, nsp2/3 and nsp3/4 cleavage sites (Harcourt et al., 2004). Downstream of PLpro are found a nucleic acid-binding domain (NAB) with a nucleic acid chaperon function (Neuman et al., 2008), which is conserved in betacoronavirus and gammacoronavirus, and one uncharacterized domain termed the marker domain (G2M) (Neuman et al., 2008). Following the G2M are two predicted double-pass transmembrane domains (TM1–2 and TM3–4) (Neuman et al., 2008, Snijder et al., 2003), a putative metal binding region (ZN) and the Y domain of unknown function (subdomains Y1–3) (Snijder et al., 2003, Thiel et al., 2003, Ziebuhr et al., 2001). Interestingly, comparative genome and proteome analyses of two bovine CoV (BCoV) isolates showed a predominant clustering of mutations within the nsp3 multi-domain (Chouljenko et al., 2001). Consequently, the multi-functionality of the nsp3, the frequency of point mutations observed in nsp3 domains, and the involvement of nsp3 in structural arrangements of the replicase complex and double-membrane vesicles may engender pleiotropic effects, not only in SARS-CoV pathogenicity, but also on future emerging coronaviruses (Snijder et al., 2003).
1.3. The SARS-CoV PLpro domain within the nsp3
The SARS-CoV PLpro catalytic domain is flanked by numerous catalytically active enzymes, transmembrane domains, and domains of unknown function, and the entire nsp3 is localized to the ER membranes where the majority of the domains reside in the cytosol of the cell (Fig. 1) (Hagemeijer et al., 2010, Oostra et al., 2008). In the cytosol, the membrane associated PLpro domain recognizes the P4–P1 consensus cleavage sequence LXGG, found in the boundaries of nsp1/2, nsp2/3 and nsp3/4 where membrane association is required for cleavage of the nsp3/4 (Han et al., 2005, Harcourt et al., 2004). Proteolytic cleavage of the peptide bond after the glycine at position P1 results in the release of nsp1, nsp2 and nsp3 from the viral polyprotein (Fig. 1, left bottom panel), a process that is essential for viral replication. Therefore, SARS-CoV PLpro is proposed to be an excellent candidate as a drug target for the development of anti-CoV therapeutics.
1.4. SARS-CoV PLpro is a protease, a deubiquitinating (DUB) and deISGylating enzyme
Reminiscent of the overall architecture of human deubiquitinating enzymes (DUBs) in the ubiquitin specific protease family (USP), the molecular structure of the PLpro catalytic domain consists of a canonical, right-handed thumb–palm–fingers architecture which is flanked at the N-terminus by an additional ubiquitin-like (UBL) domain of unknown function (Fig. 2 a and b) (Ratia et al., 2006). The in vitro characterization of PLpro enzymatic activities reveal that PLpro can recognize and hydrolyze the cellular proteins ubiquitin (Ub) (Barretto et al., 2005, Barretto et al., 2006, Lindner et al., 2005, Lindner et al., 2007) and the UBL protein ISG15 (interferon-induced gene 15) (Lindner et al., 2007, Nicholson et al., 2008, Ratia et al., 2014), both bearing the LXGG recognition motif at their C-terminus (Fig. 1, right bottom panel). Ubiquitin and ISG15 are important cellular modifiers that are covalently attached to target proteins via the formation of an isopeptide bond (Fig. 1, right bottom panel) between their C-terminus and the ε-amino group of a lysine side chain on a target protein. These isopeptide bonds can be hydrolyzed by the isopeptidase activities of DUB and deISGlating enzymes to remove Ub and ISG15 from host cell proteins.
Fig. 2.
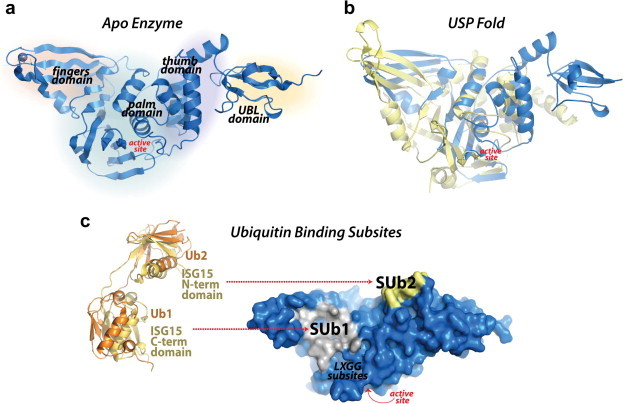
Multi-domain architecture of PLpro and its two ubiquitin binding subsites. (a) The SARS-CoV PLpro monomer (PDB: 2FE8) consists of four domains: starting from N- to the C-terminus, the extended UBL, the thumb, the palm and the fingers domain. (b) An overlay of SARS-CoV PLpro (blue) with USP7 (yellow, PDB: 4M5W) displaying the USP fold. (c) A solvent-accessible surface representation of SARS-CoV PLpro is shown in blue. A K48-linked di-Ub molecule (orange) superimposed onto an ISG15 (yellow) molecule is shown with molecules represented as ribbons. The ISG15 structure consists of two tandem UBL folds (Ub1 and Ub2). The two Ub and UBL binding subsites of SARS-CoV PLpro (Ratia et al., 2014) are shown in the solvent accessible surface representation with SUb1 shaded white and SUb2 shaded yellow.
Kinetic studies on the catalytic efficiency of SARS-CoV PLpro toward different substrates have shown that ISGylated and ubiquitinated substrates are more readily hydrolyzed than small peptide substrates such as RLRGG-AMC, which represents the C-terminus sequence of Ub and ISG15, suggesting a more complex mechanism for substrate recognition that extends well beyond the S4 to S1 enzyme recognition subsites for the LXGG peptide (Baez-Santos et al., 2014b, Lindner et al., 2005, Lindner et al., 2007, Ratia et al., 2014). More recently, a comprehensive analysis of PLpro substrate specificity by X-ray crystallography and mutational analyses demonstrated that two distinct Ub binding subsites (SUb1 and SUb2) exist distant from the catalytic site, providing SARS-CoV PLpro a unique bivalent mechanism of interaction with Ub-like modifier substrates (Ratia et al., 2014). For most USPs and SARS-CoV PLpro, the primary Ub/UBL binding subsite (SUb1) is distal to the isopeptide bond and located at the boundaries of palm domain and fingers regions (Fig. 2c). For SARS-CoV PLpro, a second, distal Ub binding subsite (SUb2) exists which is located in a ridge region of the thumb domain (Fig. 2c). This region provides interactions for a second Ub molecule on a K48-linked di-Ub chain, and for ISG15, which has structural resemblance to K48-linked di-Ub molecules (Ratia et al., 2014).
1.5. SARS-CoV PLpro innate immune functions
The DUB and deISGylating activities of SARS-CoV PLpro have significant functional implications in the innate immune response during SARS-CoV infection. Both Ub and ISG15 are important signaling elements of the host innate immune response against viral infection, which can be negatively regulated by viral DUB and deISGylating enzymes (Calistri et al., 2014). SARS-CoV PLpro has been shown to act as a strong antagonist of many Ub-dependent cellular responses to viral infection (Mielech et al., 2014a). Although the mechanism of PLpro-mediated antagonism of cellular pathways is not well understood, the evidence strongly suggests that catalytic activity is important for antagonism and, therefore, DUB and deISGylating activities have been proposed as a mechanism.
Because the multiple cellular roles of CoV PLpro DUB/deISGylating activities have been reviewed elsewhere (Mielech et al., 2014a), only a brief overview is presented here and the reader should visit other sources for a complete assessment on this topic. SARS-CoV PLpro antagonistic activities have been shown to block the production of important cytokines involved in the activation of the host’s innate immune response against viral infection, including the Type I interferon β (IFNβ) and chemokines such as CXCL10 and CCL5. Based on the in vitro and cell culture data available on SARS-CoV PLpro and other homologous coronaviral PLPs, a summary of the possible mechanisms for how PLpro can interfere with the expression of proinflamatory cytokines, such as interferon β (IFN-β), and block the establishment of the antiviral state is illustrated in Fig. 3 . Initially, SARS-CoV PLpro was shown to interfere with the activation of the transcription factors IRF3 (Devaraj et al., 2007) and NF-κB (Clementz et al., 2010, Frieman et al., 2009). Devaraj et al. showed that SARS-CoV PLpro-mediated antagonism of Type I interferon (IFN) production functions upstream of IRF3 activation by blocking IRF3 phosphorylation, homodimerization, and consequently nuclear translocation (Devaraj et al., 2007, Sun et al., 2012). On the other hand, Frieman et al. demonstrated that PLpro stabilizes the NF-κB inhibitor, IκBα, and thereby blocks the activation of the NF-κB signaling pathway (Frieman et al., 2009). More recently, Mielech et al. demonstrated SARS-CoV PLpro ability to decrease endogenous levels of proinflamatory cytokines and chemokines in activated cells (Mielech et al., 2014b).
Fig. 3.
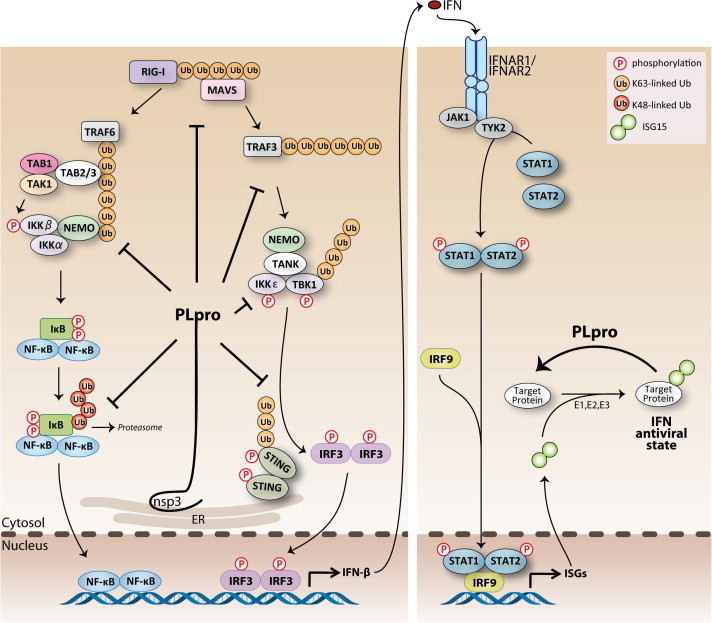
Currently proposed sites of action for SARS-CoV PLpro-mediated antagonism of the innate immune response. Viral infection is sensed by RIG-I (RIG-I-like helicase) and MDA-5 (melanoma differentiation-associated protein 5) recognition of viral RNA. Recruitment of adaptor proteins MAVS transduces signals to the downstream kinase complex, which activates the transcription factor, IRF-3 and NF-κB, which coordinates the expression of type I interferons (IFN-β and -α). Type I IFN induces the activation of STAT transcription factors resulting in the expression of ISGs (IFN-stimulated genes) and the establishment of an antiviral state in surrounding cells. PLpro can act on different branches of these pathways by interacting with or recognizing and deISGylating and/or deubiquitinating proteins within these pathways. The net effect of these different functions is to help SARS-CoV evade the host antiviral response via antagonizing the establishment of an antiviral state.
