

Abstract
Posttranslational modifications are covalent changes made to proteins that typically alter the function or location of the protein. AMPylation is an emerging posttranslational modification that involves the addition of adenosine monophosphate (AMP) to a protein. Like other, more well-studied posttranslational modifications, AMPylation is predicted to regulate the activity of the modified target proteins. However, the scope of this modification both in bacteria and in eukaryotes remains to be fully determined. In this review, we provide an up to date overview of the known AMPylating enzymes, the regulation of these enzymes, and the effect of this modification on target proteins.
Graphical Abstract
1. INTRODUCTION
Within a cell and its surrounding environment, most changes are exerted by the activity of proteins. A major effort by researchers in the life sciences is spent understanding these changes and how individual proteins regulate cellular machinery. Not only is it important to understand the structure and individual functions of proteins, it is critical to understand the ways in which these proteins are regulated. While the regulation of protein expression is a key factor in determining where and when a protein is needed, understanding how these proteins are altered through posttranslational modifications (PTMs) is critical for determining how these proteins are temporally and locally regulated. Furthermore, pathogens are known to hijack the cellular machinery of infected cells using PTMs to aid in infection.
One such PTM that has recently been gaining attention is AMPylation (also known as protein adenylylation). AMPylation is the covalent attachment of adenosine monophosphate (AMP) to a peptide. The most common and stable form of AMPylation occurs on the hydroxyl group of threonine, serine, or tyrosine through a phosphodiester bond.1 Less stable AMPylation modifications on lysine, histidine, and the carboxyl group of peptides have been reported; however, many of these reported less stable modifications are transient intermediates in which the AMP is transferred to other molecules.2–4
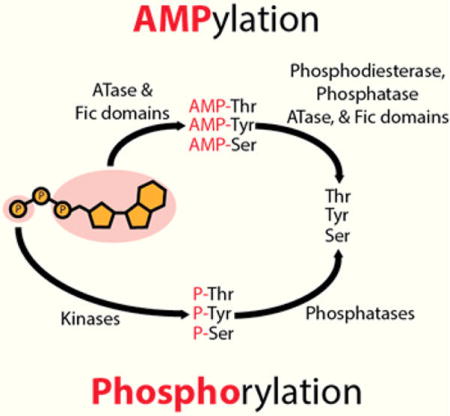
Like phosphorylation, AMPylation uses ATP as a substrate. However, instead of a nucleophilic attack occurring on the γ-phosphate as observed in phosphorylation, the α-phosphate of ATP is targeted, resulting in the release of pyrophosphate and modification with AMP5,6 (Figure 1).
Figure 1.
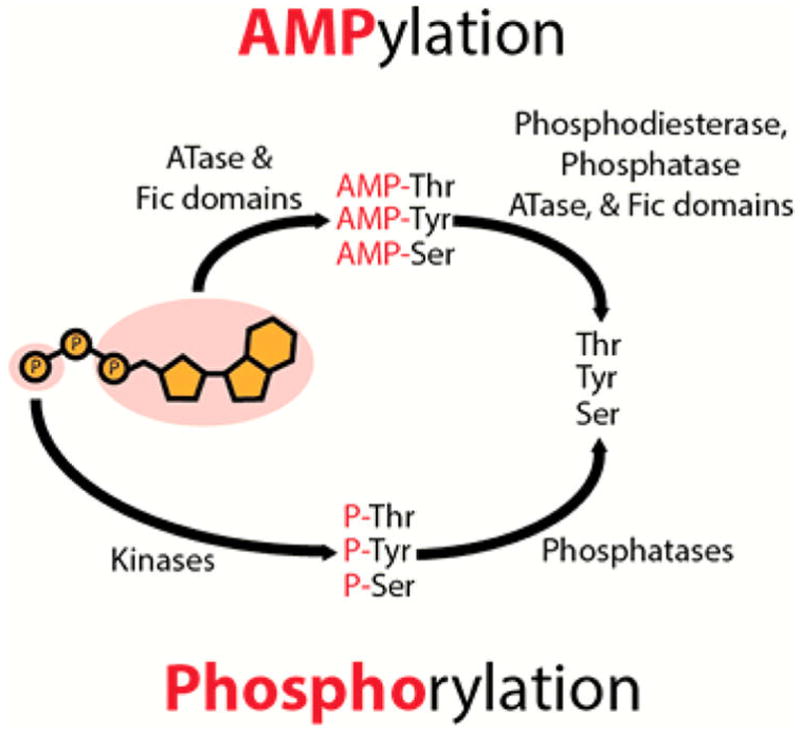
AMPylation and phosphorylation both require ATP as a substrate. Phosphorylation, addition of the γ-phosphate of ATP, is mediated by kinases and reversed by phosphatases. ATase and Fic domains mediate AMPylation: covalent attachment of AMP to the hydroxyl group of threonine, tyrosine, and serine. DeAMPylation is catalyzed by several enzyme domains (phosphodiesterase domains, phosphatase domains, ATase domains, and Fic domains).
Protein AMPylation was originally described by the Stadtman lab in 1967 when it was observed that an unknown protein transferred an AMP to glutamine synthetase in response to changes in nitrogen conditions, resulting in a change in the modified enzyme’s catalytic activity.7 Further biochemical studies identified and characterized the enzyme responsible for this AMP modification as glutamine synthetase adenyltransferase (GS-ATase).7–9 However, it was not until over 40 years later that another example of AMPylation was identified. This time the modification was mediated by the Fic (Filamentation induced by cAMP) domain of the bacterial effector VopS from the human pathogen Vibrio parahaemolyticus.10 Since the rediscovery of this PTM, this field of research has dramatically expanded and includes a growing number of enzymes from a variety of species that modify and hydrolyze novel protein PTMs, including AMPylation, UMPylation, phosphocholination, and others. Herein, we discuss the discoveries made on this new and evolving field of posttranslational regulation.
2. METHODS USED TO DETECT AMPYLATED PROTEINS
Before discussing the families of enzymes and substrates involved in AMPylation, we will review the approaches and tools that are used to identify modified substrates. As this modification is mechanistically similar to phosphorylation (Figure 1) and many useful tools have been developed to study phosphorylation, it was only logical for the study of AMPylation to follow in its footsteps.
Both kinases and AMPylators use ATP as a substrate. Therefore, the use of radiolabeled ATP to modify a substrate is reasonable; however, the position of the radiolabel must change to detect these two modifications. For phosphorylation the γ-phosphate is radiolabeled, whereas for AMPylation the α-phosphate is radiolabeled.7,10–12 For example, in an AMPylation assay the 32P-AMP from 32P-α-labeled ATP is transferred to the hydroxyl of a threonine, serine, or tyrosine, resulting in a phosphodiester bond between the 32P-AMP and the protein substrate. The radiolabeled phosphate can be detected in many ways, including phosphorimaging of radiolabeled gels and quantitation using dot blots or scintillation counters. Alternatively, 14C can be utilized to label the adenosine in the AMP transferred during AMPylation in vitro; however, these modified proteins are not as easily measured by visual means due to the limited sensitivity of 14C radioactivity detection.7
Another tool that is valuable for detecting phosphorylated proteins is phosphospecific antibodies. Soon after the discovery of AMPylation, antibodies directed at AMPylated-tyrosine and AMPylated-threonine residues were generated. Although these antibodies have limited sensitivity, they have been very useful for detecting specific AMPylated proteins in vitro and in lysates13,14 (Figure 2). The antibodies against AMPylated proteins are somewhat biased in specificity because the peptide backbone that was used to produce them came from a Rho GTPase and therefore can lead to both false positive and false negative results.
Figure 2.

AMPylated-threonine antibody is specific for AMPylated threonine residues. (A) Schematic of the antigen used to generate AMPylated-threonine antibody antigen: AMPylated peptide from the switch I region of Rac1. (B) AMPylated-threonine antibody recognizes both threonine-AMPylated Rac1 and Cdc42. Whereas low levels of AMPylated-tyrosine cross-reactivity are observed, this can be removed using immune-depletion techniques. (C) AMPylated-threonine antibody is specific for AMP modifications as competitive binding with AMP but not UMP and GMP inhibits AMPylated protein detection. Reproduced from ref 13. Copyright 2011, with permission from Elsevier.
AMPylated proteins have been observed to migrate differently in native and isoelectric focusing protein gels, similar to the changes in migration observed for phosphorylated proteins.15 However, these techniques cannot be relied upon solely to determine if a protein is AMPylated as many other modifications can cause shifts in protein mobility. Validation of these assays using another technique is needed to confirm potential AMPylation sites.
Because ATP is required in a large number of cellular processes including RNA chemistry, large amounts of radiolabeled ATP are required in cellular lysates to obtain enrichment in AMPylated proteins. Furthermore, identifying [α-32P]ATP radiolabeled peptides from complex protein samples is challenging. Overall, [α-32P]ATP labeling of AMPylated proteins is most useful in vitro with known AMPylators and protein targets where this method is sensitive enough to be used for kinetic measurements of AMPylation activity.12
Another way to observe AMPylation is by analysis of modified peptides using LC-MS/MS.16 The covalent attachment of AMP to a peptide has an observed mass increase of 329 Da5 (Figure 3). The position of this modification can be assigned by analyzing fragmentation spectra of the modified peptide generated by collision-induced dissociation (CID).16 While the CID fragmentation patterns of AMPylated peptides have been characterized previously, mass spectrometry analysis still has several drawbacks. Fragmentation spectra often require careful manual verification in addition to a sequence database search in order to accurately identify AMPylated peptides and localize modification sites (Figure 4).16 Other methods have been developed that allow AMPylated peptides to be identified in mixed protein samples using stable isotope-labeled ATP.17 By using a mixture of unlabeled ATP, [15N]ATP, and [15N,13C]ATP in AMPylation reactions, AMPylated peptides can be identified by detecting a defined triple mass shift cluster by MS analysis (Figure 5).17
Figure 3.

LC-MS/MS spectra of digested Rac1 peptides from the switch one region indicate a change in 329 Da increase when AMPylated. (A) Wild-type Rac peptide exhibits a m/z ratio of 1488.7 when z = 2. (B) Wild-type Rac peptide AMPylated by VopS exhibits a m/z ratio of 1653.3 when z = 2, a 329.2 Da increase compared to the unmodified peptide. Reproduced from ref 10. Copyright 2009, with permission from AAAS.
Figure 4.

Detection of ions of AMPylated peptide ions by ESI-MS/MS. (A) MS/MS spectrum of unmodified peptide–AAAHAAATVHV spectra analyzed by ESI-MS/MS. (B) MS/MS spectrum of AMPylated peptide–AAAHAAAT(amp)VHV spectra analyzed by ESI-MS/MS. Major unique ions detected are indicated with an asterisk (*). (C) Formation of unique ions detected of AMPylated peptides. “X” indicates the mass of the modified peptide fragment. Reproduced from ref 16. Copyright 2011, with permission from Springer.
Figure 5.

Experimental strategy for the identification of in vitro AMPylated peptides using a mixture of unlabeled ATP, [15N]ATP, and [15N,13C]ATP. (A) Workflow for experimental analysis of AMPylated peptides. Briefly, AMPylation assays are performed with [32P]-labeled ATP and analyzed by PAGE to determine the molecular weight of the target proteins. Additionally, AMPylation assays are performed with a mixture of unlabeled ATP, [15N]ATP, and [15N,13C]ATP and also separated by PAGE for analysis by LC-MS/MS. [32P] radiolabeled gel is used to identify appropriate bands for excision and analysis on the [15N]ATP/[15N,13C]ATP-labeled samples. (B) Example of [32P]-labeled AMPylated protein bands identified by autoradiography. (C) Example of a MS/MS spectrum of a peptide that has been in vitro AMPylated with a mixture of unlabeled ATP, [15N]ATP, and [15N,13C]ATP. Peaks corresponding to all three forms to ATP labeling were identified. Reproduced from ref 17. Copyright 2014, with permission from John Wiley and Sons.
