

Abstract
Hybrid incompatibilities play a critical role in the evolution and maintenance of species. We have discovered a simple genetic incompatibility that causes lethality in hybrids between two closely related species of yellow monkeyflower (Mimulus guttatus and M. nasutus). This hybrid incompatibility, which causes one sixteenth of F2 hybrid seedlings to lack chlorophyll and die shortly after germination, occurs between sympatric populations that are connected by ongoing interspecific gene flow. Using complimentary genetic mapping and gene expression analyses, we show that lethality occurs in hybrids that lack a functional copy of the critical photosynthetic gene pTAC14. In M. guttatus, this gene was duplicated, but the ancestral copy is no longer expressed. In M. nasutus, the duplication is missing altogether. As a result, hybrids die when they are homozygous for the nonfunctional M. guttatus copy and missing the duplicate from M. nasutus, apparently due to misregulated transcription of key photosynthetic genes. Our study indicates that neutral evolutionary processes may play an important role in the evolution of hybrid incompatibilities and opens the door to direct investigations of their contribution to reproductive isolation among naturally hybridizing species.
Author summary
The evolution of hybrid incompatibilities (gene interactions that cause hybrids to be sterile or inviable) is a common outcome of genomic divergence between lineages. However, evaluating the importance of hybrid incompatibilities for speciation requires that we identify the causal genes and evolutionary forces in recently diverged, wild species. We discovered that hybrid seedling lethality between two closely related sister species of yellow monkeyflower is caused by duplicate copies of a gene critical for chloroplast development. Because each lineage carries its one functional gene copy in a distinct genomic location, some hybrids inherit only inactive (or missing) alleles. We infer that hybrid lethality in this young species pair has arisen through divergent resolution of gene duplicates by degenerative mutations and (likely) genetic drift. These findings are an important step toward understanding the evolutionary dynamics of hybrid incompatibility genes in nature, as well as the role of such genes in species divergence.
Introduction
Across diverse taxa, hybrid incompatibilities arise as a byproduct of genetic divergence among incipient species. The basic genetic underpinnings of this process are well understood: two or more mutational differences between species interact epistatically to cause hybrid inviability or sterility [1–3]. However, what is less clear, and often very challenging to uncover, is the nature of the molecular changes and evolutionary forces that lead to hybrid incompatibilities. What sort of mutations are perfectly functional within species but cause reproductive failure or death in hybrids? Do such mutations accumulate within species by neutral processes or are they positively selected, perhaps providing an ecological advantage or resolving an intragenomic conflict? Addressing the first of these questions is most straightforward in systems with well-developed genetic tools that facilitate positional cloning, which explains why most progress has been made in traditional models like Drosophila, Arabidopsis, and rice. However, insight into the evolutionary forces acting on hybrid incompatibilities during the speciation process requires a focus on young species pairs with natural populations.
Over the past two decades, genetic dissection of diverse incompatibilities has provided some hints about their evolutionary origins (reviewed in [4–7]). Often, hybrid incompatibility genes show molecular signatures of positive selection [8–13], and there is suggestive evidence that incompatibility alleles can arise through ecological adaptation [14–16] or recurrent bouts of intragenomic conflict [11, 17–19]. On the other hand, there is also evidence from a handful of cases, all involving gene duplicates, that the evolution of hybrid dysfunction need not involve natural selection [20–22].
The idea that gene duplication might play a key role in hybrid incompatibilities was initially proposed by Muller as a variant of his original model [3]. He explained how gene duplication, followed by degenerative mutations and divergent copy loss, could lead to a difference in gene position between species with missing (or inactive) copies acting as recessive incompatibility alleles [3]. This same scenario was emphasized later as an explanation for defects in pollen development between subspecies of Asian cultivated rice, Oryza sativa [23], and more recently, as a general mechanism of hybrid breakdown via neutral processes [24, 25]. There have now been three empirical demonstrations of gene duplication/transposition giving rise to interspecific hybrid male sterility; one case involves Drosophila melanogaster-D. simulans hybrids [26] and the other two arise from crosses between O. sativa and wild species [21, 22]. Gene duplication also causes lethal and sterile combinations that segregate within Arabidopsis thaliana [27, 28] and O. sativa [20]. However, it is not yet clear whether divergent resolution of gene duplicates contributes to hybrid incompatibilities between wild species in the early stages of divergence. Only by identifying examples in young species pairs, particularly those with sympatric populations and still connected by some degree of gene flow, will it be possible to evaluate the contribution of such loci to speciation.