Interestingly, in some instances, mutation of the active site catalytic cysteine residue of SARS-CoV PLpro only had a small decrease in antagonism of certain pathways, suggesting that the protease activity is not the only requirement for antagonism (Devaraj et al., 2007). However, Clementz et al. demonstrated that the SARS-CoV PLpro-mediated block of NF-κB activation can be reversed by the addition of a PLpro competitive inhibitor, emphasizing the importance for catalytic activity during antagonism of this cellular pathway (Clementz et al., 2010). Nevertheless, it is important to consider that a mutation of the catalytic machinery of PLpro may not affect its ability to interact with Ub and ISG15 molecules as the competitive inhibitor does. Therefore, a protein-to-protein interaction mediated by PLpro’s strong affinity for Ub/ISG15 molecules on ubiquitinated/ISGylated proteins could still lead to the interference of the IRF3/NF-κB signaling pathways.
2. SARS-CoV PLpro structure and function
2.1. Active site structure and catalytic mechanism of SARS-CoV PLpro
2.1.1. Active site structure
SARS-CoV PLpro belongs to the peptidase clan CA (family C16). The active site contains a classic catalytic triad, composed of Cys112–His273–Asp287, that is well-aligned, functional, and located at the interface of the thumb and palm sub-domains (Fig. 4 ) (Ratia et al., 2006). The catalytic cysteine (Cys112) is situated at the foot (N-terminus) of α-helix α4 in the thumb domain. The side chain sulfur atom of Cys112 is positioned 3.7 Å from the pros(π)-nitrogen atom of the catalytic histidine (His273) (McNaught and Wilkinson, 1997), which is located at the foot of the palm domain and adjacent to the flexible loop BL2 (also called the G267–G272 loop (Ratia et al., 2008) or β-turn (Baez-Santos et al., 2014a)). One of the oxygen atoms of the side chain of catalytic aspartic acid (Asp287) is located 2.7 Å from the tele(τ)-nitrogen of the catalytic histidine at the foot of the palm domain (Fig. 4). The side chain of Trp107 is located within the oxyanion hole and the indole-ring nitrogen is proposed to participate in the stabilization of negatively-charged tetrahedral intermediates produced throughout catalysis (Ratia et al., 2014, Ratia et al., 2008). The overall tertiary structure of PLpro is remarkably similar to the cellular ubiquitin specific proteases (USP) including USP14 and USP7 (or HAUSP) (Fig. 2b). However, in their unbound states, the catalytic triad of PLpro aligns well structurally only with the catalytic triad of USP14 and not USP7. In order for the catalytic triad of USP7 to adopt the proper orientation for catalysis, a substrate-induced alignment must occur upon association with ubiquitin or other regulatory domains (Hu et al., 2002, Molland et al., 2014, Ratia et al., 2006). The fingers domain of PLpro, which contains a zinc ion that is tetrahedrally coordinated by four cysteines, is essential for catalysis because it maintains the structural integrity of PLpro (Barretto et al., 2005, Ratia et al., 2006).
Fig. 4.
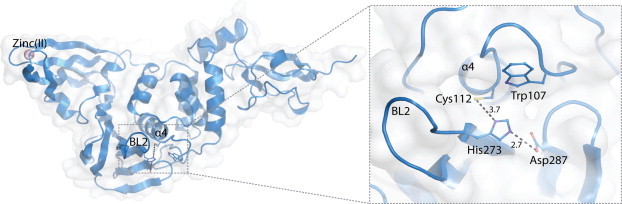
Catalytic center of the active site of SARS-CoV PLpro. Unliganded X-ray crystal structure of SARS-CoV PLpro (PDB: 2FE8). The catalytic triad residues, Cys112, His273 and Asp287 are shown along with the oxyanion hole-stabilizing residue Trp107 (right panel). The flexible BL2 loop that interacts with the substrates is indicated as are the zinc ion (purple sphere) and α-helix α4 in the thumb domain. Distances between atoms are given in units of Ångströms (Å) and are indicated by dashed lines.
2.1.2. Catalytic mechanism
SARS-CoV PLpro functions through a proposed cysteine protease catalytic cycle where Cys112 acts as nucleophile, His273 functions as a general acid-base, and Asp287 is paired with histidine helping to align it and promote deprotonation of Cys112 (Fig. 5 ). Currently, there are no experimental studies defining the protonation state of Cys112 prior to nucleophilic attack on the substrate, or whether the reactive nucleophilic species is the thiolate ion of the thiolate–histidine pair, or the neutral thiol, that acts via a mechanism analogous to that of serine peptidases where a thionium ion must be produced after reaction with the ligand (Barrett et al., 2012). Because of the extremely low pKa of the thionium intermediate that must be produced in this case, we have chosen to show the classically accepted nucleophilic thiolate form in our general mechanism.
Fig. 5.
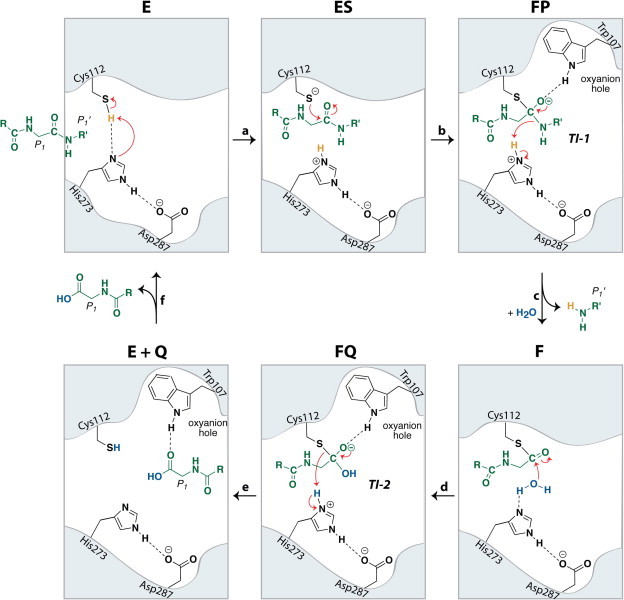
Catalytic cycle and proposed chemical mechanism of the SARS-CoV PLpro catalyzed reaction. Active site residues of the catalytic triad (Cys112, His273, Asp287) and oxyanion hole residue Trp107 are shown in black. The peptide substrate is shown in green and a catalytic water molecule is shown in blue.
In its unliganded form, “E”, the catalytic residues of SARS-CoV PLpro are within hydrogen-bonding distance of one another, indicating that the protonation state of Cys112 may exist in equilibrium with that of His273 and that the event of substrate binding drives this equilibrium toward the reactive thiolate (Fig. 4, Fig. 5, step a). Alternatively, deprotonation of Cys112 may occur during substrate binding to form the “ES” complex (Fig. 5, step a). An addition–elimination sequence follows, where attack of the thiolate of Cys112 on the carbonyl carbon of the peptide bond forms the first negatively-charged tetrahedral intermediate “TI-1” or “FP,” (Fig. 5, step b). The oxyanion of the tetrahedral intermediate is stabilized via the presence of an oxyanion hole adjacent to the PLpro enzymatic active site (Chou et al., 2014, Ratia et al., 2014, Ratia et al., 2006). This oxyanion hole contains a tryptophan residue (Trp107), which our group has confirmed is crucial for the enzymatic function of PLpro through site-directed mutagenesis (Baez-Santos, 2012). In addition, we recently showed via the X-ray structure of SARS-CoV PLpro covalently modified with ubiquitin-aldehyde that indole-ring nitrogen of Trp107 acts as a hydrogen-bond donor to the hemithioacetal intermediate (Ratia et al., 2014), confirming its function in the oxyanion hole. Furthermore, an asparagine residue (Asn110, not depicted) has been found to be highly conserved among coronaviral PLP2s and may contribute to stabilization in the oxyanion hole, as it is positioned above the catalytic cysteine. Elimination of the C-terminal amine of the substrate cleaves the peptide bond, subsequently forming the thioester intermediate “F” (Fig. 5, step c). A second addition–elimination sequence follows, where the nucleophilic addition of water to the carbonyl carbon of the thioester forms a second, negatively-charged tetrahedral intermediate “TI-2” or “FQ” (Fig. 5, step d). The oxyanion of this intermediate is again stabilized via the oxyanion hole of the PLpro active site. Elimination of cysteine from the tetrahedral intermediate results in the formation of the N-terminal carboxylic acid “EQ”, which may be transiently stabilized in the PLpro active site via a hydrogen-bond between the carbonyl carbon of the newly formed acid and the indole ring nitrogen of Trp107 (Fig. 5, step e) (Chou et al., 2014). Elimination of the cleaved N-terminus of the peptide substrate then completes the catalytic cycle where the final product “Q” is released from the active site and the free enzyme “E” is regenerated (Fig. 5, step f).
3. SARS-CoV PLpro inhibitor reactions with Cys112
Cysteine proteases are known to react with a variety of electrophilic or “warhead” functionalities within covalent inhibitor molecules. These warhead inhibitors typically function by first forming a noncovalent interaction complex within the cysteine protease active site, where the warhead group of the inhibitor is positioned in the proximity of the reactive cysteine nucleophile. The reaction proceeds via a nucleophilic attack of the thiolate on the electrophilic carbon of the warhead group, forming a covalently modified enzyme–inhibitor complex, which subsequently inactivates the enzyme. Examples of such reactive warhead groups that are known to inhibit cysteine proteases include aldehydes, epoxy-ketones, Michael acceptors, activated ketones, activated esters, vinyl sulfones, acrylamides, alkynes, alkylhalides, and nitriles (Fig. 6 ) (Ghosh et al., 2006, Ghosh et al., 2005, Jacobs et al., 2013, Jain et al., 2004, Xue et al., 2008, Yang et al., 2006, Zhang et al., 2007).
Fig. 6.