When combined with other biochemical techniques, mass spectrometry analysis can be used to identify potential AMPylated proteins for further study in a relatively unbiased way. By using a chemical ATP analog, such as N6-propargyl adenosine-5′-triphosphate (N6pATP), with click chemistry, AMPylated proteins can be tagged and enriched from complex populations such as cell lysates18–20 (Figure 6). Furthermore, utilization of click chemistry techniques can be applied to more systematic approaches such as protein arrays to identify the targets of known AMPylators18,21,22 (Figure 7). While these methods are useful in the identification of candidate AMPylation substrates, it is critical that each candidate is validated rigorously for authenticity and biological relevance.
Figure 6.

Experimental strategy for the detection and identification of AMPylated proteins using N6-propargyl adenosine-5′-triphosphate (N6pATP). Briefly, AMPylation assays are performed with N6pATP, which contains an alkynyl group. Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC), “click” chemistry, is used to label AMPylated proteins with azide fluorescent dyes or affinity enrichment tags, such as azide-biotin. These tags can be used to identify AMPylated bands by PAGE or via affinity enrichment techniques. Reproduced from ref 18. Copyright 2011 American Chemical Society.
Figure 7.
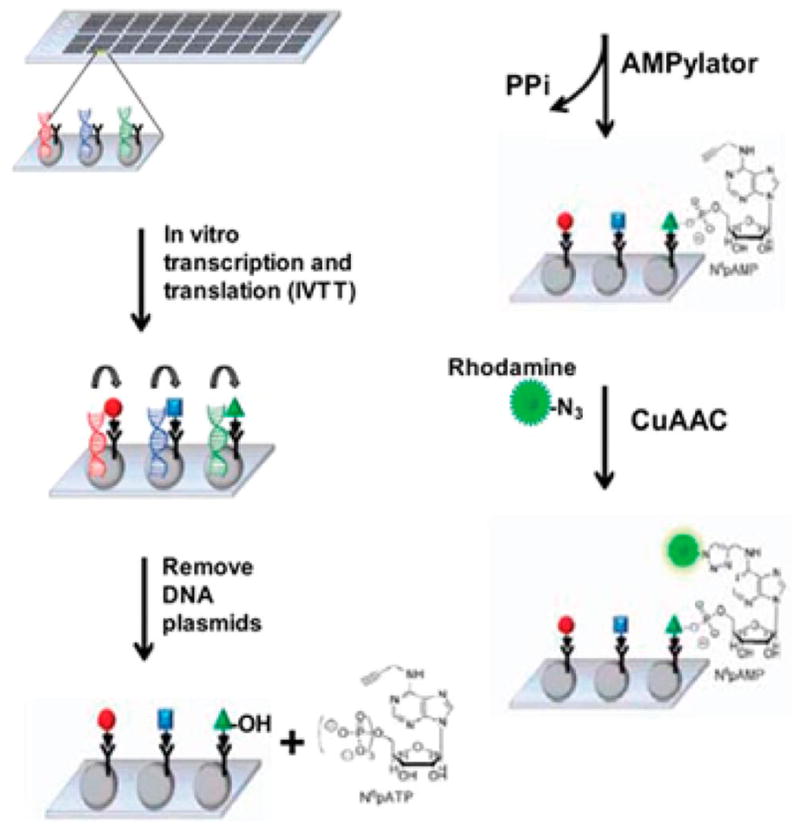
Experimental strategy for utilizing the Nucleic Acid Programmable Protein Array (NAPPA) to identify AMPylation targets. Slides printed with expression plasmids are used to produce an in vitro translated protein library. In vitro AMPylation experiments are then performed on the protein with N6pATP. AMPylated proteins are detected using CuAAC “click chemistry” with azide-fluorescent dyes. Reproduced from ref 21. Copyright 2014, with permission from American Society for Biochemistry and Molecular Biology.
3. CATALYTIC DOMAINS FOR AMPYLATORS
Currently, two different catalytic domains have been identified to have AMPylation activity: the adenylyl transferase (ATase) domain and the Fic domain. ATase domains are a subset of a larger protein family of nucleotidyl transferases and contain a 23,24 The conserved G-X11-D-X-D motif in their active site. aspartate residues are critical in the coordination of two Mg2+ ions in the active site of the ATase6,25 (Figure 8). Though this domain is found in many bacterial species, only two ATase domain-containing enzymes have been characterized to have AMPylation activity: glutamine synthetase ATase and the Legionella effector DrrA/SidM.7,9,23
Figure 8.
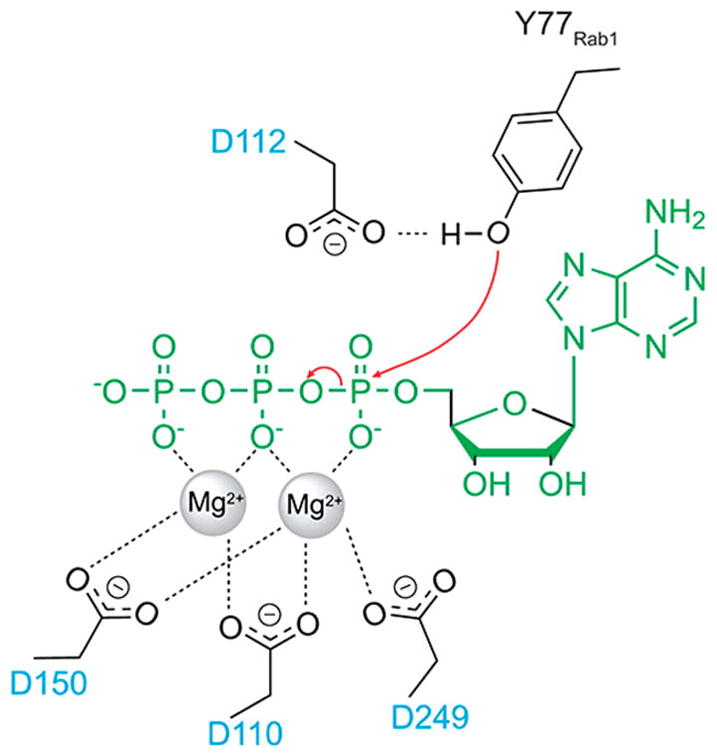
Proposed mechanism of adenylyl transferase (ATase) mediated AMPylation. Three aspartate residues coordinate two Mg2+ ions in the active site. ATP is shown in green; ATase residues are shown in black. Adapted and reproduced from ref 6. Copyright 2015 American Chemical Society.
The second protein domain with AMPylation activity is the Fic (Filamentation induced by cAMP) domain. The Fic domain was originally identified in 1982 in E. coli when a mutation was identified that lead to E. coli filamentation phenotype in the presence of cAMP.26 However, the function of this domain in E. coli remained elusive until 2009 when VopS, an effector from V. parahaemolyticus, was identified to have AMPylation activity via the Fic domain.10 Since this discovery, several Fic domain proteins have been identified and constitute the vast majority of newly discovered bona fide AMPylators.27
The Fic domain is a part of a larger Fido (Fic/Doc) superfamily.28 Fido domains share a common fold, a highly conserved active site motif, and an inhibitory α-helix motif that provides autoregulation the enzymatic activity. The Fic domain is conserved from bacteria to higher eukaryotes and is found in a diverse set of proteins. Several thousand genes containing predicted Fic domains have been identified, with the large majority of these domains found in bacterial species.29 Crystal structures of Fic domains reveal a primarily α-helical fold consisting of a core structure with six α-helices and an active site loop28,30–32 (Figure 9). The active site contains a highly conserved HPFx[D/E]GN[G/K]R motif.12,33 Kinetic analyses of Fic domain-mediated AMPylation support a ternary complex mechanism where the enzyme coordinates a direct transfer of AMP to the protein substrate.12 Structural and biochemical studies have since confirmed the conserved motif coordinates Mg2+ and ATP in the binding pocket12,28,34 (Figure 10). It is proposed that upon binding to ATP, Fic domains induce a conformational change in the switch II binding site involved in recognizing substrate proteins.34 The conserved histidine residue within this catalytic loop is responsible for the deprotonation of serine, tyrosine, or threonine during AMPylation.12,34 Many Fic domains also contain a β-hairpin near the active site that is predicted to play a role in substrate recognition via physical interactions.27,35
Figure 9.

Structure of VopS at 1.8 Å resolution. (A) Ribbon diagram of VopS. Helices in green and white represent the conserved Fic domain of VopS. Conserved catalytic histidine 348 is represented in blue. Helices in red are specific to VopS. (B) Detailed view of VopS fic domain. Residues in the conserved HPFx[D/E]GN[G/K]R domain are labeled and displayed. Reproduced from ref 12. Copyright 2010, with permission from the American Society for Biochemistry and Molecular Biology.
Figure 10.
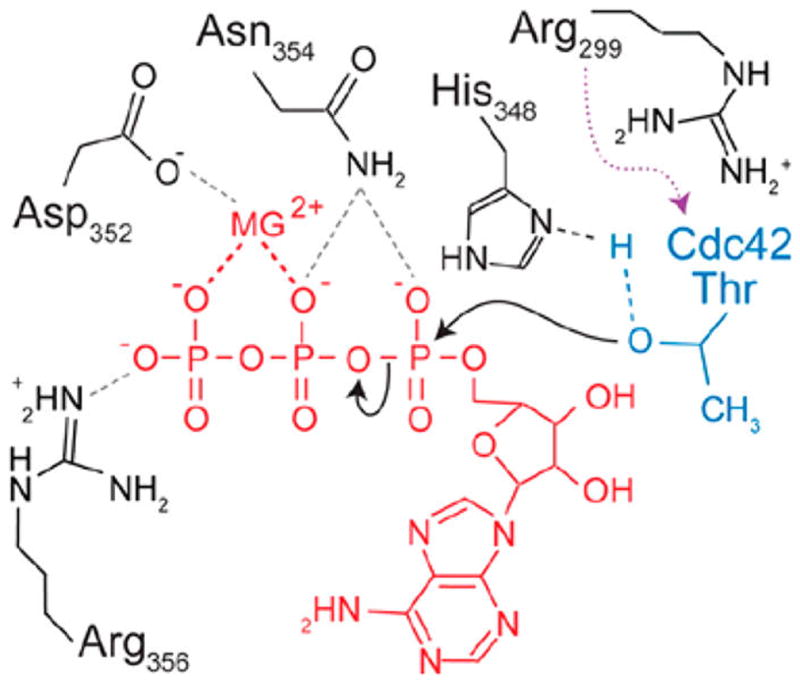
Proposed mechanism of VopS Fic domain-mediated AMPylation. Histidine 348 acts as a general base to deprotonate the hydroxyl residue of the target protein, shown in blue. Aspartic acid residue coordinates the Mg2+ ion, while asparagine and arginine residues coordinate phosphates of ATP, shown in red. Adapted and reproduced from ref 12. Copyright 2010, with permission from the American Society for Biochemistry and Molecular Biology.
4. CLASSES OF FIC DOMAINS
Most Fic domains can be separated into three distinct classes based on species and distinct topology of the inhibitory α-helix present in most Fic domain proteins31 (Figure 11). For class I Fic domains, the inhibitory α-helix is provided by a separate protein, forming a toxin–antitoxin module. This class of Fic domains is only found in bacteria, and because of this topology, these proteins are sometimes called FicT toxins.36,37 When expressed without the corresponding antitoxin, the expression of a FicT protein is toxic to bacterial cells.37
Figure 11.