In this study we investigate the molecular genetic basis of hybrid seedling lethality between two closely related sister species of yellow monkeyflower, Mimulus guttatus and M. nasutus. These recently diverged species (200-500kya; [29]) co-occur throughout much of their shared range in western North America, where reproductive isolation between sympatric populations occurs through a number of prezygotic [30–33] and postzygotic barriers [34–40]. Despite substantial reproductive isolation, patterns of shared variation across their genomes indicate historical and ongoing gene flow between the two species [29, 31, 41]. Here we focus on sympatric populations of M. guttatus and M. nasutus located at Don Pedro Reservoir (DPR) in central California, where both species coexist within centimeters of one another. Species at DPR are strongly isolated by divergence in flowering time and mating system [33]; nevertheless, studies have shown low levels of hybridization [33] and a clear signal of introgression [29]. Using high-resolution genetic mapping and genome-wide expression analyses we identify a duplicate gene pair as the cause of Mimulus hybrid lethality. As the first case of hybrid incompatibility genes identified between naturally hybridizing species, this study opens the door to direct investigations of their evolutionary dynamics and contribution to reproductive isolation.
Results
Hybrid lethality is caused by a two-locus incompatibility
Hybrid lethality occurs in the hybrid progeny of M. guttatus and M. nasutus from the sympatric DPR site and is easily characterized by seedlings that completely lack chlorophyll (white seedlings, see S1 Fig). As a first step toward investigating the genetic basis of this phenotype, we self-fertilized and intercrossed the inbred lines DPR102-gutt (M. guttatus) and DPR104-nas (M. nasutus). We then examined phenotypic ratios of white and green seedlings among their selfed progeny and reciprocal F1 and F2 hybrids (S1 Fig, S1 Table). Although we never observed white seedlings in the selfed progeny of parental lines or in F1 hybrids, we discovered that roughly 1/16 of F2 hybrid seedlings were white (maternal parent listed first: DPR102-gutt x DPR104-nas, N = 516, 7.36% white seedlings; DPR104-nas x DPR102-gutt, N = 661, 5.75% white seedlings). Segregation of white seedlings in reciprocal F2 hybrids suggests a nuclear, rather than cyto-nuclear, genetic incompatibility. Chi-squared tests rejected several genetic models that could potentially explain the observed phenotypic ratios, but could not reject a two-locus model involving only recessive alleles in either F2 population, or when their ratios were combined (S1 Table). These results suggest that hybrid lethality between sympatric M. guttatus and M. nasutus is caused by a two-locus, recessive-recessive hybrid incompatibility.
To genetically map Mimulus hybrid lethality, we performed two rounds of bulked segregant analysis (BSA). In the first round, we pooled DNA from green and white F2 seedlings into eight separate tubes (six individuals per pool, four replicates each for green and white). Because incompatibility alleles act recessively, our expectation was that pooled white seedlings should be homozygous (for either DPR102-gutt or DPR104-nas alleles) at markers linked to hybrid lethality loci, whereas green seedlings should segregate 1:2:1 (for DPR102-gutt homozygotes: heterozygotes: DPR104-nas homozygotes). Of the 126 size-polymorphic markers (spanning much of the Mimulus genome) that we used for genotyping, four showed an association with seedling phenotype: the four tubes with white seedlings carried only (or mostly) DPR104-nas alleles, whereas green seedlings carried both parental alleles. All four markers map to a region of roughly 40 cM on linkage group 14 (inferred by marker position in [35]), which we named hybrid lethal 14 (hl14).