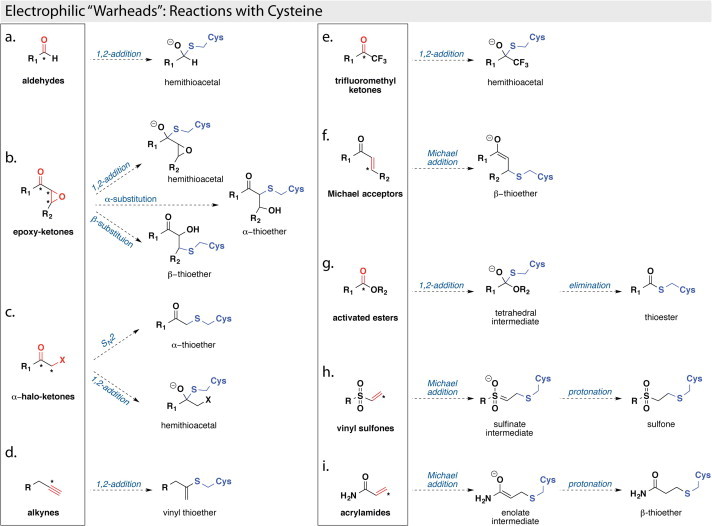
Examples of cysteine protease inhibitors with covalent “warheads.” The chemical “warhead” groups are shown in boxes and the reactive, electrophilic portions are colored in red, with the corresponding electrophilic “warhead” carbon indicated with an asterisk. The resulting covalent inhibitor–cysteine complexes are shown outside the boxes and the cysteine portions of the covalent adducts are colored in blue.
Our lab successfully solved the first X-ray crystal structure of SARS-CoV PLpro covalently modified with an electrophilic “warhead.” (Ratia et al., 2014). The SARS-CoV PLpro enzyme was co-crystallized with a semisynthetic version of ubiquitin, where the C-terminus of ubiquitin was modified with an aldehyde functional group (Ubal). This intentional modification of ubiquitin was introduced as a way to modify the catalytic cysteine with a covalent-reversible group thereby trapping the PLpro–Ubal complex and mimicing the reaction intermediate TI-2 in Fig. 5. The SARS-CoV PLpro–Ubal structure was determined to a resolution of 2.5 Å and is the first X-ray crystal structure of the hemithioacetal, tetrahedral intermediate in SARS-CoV PLpro (Fig. 7 a and b). The catalytic cysteine is positioned 1.6 Å from the carbon of the tetrahedral intermediate, a distance irrefutably indicating a covalent C–S bond. The tetrahedral, hemithioacetal intermediate is stabilized via the oxyanion hole of SARS-CoV PLpro by a hydrogen bond to the indole nitrogen of Trp107, and is also within hydrogen-bonding distance to the pros(π)-nitrogen atom of His273.
Fig. 7.
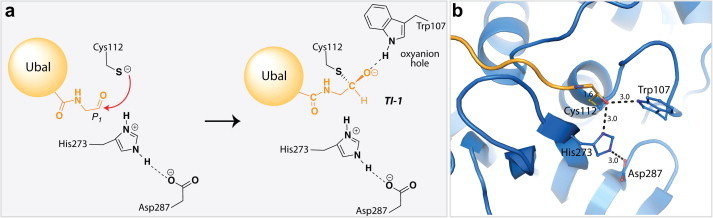
X-ray structure of SARS-CoV PLpro in complex with ubiquitin-aldehyde (Ubal). (a) Pictorial representation of the tetrahedral hemithioaminal intermediate generated upon PLpro–Ubal complex formation, where the Ubal C-terminal residues are shown in orange and the SARS-CoV PLpro residues are shown in black. (b) X-ray crystal structure of PLpro–Ubal complex (PDB: 4MM3 (Ratia et al., 2014)) where the PLpro is shown in blue and the residues His273, Cys112, Asp287, and Trp107 are shown in ball-and-stick and colored according to atom type, and Ubal is shown in orange and colored according to atom type. Distances between atoms are given in units of ångströms (Å) and are indicated by dashed lines.
4. SARS-CoV PLpro inhibitor classes
4.1. Inhibitors identified via a designed yeast-based screen
In 2011, two new PLpro inhibitors were reported by Frieman and coworkers (Frieman et al., 2011). These inhibitors were identified via a yeast-based screening assay that was developed to discover inhibitors of SARS-CoV PLpro. The assay involved the expression of SARS-CoV PLpro in S accharomyces cerevisiae that results in a slow-growth phenotype. A 2000 compound NIH Diversity Set library was screened for molecules capable of reversing the PLpro-induced slow-growth phenotype. Compounds identified as hits in this primary assay were then tested for efficacy against SARS-CoV replication and inhibition of PLpro function in cell culture models. This screening method resulted in the identification of NSC158362, a compound capable of specifically inhibiting SARS-CoV replication in cell culture without cytotoxic effects. However, NSC158362 was found incapable of inhibiting the protease, deubiquitinase and anti-interferon functions of PLpro – pointing to a potential new mechanism for inhibition of SARS-CoV replication. A second compound, NSC158011 was found capable of inhibiting the protease activity of PLpro in cell-based assays; however, it was unsuccessful at blocking SARS-CoV viral replication. The structures of NSC15832 and NSC158011 are shown in Fig. 8 a (Frieman et al., 2011).
Fig. 8.
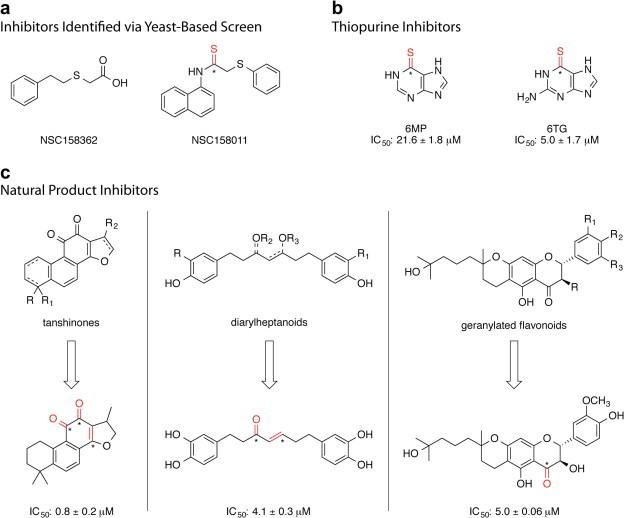
SARS-CoV PLpro inhibitors identified through small-scale screening efforts. Positions within each molecule that are susceptible to covalent modification through nucleophilic attack (“warhead” positions) are indicated in red where the electrophilic carbons are indicated with an asterisk. (a) Inhibitors identified via yeast-based screen (Frieman et al., 2011), (b) thiopurine inhibitors (Chen et al., 2009, Chou et al., 2008), (c) natural product inhibitors (Cho et al., 2013, Park et al., 2012a, Park et al., 2012b), where the most potent natural product of each class is shown and the IC50 detailed.
4.2. Thiopurine compounds
Chou and coworkers screened a small library of 960 compounds for the inhibition of SARS-CoV PLpro using a FRET-based assay and purified SARS-CoV PLpro enzyme (Chen et al., 2009, Chou et al., 2008). Two thiocarbonyl-containing compounds, 6MP and 6TG, were found to be effective inhibitors of PLpro with IC50 values of 21.6 and 5 μM, respectively. In their assay, zinc ion (Zn2+) was also found to be an effective inhibitor of PLpro (vide infra). Both 6MP and 6TG were found to be slow-binding, competitive, reversible, and selective inhibitors of SARS-CoV PLpro. Through comparison to molecular analogs of 6MP and 6TG that lacked the thiocarbonyl functionality, the thiocarbonyl was found to be the crucial moiety for these compound’s inhibition of SARS-CoV PLpro and it is postulated to react covalently with the active site cysteine. Currently, both 6MP and 6TG are used clinically in the treatment of leukemia, and the anti-cancer action of 6TG is dependent upon its conversion to 6TG nucleotides and incorporation into DNA, preventing replication. This, and the acute toxicities associated with both 6MP and 6TG, makes them feasible as leads for future PLpro inhibitors, but not as SARS-CoV drugs themselves (Fig. 8b) (Chen et al., 2009, Chou et al., 2008).
4.3. Natural product inhibitors
A variety of natural products have been found to inhibit SARS-CoV PLpro activity including tanshinones derived from Salvia miltiorrhiza (Park et al., 2012b), diarylheptanoids derived from Alnus japonica (Park et al., 2012a), and geranylated flavonoids isolated from the fruits of the Paulownia tomentosa tree (Cho et al., 2013).
4.3.1. Tanshinones
Park and coworkers assayed seven different tanshinones for SARS-CoV PLpro inhibition using a continuous fluorometric assay (Park et al., 2012b). The tanshinones were found to inhibit SARS-CoV PLpro activity with IC50 values ranging from 0.8 to 30.0 μM. Interestingly, the authors found SARS-CoV PLpro inhibition increased as the pre-incubation time of PLpro with tanshinone increased, suggesting a slow-binding mechanism where inhibition of PLpro occurs through the rapid formation of an enzyme–inhibitor complex that slowly isomerizes to form a modified enzyme complex and possible covalent inhibition. The most potent tanshinone inhibitor of SARS-CoV PLpro enzymatic activity is shown in Fig. 8c, left panel (Park et al., 2012b).
4.3.2. Diarylheptanoids
The diarylheptanoid inhibitors were identified via activity-guided fractionation of the ethanolic extracts from the air-dried bark of A. japonica and assayed for inhibition of SARS-CoV PLpro using a continuous fluorometric assay (Park et al., 2012a). The most active diarylheptanoid inhibitor was found to contain an α,β-unsaturated carbonyl group – suggesting that inhibition of SARS-CoV PLpro may occur through formation of a covalent bond with the active site cysteine (vide supra). The most potent diarylheptanoid inhibitor of SARS-CoV PLpro enzymatic activity is shown in Fig. 8c, middle panel (Park et al., 2012a).
4.3.3. Geranylated flavonoids
Using activity-guided fractionation of the methanolic extracts of P. tomentosa fruits, Cho and coworkers identified five new geranylated flavonoid derivatives and seven known flavonoids that exhibit micromolar inhibition of SARS-CoV PLpro (Cho et al., 2013). Using a fluorogenic assay, the 12 flavonoids were found to inhibit SARS-CoV PLpro with IC50 values between 5.0 and 14.4 μM. The most active inhibitor was found to be a novel geranylated flavonoid. In general, it was shown that compounds containing the dihydro-2H-pyran moiety displayed better PLpro inhibition than their parent compounds. The most potent geranylated flavonoid inhibitor of SARS-CoV PLpro enzymatic activity is shown in Fig. 8c, right panel (Cho et al., 2013).