Classifications of Fic domains. (A–C) Ribbon diagrams of Fic domain structures. Fic domain core shown in red, active site loop shown in yellow, inhibitory α-helix shown in green. (A) Class I Fic domain VbhT shown in complex with antitoxin VbhA. (B) Class II Fic domain SoFic (from S. oneidensis). (C) Class III Fic domain from NmFic. (D) Protein domain maps of VbhT/VbhA, SoFic, and NmFic. Adapted and reproduced from ref 31. Copyright 2012, with permission from Nature Publishing Group.
The vast majority of identified Fic domain proteins are class II in which an inhibitory α-helix is found N-terminal to the Fic domain.38 The class II of Fic domain proteins is also the most diverse, with many subclasses of proteins identified with distinct topologies containing a variety of different additional domains. Little is known about many of these enzymes and their substrates due in part to the sheer number and diversity of this class of Fic domains.38 Class III of Fic domain proteins is highly conserved and found widely throughout all classes of Proteobacteria. These single-domain proteins contain a C-terminal inhibitory α-helix after the Fic domain.31
Approximately 10% of conserved Fic domains do not contain an inhibitory α-helix and thus do not belong to any of these described classes.31 Primarily, these unclassified Fic domains are bacterial effectors, such as VopS and IbpA.31 Because these secreted effectors recognize and AMPylate eukaryotic proteins during infection, this level of recognition is not needed.
5. REGULATION OF FIC DOMAINS
As more Fic domain AMPylators are identified, scientists have discovered several different mechanisms that add layers of regulation to these enzymes. First, almost all Fic domains contain an inhibitory helix with a conserved [S/T]xxxE[G/N] motif required for the proper catalytic regulation of these enzymes.31,39 The conserved glutamate in this inhibitory α-helix is critical for this regulation.14,31,39 The glutamate in the inhibitory α-helix forms a salt bridge with a conserved arginine in the active site of the Fic domain. When the salt bridge is formed, ATP is unable to bind in the catalytic pocket40 because the arginine from the salt bridge has moved into a space required for binding the γ-phosphate of ATP31,40 (Figure 12). Additionally, comparative crystal structures of inhibited wild-type Fic domains and glutamate mutated to a glycine Fic domains show changes in γ-phosphate coordination, allowing for recognition of the ATP substrate.40 It is not well understood how the binding of the inhibitory helix is altered during enzyme activation in vivo. Mutations in the helix that prevent salt bridge formation typically result in a constitutively active AMPylator with increased catalytic activity, increased auto-AMPylation, and reduced substrate specificity.11,14,15,19 The mechanisms by which the inhibitory α-helix is regulated remain unknown and could likely vary between different Fic domain proteins.
Figure 12.

Model of inhibitory α-helix regulation of Fic domain-mediated AMPylation. Salt bridge formation between the conserved glutamate in the inhibitory α-helix prevents proper coordination of the γ-phosphate of ATP. When the salt bridge is removed and the inhibitory α-helix is disengaged, the γ-phosphate of ATP is properly coordinated and the α-phosphate is positioned appropriately for AMPylation to occur.
There are exceptions to the inhibitory helix form of regulation. Structural analysis of several Fic AMPylators has revealed that a subset of Fic domains has lost the conserved glutamate in the inhibitory loop and retained a positively charged lysine or arginine residue at this location34 (Figure 13). Intriguingly, this mutation frequently correlates with changes in the conserved Fic motif for which the second conserved arginine is replaced with a threonine.34 In these cases, the positively charged lysine or arginine in the inhibitory helix is thought to facilitate rather than inhibit ATP binding.34
Figure 13.

Model of inhibitory α-helix coordination of ATP binding in a subset of Fic domains which retain a positively charged lysine or arginine in the inhibitory α-helix and also contain mutations in the second conserved arginine in the Fic motif. γ-Phosphate of ATP is coordinated by the inhibitory α-helix lysine or arginine.
Other Fic domains have evolved alternative strategies to bypass regulation of the inhibitory helix. One example is CdFic, an AMPylator found in virulent strains of Clostridium difficile.41,42 While there is currently no identified target known for CdFic, its autoAMPylation is not inhibited by the autoinhibitory helix motif as observed with other Fic AMPylators.42 Structural analysis also revealed that the ineffectiveness of the autoinhibitory helix of CdFic is due to structural changes in the active site of the Fic domain. This results in changes to the positioning of ATP in the ATP binding pocket42 (Figure 14). The α and β-phosphates of ATP are repositioned by formation of a new salt bridge formation between Arg-200 of CdFic and the α-phosphate of ATP.42 In other known Fic domains, this position is usually a hydrophobic residue. This repositioning allows for the accommodation of the γ-phosphate in the anion hole despite the presence of the conserved autoinhibitory helix, supporting the idea that AMPylation of other targets may not be inhibited.42
Figure 14.
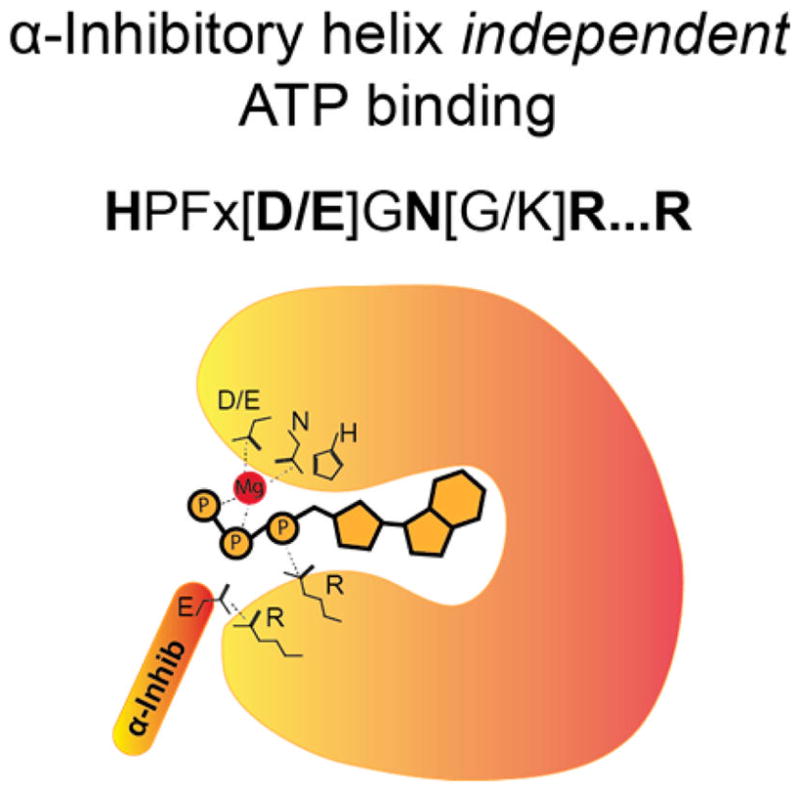
Model of inhibitory α-helix-independent ATP binding in a subset of Fic domains. Some Fic domains, such as CdFic, contain additional residues in the Fic domain binding pocket that aid in the coordination of ATP. These additional residues bypass the requirement for the second conserved arginine coordination of the γ-phosphate of ATP.
Other levels of regulation for AMPylation activity involve the self-association of Fic domains. One possible mode of regulation is oligomeric state. Many Fic domains have been observed to form dimers both by crystallization and by size exclusion/chromatography techniques.19,39,42 One such Fic AMPylator is CdFic, which forms a dimer via interactions in a loop between α′1 helix and α1 helix of the Fic domain core.42 Mutations in this dimerization interface lead to significantly increased in vitro AMPylation activity compared to wild-type CdFic.42
Dimerization is also observed to have an activating role in the metazoan Fic protein.19 In Caenorhabditis elegans, dimerization of Fic-1 is mediated by two interaction domains, including conserved residues in a loop between α′1 helix and α 1 helix of the Fic domain core, similar to CdFic in its location.19 However, mutations resulting in loss of Fic-1 dimerization lead to decreased AMPylation activity, even when the conserved inhibitory glutamate is also mutated.19 Likewise, mutations in the observed dimerization interface of human FicD result in reduced in vitro AMPylation activity and a monomeric protein.39 These mutations may simply cause an indirect destabilization of the active site, resulting in reduced enzymatic efficiency.
In Neisseria meningitidis NmFic, the homologue of a highly conserved class III Fic protein found in many bacteria is regulated by self-association.43 NmFic forms a stable tetramer via interactions on two highly conserved surfaces, leading to inactivation of NmFic.43 Formation of the NmFic dimer and tetramer is tightly controlled by protein concentration. At concentrations of 250 nM and below, NmFic behaves primarily as a monomer and exhibits much higher AMPylation activity.43 In contrast to the aforementioned Fic domain proteins, mutations in the conserved interaction surfaces result in oligomerization-deficient NmFic with increased AMPylation activity.43
Most Fic domains have some level of autoAMPylation that correlates with the AMPylation activity. For example, autoAMPylation of NmFic on a cluster of tyrosines (Y183, Y184, Y185, and Y188) results in the partial unfolding of the NmFic inhibitory helix.43 As a consequence, auto-AMPylation of NmFic leads to a change in positioning of the α-inhibitory loop and release of autoinhibition of catalytic activity.43 AutoAMPylation occurs primarily in the monomeric form of this protein, indicating that both concentration-dependent self-association and auto-AMPylation play a cooperative role in the regulation of NmFic.43 Because NmFic is highly conserved in many bacteria, it is predicted that this form of regulation is also conserved in this subset of Fic domains. Similarly, mutations of the autoAMPylation sites in class I FicT enzymes have been shown to reduce AMPylation of target proteins.37
Though AMPylation sites for many Fic AMPylators have been identified, the molecular effects of most of these sites have yet to be elucidated. Because no target substrate has been identified for CdFic other than autoAMPylation on the inhibitory α-helix, it is inconclusive if mutations to the CdFic AMPylation site change its enzymatic activity.42 Although autoAMPylation sites for the human FicD have been identified at residues T183, S79, and T80, mutagenesis and biochemical analysis of these sites indicate that autoAMPylation is not required for efficient AMPylation activity.44
6. AMPYLATION AND BACTERIAL HOMEOSTASIS
Many well-studied bacterial AMPylators are involved in cellular homeostasis. One of the first identified AMPylating enzymes was GS-ATase (Gln E), which plays a key role in complex regulation of nitrogen metabolism through its AMPylation of glutamine synthetase.7–9,45,46 Glutamine synthetase catalyzes the synthesis of L-glutamine from glutamate and ammonia, a critical step to the production of several other amino acids and nucleotides. GS-ATase contains both an AMPylation domain and a separate deAMPylation domain.47 The activity of GS-ATase is modulated by a signal-transducing protein trimer called PII that exists in two distinct forms8,24 (Figure 15). When nitrogen levels are high, the PII protein interacts with GS-ATase, stimulating its AMPylation activity. GS-ATase AMPylates glutamine synthetase at tyrosine 397, resulting in inactivation of glutamine synthesis.7,9,48 When nitrogen levels are low, the PII trimer is modified by the covalent attachment of uridine monophosphate (UMP) (referred to as UMPylation) by the enzyme uridylyl transferase (UTase).8,49,50 UMPylated PII interacts with GS-ATase, stimulating the deAMPylation activity of this enzyme. This results in the deAMPylation and activation of glutamine synthetase. Because glutamine synthetase and PII are made up of several identical subunits, the regulatory modifications made on these enzymes during changes in nitrogen availability are predicted to act as a molecular rheostat that allows for the activity of GS to be finely tuned to the cells’ needs.51
Figure 15.