To identify the partner locus, we performed a second round of BSA controlling for genotype at hl14. We generated 60 F3 families by self-fertilizing green F2 hybrids that were homozygous for DPR104-nas alleles at hl14 (determined by genotyping flanking markers); these F3 families segregated green and white seedlings in ratios of either 3:1 or 1:0. We reasoned that if hybrid lethality is caused by hl14 and a single interacting locus, white F3 hybrids should be homozygous for DPR102-gutt alleles at the partner, whereas green F3 families that do not segregate white seedlings should be homozygous for DPR104-nas alleles. Based on this logic, we formed two separate pools of DNA from F3 hybrids: one with 34 white seedlings and one with 26 green seedlings from non-segregating families. Note that each F3 seedling was derived from a different family (i.e., from a unique F2 maternal parent) so that at markers unlinked to hybrid lethality, both pools should carry each of the two parental alleles at ~50% frequency. For the two pools, we performed whole genome sequencing, generated a genome-wide SNP dataset, and calculated average allele frequency difference in 200-SNP sliding windows (100-SNP overlap between windows). Using this approach, we discovered that the top 5% most divergent windows were located in contiguous windows along the distal end of chromosome 13 (S2 Fig), which we named hybrid lethal 13 (hl13).
To fine-map hl13 and hl14, we generated a large DPR104-nas x DPR102-gutt F2 mapping population, oversampling white seedlings to roughly equalize frequencies of the two phenotypes (white = 44%, green = 56%, N = 2,652). Each F2 individual was genotyped with two pairs of markers targeted just outside of the hybrid lethality loci: M208 and M236 at hl13, and M241 and M132 hl14. As expected, nearly all white seedlings were homozygous for DPR102-gutt alleles at the hl13-linked markers and homozygous for DPR104-nas alleles at the hl14 markers (92%, N = 1174), whereas green seedlings never carried this genotype (N = 1478) (Fig 1). Because we later discovered that both M208 and M236 are proximal to hl13, an additional 2,182 F2 hybrids were screened with one of two more distal markers (M263 or M255) that allowed us to flank the locus. We genotyped informative recombinants at additional size-polymorphic and SNP-based markers designed in each interval. Although white seedlings must be destructively sampled for DNA, green seedlings were allowed to grow into adult plants so that informative recombinants could be self-fertilized and phenotyped via progeny testing. In this way, we determined if green F2 hybrids were heterozygous for hl13 and/or hl14 (versus homozygous for compatible alleles). Using this strategy, we mapped the hl13 locus to a 72.2 kb-region at the distal end of chromosome 13 that contains 24 genes (Fig 2A). At the same time, we mapped the hl14 locus to a 51.6 kb-region of chromosome 14 that contains six genes, as well as a gap of unknown size in the M. guttatus IM62 reference genome (Fig 2B). For each hl13 and hl14 candidate gene, we identified its top blast hit(s) in Arabidopsis thaliana, gene ontology terms, known mutant phenotypes, and predicted functions (S2 Table).
Fig 1. Two-locus genotypes of green and white seedlings at markers linked to hl13 and hl14.
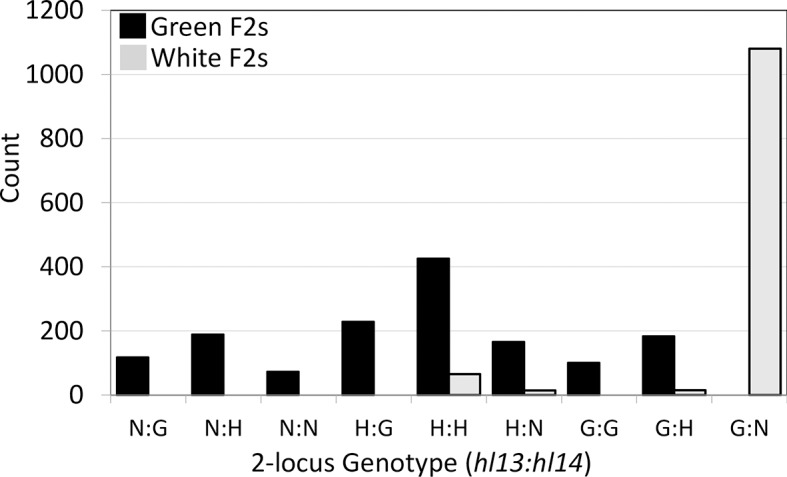
Count represents the number of F2s with a given genotype. Genotypes are homozygous M. guttatus (‘G’), homozygous M. nasutus (‘N’), and heterozygous (‘H’). The vast majority (92%) of white F2s carry the G:N genotype, whereas green F2s carry all genotypes except G:N. Sample size is 2,652, which represents the subset of our mapping population that was genotyped with markers M208, M236, M241, and M132.