4.4. Zinc Ion (Zn2+) and zinc conjugate inhibitors
Using a fluorogenic inhibitor-screening platform that utilized nanomolar concentrations of SARS-CoV PLpro, Han and coworkers established that zinc ion (Zn2+) was capable of inhibiting PLpro protease activity with an IC50 value of 1.3 μM (Han et al., 2005). Additionally, they found the zinc conjugates N-ethyl-N-phenyldithiocarbamic acid–Zn(II) and hydroxypyridine-2-thione–Zn(II) to inhibit PLpro protease activity with IC50 values of 3.3 and 3.7 μM, respectively. In this study, inhibition of PLpro activity by Zn(II) appeared to be specific as the other divalent metals tested (Mg(II), Mn(II), Ca(II), Ni(II), and Co(II)) showed no inhibitory effect on the activity of SARS-CoV PLpro when tested at a concentration of 10 μM; though Cu(II) was found to inhibit SARS-CoV PLpro protease activity to 70% at a concentration of 10 μM (Lindner et al., 2005).
4.5. Naphthalene inhibitors
4.5.1. Compound 24
The class of naphthalene inhibitors was first reported in 2008, and was discovered by our group through the implementation of a high-throughput screen (HTS) of 50,080 compounds for inhibitors of SARS-CoV PLpro (Ratia et al., 2008). Two promising SARS-CoV PLpro inhibitors were identified, 7724772 and 6577871, having IC50 values of 20.1 μM and 59 μM, respectively (Fig. 9 a1 and a2). Interestingly, both of these hits from the primary HTS contained a naphthylmethylamine moiety. The more potent naphthyl inhibitor identified, compound 7724772, was identified as a racemate, and upon further investigation the (R)-enantiomer was found to be a more potent inhibitor, having and IC50 value of 8.7 ± 0.7 μM (Fig. 9a1). Analogs of (R)-7724772 were synthesized to explore the effects of adding different substituents at the ortho- and meta-positions of the benzene ring and the effects of changing the linkage to either the 1- or 2-naphthyl position. Preliminary structure–activity relationship (SAR) analysis indicated that substitutions at the ortho-benzene position other than a methyl group were unfavorable and decreased binding affinity, while changing the linkage to the 1-napthyl position increased inhibitor potency by 4-fold. Addition of a nitro-substituent at the meta-position to the amide on the benzene ring decreased inhibitory potency, while addition of an amine at that same position increased inhibition of PLpro by 4-fold (Fig. 9b1). These SAR studies guided the construction of inhibitor 24, which has a significantly increased inhibitory potency against SARS-CoV PLpro relative to 7724772 (IC50 = 0.6 ± 0.1 μM vs. 20.1 ± 1.1 μM). Compound 24 was also found to inhibit SARS-CoV viral replication in Vero cells with an EC50 of 14.5 ± 0.8 μM (Fig. 9c1) (Ghosh et al., 2009, Ratia et al., 2008). More in depth kinetic and biochemical studies demonstrated 24 to be a potent, noncovalent, competitive inhibitor of SARS-CoV PLpro (K i = 0.49 ± 0.08 μM).
Fig. 9.
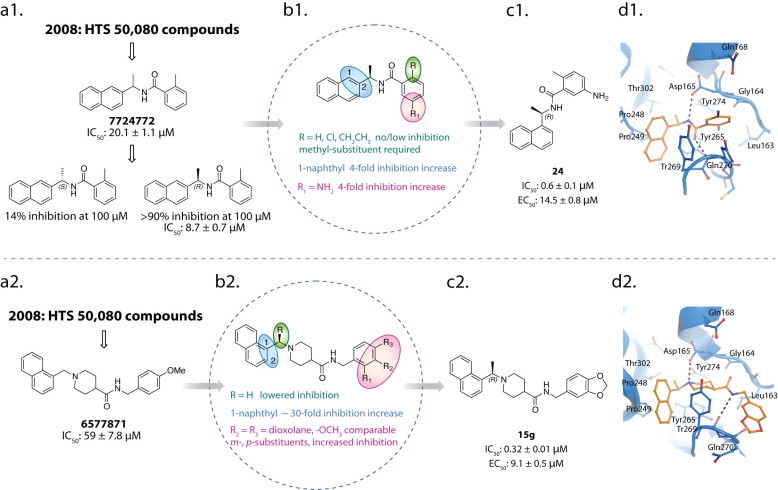
Discovery and design pipeline for SARS-CoV PLpro inhibitors 24 and 15g. Results of the primary high-throughput screen identified hit compounds 7724772 (a1) and 6577871 (a2). SAR diagrams for analogs of 7724772 (b1) and 6577871 (b2) (Ghosh et al., 2010, Ratia et al., 2008). Structures of potent, first-generation leads 24 (c1) and 15g (c2). X-ray crystal structures of 24 (d1) (PDB: 3E9S) and 15g (d2) (PDB: 3MJ5) bound to SARS-CoV PLpro highlighting the residues involved in binding each inhibitor. Hydrogen bonds are shown as dashed lines.
The X-ray structure of 24 in complex with SARS-CoV PLpro was determined to a resolution of 2.5 Å, where 24 was found to bind to SARS-CoV PLpro within the S3 and S4 subsites in a cleft leading to the active site (Ratia et al., 2008). The amide group of 24 forms two hydrogen bonds with the Asp165 and Gln270 residues of SARS-CoV PLpro, while the rest of the interactions between the inhibitor and SARS-CoV PLpro were hydrophobic. The naphthyl group of the inhibitor forms hydrophobic interactions with the aromatic rings of the Tyr265 and Tyr269 residues and with the side chains of Pro248 and Pro249. The (R)-methyl group of 24 is positioned directly into a polar cavity, between residues Tyr265 and Thr302. The ortho-methyl substituent on the benzene ring is directed toward the floor of this cavity, and surrounded by residues Tyr265, Tyr274, and Leu163. The amino-substituent of the benzene ring is surrounded by several polar groups that may serve as hydrogen-bond acceptors (Fig. 9d1).
4.5.2. Compound 15g
The second, less potent naphthylmethylamine hit identified from our primary HTS campaign (6577871, Fig. 9a2) was also found to be a competitive, noncovalent inhibitor of SARS-CoV PLpro (Ratia et al., 2008). Interestingly, 6577871 had a 1-substituted naphthalene ring, which was found to be the more active linkage in the 7724772 series (vide supra). A small library of 6577871 analogs was synthesized to investigate the effect of substitution at the 1- or 2-naphthyl positions, at the benzylic-naphthyl position, and at positions on the benzene ring (Ghosh et al., 2010). Similar to the SAR trends found for 7724772, a 1-napthyl linkage was found to be most favorable for increased PLpro inhibition. Again, substitution of a methyl group at the benzylic-naphthyl position was found to increase inhibition; however, in the case of the 6577871 series, the stereochemistry of this chiral center had a much less dramatic effect, where the preference for an (R)- or (S)-stereochemistry changed depending upon the substituents on the benzene ring. A 2-methoxy substituent on the benzene ring was found to have a 3.5-fold decrease in inhibition, while positioning the methoxy substituent at the 3- or 4-positions on the benzene ring did not measurably change the IC50 values. A series of benzodioxole analogs was also investigated and found to have minimally increased inhibitory potency relative to the methoxy-substituted benzene analogs (Fig. 9b2). These studies and SAR analyses ultimately led to the generation of inhibitor 15g, which has a significantly increased inhibitory potency against SARS-CoV PLpro relative to 6577871 (IC50 = 0.32 ± 0.01 μM vs. 59 ± 7.8 μM). Compound 15g was found to inhibit SARS-CoV viral replication in Vero cells with an EC50 = 9.1 ± 0.5 μM. Interestingly, the (S)-enantiomer of 15g, compound 15h, was found to have only slightly decreased inhibition of SARS-CoV PLpro, having an IC50 value of 0.56 ± 0.03 μM. Moreover, 15g showed better inhibition of SARS-CoV PLpro and better inhibition of SARS-CoV replication than the previously identified compound, 24 (Fig. 9c2). Additional kinetic and biochemical studies demonstrated that 15g is a potent, noncovalent, competitive inhibitor of PLpro (Ghosh et al., 2010).
The X-ray structure of 15g in complex with SARS-CoV PLpro was determined to a resolution of 2.63 Å and 15g was found to bind in the same cleft leading to the active site as 24 (Fig. 9d1 and d2). Both compounds, 15g and 24, bind with their napthylmethylamine moieties positioned in the same orientation and location within the enzyme active site. The naphthyl moieties sit in a hydrophobic pocket surrounded by PLpro residues Tyr269, Tyr265, Pro248, Pro249, and Thr302. The (R)-configured methyl group of 15g extends into a small pocket that is lined with both hydrophobic and polar groups that are bound to water molecules. The piperidine nitrogen of 15g is located in an analogous position to the carboxamide nitrogen of 24, and at physiological pH the piperidine nitrogen acts as a nitrogen-centered hydrogen-bond donor, forming a hydrogen-bond with the side chain carboxylate of Asp165. The remaining portion of the 15g molecule, containing the amide and benzodioxole, extends to occupy space within the binding cleft not utilized by 24, where the amide nitrogen of 15g makes a hydrogen bond with the backbone carbonyl oxygen of Tyr269, further anchoring the inhibitor in the binding site. The presence of these additional interactions within the PLpro binding cleft is likely responsible for the increased inhibitory ability of 15g relative to 24 (Fig. 9d2).
4.6. Second generation naphthalene inhibitors
4.6.1. Compounds 3k and 3j
Though compounds 24 and 15g had low micromolar inhibitory potency and minimal cytotoxicity in SARS-CoV infected Vero E6 cells, further optimization of their antiviral potencies and physiochemical properties was still required to make them therapeutically viable. Using the SARs previously described, and the structural information obtained from the X-ray crystal structures of 24 and 15g, a second generation of 30 inhibitors was synthesized and evaluated for SARS-CoV PLpro inhibition. This second generation library was designed to probe the steric and electronic demands of the benzylic-naphthyl position, benzylic position, substituent and bioisoster tolerance of the benzyl and naphthyl rings, and the effect of distance between the aromatic rings. The common features of 15g and 24 required for good PLpro were held constant, namely the 1-naphthyl substitution and position of the hydrogen-donating nitrogen (Fig. 10 a).
Fig. 10.
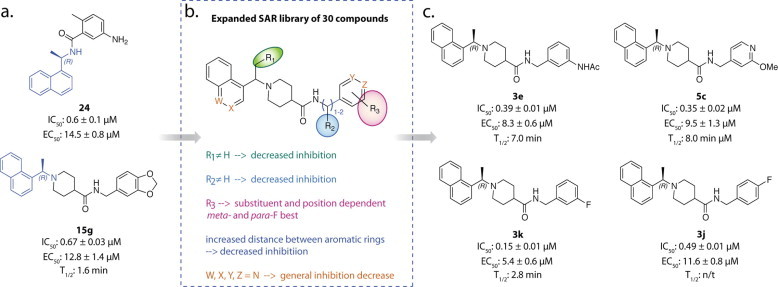
Discovery and design pipeline of compounds 3k, 3j, 3e, and 5c. (a) Initial SARS-CoV PLpro inhibitor leads, 15g and 24, (b) SARs derived from small 15g analog library, (c) top SARS-CoV PLpro inhibitor candidates, 3k, 3j, 3e, and 5c (Baez-Santos et al., 2014a).