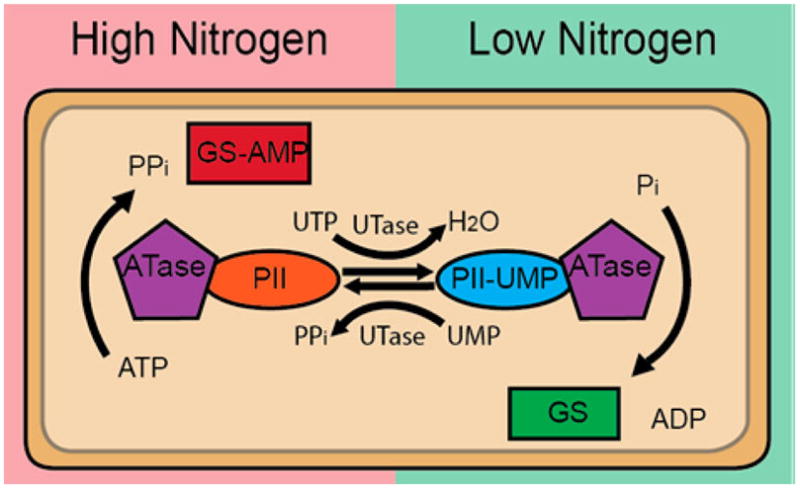
Schematic of the signaling that regulates the activity of glutamine synthetase (GS). Briefly, GS activity is regulated by a bifunctional ATase. When nitrogen levels are high, this ATase, in complex with the PII regulatory protein, AMPylates and inactivates GS. When nitrogen levels are low, PII is UMPylated. Subsequently, ATase, in complex with UMPylated PII regulator protein, deAMPylates and thus activates GS.
Another characterized group of bacterial AMPylators involved in bacterial homeostasis is the class I Fic AMPylators for which the Fic domain was originally named.26 Since the original identification of Fic-1 in E. coli,26,52,53 several Fic-1 homologues have recently been identified and characterized in other bacterial species.31,36,37,43,54,55 The FicT toxins are typically coexpressed with their antitoxin-containing inhibitory α-helix as part of a toxin–antitoxin system in the cells.31,36 When expressed without the corresponding antitoxin anti-Fic-1 (AntF), cellular growth is arrested31,37 (Figure 16). FicT AMPylators modify the GyrB subunit of DNA gyrase and its paralog, the ParE subunit of Topoisomerase IV, at a conserved tyrosine residue needed for ATP binding.36,37 This inactivation of DNA Gryase by AMPylation leads to the activation of a global cellular response to DNA damage, called the SOS response.36,37 In cells with a defective or blocked SOS response, FicT-mediated AMPylation of GyrB leads to elongated (filamented) cells for which the Fic phenotype was originally named.37 Interestingly, the effects of FicT AMPylation are reversible and do not lead to cell death but arrested cell growth. In Wolbachia bacteria, levels of FicT were shown to be upregulated in response to doxycycline, indicating that FicT AMPylation may be a regulator of bacterial cell stress;55 however, the conditions that result in FicT activation remain to be elucidated. Current models of FicT activation predict that either competitive binding for AntF or AntF degradation will result in the activation of FicT AMPylation (Figure 16).36,37
Figure 16.
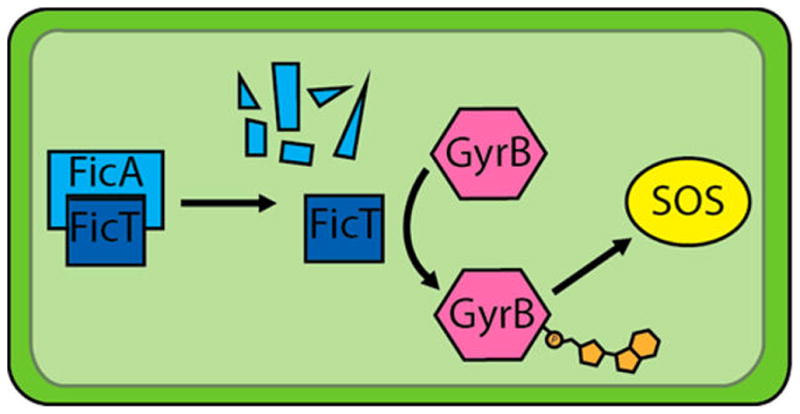
Model of Fic-1 mediation of Gyrase B (GyrB) activity. Briefly, FicT and FicA form a complex that inhibits FicT AMPylator activity. When the FicA toxin is sequestered, or degraded, FicT AMPylates and inactivates Gyrase B. This leads to DNA knotting, stalls in DNA replication, and induction of the SOS response.
Interestingly, a Class III Fic AMPylator NmFic from N. meningitidis has also been found to AMPylate GyrB at the same conserved tyrosine required for ATP binding.43 Furthermore, the highly conserved nature of all Class III Fic domains strongly indicates that the AMPylation and inactivation of DNA Gyrase must be a critical cellular strategy for bacteria to regulate stress. As described above, the regulation of NmFic is very different from the Class I FicT enzymes, as it involves concentration-dependent monomerization and autoAMPylation for activation of NmFic activity.43 However, the cellular mechanism by which the regulation is modulated has yet to be determined.
7. AMPYLATION AND BACTERIAL PATHOGENICITY
The study of viral and bacterial virulence factors has led to many discoveries including previously unknown mechanisms used in eukaryotic signaling pathways.56 Typically, these factors manipulate normal cellular processes in the infected host for the advantage of the pathogen. Of note, AMPylation is used by pathogens to manipulate small GTPases that are involved in regulating actin cytoskeleton dynamics, phagocytosis, innate immunity, and vesicle trafficking.10,57
The first identified AMPylator was VopS, a type III secretion system effector from the pathogenic V. parahaemolyticus10 (Figure 17). Once translocated into the host cell, VopS AMPylates Rho family GTPases at a conserved threonine residue on the switch 1 region of the protein.10 AMPylation at this site prevents these Rho family GTPases from associating with downstream effectors, leading to a deregulation of the host actin cytoskeleton and cell rounding10 (Figure 17).
Figure 17.
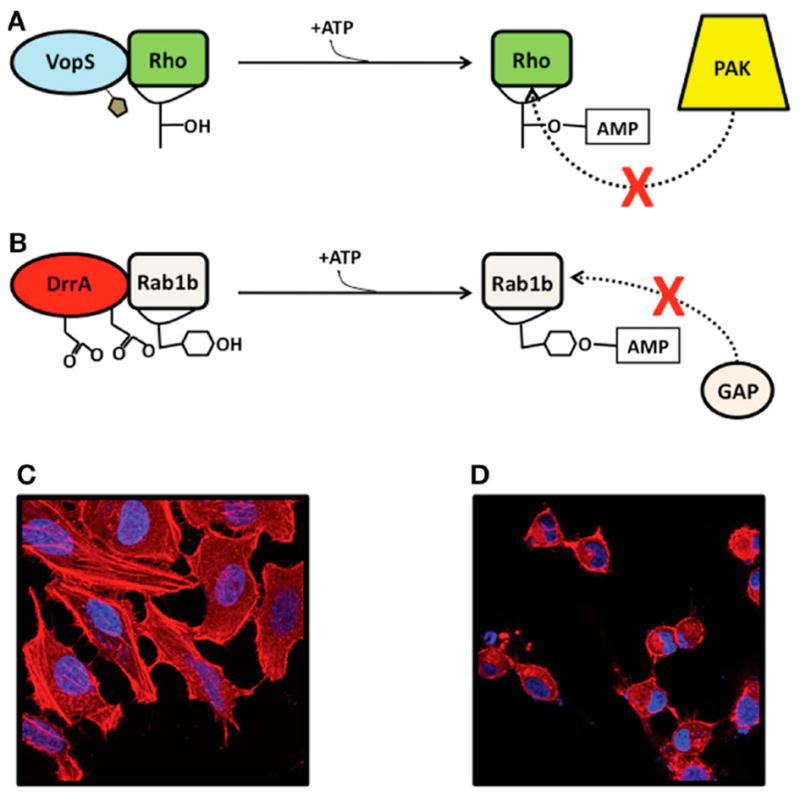
(A) VopS AMPylates a conserved threonine of Rho family GTPases. (B) DrrA AMPylates a tyrosine on Rab1b, which prevents association of Rab1b with its GAP. (C) HeLa cells mock transfected. (D) HeLa cells transfected with active VopS. Reproduced from ref 1. Copyright 2010, with permission from Frontiers.
Another bacterial AMPylator IbpA is from Histophilus somni, a Gram-negative bacterium that infects the respiratory tracts of cattle and can lead to bovine respiratory disease complex, uses a similar strategy. IbpA is a very large secreted protein (4095 amino acids) that contains an N-terminus with adhesion domains used to help its internalization into the host cell as well as two Fic domains at its C-terminus.58 These Fic domains have been shown in vitro to AMPylate the Rho GTPases RhoA, Rac, and Cdc42 but on a conserved tyrosine reside in the GTPase switch 1 region.11,58–60 The result of this AMPylation is similar to that of VopS as AMPylation of tyrosine also blocks association of downstream effectors, leading to cell rounding and cytotoxicity.58
Another set of effectors worthy of increased study are found in several species of Bartonella, mammalian pathogens frequently transmitted from blood-sucking arthropods.61 These bacteria rely on a VirB/D4 T4SS to secret a large variety of Bartonella effector proteins (Beps) into the host. Interestingly, most Beps have an N-terminal canonical FIC motif indicative of an AMPylator and a C-terminal BID (Bartonella intracellular delivery) domain; furthermore, all Beps are predicted to evolve from the same VbhT ancestor.31,38,61 Confirmation of AMPylation activity for many of these Beps has yet to be performed.61 However, some intriguing bacterial studies have successfully validated the AMPylation activity of a few individual Beps. BepA from B. henselae has confirmed AMPylation activity and been shown to AMPylate at least two unidentified host proteins (40 and 50 kDa) in HeLa cell lysates.35 Another Bep, Bep2 from B. rochalimae, was also shown to AMPylate the host protein vimentin, an intermediate filament protein. However, the biological consequence of vimentin AMPylation remains to be elucidated.17 The role of AMPylation in Bartonella pathogenicity remains to be determined, but the diversity and scale of Fic domains within this family of effectors strongly suggest more mechanisms in pathogenicity exists.
C. difficile, a causal agent of nosocomial antibiotic-associated diarrhea and colitis, contains a Fic domain AMPylator.41 Though no substrate for CdFic has been identified, the virulence factor is predicted to be similar to VopS and constitutively active.42 As pathogens use AMPylators to target critical components of host-signaling pathways, identifying their substrates is important for our understanding of pathogenesis and host signaling.
The intracellular pathogen and causal agent of pneumonia in humans, Legionella pneumophila, uses both ATase domains and Fic domains to modulate host signaling during infection. The virulence factor DrrA, encoding an ATase domain, AMPylates another type of small GTPase, Rab1b associated with host vacuoles (Figure 17). L. pneumophila is an intracellular pathogen and resides in Legionella-containing vacuoles (LCVs), an environment similar to the host ER.23,62 In addition to its ATase domain, DrrA contains a guanine nucleotide exchange factor (GEF) domain for activation of GTPases.23,63 DrrA targets Rab1b by converting it into its active state first with its GEF domain. The ATase domain of DrrA then AMPylates the tyrosine residue 77 on its switch 1 region to block interactions between Rab1b and host GTPase-activating proteins (GAPs) that might inactivate Rab1b.23,64 AMPylation at Y77 prolongs the active GTP-bound state of Rab1b and blocks GAP-stimulated inactivation via GTP hydrolysis.23 L. pneumophila efficiently hijacks the activity of Rab1b to target ER vesicles resulting in their association with the LCV.65,66 This recruitment of vesicles facilitates the formation of the LCV and is required for Legionella to form a proliferative intracellular niche.62
AMPylation by DrrA is just one layer of the complex regulation used in the formation and maintenance of the LCV because the Legionella control of Rab1b is further regulated spatially and temporally by deAMPylation and phosphocholination, the covalent attachment of phosphocholine.67–70 Interestingly, the phosphocholination of Rab1b is catalyzed by a Fic protein, AnkX.67 This is just one example of several Fic domains that catalyze different modifications other than AMPylation. Though most Fic domains are not bacterial effectors, it is through the study of these Fic effectors that these diverse activities performed by Fic domains were identified and the molecular plasticity of Fic domains was revealed.