Fig 2. Fine-mapping localizes hybrid lethality loci to small genomic intervals.

(A) hl13 maps to a 72kb region on scaffold 115 containing 22 genes. (B) hl14 maps to a 51.6kb region on scaffold 291 containing 6 genes and a gap in the reference genome (red box) of unknown size. Horizontal bars represent F2 recombinants that were informative for mapping hl13 (between M208 and M255) and hl14 (between M280 and M241), where marker genotypes are yellow (homozygous for DPR102-gutt alleles), blue (homozygous for DPR104-nas alleles), and green (heterozygous). F2 individual used for mapping, F2 phenotype (white or green), genotype at partner locus (DPR102-gutt, DPR104-nas, or heterozygous), and green:white ratio of F3 progeny are given for each recombinant. Vertical and diagonal hatch marks are marker positions and breaks within/between scaffolds, respectively. Scaffolds from v1.1 of the reference genome are included in figure, though all gene annotation and naming is based on the updated v2.0 assembly (phytozome.org).
Gene duplicates map to hybrid lethality loci
In the hl13 interval, we discovered several strong functional candidates for hybrid lethality including Migut.M02023, a homolog of pTAC14 (PLASTID TRANSCRIPTIONALLY ACTIVE CHROMOSOME 14). In A. thaliana, pTAC14 is essential for proper chloroplast development and mutants show a chlorotic lethal phenotype [42] that appears identical to DPR102-gutt x DPR104-nas F2 hybrid lethality. In addition to Migut.M02023 on chromosome 13, we also identified a highly similar and slightly truncated protein homolog of pTAC14 (99.1% amino acid similarity along length of truncated homolog), Migut.O00467, located on an unmapped scaffold of the IM62 M. guttatus reference genome (v2.0 scaffold_193). To investigate the possibility that this additional copy of Mg.pTAC14 resides on chromosome 14, we turned to several large-insert IM62 genomic libraries (six fosmid and two BAC libraries) that were generated and end-sequenced as part of the reference genome assembly effort [43]. Among these libraries, only a single end-sequence of one fosmid blasts to v2.0 scaffold_193. Intriguingly, the other end-sequence of this same fosmid blasts to the first exon of Migut.N01489, a gene within the mapped interval of hl14. This finding provides evidence that a second copy of Mg.pTAC14 is located on chromosome 14 in IM62, despite it being absent from the current genome assembly. Using PCR, we confirmed that the DPR102-gutt genome also contains two copies of pTAC14 (S3 Fig). However, despite exhaustive PCR and cloning efforts (using many different primer combinations), we recovered only one copy of pTAC14 from DPR104-nas genomic DNA.
To determine if Mimulus pTAC14 duplicates genetically map to hybrid lethality loci, we obtained a set of 96 DPR102-gutt x DPR104-nas F2 hybrids carrying each of the nine possible two-locus genotypes at hl13 and hl14 (10 replicates for each green genotype and 16 replicates of the white seedling genotype, see Fig 3). Using a set of conserved primers spanning exons 6–8, we PCR-amplified and sequenced Mimulus pTAC14 from each of these F2 hybrids. Across this region, 10 SNPs define three distinct haplotypes of pTAC14: “G1” and “G2” from DPR102-gutt and “N1” DPR104-nas (Fig 3). Remarkably, we discovered a perfect association between pTAC14 haplotype and hl13/hl14 genotype: G1 is present in all individuals with DPR102-gutt alleles at hl13, G2 is in all individuals with DPR102-gutt alleles at hl14, and N1 is in all individuals with DPR104-nas alleles at hl13. From this pattern, we infer that both DPR102-gutt and DPR104-nas carry copies of pTAC14 at hl13 (hereafter referred to as Mg.pTAC14_1 and Mn.pTAC14_1, respectively), but that only DPR102-gutt carries a copy at hl14 (referred to as Mg.pTAC14_2).
Fig 3. Duplicate copies of pTAC14 map to both hybrid lethality loci in M. guttatus.