Increasing the size of the benzylic-naphthyl substituent from a methyl group to anything with greater steric demands decreased SARS-CoV PLpro inhibition in a trend proportional to substituent size, indicating that the pocket accessed by the (R)-methyl group was less accessible than initially indicated by the crystal structures of 24 and 15g (Fig. 10b, position R1). Adding a variety of substituents to the benzylic position did not increase inhibitory potency beyond that of 15g, indicating that additional functionalization at this position does not further engage the residues of the PLpro binding cavity (Fig. 10b, position R2). The effect of substitution on the benzene ring was found to be completely substituent and position dependent, with no clear SAR trend; however two analogs with meta- and para-fluoro substituents were found to have the greatest PLpro inhibition, 3k and 3j (Fig. 10b, position R3). The effect of extending the linkage between the amide and benzene ring was examined and lengthening the chain by even one carbon atom was found to weaken PLpro inhibition. A variety of bioisosteres were also investigated where the W, X, Y, and Z positions (Fig. 10b) were substituted with nitrogen. In general, these bioisosteres did not increase the inhibitory potency of the compounds, with the exception of 5c, which had a similar IC50 and EC50 to 15g (Fig. 10b and c).
The X-ray structures of SARS-CoV PLpro in complex with lead inhibitors 3k and 3j were determined to resolutions of 2.1 Å and 2.5 Å, respectively (Baez-Santos et al., 2014a). Similar to the X-ray structures of SARS-CoV PLpro in complex with lead inhibitors 24 and 15g, 3k and 3j were found to bind adjacent to the active site, within the S3 and S4 sub-sites of the SARS-CoV PLpro enzyme and in the same orientation. The inhibitors bind to SARS-CoV PLpro through an induced-fit mechanism, where the BL2 loop, which contains Tyr269, adopts a closed conformation in order to interact strongly with the inhibitors. The interactions between the BL2 loop and the inhibitor occur through different bonds including a hydrogen-bond between the backbone carbonyl of Tyr269 and the amide nitrogen of the inhibitors. The motions associated with the BL2 loop and its different conformations are further discussed below in Section 5.
The naphthyl-rings of 3k and 3j are located in identical chemical space to the inhibitors 15g and 24 within the hydrophobic pocket hedged by Pro248 and Pro249 and lined by Tyr265, Tyr269, and Thr302 (Fig. 11 a). The higher resolution X-ray structures of 3k and 3j allowed for a more definitive placement of three conserved water molecules present in the small cavity occupied by the (R)-methyl substituent. These water molecules decrease the effective size of the cavity and support our previously described SAR for substituents at the benzylic-naphthyl position. The superposition of compounds 3k, 3j, and 15g shows a 1.0 Å difference in the position of the amide nitrogen of 15g compared to 3k and 3j, allowing a slight tilt in the orientation of the benzene ring to accommodate the different substituents (Fig. 11b). This tilt allows 3k to form stronger interactions with Tyr269 and Gln270 and is postulated to explain the increased inhibitory potency of 3k.
Fig. 11.
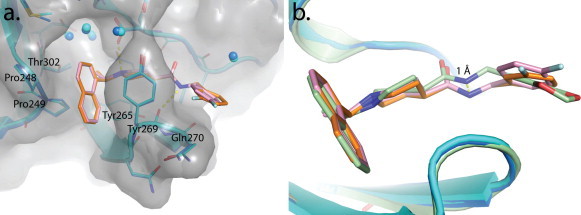
X-ray crystal structures of SARS-CoV PLpro in complex with inhibitors 3k and 3j and comparison to 15g. (a) An overlay of SARS-CoV PLpro (blue cartoon, gray surface) in complex with 3k (orange stick, PDB: 4OW0) and PLpro (cyan cartoon, gray surface) in complex with 3j (pink sticks, PDB: 4OVZ) depicting amino acids important for inhibitor binding (Baez-Santos et al., 2014a). (b) An overlay of PLpro–3k (PLpro in blue cartoon, 3k in orange sticks), PLpro–3j (PLpro in cyan cartoon, 3j in pink sticks) and PLpro–15g (green cartoon and sticks, (PDB: 3MJ5, (Ghosh et al., 2010)) complexes.
4.6.2. Metabolically stable naphthalene-based SARS-CoV PLpro inhibitors
The most potent identified inhibitor of PLpro, compound 3k, was expected to have greater metabolic stability than the previously identified lead compound, 15g, as the metabolically labile dioxolane of 15g had been replaced with a fluoro-substituent (Baez-Santos et al., 2014a). Additionally, as 3k has a lower topological polar surface area relative to 15g, it was expected to have increased cell permeability and subsequently, greater antiviral activity in comparison to 15g. The most potent analogs of 15g were subjected to anti-SARS-CoV assays in cell culture to determine EC50 values, which resulted in the identification of four top compounds with good antiviral activity: 3k, 3j, 3e, and 5c (Baez-Santos et al., 2014a). To assess the potential stability of these compounds in vivo, their stability to Phase I metabolism by mouse liver microsomes was evaluated. Surprisingly, 3k was found to be rapidly metabolized; however, the compounds 3e and 5c showed significantly improved metabolic stability relative to the best first generation inhibitor, 15g (Baez-Santos et al., 2014a). Because of their improved metabolic stability, inhibitors 3e and 5c are promising candidates for advancing to animal efficacy models.
4.6.3. Selectivity of naphthalene-based SARS-CoV PLpro inhibitors
There are at least 98 functional deubiquitinating enzymes in the human genome which fall into one of 6 different classes, where the number of members within each class are indicated in parenthesis; USP (56), OTU (15), JAMM (12), MCPIP (7), MJD (4) and UCH (4) (Fraile et al., 2012). The USP class, to which coronaviral PLpro’s belong, represent the largest class with 56 members. As the catalytic domains among the USP class are highly conserved, the question immediately arises as to how selective the designed inhibitors are for SARS-CoV PLpro and against human USPs? At least two approaches have been used so far in an attempt to answer this question. The first was a chemical biology approach pioneered by Hidde Ploeugh and colleagues (Borodovsky et al., 2001, Borodovsky et al., 2002, Love et al., 2007). In this approach, the active-site-directed probe HA–Ub–vinyl sulfone (VS) was used to treat cellular lysates of untransfected cells and cells transfected with SARS-CoV PLpro both in the absence and presence of inhibitors (Ratia et al., 2008). Since HA–Ub–VS potently inhibits a number of human USPs, the ability of compounds to block modification of USPs or PLpro from cellular lysates can be determined via Western blot analysis by using an anti-HA antibody. Using this approach, we found that the naphthalene-based class of inhibitors do not compete against HA–Ub–VS modification of cellular DUBs but does compete against modification of SARS-CoV PLpro.
A second approach for testing selectivity against USPs, other DUBs and cysteine proteases is to test for compound inhibition against purified enzymes, as many of these enzymes are now commercially available. We used this approach to test the naphthalene-based inhibitors against purified USP2, USP7, USP8, USP18, USP20, USP21, USP28, UCH-L1, UCH-L3, DEN1, caspase 3 and cathepsin K in biochemical-based assays (Baez-Santos et al., 2014a, Ratia et al., 2008). We found that none of these human enzymes was significantly inhibited by the naphthalene-based PLpro inhibitors up to concentrations as high as 100 μM. Importantly, the selective inhibition of SARS-CoV PLpro by designed, non-covalent inhibitors is the first example demonstrating that it is possible to design selective inhibitors against the USP class of enzymes which is of universal importance for selectively targeting DUBs involved in promoting different diseases.
5. X-ray crystal structures of SARS-CoV PLpro
5.1. Structure of SARS-CoV PLpro in an unbound state
The X-ray structure of the 35-kDa catalytic domain of PLpro (residues 1541–1855 of the SARS-CoV nsp3 polyprotein domain) was originally determined to a resolution of 1.85 Å in its unbound state, providing the first structure of a coronaviral PLpro (Ratia et al., 2006). Although gel filtration analyses indicate that PLpro is predominately monomeric in solution (Han et al., 2005), the unliganded structure of PLpro crystallized in space group C2 as three closely packed monomers in the crystallographic asymmetric unit (Fig. 12 a, left panel). Structural alignment of the three monomers shows no significant conformational differences in the central body (palm and thumb domains) of the enzyme or within the active site. However, significant conformational differences between all three monomers were detected at the fingers domain and the BL2 loop of the palm domain at the entrance of the active site, suggesting high plasticity at these regions (Fig. 12a, right panel).
Fig. 12.
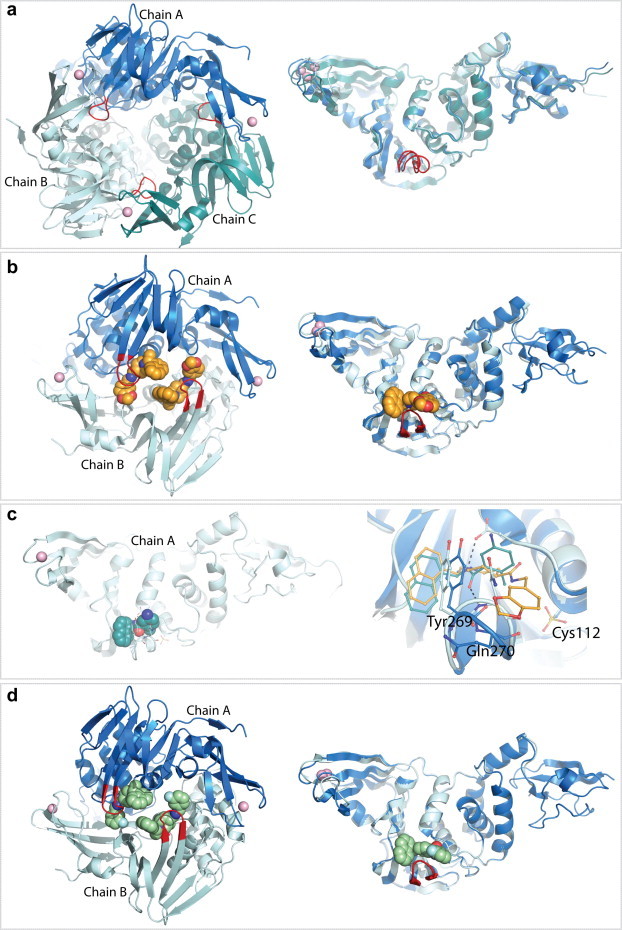
X-ray crystal structures of SARS-CoV PLpro in different asymmetric units. PLpro is shown as a ribbon representation with each monomer within an asymmetric unit colored in a different shade of blue. The BL2 loop is highlighted in red and the zinc ion is shown as a pink sphere. (a) The crystallographic trimer of unliganded PLpro (PDB: 2FE8, (Ratia et al., 2006)) is shown in the left panel and a structural superposition of the three monomers is shown in the right panel. (b) The crystallographic dimer of the SARS-CoV PLpro–15g inhibitor complex (PDB: 3MJ5, (Ghosh et al., 2010)) is shown in the left panel and a structural superposition of the two monomers is shown in the right panel. Compound 15g is shown in orange. (c) The crystallographic monomer of the SARS-CoV PLpro–24 inhibitor complex (PDB: 3E9S, (Ratia et al., 2008)) is shown in the left panel. Compound 24 is shown in green. The right panel depicts the structural superposition of the SARS-CoV PLpro–15g and SARS-CoV PLpro–24 inhibitor complexes. (D) The crystallographic dimer of the SARS-CoV PLpro–3k inhibitor complex (PDB: 4OW0, (Baez-Santos et al., 2014a)) is shown in the left panel and a structural superposition of the two monomers in the asymmetric unit is shown in the right panel.