The Fic effector CBU2078 of Coxiella burnetti also has observed phosphocholination activity. When overexpressed in human cells, CBU2078 caused the phosphocholination of proteins; however, these targets have yet to be identified.67 Another example is AvrAC, a Fic effector of the plant pathogen Xanthomonas campestris. AvrAC catalyzes the UMPylation, the covalent attachment of uridine monophosphate (UMP), of BIK1 and RIPK.71
Additionally, AvrB, a Fic effector of the plant pathogen Pseudomonas syringae, is predicted to have kinase activity and phosphorylate RIN4, a regulator of the innate immune response in plants.72 Another large family of Fic domain-related effectors is the toxin Doc toxin family. Doc is a kinase and has been shown to phosphorylate the bacterial translation factor EF-Tu. This change from AMPylator to kinase is possible because of a single residue change in the active site that inverts the binding of ATP. Of note, Doc can also catalyze the dephosphoryation of EF-Tu, indicating a dual function for this enzyme.73 Interestingly, the Doc-mediated dephosphorylation of EF-Tu occurs through the reversibility of Doc’s kinase activity and is dependent on increased levels of ADP.
In Bartonella, several Beps contain noncanonical Fic domains with unknown activities.61 Further study of these bep effectors along with many other uncharacterized Fic effectors may reveal an even wider variety of modifications catalyzed by Fic domains.
8. AMPYLATION IN EUKARYOTES
Though several bacterial effectors have been shown to AMPylate eukaryotic host targets, only one eukaryotic AMPylator has been identified.11 In humans, this AMPylator is known as FicD (also known as HYPE or hFic) and is conserved in most metazoan species. FicD was identified in a yeast two-hybrid screen for physical interactions with Huntingtin protein and therefore is also referred to as HYPE (Huntingtin yeast interacting protein E).74 FicD and its orthologs contain a conserved topology consisting of a type II N-terminal transmembrane domain, tetratricopeptide repeat (TPR) domain, and a C-terminal class II Fic domain (Figure 18). FicD localizes to the ER, with the TPR domain and Fic domain facing the ER lumen.14,44,75 Of note, FicD and its orthologs are the only Fic domain containing proteins identified in eukaryotes.28 Expression of FicD and its orthologs is ubiquitous but at very low levels.19,44 Endogenous protein detection in most eukaryotes has been a challenge;15,19,44,76 however, the expression of FicD is increased during ER stress.14,15,44
Figure 18.

Crystal structure of FicD (HYPE/hFic). (A) Protein domain maps of FicD, full length, and truncation used for crystallization. (B) Ribbon diagram (left) and surface structure (right) of FicD. TPR domains shown in orange, linker domain shown in yellow, and Fic domain shown in gray. Red asterisk indicates ATP binding pocket. Reproduced from ref 39. Copyright 2014, with permission from Elsevier Inc.
FicD and its orthologs are predicted to function as a dimer both by crystal structures and by size exclusion chromatography19,39 (Figure 19). The AMPylation activity of FicD and its orthologs is highly regulated both in vivo and in vitro by the conserved autoinhibitory α-helix, as discussed above for class II Fic domains. Because of this regulation, overexpression of wild-type FicD does not lead to detectible increases in AMPylation.15 Mutations in the conserved inhibitory α-helix result in increased AMPylation activity. Mutations that perturb stable dimerization of FicD result in reduced AMPylation activity, even in mutants where the conserved α-inhibitory glutamate is also mutated.19,39 Currently, it is unclear how the autoinhibition is released in vivo.15,19,77,78
Figure 19.

Crystal structure of FicD (HYPE/hFic) dimerization. (A) Ribbon diagram (left) and surface structure (right) of FicD dimers. Monomer A of FicD: TPR domains are shown in orange, linker domain is shown in yellow, Fic domain is shown in gray. Monomer B of FicD is shown in green. (B) Ribbon diagram of FicD dimer interface. Key residues for dimer interface shown are modeled as sticks and labeled. Reproduced from ref 39. Copyright 2014, with permission from Elsevier Inc.
Because of these limitations, researchers have typically taken advantage of overactive FicD mutants in which the required glutamate in the inhibitory α-helix is mutated.14,15,19,44,77 However, these overactive mutants may lead to changes in substrate specificity and off target effects. Overexpression of wild-type FicD and its orthologs appears to cause no adverse effects on the health of a cell, likely due to strong catalytic regulation by the inhibitory α-helix.14,15,44 Consequently, overexpression of a mutant FicD with the conserved glutamine in the autoinhibitory helix changed to a glycine (FicDE234G) causes cytotoxicity and induction of apoptosis.15,44
Initial studies in Drosophila found that the Drosophila homologue of FicD, dFic, was required for proper vision neurotransmission in flies (Figure 20). dFic null flies are viable and healthy, but in the eye, they display defects in synaptic transmission to laminal neurons.75 Immunohistochemistry staining of fly eye sections showed that fic null flies also exhibit defects in histamine neurotransmitter recycling, a process mediated by glial cells.75 Furthermore, expression of dFic specifically in glial cells was sufficient to rescue the vision defects observed in the flies.75 This indicates that, while not essential, dFic AMPylation is important for glial cell function.
Figure 20.
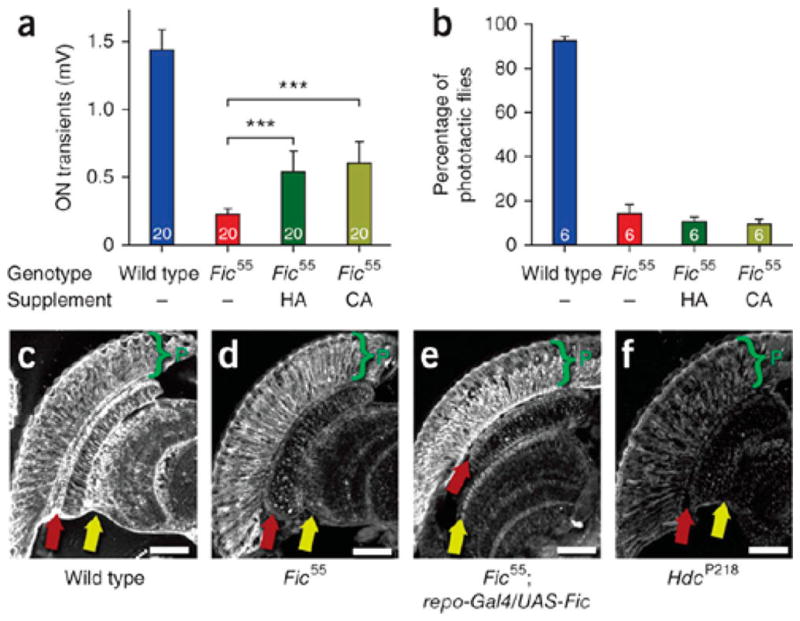
dFic is required for histamine recycling in the Drosophila eye. (A) Quantification of ON transients from electroretinograms (ERGs) indicates dFic mutant (Fic55) flies have defects in synaptic transmission to laminal neurons. These defects can be partially rescued by supplementing flies with additional histamine (HA) or carcinine (CA) to their diet. (B) dFic mutant flies also display defects in fast phototaxis response. (C–F) Histamine staining of head sections indicates that compared to wild-type (C), dFic mutant (D) flies have reduced histamine enrichment in glial cells (red arrows) proximal to the photoreceptor cells and synapses of the lamina and medulla (yellow arrows). Overexpression of dFic in glial cells (E) partially rescues histamine staining in dfic mutant flies. (F) HdcP218 control for complete loss of histamine staining. Reproduced from ref 75. Copyright 2012, with permission from Nature Publishing Group.
Subsequent studies of dFic in Drosophila identified BiP as a major target of AMPylation by dFic during ER homeostasis14 (Figure 21). Since this initial discovery, several studies across multiple species have revealed the major target of FicD to be BiP (also known as Grp78 and HSPA5 humans),14,15,19,44 a HSP70 family chaperone in the ER with a critical role in the unfolded protein response (UPR).79 BiP is an ATPase-dependent chaperone that aids in the folding of protein within the ER lumen, modulation of the UPR pathway, and activation of the ERAD (ER-associated protein decay) pathway.79,80 Several layers of regulation, including transcriptional induction, cofactor binding, oligomerization of BiP, and PTMs, modulate the activity of BiP.78,79,81 FicD is responsible for the reversible AMPylation of BiP, a posttranslational modification that is regulated by induction of ER stress.14,44
Figure 21.

FicD-mediated AMPylation of BiP is regulated by ER stress. (A) Drosophila BiP is AMPylated in vitro by wild-type dFic (dFic Wt) but not the catalytically dead dFicH375A (dFic H/A). (B) Human BiP (hBiP) is AMPylated in vitro by overactive hFicE234G. (C and D) AMPylation of BiP is altered by ER stress. S2 cells were treated with 5 mM DTT and 100 μg/mL cycloheximide (CHX). (C) Whole cell lysates were analyzed with anti-AMPylated threonine, anti-BiP, and anti-tubulin antibodies. (D) Cells were transfected with BiP-FLAG. BiP was immunoprecipitated with anti-FLAG resin and analyzed by western with anti-AMPylated threonine and anti-BiP antibodies. Adapted and reproduced from ref 14. Copyright 2014, with permission from The American Society for Biochemistry and Molecular Biology.
Despite the conserved nature of both FicD and BiP, studies in different metazoan species have led to some conflicting results that have yet to be addressed in the field. One of the major conflicts is the site(s) of BiP AMPylation. The first reports of BiP AMPylation from in vitro studies with Drosophila and human proteins indicated that AMPylation occurred on T366, a conserved threonine in the ATPase domain of BiP.14,44 However, recent in vivo and in vitro studies identified T518, a conserved threonine in the substrate-binding domain as the primary AMPylation site in mammalian BiP.15,77 Furthermore, in vitro AMPylation studies in C. elegans reports AMPylation of T176, a different threonine in the ATPase domain.19 These observed differences in modification sites of BiP in different species may indicate evolutionary changes to FicD substrate recognition and role in BiP regulation or potential variability in overactive FicD enzyme specificity in different species homologues.
It is also uncertain how AMPylation of BiP regulates its function in the UPR. When levels of ER stress are low, FicD AMPylates a subset of BiP.14,15,44 Consistent with observations of Drosophila dFic and BiP, studies from the laboratory of David Ron indicate that AMPylated BiP has reduced ATPase activity and decreased substrate-binding affinity, suggesting that the pool of AMPylated BiP is inactive.14,15 When levels of ER stress are high, BiP is deAMPylated and predicted to return to the active pool of BiP.14 Study of the UPR response in mammalian FicD CRISPR nulls also indicates that loss of BiP AMPylation results in a less responsive UPR.15 These data support a model in which AMPylation of BiP during ER homeostasis allows for the ER to provide a sensitive UPR response but still maintains an excess pool of inactive yet readily available BiP (Figure 22). This could be especially critical in cell types that require sensitive UPR responses such as neurons and supporting cells.82
Figure 22.
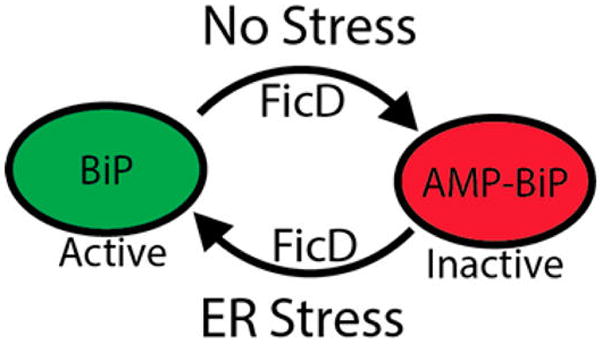
Model of AMPylation-mediated inactivation of BiP during ER homeostasis.