(A) Ten SNPs differentiate Mg.pTAC14_1 (G1), Mg.pTAC14_2 (G2), and Mnas.pTAC14 (N1). (B) Phenotype: green (GRN) or white (WHT) F2. hl13 and hl14: genotype at flanking markers [G (DPR102-gutt), N (DPR104-nas, and G/N (heterozygous)]. N is sample size. Observed: Copies of pTAC14 observed in each F2, consistent across all F2s for a given genotype. Note that individuals with DPR102-gutt alleles at hl13 always carry Mg.pTAC14_1, individuals with DPR102-gutt alleles at hl14 always carry Mg.pTAC14_2, and individuals with DPR104-nas alleles at hl13 always carry Mnas.pTAC14. (C) Location of different copies of pTAC14 based on our mapping experiment.
To examine sequence similarity among Mimulus pTAC14 genes, we obtained full-length genomic sequences from both DPR parents and generated a neighbor-joining tree (Fig 4, S3 Fig). As expected, chromosome 13 copies of pTAC14 from DPR102-gutt and IM62 cluster together (Mg.pTAC14_1 and Migut.M02023). Likewise, chromosome 14 copies of pTAC14 from DPR102-gutt and IM62 cluster together (Mg.pTAC14_2 and Migut.O00467). However, somewhat counterintuitively, the DPR104-nas copy (Mn.pTAC14_1), which is located on chromosome 13, clusters more closely with M. guttatus copies on chromosome 14 than with copies on chromosome 13 (Fig 4B).
Fig 4. pTAC14 gene structure and neighbor-joining tree.

(A) Gene model of pTAC14 in Mimulus is shown along the top. A frameshift mutation in the G1 copy of pTAC14 (DPR102-gutt, Mg.pTAC14_1) is caused by the insertion of an adenine in the 7th exon, highlighted with a box. (B) Unrooted neighbor-joining tree of pTAC14 genes from DPR102-gutt, DPR104-nas, and the IM62 reference genome. Bootstrap support given at node and substitution rate shown for scale.
Mimulus pTAC14 duplicates are nonfunctional in hybrid lethal seedlings
Consistent with a causal role for Mimulus pTAC14 duplicates in hybrid lethality, we discovered several lines of evidence that suggest only one of the two copies is functional in DPR102-gutt. First, Mg.pTAC14_1 (at hl13) contains a frameshift mutation in exon 7, which results in the production of numerous premature stop codons in downstream sequence (Fig 4A, S3 Fig). Second, from each inbred line, we PCR-amplified only a single copy of the gene from leaf cDNA: Mg.pTAC14_2 in DPR102-gutt and Mn.pTAC14_1 in DPR104-nas. Third, pTAC14 expression is nearly absent in white F2 hybrid seedlings, which inherit hl13 from DPR102-gutt (containing Mg.pTAC14_1) and hl14 from DPR104-nas (containing no copy of pTAC14). Using qPCR and primers that amplify both Mimulus pTAC14 duplicates, we found strong expression in green parental and F2 seedlings, but not in white F2 seedlings (qPCR on eight additional functional candidates in the hl13 and hl14 intervals showed no association between expression and seedling phenotype; S2 Table, S4 Fig). Additionally, we performed RNAseq on DPR102-gutt, DPR104-nas, green F2, and white F2 seedlings. Consensus sequences generated from de novo assemblies of DPR102-gutt reads that align to Migut.M02023 and/or Migut.O00467 (high sequence similarity between pTAC14 duplicates means that reads align equally well to both copies) correspond to Mg.pTAC14_2; consensus sequences generated in the same manner from DPR104-nas reads correspond to Mn.pTAC14_1. Moreover, RNAseq SNP variation in green F2 seedlings suggests they express only Mg.pTAC14_2 from DPR102-gutt and/or Mn.pTAC14_1 from DPR104-nas. In contrast, read coverage of pTAC14 transcripts in white F2 seedlings is exceptionally low: of the 1,092 genes that are significantly differentially expressed between white F2 seedlings and green seedlings (DPR102-gutt, DPR104-nas, green F2 hybrids), the duplicate copies of pTAC14 are the two most underexpressed (Fig 5). Taken together, these results provide strong evidence that Mimulus hybrid lethality is caused by nonfunctional pTAC14 duplicates: white hybrid seedlings carry unexpressed Mg.pTAC14_1 alleles at hl13 and are missing pTAC14 alleles altogether at hl14.
Fig 5. Genome-wide patterns of differential expression.