5.2. Structure of SARS-CoV PLpro in complex with inhibitors
Generally, SARS-CoV PLpro has been highly challenging to crystallize. The crystallization conditions that yielded the SARS-CoV PLpro crystals in its unliganded state were often irreproducible or did not to produce diffraction quality crystals, preventing the crystallization of SARS-CoV PLpro in complex with inhibitors via soaking experiments. Attempts to co-crystallize SARS-CoV PLpro with inhibitors using the crystallization conditions identified for the unliganded SARS-CoV PLpro enzyme were also unsuccessful, presumably due to the tight packing interactions of SARS-CoV PLpro monomers and the positioning of the active site on the interior of the crystallographic trimer (Fig. 12a). Eventually, co-crystals of SARS-CoV PLpro–inhibitor complexes were achieved by the screening of thousands of crystallization conditions using a crystallization robot. Ultimately, the SARS-CoV PLpro–inhibitor complexes were found to crystallize in different crystallization conditions (Table 1 ). The co-crystal of SARS-CoV PLpro–15g complex diffracted to a slightly lower resolution of 2.63 Å, and similarly to unliganded SARS-CoV PLpro, it belongs to the space group C2 but with two monomers per asymmetric unit (Ghosh et al., 2010) (Fig. 12b). Crystals of the SARS-CoV PLpro–inhibitor 24 complex (also known as compound GRL0617) grew in space group I222 with one monomer per asymmetric unit and diffracted to a resolution of 2.5 Å (Ghosh et al., 2009, Ratia et al., 2008) (Fig. 12c). Unfortunately, as observed before for the unliganded SARS-CoV PLpro crystals, the crystallization conditions that yielded the inhibitor-bound SARS-CoV PLpro crystals were irreproducible and not suitable for the crystallization of SARS-CoV PLpro in complex with newly developed inhibitors.
Table 1.
Summary of SARS-CoV PLpro crystallization conditions and crystal properties.
| Protein form | PDB code | Resolution range (Å) | Crystallization conditions | Crystallization method | Space group | Molecules per asymmetric unit | Reference |
|---|---|---|---|---|---|---|---|
| Apo | 2FE8 | 1.85 | 1.1–20 mg/mL PLpro (in 20 mM Tris pH 7.5, 10 mM DTT), 100 mM sodium citrate, pH 5.2, 3 M ammonium sulfate | Vapor diffusion, sitting drop | C2 | 3 | Ratia et al. (2014) |
| Inhibitor 15g complex | 3MJ5 | 2.63 | 5 mg/mL PLpro (20 mM Tris, pH 7.5, 10 mM DTT), 1 mM inhibitor, 1 M (NH4)2SO4, 50 mM MES, pH 6.5, and 2.5% PEG 400. Cryo solution: well solution, 400 μM inhibitor, 16% glycerol | Vapor diffusion, sitting drop | C2 | 2 | Ghosh et al. (2010) |
| Inhibitor 24 complex | 3E9S | 2.5 | 8 mg/mL PLpro (20 mM Tris, pH 7.5, 10 mM DTT) with 200 uM inhibitor, 1 M LiCl2, 0.1 M MES pH 6.0, 30% PEG 6000. Cryo solution: well solution, 400 μM inhibitor, 16% glycerol | Vapor diffusion, sitting drop | I 2 2 2 | 1 | Ghosh et al. (2009) and Ratia et al. (2008) |
| Inhibitor 3k and 3j complex | 4OW0 and 4OVZ | 2.1 and 2.5 | 6 and 12 mg/mL PLpro (25 mM Tris, pH 7.5, 100 mM NaCl, 10 mM DTT) with 200 μM inhibitor, 100 mM sodium citrate, pH 5.5, 40% (v/v) PEG 600. Cryo solution: well solution, 400 μM inhibitor, 20% glycerol | Vapor diffusion, sitting drop | C2 | 2 | Baez-Santos et al. (2014a) |
| Ubiquitin complex | 4MM3 | 2.75 | 3–12 mg/mL PLpro–Ubals (20 mM Tris, pH 7.5) | Vapor diffusion, hanging drop | P 3121 | 1 | Ratia et al. (2014) |
| Ubiquitin-catalytic mutant complex | 4M0 W | 1.4 | 8 mg/mL PLproC112S-Ub (0.1 M CHES, pH 9.5, 18% PEG 3000) | Vapor-diffusion, sitting-drop | P 21 | 1 | Chou et al. (2014) |
As more SARS-CoV PLpro inhibitors were developed, the need for less costly and labor-intensive crystallization methods increased. Therefore, new crystallization efforts were centered on finding conditions for the rapid and reliable co-crystallization of SARS-CoV PLpro–inhibitor complexes. These efforts included developing a new approach for enzyme preparation and crystallization screening and ultimately resulted in the identification of a new crystallization condition, which after optimization, yielded crystals with improved reproducibility and increased diffraction ability. The structure of SARS-CoV PLpro in complex with compounds 3k and 3j resulted from these new crystallization conditions (Baez-Santos et al., 2014a) (Fig. 12d). As observed for the SARS-CoV PLpro–15g complex, the co-crystals of PLpro–3k/3j complexes belong to space group C2 with two molecules per asymmetric unit. Although poorer electron density is observed for the UBL domain, the SARS-CoV PLpro–3k/3j crystals diffracted to improved resolutions of 2.1 Å and 2.5 Å, respectively, and provided the basis for the crystallization and structure determination of SARS-CoV PLpro in complex with inhibitors from the same series of compounds. A summary of all SARS-CoV PLpro crystal structures and crystallization conditions reported to date is provided in Table 1.
The multiple X-ray crystal structures of SARS-CoV PLpro have consistently shown multiple conformations at the fingers domain and the BL2 loop (Fig. 12). The BL2 loop, which is observed in an open conformation in the unliganded SARS-CoV PLpro structure, closes upon inhibitor binding toward the catalytic cleft allowing the formation of intermolecular interactions with the inhibitors. The more closed conformation is observed with the smallest compound 24, in which a flip of the peptide bond between Gln270 and Tyr269 allows the formation of a hydrogen bond between the backbone amide of Gln270 and the carbonyl at the center of the inhibitor (Ratia et al., 2008) (Fig. 12c, right panel). A more open conformation is observed upon binding of the larger compounds 15g, 3k and 3j, in which the BL2 loop retains the backbone orientation previously observed in the unliganded PLpro structure (Fig. 12c, right panel). While the closing motions of the BL2 loop are driven by the induced-fit mechanism of inhibitor binding, other inhibitor-independent motions are observed at the tip of the fingers domain, which may account for PLpro’s ability to interact with topologically different poly-Ub chains and the UBL modifier ISG15 (see Section 2, Fig. 2c).
In contrast to the motions observed outside the catalytic site, the catalytic triad undergoes limited movement. However, the active site cysteine Cys112 is highly reactive, as it has been observed in a fully oxidized state in the inhibitor bound structures (Fig. 12c, right panel). It remains unknown whether oxidation of Cys112 is enhanced by inhibitor-induced closure of the BL2 loop, which may restrict access of the active site to reducing agents but still allow molecular oxygen to enter the active site, or whether the inhibitor-induced conformation increases the reactivity of the cysteine to molecular oxygen. Nevertheless, the observed oxidation of SARS-CoV PLpro Cys112 in various crystal structures demonstrates the high reactivity of the SARS-CoV PLpro active site.
5.3. Structure of SARS-CoV PLpro in complex with ubiquitin
Recently, crystal structures of SARS-CoV PLpro in complex with Ub were determined to resolutions of 1.4 Å (Chou et al., 2014) and 2.75 Å (Ratia et al., 2014) (Fig. 13 a). Two different approaches were used to stabilize the SARS-CoV PLpro ubiquitin complex for co-crystallization. One approach used an active site cysteine mutant, C112S, to form a noncovalent complex with ubiquitin. The C112S-PLpro–Ub complex crystallized in the monoclinic space group P 21 with one PLpro–Ub complex per asymmetric unit (Chou et al., 2014). The wild type SARS-CoV PLpro–ubiquitin complex is very weak as the Ki value of free ubiquitin cannot be determined from competitive inhibition studies (Baez-Santos et al., 2014b, Ratia et al., 2014). The introduction of the serine side chain however appears to stabilize the interaction of ubiquitin with SARS-CoV PLpro through a hydrogen bond between the hydroxy group of serine and the carboxy terminus of ubiquitin. In the mechanism illustrated in Fig. 5, the C112S complex would represent the product (“E+Q”) complex.
Fig. 13.
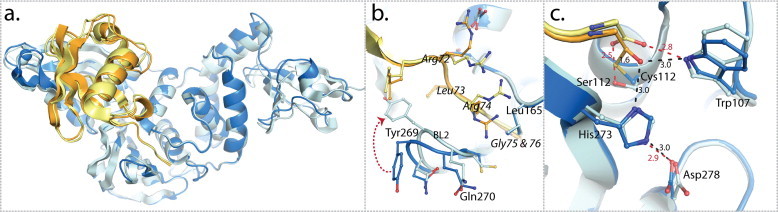
X-ray crystal structures of SARS-CoV PLpro in complex with ubiquitin. (a) An overlay of the crystallographic monomer from the two PLpro–Ub complex (PDB: 4M0W, (Chou et al., 2014) and 4MM3 (Ratia et al., 2014)) crystal structures. The PLpro–Ubal complex is shown in blue with Ubal in orange and the PLproC112S–Ub complex is shown in light blue with Ub in yellow. (b) The conformational difference of the BL2 loop and the different orientations of PLpro BL2 residues and the C-terminal residues (72–76) of Ub (italics labels). (c) The different orientations of the active site residues with the distances between the atoms indicated in angstrom (black for PLpro–Ubal and red for PLproC112S–Ub).