While this model is compelling, other studies provide conflicting results. Studies in C. elegans fic-1 CRISPR null animals indicate no discernible defect in UPR induction using hsp-4::GFP reporter induction as a readout.19 Furthermore, survival and development of animals was not affected in fic-1 CRISPR nulls or overactive Fic-1E274G mutants under UPR stress.19 However, fic-1 null animals were more susceptible to death by P. aeruginosa infection.19 These conflicting studies also differ in the identified AMPylation sites of BiP, which could suggest distinct modes of regulation. More careful analysis of the differences in AMPylation and the effect on BiP function in these models is needed to determine if one or multiple mechanisms of FicD-mediated regulation of BiP are present.
Of note, there is no known Fic domain-containing protein in yeast. Because the UPR is much simpler in yeast, such regulation of BiP may not be required. Recent studies from the Ploegh laboratory have shown that overexpression of overactive C. elegans Fic-1E274G is lethal in yeast.83 Furthermore, in mammalian cells, overexpression of overactive FicDE234G is also lethal in cells where endogenous FicD is also knocked out.84 This is most likely due to the unrestrained AMPylation activity of these mutant enzymes and a lack of deAMPylation in the ER (see Regulation of deAMPylation below).84
Interestingly, overexpression of overactive Fic-1E274G and FicDE234G was found to induce the heat shock response (HSR) and the cytosolic stress response (CSR) in yeast.83 Furthermore, overexpression of the cytosolic HSP70 family chaperone SSA2 partially rescued lethality of overactive Fic-1E274G expression.83 This indicates that lethality of Fic-1E274G may be due to activation of the heat shock response.83 Previous work has shown that the toxic effects of upregulation of the UPR in IRE1 mutants can be counterbalanced by upregulation of the HSP in yeast.85 Furthermore, other studies have revealed that activation of the UPR in yeast leads to the upregulation of the transcription factor Sir2, which both repressed UPR-induced genes while activating the HSR through activation of Hsf1.86 While a direct effect of Fic-1 and FicD overexpression on cytosolic chaperones is highly unlikely due to the compartmentalization of the enzymes and substrates, additional studies are needed to fully evaluate the effects of endogenous Fic-1 and FicD activity.
Analysis of overactive FicDE234G mutant and the C. elegans overactive Fic-1E274G mutant with lysates identified several “artificial” substrates, including histones, translation elongation factors (eEF-1A, eEF-1G, and eEF-2), HSP40, HSP90, and HSP70 family members including HSP-1 and HSP-3 (BiP).19,83 Similarly, FicD was shown to AMPylate RhoA, Rac, and Cdc42 in vitro.11 However, the physiological relevance of these findings is lacking as these substrates do not appear to be relevant targets in vivo since they are cytoplasmic proteins.44 While the premise of an AMPylator in the cytoplasm is intriguing, there is currently very little data that suggests endogenous FicD plays that role outside the ER.75,77
9. REGULATION OF DEAMPYLATION
Little is known about the scope of AMPylation in protein regulation, and less is known about deAMPylation of proteins. Only a few proteins with deAMPylating activity have been identified. The first identified deAMPylator is also the first identified AMPylator: GS-ATase (Figure 15). As described previously, this protein contains a separate deAMPylation domain and its activity is modulated by the binding of PII and associated PTMs of PII.8,47,49,50 To remove the AMP from glutamine synthetase, the adenyl–tyrosine bond is phosphorolyzed using orthophosphate to produce ADP.24,48,87
Only one bacterial effector has been identified with deAMPylating activity. As described above, in L. pneumophila the recruitment of ER vesicles to form the LCV requires AMPylation of Rab1b at Y77, locking Rab1b into an ATP bound state.23,64,65 Whereas this is critical for early LCV formation, inactivation of Rab1b is preferred after establishment of the LCV. Expression of the Legionella deAMPylator SidD is temporally regulated so that deAMPylation of Rab1b occurs after the LCV is established.70 The catalytic domain of SidD resembles a metal-dependent phosphatase domain and catalyzes deAMPylation of Y77 of Rab1b without the need for phosphate, resulting in the release of AMP.70 Furthermore, SidD deAMPylation is highly specific to AMPylated Rab1, as other AMPylated Rho GTPases cannot be deAMPylated by this enzyme.69 Once deAMPylated, Rab1b is then deactivated by a different Legionella effector, LepB, at a later stage in infection.65,70
The most recently identified deAMPylator is the conserved eukaryotic AMPylator FicD.84 Previously, it was known that FicD AMPylated BiP when levels of ER stress are low; however, the mechanism by which BiP was deAMPylated during UPR induction was unknown.14,15,44 Recent studies in mammalian and Drosophila show that FicD can also deAMPylate BiP both in vitro and in vivo.84,88 Furthermore, this reaction is catalyzed by the same conserved Fic domain that catalyzes AMPylation.84,88 However, this reaction is not the exact reverse of AMPylation, as deAMPylation of BiP results in the release of AMP not the production of ATP.84 Thus, the active site of FicD is capable of catalyzing two distinct reactions: AMP transferase and phosphodiesterase.
It is still unknown how the activity of FicD switches between AMPylator and deAMPylator. However, the conservation of this activity in mammals and Drosophila suggests the deAMPylation activity of FicD is critical for regulation of BiP activity. Of note, mutation of the residues in the regulatory salt bridge of FicD, such as FicDE234G, results in a mutant enzyme that can no longer deAMPylate substrates but contains very high AMPylation activity.84,88 This could indicate that a structural change in the inhibitory α-helix is involved in producing the different enzymatic activities of FicD. One possibility is that FicD would preferentially bind to AMPylated proteins when the inhibitory α-helix is engaged but would preferentially bind ATP when the inhibitory α-helix is disengaged84 (Figure 23). Whereas it is known that FicD can both AMPylate and deAMPylate targets, it is still unknown how these opposing activities of endogenous wild-type FicD are regulated in the cell in the presence or absence of UPR. Understanding the regulation of the FicD enzyme within the ER is a critical question that must be addressed.
Figure 23.
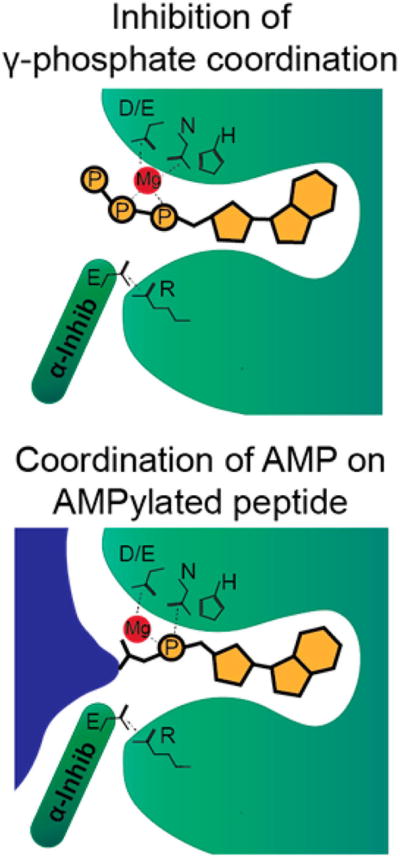
Model of AMPylated peptide recognition by FicD.
Of note, the currently known deAMPylators have little in common structurally, indicating a much broader evolutionary history when compared to known AMPylators. Other Fic AMPylators in bacteria could exhibit deAMPylation activity similar to the eukaryotic FicD. Previously, snake venom phosphodiesterases were also shown to deAMPylate proteins.7,11 Though the deAMPylation by snake venom phosphodiesterases is not predicted to be biologically relevant, it does suggest that other phosphodiesterases could deAMPy-late proteins in cells as well.
10. CONCLUDING REMARKS
Herein we have described recent findings on a novel posttranslational modification called AMPylation. This modification, the covalent attachment of AMP to a protein, has gone from relative obscurity to a well-established modification in less than a decade. Currently, only a handful of predicted AMPylating enzymes have been characterized, many playing a role in cellular homeostasis and pathogenesis.
While both ATase and Fic domains have been found to catalyze AMPylation, these domains can also catalyze other modifications with similar chemistry, such as UMPylation, phosphocholination, and phosphorylation. The prevalence and variety of these other modifications have yet to be determined. Additional studies are needed to fully determine the scope of chemical modifications these domains can mediate.
In bacteria, several protein AMPylators have recently been identified with a variety of protein targets, both in prokaryotic homeostaisis and in bacterial pathogenesis. However, to date only a single eukaryotic AMPylator has been identified. Common chemical modifications are frequently repeated throughout the tree of life in diverse ways to produce different effects. As this field expands, it is likely that more AMPylating enzymes will be identified with a variety of target pathways, both in bacteria and in eukaryotes. It is also possible that additional domains with AMPylation activity have yet to be discovered that may further expand this field. As the fields of AMPylation and related PTMs continue to develop, we will continue to learn more about the prevalence of these modifications, how they impact protein function and structure, and their roles in diverse biological processes.
Acknowledgments
This work was funded by National Institutes of Health (NIH), grant R01 GM115188, the Welch Foundation, grant I-1561, and Once Upon a Time... Foundation. K.O. is a Burroughs Welcome Investigator in Pathogenesis of Infectious Disease, a Beckman Young Investigator, and a W. W. Caruth, Jr., Biomedical Scholar and has an Earl A. Forsythe Chair in Biomedical Science.
Biographies
Amanda K. Casey received her B.A. degree in Biochemistry and Molecular Biology from Hendrix College and her Ph.D. degree in Cell and Developmental Biology from Vanderbilt University under the mentorship of Susan Wente. She is now a postdoctoral fellow with Kim Orth at the University of Texas Southwestern Medical Center as a postdoctoral researcher.