Average log2 fold-change among the 1,092 genes (representing 3% of annotated genes) that are significantly differentially expressed in all pairwise comparisons of white (F2) and green (DPR102-gutt, DPR104-nas, F2) seedlings. Genes above and below line are over- and underexpressed in white seedlings, respectively. Annotated pTAC14 duplicates are shown as red dots.
Genome-wide misexpression in hybrid lethal seedlings
Comparison of genome-wide RNAseq patterns among DPR102-gutt, DPR104-nas, green F2, and white F2 seedlings provides additional support for disrupted pTAC14 function as a cause of hybrid lethality. White F2 seedlings show a strong signature of genome-wide misexpression: of 27,948 annotated genes, 1,092 (3%) are significantly misexpressed in all three pairwise comparisons between white and green seedlings (Fig 5). Among transcripts that are underexpressed in white seedlings (N = 209), we found a significant enrichment of genes involved in photosynthesis and/or located within the thylakoid and photosynthetic membranes. Among overexpressed transcripts (N = 883), we observed an enrichment of heat shock proteins and glutathione peroxidase proteins (S3 Table). Furthermore, consistent with disrupted pTAC14 function, we discovered evidence for severe misexpression of chloroplast-encoded genes in white seedlings (Fig 6, S5 Fig). In A. thaliana, knockouts of pTAC14 disable the PEP (plastid-encoded bacterial type) RNA polymerase, which leads to reduced transcription of some chloroplast-encoded genes, particularly those involved in photosynthesis (e.g., photosystem I, photosystem II, and cytochrome b6f), and increased transcription of others such as the rpo genes [42]. Of 52 putative Mimulus chloroplast genes, those involved in photosynthesis, and thus likely to be transcribed by PEP RNA polymerase, were often significantly underexpressed in white F2 seedlings. In contrast, homologs of A. thaliana rpo genes were significantly overexpressed. Several additional putative chloroplast genes (e.g., ATP synthase, NADH Dehydrogenase, ribosomal proteins) that are likely transcribed by both PEP and the nuclear-encoded phage-type (NEP) RNA polymerase [44–50] were also significantly misexpressed (both up- and downregulated) in white seedlings. Taken together, these patterns of gene misexpression in Mimulus F2 white seedlings, which show a remarkable similarity to patterns observed in A. thaliana pTAC14 knockouts [42], provide additional support for a causal role of pTAC14 duplicates in Mimulus hybrid lethality.
Fig 6. Misexpression of chloroplast genome in white seedlings.

Heat map displaying expression patterns of 52 chloroplast genes from seven chloroplast gene families in seedlings from DPR102-gutt, DPR104-nas, Green F2s and White F2s. Z-scores of normalized FPKM values were calculated for each row to illustrate relative expression differences among genes. Bars on left of heatmap indicate whether genes are primarily transcribed by PEP, PEP and NEP, or NEP RNA polymerases. *denotes genes that are significantly differentially expressed between white seedlings and all green seedlings (DPR102-gutt, DPR104-nas, and Green F2s).
Discussion
Identifying the molecular genetic basis of hybrid incompatibilities between recently diverged, wild species is a critical first step toward understanding their evolutionary origins and role in speciation. We have shown that duplicate copies of Mimulus pTAC14, a gene critical for chloroplast development in A. thaliana [42], causes hybrid lethality between sympatric M. guttatus and M. nasutus at the DPR site. We fine-mapped hybrid lethality to hl13 and hl14, two small nuclear genomic regions on chromosomes 13 and 14. In DPR102-gutt (M. guttatus), pTAC14 is present in each of these genomic intervals, but only the hl14 copy is expressed. In DPR104-nas (M. nasutus), pTAC14 is present only in the hl13 interval, consistent with either of two possibilities: the hl13 copy is ancestral and this line lacks the duplication, or a large deletion has removed all trace of the gene from hl14. As a consequence of divergent resolution of these duplicate genes, F2 hybrids that are homozygous for DPR102-gutt alleles at hl13 and homozygous for DPRG102-nas alleles at hl14 contain no functional copy of Mimulus pTAC14. These hybrids fail to produce chlorophyll and die in the cotyledon stage of development, remarkably similar to what is observed in pTAC14 knockouts in A. thaliana [42]. To our knowledge, this is the first pair of hybrid incompatibility genes identified between naturally hybridizing species.