Our lab utilized a different approach to capture a transient, covalent intermediate complex (“FQ”) that forms during the SARS-CoV PLpro catalyzed reaction cycle (Fig. 5). Ubiquitin-aldehyde (Ubal) was synthesized, using methodologies developed by Wilkinson et al. (2005), to form a stable, wild type SARS-CoV PLpro–Ubal complex (Ratia et al., 2014). Ubiquitin-aldehyde can form a covalent but reversible complex that can increase the binding affinity of a protein for ubiquitin by as much as 300,000-times relative to unmodified ubiquitin (Melandri et al., 1996). The SARS-CoV PLpro–Ubal complex crystallized in the trigonal space group P 3121 with one PLpro–Ubal complex per asymmetric unit. The crystals diffracted to well beyond 2.75 Å but the 342 Å unit cell dimension in the c-dimension and the synchrotron setup prohibited data collection beyond this resolution. Nonetheless, the X-ray structure of the SARS-CoV PLpro–Ubal complex clearly revealed the covalent bond between the cysteine 112 sulfur atom and the c-terminal carbonyl carbon of ubiquitin (Fig. 5, Fig. 7).
A structural superposition of the wild type PLpro–Ubal and C112S PLpro–Ub complexes is shown in Fig. 13 and both structures reveal that the binding of ubiquitin induces a significant shift in the zinc-binding motif toward the Ub molecule and alters the conformation of the BL2 loop so that it interacts with the C-terminal residues (residues 72–76) of Ub (Fig. 13b). The BL2 loop contributes the majority of PLpro–Ub intermolecular hydrogen bonds and plays an important role in aligning and orienting the C-terminus of ubiquitin toward the active site of PLpro. There are observable conformational differences of the BL2 loop between the two crystal structures of PLpro–Ubal and C112S–PLpro–Ub. For example, different orientations are observed for the side chain of Tyr269 (Fig. 13b). In addition, Arg72, Arg74 and Gly76 of ubiquitin are found in different positions (Fig. 13b and c). In the C112S PLpro–Ub complex, Gly76 is found within hydrogen bond distance of C112S and Trp107, an interaction that would not naturally occur with C112. However, in the PLpro–Ubal complex, Ub C-terminal Gly76 is found covalently bonded to Cys112 and hydrogen bonded to Typ107 and His273, corresponding to the intermediate (“FQ”) state (see also Fig. 5, Fig. 7). The subtle shift in the positioning of the Ub C-terminal Gly76 results from the formation of a covalent (1.6 Å) versus a hydrogen bond (2.5 Å) with PLpro, and perhaps, this shift translates to more prominent differences in the orientations of the preceding C-terminal residues Arg72 and Arg74 of Ub. Thus, these two PLpro–Ub complex crystal structures may provide a snapshot of the transition of PLpro from the “FQ” to the “E+Q” states revealing once again the high plasticity of the BL2 loop and its interactions with substrates, intermediates, products and inhibitors.
6. Design of antiviral inhibitors targeting PLpro from other coronaviruses
The success of designing inhibitors against SARS-CoV PLpro with antiviral activity and selectivity suggests that it will be possible to design inhibitors with similar properties against other coronaviruses. In addition, since the naphthalene-based inhibitors are potent, competitive inhibitors and bind within the active site of SARS-CoV PLpro, these scaffolds would appear to be excellent starting points. Toward this goal, we tested over 30 of the napthalene-based inhibitors (Fig. 9, Fig. 10) for their ability to inhibit the papain-like protease two (PLP2) from the human coronavirus (HCoV) NL63 and PLpro from the human Middle East Respiratory Syndrome coronavirus (MERS-CoV). We found that 6 compounds are able to inhibit the HCoV-NL63 PLP2 enzyme with potencies ranging from 18 to 60 μM (Baez-Santos et al., 2014a). However, none of the compounds inhibited PLpro from MERS-CoV (Baez-Santos et al., 2014b).
The lack of inhibition of the naphthalene-based inhibitors toward MERS-CoV PLpro and the lower inhibitory potency toward HCoV-NL63 PLP2 may be explained in part by the structural differences within the BL2 loop of these enzymes. In Fig. 14 we show the X-ray structure of SARS-CoV PLpro in complex with inhibitor 3k (Baez-Santos et al., 2014a), which has been superimposed onto the homology models of HCoV-NL63 PLP2 and MERS-CoV PLpro (Baez-Santos et al., 2014b). Although the X-ray structure of unliganded MERS-CoV PLpro was recently determined by Rolf Hilgenfeld’s group (Lei et al., 2014), a number of amino acids in the β-turn/loop were not modeled in because of missing electron density. However, our energy-minimized homology models of MERS-CoV PLpro and HCoV-NL63 PLP2 included the BL2 β-turn/loop and a comparison of our MERS-CoV PLpro homology model and the X-ray crystal structure show that the overall structures were very similar (Baez-Santos et al., 2014b).
Fig. 14.
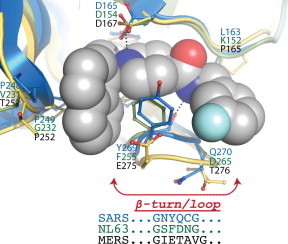
Structural comparisons of the active sites of SARS-CoV PLpro, HCoV-NL63 PLP2 and MERS-CoV PLpro. The X-ray structure of SARS-CoV PLpro in complex with compound 3k (orange ball and sticks, PDB: 4OW0) is shown superimposed with homology models of MERS-CoV PLpro and HCoV-NL63 PLP2 (green). The amino-acid residues important for SARS-CoV PLpro–inhibitor interactions are shown (blue front) along with the predicted corresponding amino acid in HCoV-NL63 PLP2 (green font) and MERS-CoV PLpro (black font). Highlighted in bold are the non-conserved substitutions in MERS-CoV PLpro sequence. Shown at the bottom of the figure is a comparison between SARS-CoV, HCoV-NL63 and MERS-CoV PLpro’s amino acid composition of the BL2 β-turn/loop (highlighted with an arrow). This loop is essential for the inhibitor induced fit mechanism of association of compound 3k and related inhibitors with SARS-CoV PLpro. This figure has been adapted from Baez-Santos et al. (2014b).
The structural models reveal that Y269 in SARS-CoV PLpro, an essential residue for binding of the naphthalene-based inhibitors, is absent in MERS-CoV PLpro resulting in the removal of a T-shaped π–π interaction with the naphthalene group of the inhibitor as well as other van der Waals interactions (Fig. 14). In addition, the BL2 β-turn/loop of MERS-CoV PLpro is one residue longer which may alter the hydrogen-bonding interactions between the inhibitor and backbone atoms of Y269. In contrast, the length of the BL2 β-turn/loop is identical for HCoV-NL63 PLP2 and F271 is a conservative substitution for Y269 in SARS-CoV PLpro. The benzyl group of F271 is still fully capable of forming a T-shaped π–π interaction with the naphthalene group and the van der Waals interactions. However, subtle structural shifts surrounding the binding pocket are likely to cause the decreased inhibitory potency of the naphthalene-based series for HCoV-NL63 PLP2. The sequence and structural variability of the BL2 β-turn/loop among coronavirus suggests that design of a broad-spectrum coronavirus antiviral compound based on the naphthalene scaffold will be difficult, but a more narrow spectrum antiviral may be more readily achievable.
7. Conclusions
Over the past 10 years, the papain-like protease domain of SARS-CoV nsp3 has emerged as a viable drug target for the development of anti-SARS therapeutics. The PLpro enzyme is essential for viral replication and small molecule inhibitors of this enzyme are capable of eliciting a SARS-CoV antiviral response in cell culture. The X-ray structures of SARS-CoV PLpro in complex with inhibitors provide a detailed map of the active site, which can be exploited for structure-based drug design (SBDD) of potent inhibitors. Analyses of the structural differences surrounding the active site BL2 loop indicate that the plasticity of this loop and the amino acid side chains within this loop will have a profound influence on the design of inhibitors against SARS-CoV PLpro and other coronavirus PLpro enzymes including the enzyme from the Middle East Respiratory Syndrome coronavirus. Finally, the SBDD approaches and studies that we have applied to SARS-CoV PLpro prove that it is entirely possible to develop highly-selective, non-covalent inhibitors against other USPs involved in disease processes.
Acknowledgements
The authors wish to express their gratitude to Dr. Kiira Ratia from the Department of Medicinal Chemistry and Pharmacognosy, University of Illinois, Chicago, Prof. Susan Baker from the Department of Microbiology & Immunology, Loyola University Chicago Stritch School of Medicine, Prof. Arun Ghosh from the Department of Chemistry, Purdue University, and Prof. Scott Larsen from the Department of Medicinal Chemistry, University of Michigan for their significant scientific contributions and collaboration in developing SARS-CoV PLpro inhibitors over the past decade. In addition, we would like to acknowledge financial support over the years for this project from the National Institute of Allergy and Infections Diseases (NIAID) via R01AI085089 and P01AI060915. ADM is supported by a grant from the Walther Cancer Foundation. Support for continued crystallographic and computational modeling of this project is being provided in part by the Purdue Center for Cancer Research, which is supported by a grant from the NCI P30 CA023168. Finally, we would like to acknowledge the beamline support staff at LS-CAT, SER-CAT and LRL-CAT at the Advanced Photon Source, Argonne National Lab over the years for their assistance in our X-ray work, which provided the structures of SARS-CoV PLpro in complex with the antiviral inhibitors.