Kim Orth received her B.S. degree in Biochemistry from Texas A&M University, followed by her M.S. degree in Biological Chemistry from the University of California, Los Angeles, and her Ph.D. degree in Biochemistry and Molecular Biology from UT Southwestern Medical Center. After a postdoctoral fellowship at the University of Michigan, she returned to the UT Southwestern Medical Center, where she is now a Professor and HHMI Investigator.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Woolery AR, Luong P, Broberg CA, Orth K. AMPylation: Something Old is New Again. Front Microbiol. 2010;1:113. doi: 10.3389/fmicb.2010.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner C. Hint, Fhit, and GalT: Function, Structure, Evolution, and Mechanism of Three Branches of the Histidine Triad Superfamily of Nucleotide Hydrolases and Transferases. Biochemistry. 2002;41:9003–9014. doi: 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gumport RI, Lehman IR. StructureoftheDNALigase-AdenylateIntermediate: Lysine (eamino)-Linked Adenosine Monophosphoramidate. Proc Natl Acad Sci U S A. 1971;68:2559–2563. doi: 10.1073/pnas.68.10.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmelz S, Naismith JH. Adenylate-Forming Enzymes. Curr Opin Struct Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yarbrough ML, Orth K. AMPylation is a New Post-translational modiFICation. Nat Chem Biol. 2009;5:378–379. doi: 10.1038/nchembio0609-378. [DOI] [PubMed] [Google Scholar]
- 6.Hedberg C, Itzen A. Molecular Perspectives on Protein Adenylylation. ACS Chem Biol. 2015;10:12–21. doi: 10.1021/cb500854e. [DOI] [PubMed] [Google Scholar]
- 7.Kingdon HS, Shapiro BM, Stadtman ER. Regulation of Glutamine Synthetase, VIII. ATP: Glutamine Synthetase Adenylyltransferase, an Enzyme that Catalyzes Alterations in the Reguatory Properties of Glutamine Synthetase. Proc Natl Acad Sci U S A. 1967;58:1703–1710. doi: 10.1073/pnas.58.4.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MS, Segal A, Stadtman ER. Modulation of Glutamine Synthetase Adenylylation and Deadenylylation is Mediated by Metabolic Transformation of the P11-Regulatory Protein. Proc Natl Acad Sci U S A. 1971;68:2949–2953. doi: 10.1073/pnas.68.12.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hennig SB, Anderson WB, Ginsburg A. Adenosine Triphosphate: Glutamine Synthetaes Adenylyltransferase of Escherichia coli: Twho Active Molecular Forms. Proc Natl Acad Sci U S A. 1970;67:1761–1768. doi: 10.1073/pnas.67.4.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yarbrough ML, Li Y, Kinch LN, Grishin NV, Ball HL, Orth K. AMPylation of Rho GTPases by Vibrio VopS Disrupts Effector Binding and Downstream Signaling. Science. 2009;323:269–272. doi: 10.1126/science.1166382. [DOI] [PubMed] [Google Scholar]
- 11.Worby CA, Mattoo S, Kruger RP, Corbeil LB, Koller A, Mendez JC, Zekarias B, Lazar C, Dixon JE. The Fic Domain: Regulation of Cell Signaling by Adenylylation. Mol Cell. 2009;34:93–103. doi: 10.1016/j.molcel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luong P, Kinch LN, Brautigam CA, Grishin NV, Tomchick DR, Orth K. Kinetic and Structural Insights into the Mechanism ofAMPylation by VopS Fic Domain. J Biol Chem. 2010;285:20155–20163. doi: 10.1074/jbc.M110.114884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hao YH, Chuang T, Ball HL, Luong P, Li Y, Flores-Saaib RD, Orth K. Characterization of a Rabbit Polyclonal Antibody Against Threonine-AMPylation. J Biotechnol. 2011;151:251–254. doi: 10.1016/j.jbiotec.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ham H, Woolery AR, Tracy C, Stenesen D, Kramer H, Orth K. Unfolded Protein Response-regulated Drosophila Fic (dFic) Protein Reversibly AMPylates BiP Chaperone during Endoplasmic Reticulum Homeostasis. J Biol Chem. 2014;289:36059–36069. doi: 10.1074/jbc.M114.612515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Preissler S, Rato C, Chen R, Antrobus R, Ding S, Fearnley IM, Ron D. AMPylation Matches BiP Activity to Client Protein Load in the Endoplasmic Reticulum. eLife. 2015;4:e12621. doi: 10.7554/eLife.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Al-Eryani R, Yarbrough ML, Orth K, Ball HL. Characterization of AMPylation on Threonine, Serine, and Tyrosine Using Mass Spectrometry. J Am Soc Mass Spectrom. 2011;22:752–761. doi: 10.1007/s13361-011-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pieles K, Glatter T, Harms A, Schmidt A, Dehio C. An Experimental Strategy for the Identification of AMPylation Targets from Complex Protein Samples. Proteomics. 2014;14:1048–1052. doi: 10.1002/pmic.201300470. [DOI] [PubMed] [Google Scholar]
- 18.Grammel M, Luong P, Orth K, Hang HC. A Chemical Reporter for Protein AMPylation. J Am Chem Soc. 2011;133:17103–17105. doi: 10.1021/ja205137d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Truttmann MC, Cruz VE, Guo X, Engert C, Schwartz TU, Ploegh HL. The Caenorhabditis elegans Protein FIC-1 Is an AMPylase That Covalently Modifies Heat-Shock 70 Family Proteins, Translation Elongation Factors and Histones. PLoS Genet. 2016;12:e1006023. doi: 10.1371/journal.pgen.1006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewallen DM, Steckler CJ, Knuckley B, Chalmers MJ, Thompson PR. Probing Adenylation: Using a Fluorescently Labelled ATP Probe to Directly Label and Immunoprecipitate VopS Substrates. Mol BioSyst. 2012;8:1701–1706. doi: 10.1039/c2mb25053e. [DOI] [PubMed] [Google Scholar]
- 21.Yu X, Woolery AR, Luong P, Hao YH, Grammel M, Westcott N, Park J, Wang J, Bian X, Demirkan G, et al. Copper-Catalyzed Azide-Alkyne Cycloaddition (Click Chemistry)-Based Detection of Global Pathogen-host AMPylation on Self-assembled Human Protein Microarrays. Mol Cell Proteomics. 2014;13:3164–3167. doi: 10.1074/mcp.M114.041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewallen DM, Sreelatha A, Dharmarajan V, Madoux F, Chase P, Griffin PR, Orth K, Hodder P, Thompson PR. Inhibiting AMPylation: a Novel Screen to Identify the First Small Molecule Inhibitors of Protein AMPylation. ACS Chem Biol. 2014;9:433–442. doi: 10.1021/cb4006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller MP, Peters H, Blümer J, Blankenfeldt W, Goody RS, Itzen A. The Legionella Effector Protein DrrA AMPylates the Membrane Traffic Regulator Rab1b. Science. 2010;329:946–949. doi: 10.1126/science.1192276. [DOI] [PubMed] [Google Scholar]
- 24.Jiang P, Mayo AE, Ninfa AJ. Escherichia coli Glutamine Synthetase Adenylyltransferase (ATase, EC 2.7.7. 49): Kinetic Characterization of Regulation by PII, PII-UMP, Glutamine, and R-Ketoglutarate†. Biochemistry. 2007;46:4133–4146. doi: 10.1021/bi0620510. [DOI] [PubMed] [Google Scholar]
- 25.Gavriljuk K, Schartner J, Itzen A, Goody RS, Gerwert K, Kötting C. Reaction Mechanism of Adenylyltransferase DrrA from Legionella pneumophila Elucidated by Time-Resolved Fourier Transform Infrared Spectroscopy. J Am Chem Soc. 2014;136:9338–9345. doi: 10.1021/ja501496d. [DOI] [PubMed] [Google Scholar]
- 26.Utsumi R, Nakamoto Y, Kawamukai M, Himeno M, Komano T. Involvement of Cyclc AMP and Its Receptor Protein in Filamentation of an Escherichia coli fic Mutant. J Bacteriol. 1982;151:807–812. doi: 10.1128/jb.151.2.807-812.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harms A, Stanger FV, Dehio C. Biological Diversity and Molecular Plasticity of FIC Domain Proteins. Annu Rev Microbiol. 2016;70:341–360. doi: 10.1146/annurev-micro-102215-095245. [DOI] [PubMed] [Google Scholar]
- 28.Kinch LN, Yarbrough ML, Orth K, Grishin NV. Fido, a Novel AMPylation Domain Common to Fic, Doc, and AvrB. PLoS One. 2009;4:e5818. doi: 10.1371/journal.pone.0005818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das D, Krishna SS, McMullan D, Miller MD, Xu Q, Abdubek P, Acosta C, Astakhova T, Axelrod HL, Burra P, et al. Crystal Structure of the Fic (Filamentation induced by cAMP) Family Protein SO4266 (gi|24375750) from Shewanella oneidensis MR-1 at 1.6 A Resolution. Proteins: Struct, Funct Genet. 2009;75:264–271. doi: 10.1002/prot.22338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engel P, Goepfert A, Stanger FV, Harms A, Schmidt A, Schirmer T, Dehio C. Adenylylation Control by Intra- or Intermolecular Active-site Obstruction in Fic Proteins. Nature. 2012;482:107–110. doi: 10.1038/nature10729. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Pino A, Zenkin N, Loris R. The Many Faces of Fic: Structural and Functional Aspects of Fic Enzymes. Trends Biochem Sci. 2014;39:121–129. doi: 10.1016/j.tibs.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Pino A, Christensen-Dalsgaard M, Wyns L, Yarmolinsky M, Magnuson RD, Gerdes K, Loris R. Doc of Prophage P1 Is Inhibited by Its Antitoxin Partner Phd through Fold Complementation. J Biol Chem. 2008;283:30821–30827. doi: 10.1074/jbc.M805654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khater S, Mohanty D. Deciphering the Molecular Basis of Functional Divergence in AMPylating Enzymes by Molecular Dynamics Simulations and Structure Guided Phylogeny. Biochemistry. 2015;54:5209–5224. doi: 10.1021/acs.biochem.5b00351. [DOI] [PubMed] [Google Scholar]
- 35.Palanivelu DV, Goepfert A, Meury M, Guye P, Dehio C, Schirmer T. Fic Domain-Catalyzed Adenylylation: Insight Provided by the Structural Analysis of the Type IV Secretion System Effector BepA. Protein Sci. 2011;20:492–499. doi: 10.1002/pro.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harms A, Stanger FV, Scheu PD, de Jong IG, Goepfert A, Glatter T, Gerdes K, Schirmer T, Dehio C. Adenylylation of Gyrase and Topo IV by FicT Toxins Disrupts Bacterial DNA Topology. Cell Rep. 2015;12:1497–1507. doi: 10.1016/j.celrep.2015.07.056. [DOI] [PubMed] [Google Scholar]
- 37.Lu C, Nakayasu ES, Zhang L, Luo Z. Identification of Fic-1 as an Enzyme that Inhibits Bacterial DNA Replication by AMPylating GyrB, Promoting Filament Formation. Sci Signaling. 2016;9:ra11. doi: 10.1126/scisignal.aad0446. [DOI] [PubMed] [Google Scholar]