Using complementary genetic mapping and functional genomics approaches, our study provides strong evidence that nonfunctional Mimulus pTAC14 is the cause of hybrid lethality between DPR M. guttatus and M.nasutus. But what causes the lack of pTAC14 expression in white hybrid seedlings? In M. nasutus (DPR104-nas), because pTAC14 is missing entirely from the hl14 interval, the hl13 copy (Mn.pTAC14_1) is the only one expressed. In M. guttatus (DPR102-gutt), the situation is less clear. Although both copies (Mg.pTAC14_1 at hl13 and Mg.pTAC14_2 at hl14) are present and highly similar in exons (S3 Fig), our qPCR and RNAseq experiments demonstrate that only one of them–Mg.pTAC14_2 –is expressed. Further work will be required to determine the molecular nature of this change in gene expression. The most obvious possibility is that non-sense mediated decay has efficiently targeted Mg.pTAC14_1, which carries a series of premature stop codons. Another possibility, is that a cis-regulatory mutation disrupts Mg.pTAC14_1 transcription in DPR102-gutt. Alternatively, expression might be prevented by the epigenetic silencing of one duplicate by the other, as was recently shown for sterile and lethal combinations segregating within A. thaliana [28, 51]. Whatever its cause, disrupted expression is not the only problem with DPR102-gutt Mg.pTAC14_1; this gene copy also carries a 1-bp insertion that, if transcribed, would result in a truncated, and potentially nonfunctional, protein. We do not yet know which of these two functional changes to DPR102-gutt Mg.pTAC14_1 arose first.
The evolution of hybrid lethality in this system thus appears entirely consistent with a scenario of duplication and neutral non-functionalization within M. guttatus. Given the ubiquity of gene duplications in plant and animal genomes, divergent resolution of paralogs due to degenerative mutation and genetic drift has been proposed as a major source of hybrid incompatibilities [24, 25, 52]. Although initially redundant duplicate genes might sometimes evolve new or partial functions favored by selection [53], our study and others suggest that duplicates involved in hybrid incompatibilities are more often subject to mutations that disable function in one copy. Within A. thaliana and between closely related Oryza species, divergent resolution of duplicates has occurred through nonsense mutations [20, 22] and disruptions to expression [21, 27, 51]. In a more distantly related species pair of Drosophila, hybrid sterility is caused by a gene transposition, with degenerative mutations having presumably removed any remnant of the duplication that likely preceded its evolution [26]. Remarkably, then, to explain the evolution of hybrid dysfunction in Mimulus and several other diverse systems, there is no need to invoke processes beyond mutation and genetic drift.
In addition to showing that Mimulus hybrid lethality is due to nonfunctional pTAC14, our analyses have begun to provide some insight into the duplication history of this gene. As might be expected, within M. guttatus (DPR102-gutt and IM62), pTAC14 copies on chromosome 13 are most related and pTAC14 copies on chromosome 14 are most related (Fig 4). However, somewhat counterintuitively, pTAC14 from DPR104-nas, which is located on chromosome 13, is most closely related to the M. guttatus copies on chromosome 14. We interpret this finding, along with the fact that we find no trace of pTAC14_2 at hl14 in DPR104-nas, as evidence that Mimulus pTAC14_1 on chromosome 13 is the ancestral copy. Under this scenario, both the duplicate copy on chromosome 14 (Mg.pTAC14_2) and the M. nasutus copy on chromosome 13 (Mn.pTAC14_1) would have arisen from a similar genetic variant (Fig 7). Standing genetic variation within and between populations of M. guttatus is high [29, 54–57], so it is likely this ancestral populations carried multiple variants of pTAC14_1. Both the duplicated copy in M. guttatus (Mg.pTAC14_2) and the ancestral copy in the selfing M. nasutus (Mn.pTAC14_1) would be expected to carry only a small subset of ancestral variation. Unfortunately, we have not yet been able to assess molecular patterns of Mimulus pTAC14 variation in a wider sample of M. guttatus and M. nasutus. Although whole genome resequence data are available from a number of lines [29, 54, 57], short-read sequences of Mimulus pTAC14 align equally well to both annotated copies in the IM62 reference genome (Migut.M02023 and Migut.O00467). Once Mimulus pTAC14 is sequenced from a broader sample of individuals, we speculate that Mn.pTAC14_1 from M. nasutus and Mg.pTAC14_2 from M. guttatus will be included in distinct monophyletic groups nested within the greater diversity of sequences present at the ancestral Mg.pTAC14_1 from M. guttatus. Interestingly, white seedlings are often observed segregating at low frequencies within M. guttatus populations, which manifest as epistatic inbreeding depression [58, 59] that may be due to divergent resolution of duplicate genes similar to the ones characterized here. However, future studies investigating natural variation for functional and non-functional pTAC14 alleles within species will be required to determine whether the hl13-hl14 incompatibility plays a role in such phenotypes outside of the DPR population.