References
- Angelini M.M., Akhlaghpour M., Neuman B.W., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4 and 6 induce double-membrane vesicles. MBio. 2013;4 doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M. University of Illinois; Chicago: 2012. Insight into the Substrate Specificity and Inhibition of Human Coronavirus Papain-Like Proteases, Pharmacy. pp. 1–230. [Google Scholar]
- Baez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M.P., Mielech A.M., Davis N.M., Baker S.C., Larsen S.D., Mesecar A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., Mielech A.M., Deng X., Baker S., Mesecar A.D. Catalytic function and substrate specificity of the PLpro domain of nsp3 from the Middle East Respiratory Syndrome Coronavirus (MERS-CoV) J. Virol. 2014 doi: 10.1128/JVI.01294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett A.J., Rawlings N.D., Woessner J.F. Handbook of Proteolytic Enzymes. third ed. vol. 1. Elsevier Science; 2012. (Catalytic Mechanisms of Cysteine Peptidases). (chapter 404) [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. Deubiquitinating activity of the SARS-CoV papain-like protease. Adv. Exp. Med. Biol. 2006;581:37–41. doi: 10.1007/978-0-387-33012-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A., Kessler B.M., Casagrande R., Overkleeft H.S., Wilkinson K.D., Ploegh H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A., Ovaa H., Kolli N., Gan-Erdene T., Wilkinson K.D., Ploegh H.L., Kessler B.M. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- Bredenbeek P.J., Pachuk C.J., Noten A.F., Charite J., Luytjes W., Weiss S.R., Spaan W.J. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus MHV-A59: a highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res. 1990;18:1825–1832. doi: 10.1093/nar/18.7.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calistri A., Munegato D., Carli I., Parolin C., Palu G. The ubiquitin-conjugating system: multiple roles in viral replication and infection. Cells. 2014;3:386–417. doi: 10.3390/cells3020386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A., Johnson M.A., Serrano P., Pedrini B., Joseph J.S., Neuman B.W., Saikatendu K., Buchmeier M.J., Kuhn P., Wuthrich K. Nuclear magnetic resonance structure shows that the severe acute respiratory syndrome coronavirus-unique domain contains a macrodomain fold. J. Virol. 2009;83:1823–1836. doi: 10.1128/JVI.01781-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Chou C.Y., Chang G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Cho J.K., Curtis-Long M.J., Lee K.H., Kim D.W., Ryu H.W., Yuk H.J., Park K.H. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg. Med. Chem. 2013;21:3051–3057. doi: 10.1016/j.bmc.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Chien C.H., Han Y.S., Prebanda M.T., Hsieh H.P., Turk B., Chang G.G., Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75:1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Lai H.Y., Chen H.Y., Cheng S.C., Cheng K.W., Chou Y.W. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr. D Biol. Crystallogr. 2014;70:572–581. doi: 10.1107/S1399004713031040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouljenko V.N., Lin X.Q., Storz J., Kousoulas K.G., Gorbalenya A.E. Comparison of genomic and predicted amino acid sequences of respiratory and enteric bovine coronaviruses isolated from the same animal with fatal shipping pneumonia. J. Gen. Virol. 2001;82:2927–2933. doi: 10.1099/0022-1317-82-12-2927. [DOI] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Malet H., Putics A., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J., Ahola T., Canard B. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J. Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraile J.M., Quesada V., Rodriguez D., Freije J.M., Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–2388. doi: 10.1038/onc.2011.443. [DOI] [PubMed] [Google Scholar]
- Frieman M., Basu D., Matthews K., Taylor J., Jones G., Pickles R., Baric R., Engel D.A. Yeast based small molecule screen for inhibitors of SARS-CoV. PLoS One. 2011;6:e28479. doi: 10.1371/journal.pone.0028479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Takayama J., Aubin Y., Ratia K., Chaudhuri R., Baez Y., Sleeman K., Coughlin M., Nichols D.B., Mulhearn D.C., Prabhakar B.S., Baker S.C., Johnson M.E., Mesecar A.D. Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome-coronavirus papain-like protease. J. Med. Chem. 2009;52:5228–5240. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Takayama J., Rao K.V., Ratia K., Chaudhuri R., Mulhearn D.C., Lee H., Nichols D.B., Baliji S., Baker S.C., Johnson M.E., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 2010;53:4968–4979. doi: 10.1021/jm1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Xi K., Johnson M.E., Baker S.C., Mesecar A.D. Progress in anti-SARS coronavirus chemistry, biology and chemotherapy. Annu. Rep. Med. Chem. 2006;41:183–196. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W.T., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeijer M.C., Verheije M.H., Ulasli M., Shaltiel I.A., de Vries L.A., Reggiori F., Rottier P.J., de Haan C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010;84:2134–2149. doi: 10.1128/JVI.01716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.S., Chang G.G., Juo C.G., Lee H.J., Yeh S.H., Hsu J.T., Chen X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry. 2005;44:10349–10359. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- Imbert I., Snijder E.J., Dimitrova M., Guillemot J.C., Lecine P., Canard B. The SARS-Coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 2008;133:136–148. doi: 10.1016/j.virusres.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. Discovery, synthesis, and structure-based optimization of a series of n-(tert-butyl)-2-(n-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R.P., Pettersson H.I., Zhang J.M., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N.G., Wishart D.S., Vederas J.C. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CL(pro) J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- Johnson M.A., Chatterjee A., Neuman B.W., Wuthrich K. SARS coronavirus unique domain: three-domain molecular architecture in solution and RNA binding. J. Mol. Biol. 2010;400:724–742. doi: 10.1016/j.jmb.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanjanahaluethai A., Chen Z., Jukneliene D., Baker S.C. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology. 2007;361:391–401. doi: 10.1016/j.virol.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Mesters J.R., Drosten C., Anemuller S., Ma Q., Hilgenfeld R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014;109:72–82. doi: 10.1016/j.antiviral.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Menard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Lytvyn V., Qi H., Lachance P., Ziomek E., Menard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007;466:8–14. doi: 10.1016/j.abb.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love K.R., Catic A., Schlieker C., Ploegh H.L. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat. Chem. Biol. 2007;3:697–705. doi: 10.1038/nchembio.2007.43. [DOI] [PubMed] [Google Scholar]
- McNaught A.D., Wilkinson A. second ed. IUPAC; 1997. Compendium of Chemical Terminology. (the “Gold Book”) [Google Scholar]
- Melandri F., Grenier L., Plamondon L., Huskey W.P., Stein R.L. Kinetic studies on the inhibition of isopeptidase T by ubiquitin aldehyde. Biochemistry. 1996;35:12893–12900. doi: 10.1021/bi9612935. [DOI] [PubMed] [Google Scholar]
- Mielech A.M., Chen Y., Mesecar A.D., Baker S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014 doi: 10.1016/j.virusres.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A.M., Kilianski A., Baez-Santos Y.M., Mesecar A.D., Baker S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology. 2014;450–451:64–70. doi: 10.1016/j.virol.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molland K., Zhou Q., Mesecar A.D. A 2.2 Å resolution structure of the USP7 catalytic domain in a new space group elaborates upon structural rearrangements resulting from ubiquitin binding. Acta Crystallogr. F Struct. Biol. Commun. 2014;70:283–287. doi: 10.1107/S2053230X14002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Joseph J.S., Saikatendu K.S., Serrano P., Chatterjee A., Johnson M.A., Liao L., Klaus J.P., Yates J.R., 3rd, Wuthrich K., Stevens R.C., Buchmeier M.J., Kuhn P. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 2008;82:5279–5294. doi: 10.1128/JVI.02631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B., Leach C.A., Goldenberg S.J., Francis D.M., Kodrasov M.P., Tian X., Shanks J., Sterner D.E., Bernal A., Mattern M.R., Wilkinson K.D., Butt T.R. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008;17:1035–1043. doi: 10.1110/ps.083450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M., Hagemeijer M.C., van Gent M., Bekker C.P., te Lintelo E.G., Rottier P.J., de Haan C.A. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 2008;82:12392–12405. doi: 10.1128/JVI.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M., te Lintelo E.G., Deijs M., Verheije M.H., Rottier P.J., de Haan C.A. Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J. Virol. 2007;81:12323–12336. doi: 10.1128/JVI.01506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.Y., Jeong H.J., Kim J.H., Kim Y.M., Park S.J., Kim D., Park K.H., Lee W.S., Ryu Y.B. Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biol. Pharm. Bull. 2012;35:2036–2042. doi: 10.1248/bpb.b12-00623. [DOI] [PubMed] [Google Scholar]
- Park J.Y., Kim J.H., Kim Y.M., Jeong H.J., Kim D.W., Park K.H., Kwon H.J., Park S.J., Lee W.S., Ryu Y.B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012;20:5928–5935. doi: 10.1016/j.bmc.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A.D. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., Baker S.C., Ghosh A.K., Mesecar A.D. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikatendu K.S., Joseph J.S., Subramanian V., Clayton T., Griffith M., Moy K., Velasquez J., Neuman B.W., Buchmeier M.J., Stevens R.C., Kuhn P. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure (Camb) 2005;13:1665–1675. doi: 10.1016/j.str.2005.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P., Johnson M.A., Almeida M.S., Horst R., Herrmann T., Joseph J.S., Neuman B.W., Subramanian V., Saikatendu K.S., Buchmeier M.J., Stevens R.C., Kuhn P., Wuthrich K. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:12049–12060. doi: 10.1128/JVI.00969-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P., Johnson M.A., Chatterjee A., Neuman B.W., Joseph J.S., Buchmeier M.J., Kuhn P., Wuthrich K. Nuclear magnetic resonance structure of the nucleic acid-binding domain of severe acute respiratory syndrome coronavirus nonstructural protein 3. J. Virol. 2009;83:12998–13008. doi: 10.1128/JVI.01253-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C., Chen Z. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Kusov Y., Mutschall D., Tech S., Nagarajan K., Hilgenfeld R., Schmidt C.L. The “SARS-unique domain” (SUD) of SARS coronavirus is an oligo(G)-binding protein. Biochem. Biophys. Res. Commun. 2007;364:877–882. doi: 10.1016/j.bbrc.2007.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Vonrhein C., Smart O.S., Bricogne G., Bollati M., Kusov Y., Hansen G., Mesters J.R., Schmidt C.L., Hilgenfeld R. The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog. 2009;5:e1000428. doi: 10.1371/journal.ppat.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Tong T.R. Drug targets in severe acute respiratory syndrome (SARS) virus and other coronavirus infections. Infect. Disord. Drug Targets. 2009;9:223–245. doi: 10.2174/187152609787847659. [DOI] [PubMed] [Google Scholar]
- van Hemert M.J., van den Worm S.H., Knoops K., Mommaas A.M., Gorbalenya A.E., Snijder E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008;4:e1000054. doi: 10.1371/journal.ppat.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson K.D., Gan-Erdene T., Kolli N. Derivitization of the C-terminus of ubiquitin and ubiquitin-like proteins using intein chemistry: methods and uses. Methods Enzymol. 2005;399:37–51. doi: 10.1016/S0076-6879(05)99003-4. [DOI] [PubMed] [Google Scholar]
- Xue X.Y., Yu H.W., Yang H.T., Xue F., Wu Z.X., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.J.C., Liao M., Bartlam M., Rao Z. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T.K., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H.J., Hsu M.C. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- Zhang J.M., Pettersson H.I., Huitema C., Niu C.Y., Yin J., James M.N.G., Eltis L.D., Vederas J.C. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004;7:412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]