- 38.Engel P, Salzburger W, Liesch M, Chang CC, Maruyama S, Lanz C, Calteau A, Lajus A, Medigue C, Schuster SC, et al. Parallel Evolution of a Type IV Secretion System in Radiating Lineages of the Host-Restricted Bacterial Pathogen Bartonella. PLoS Genet. 2011;7:e1001296. doi: 10.1371/journal.pgen.1001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bunney TD, Cole AR, Broncel M, Esposito D, Tate EW, Katan M. Crystal Structure of the Human, FIC-Domain Containing Protein HYPE and Implications for its Functions. Structure. 2014;22:1831–1843. doi: 10.1016/j.str.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goepfert A, Stanger FV, Dehio C, Schirmer T. Conserved Inhibitory Mechanism and Competent ATP Binding Mode for Adenylyltransferases with Fic Fold. PLoS One. 2013;8:e64901. doi: 10.1371/journal.pone.0064901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welner D, Dedic E, van Leeuwen HC, Kuijper E, Bjerrum MJ, Ostergaard O, Jorgensen R. Protein Expression, Characterization, Crystallization and Preliminary X-ray Crystallographic Analysis of a Fic Protein from Clostridium difficile. Acta Crystallogr, Sect F: Struct Biol Commun. 2014;70:827–831. doi: 10.1107/S2053230X1400987X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dedic E, Alsarraf H, Welner DH, Ostergaard O, Klychnikov OI, Hensbergen PJ, Corver J, van Leeuwen HC, Jorgensen R. A Novel Fic (Filamentation Induced by cAMP) Protein from Clostridium difficile Reveals an Inhibitory Motif-independent Adenylylation/AMPylation Mechanism. J Biol Chem. 2016;291:13286–13300. doi: 10.1074/jbc.M115.705491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanger FV, Burmann BM, Harms A, Aragao H, Mazur A, Sharpe T, Dehio C, Hiller S, Schirmer T. Intrinsic Regulation of FIC-Domain AMP-transferases by Oligomerization and Automodification. Proc Natl Acad Sci U S A. 2016;113:E529–E537. doi: 10.1073/pnas.1516930113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanyal A, Chen AJ, Nakayasu ES, Lazar CS, Zbornik EA, Worby CA, Koller A, Mattoo S. A Novel Link between Fic (Filamentation Induced by cAMP)- mediated Adenylylation/AMPylation and the Unfolded Protein Response. J Biol Chem. 2015;290:8482–8499. doi: 10.1074/jbc.M114.618348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll P, Pashley CA, Parish T. Functional Analysis of GlnE, an Essential Adenylyl Transferase in Mycobacterium tuberculosis. J Bacteriol. 2008;190:4894–4902. doi: 10.1128/JB.00166-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Zhang R, Joachimiak A, Carr PD, Huber T, Vasudevan SG, Ollis DL. Structure of the N-Terminal Domain of Escherichia coli Glutamine Synthetase Adenylyltransferase. Structure. 2004;12:861–869. doi: 10.1016/j.str.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 47.Jaggi R, van Heeswijk C, Westerhoff HV, Ollis DL, Vasudevan SG. The Two Opposing Activities of Adenylyl Transferase Reside in Distinct Homologous Domains, with Intra-molecular Signal Transduction. EMBO J. 1997;16:5562–5571. doi: 10.1093/emboj/16.18.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson WB, Stadtman ER. Glutamine Synthetase Deadenylylation: A Phosphorolytic Reaction Yielding ADP as Nucleotide Product. Biochem Biophys Res Commun. 1970;41:704–709. doi: 10.1016/0006-291x(70)90070-7. [DOI] [PubMed] [Google Scholar]
- 49.Son HS, Rhee SG. Cascade Control of Escherichia coli Glutamine Synthetase Purification and Properties of PII Protein and Nucleotide Sequene of Its Structural Gene. J Biol Chem. 1987;262:8690–8695. [PubMed] [Google Scholar]
- 50.Jaggi R, Ybarlucea W, Cheah E, Carr PD, Edwards KJ, Ollis DL, Vasudevan SG. The Role of the T-loop of the Signal Transducing Protein PII from Escherichia coli. FEBS Lett. 1996;391:223–228. doi: 10.1016/0014-5793(96)00737-5. [DOI] [PubMed] [Google Scholar]
- 51.Itzen A, Blankenfeldt W, Goody RS. Adenylylation: Renaissance of a Forgotten Post-Translational Modification. Trends Biochem Sci. 2011;36:221–228. doi: 10.1016/j.tibs.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Kawamukai M, Matsuda H, Fujii W, Nishida T, Izumoto Y, Himeno M, Utsumi R, Komano T. Cloning of the fic-1 Gene Involved in Cell Filamentation Induced by Cyclic AMP and Construction of a Δfic Escherichia coli Strain. J Bacteriol. 1988;170:3864–3869. doi: 10.1128/jb.170.9.3864-3869.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komano T, Utsumi R, Kawamukai M. Functional Analysis of the fic Gene Involved in Regulation of Cell Division. Res Microbiol. 1991;142:269–277. doi: 10.1016/0923-2508(91)90040-h. [DOI] [PubMed] [Google Scholar]
- 54.Mishra S, Bhagavat R, Chandra N, Vijayarangan N, Rajeswari H, Ajitkumar P. Cloning, Expression, Purification, and Biochemical Characterisation of the FIC Motif Containing Protein of Mycobacterium tuberculosis. Protein Expression Purif. 2012;86:58–67. doi: 10.1016/j.pep.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 55.Darby AC, Gill AC, Armstrong SD, Hartley CS, Xia D, Wastling JM, Makepeace BL. Integrated Transcriptomic and Proteomic Analysis of the Global Response of Wolbachia to Doxycycline-induced Stress. ISME J. 2014;8:925–937. doi: 10.1038/ismej.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alto NM, Orth K. Subversion of Cell Signaling by Pathogens. Cold Spring Harbor Perspect Biol. 2012;4:a006114. doi: 10.1101/cshperspect.a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Souza Santos M, Orth K. Subversion of the Cytoskeleton by Intracellular Bacteria: Lessons from Listeria, Salmonella and Vibrio. Cell Microbiol. 2015;17:164–173. doi: 10.1111/cmi.12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zekarias B, Mattoo S, Worby C, Lehmann J, Rosenbusch RF, Corbeil LB. Histophilus somni IbpA DR2/Fic in Virulence and Immunoprotection at the Natural Host Alveolar Epithelial Barrier. Infect Immun. 2010;78:1850–1858. doi: 10.1128/IAI.01277-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin SW, Harland RJ, Bateman KG, Nagy E. The Association of Titers to Haemophilus somnus, and other Putative Pathogens, with the Occurrence of Bovine Respiratory Disease and Weight Gain in Feedlot Calves. Can J Vet Res. 1998;62:262–267. [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao J, Worby CA, Mattoo S, Sankaran B, Dixon JE. Structural Basis of Fic-Mediated Adenylylation. Nat Struct Mol Biol. 2010;17:1004–1010. doi: 10.1038/nsmb.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siamer S, Dehio C. New Insights into the Role of Bartonella Effector Proteins in Pathogenesis. Curr Opin Microbiol. 2015;23:80–85. doi: 10.1016/j.mib.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Isberg RR, O’Connor T, Heidtman M. The Legionella pneumophila Replication Vacuole: Making a Cozy Niche Inside Host Cells. Nat Rev Microbiol. 2009;7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Treacy-Abarca S, Mukherjee S. Legionella Suppresses the Host Unfolded Protein Response via Multiple Mechanisms. Nat Commun. 2015;6:7887. doi: 10.1038/ncomms8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luitz MP, Bomblies R, Ramcke E, Itzen A, Zacharias M. Adenylylation of Tyr77 Stabilizes Rab1b GTPase in an Active State: A Molecular Dynamics Simulation Analysis. Sci Rep. 2016;6:19896. doi: 10.1038/srep19896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michard C, Doublet P. Post-translational Modifications are Key Players of the Legionella pneumophila Infection Strategy. Front Microbiol. 2015;6:87. doi: 10.3389/fmicb.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hardiman CA, Roy CR. AMPylation Is Critical for Rab1 Localization to Vacuoles Containing Legionella pneumophila. mBio. 2014;5:e01035–13. doi: 10.1128/mBio.01035-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukherjee S, Liu X, Arasaki K, McDonough J, Galan JE, Roy CR. Modulation of Rab GTPase Function by a Protein Phosphocholine Transferase. Nature. 2011;477:103–106. doi: 10.1038/nature10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Campanacci V, Mukherjee S, Roy CR, Cherfils J. Structure of the Legionella Effector AnkX Reveals the Mechanism of Phosphocholine Transfer by the FIC Domain. EMBO J. 2013;32:1469–1477. doi: 10.1038/emboj.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Tascon I, Neunuebel MR, Pallara C, Brady J, Kinch LN, Fernandez-Recio J, Rojas AL, Machner MP, Hierro A. Structural Basis for Rab1 De-AMPylation by the Legionella pneumophila Effector SidD. PLoS Pathog. 2013;9:e1003382. doi: 10.1371/journal.ppat.1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neunuebel MR, Chen Y, Gaspar AH, Backlund PS, Yergey A, Machner MP. De-AMPylation of the Small GTPase Rab1 by the Pathogen Legionella pneumophila. Science. 2011;333:453–456. doi: 10.1126/science.1207193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng F, Yang F, Rong W, Wu X, Zhang J, Chen S, He C, Zhou JM. A Xanthomonas Uridine 5′-Monophosphate Transferase Inhibits Plant Immune Kinases. Nature. 2012;485:114–118. doi: 10.1038/nature10962. [DOI] [PubMed] [Google Scholar]
- 72.Desveaux D, Singer AU, Wu AJ, McNulty BC, Musselwhite L, Nimchuk Z, Sondek J, Dangl JL. Type III Effector Activation via Nucleotide Binding, Phosphorylation, and Host Target Interaction. PLoS Pathog. 2007;3:e48. doi: 10.1371/journal.ppat.0030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castro-Roa D, Garcia-Pino A, De Gieter S, van Nuland NA, Loris R, Zenkin N. The Fic Protein Doc uses an Inverted Substrate to Phosphorylate and Inactivate EF-Tu. Nat Chem Biol. 2013;9:811–817. doi: 10.1038/nchembio.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin Interacts with a Family of WW Domain Proteins. Hum Mol Genet. 1998;7:1463–1474. doi: 10.1093/hmg/7.9.1463. [DOI] [PubMed] [Google Scholar]
- 75.Rahman M, Ham H, Liu X, Sugiura Y, Orth K, Kramer H. Visual Neurotransmission in Drosophila Requires Expression of Fic in Glial Capitate Projections. Nat Neurosci. 2012;15:871–875. doi: 10.1038/nn.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Truttmann MC, Wu Q, Stiegeler S, Duarte JN, Ingram J, Ploegh HL. HypE-Specific Nanobodies as Tools to Modulate HypE-Mediated Target AMPylation. J Biol Chem. 2015;290:9087–9100. doi: 10.1074/jbc.M114.634287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Broncel M, Serwa RA, Bunney TD, Katan M, Tate EW. Global profiling of HYPE Mediated AMPylation through a Chemical Proteomic Approach. Mol Cell Proteomics. 2016;15:715–725. doi: 10.1074/mcp.O115.054429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Preissler S, Chambers JE, Crespillo-Casado A, Avezov E, Miranda E, Perez J, Hendershot LM, Harding HP, Ron D. Physiological Modulation of BiP Activity by Trans-Protomer Engagement of the Interdomain Linker. eLife. 2015;4:e08961. doi: 10.7554/eLife.08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harbor Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hegde NR, Chevalier MS, Wisner TW, Denton MC, Shire K, Frappier L, Johnson DC. The Role of BiP in Endoplasmic Reticulum-associated Degradation of Major Histocompatibility Complex Class I Heavy Chain Induced by Cytomegalovirus Proteins. J Biol Chem. 2006;281:20910–20919. doi: 10.1074/jbc.M602989200. [DOI] [PubMed] [Google Scholar]
- 81.Wang J, Pareja KA, Kaiser CA, Sevier CS. Redox Signaling via the Molecular Chaperone BiP Protects Cells Against Endoplasmic Reticulum-Derived Oxidative Stress. eLife. 2014;3:e03496. doi: 10.7554/eLife.03496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Godin JD, Creppe C, Laguesse S, Nguyen L. Emerging Roles for the Unfolded Protein Response in the Developing Nervous System. Trends Neurosci. 2016;39:394–404. doi: 10.1016/j.tins.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 83.Truttmann MC, Zheng X, Hanke L, Damon JR, Grootveld M, Krakowiak J, Pincus D, Ploegh HL. Unrestrained AMPylation Targets Cytosolic Chaperones and Activates the Heat Shock Response. Proc Natl Acad Sci U S A. 2017;114:E152–E160. doi: 10.1073/pnas.1619234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Preissler S, Rato C, Perera LA, Saudek V, Ron D. FICD acts Bifunctionally to AMPylate and de-AMPylate the Endoplasmic Reticulum Chaperone BiP. Nat Struct Mol Biol. 2017;24:23–29. doi: 10.1038/nsmb.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Y, Chang A. Heat Shock Response Relieves ER Stress. EMBO J. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weindling E, Bar-Nun S. Sir2 Links the Unfolded Protein Response and the Heat Shock Response in a Stress Response Network. Biochem Biophys Res Commun. 2015;457:473–478. doi: 10.1016/j.bbrc.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 87.Rhee SG, Bang WG, Koo JH, Min KH, Park SC. In: Enzyme dynamics and regulation. Chock PB, Huang CY, Tsou CL, Wang JH, editors. Springer-Verlag; Berlin: 1988. [Google Scholar]
- 88.Casey A, Moehlman A, Zhang J, Servage K, Kramer H, Orth K. Fic-mediated deAMPylation is not Dependent on Homodimerization and Rescues Toxic AMPylation in Flies. J Biol Chem. doi: 10.1074/jbc.M117.799296. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]