Fig 7. Hypothetical model for the evolution of pTAC14.

Prior to gene duplication the ancestral M. guttatus-like population harbored genetic variation for pTAC14 at hl13 (different colored boxes represent genetic variation at pTAC14). During speciation, M. nasutus acquired a copy of pTAC14 harboring ‘blue’ variation (left). Similarly, the M. guttatus-specific duplication involved a closely related ‘blue’ pTAC14 variant. Contemporary variation within M. nasutus and pTAC14 variants located at hl14 resemble one another genetically due to common ancestry, while pTAC14 variants at hl13 in M. guttatus likely continue to harbor considerable genetic variation, including variants that contain functional (IM62) and non-functional (DPR102-gutt) variants.
Our study provides the first detailed study of hybrid incompatibility genes from naturally hybridizing species and contributes to a growing body of literature that suggests hybrid incompatibilities might result from neutral evolutionary change within species. Going forward, it will be important to address whether these barriers can persist in the face of ongoing gene flow. Theoretical treatments of this question have consistently concluded that the maintenance of hybrid incompatibility alleles between hybridizing populations relies heavily on a selective advantage within species [60–64]. If so, neutrally evolving hybrid incompatibility alleles might be precluded from affecting reproductive isolation in any more than a transient fashion, with gene flow temporarily constrained until the hybrid incompatibility degrades with time. Nevertheless, other factors such as strong linkage to selected alleles (e.g., [16]) and constraints on gene dosage (e.g., [65]) may play an important role in such incompatibilities. By showing that the duplication of pTAC14 underlies hybrid lethality among sympatric Mimulus species, we now have a natural system in place to test broader questions regarding the evolutionary significance of hybrid incompatibilities in speciation.
Materials and methods
Mimulus lines and genetic crosses
In this study, we used inbred lines of M. guttatus and M. nasutus derived from wild individuals collected from Don Pedro Reservoir (DPR) in central California [33]. Wild-collected seed was sown on moist Fafard 3-B potting soil in 2.5” pots, cold-stratified in the dark at 4C for two weeks, and moved to the UGA greenhouses to germinate under 16 hour days at 23°C (growth conditions were constant across all experiments with the exception of plants used in RNAseq, which were grown in a growth chamber). Because M. nasutus is a selfer, the DPR104-nas line used in this study was naturally inbred. To generate the DPR102-gutt inbred line, we self-fertilized M. guttatus for three generations, which is expected to result in a genome that is 87.5% homozygous. The DPR102-gutt and DPR104-nas inbred lines were intercrossed to generate reciprocal F1 and F2 hybrids (maternal parent always listed first in crosses). To ensure that both inbred lines were homozygous at hl13 and hl14, we analyzed white:green seedling ratios in 32 F2 families (generated from distinct F1 hybrids, 16 from each cross direction); indeed, all F2 families segregated white seedlings at a frequency of one sixteenth, confirming homozygosity at both hybrid lethality loci.
Molecular analyses and whole genome sequencing
DNA was extracted from seedlings and adult leaf tissue using a standard CTAB-chloroform protocol [66] modified for use in a 96-well format. Genotyping was performed using a combination of exon-primed intron-spanning size polymorphic markers containing 5’ fluorescent tags (6-FAM or HEX) and SNP-containing gene fragments that were analyzed through Sanger sequencing. We designed size-polymorphic and SNP markers from polymorphisms observed in whole genome re-sequence data of multiple lines of M. guttatus and M. nasutus [29, 67, 68] and confirmed polymorphisms by genotyping parental lines used in our study. A standard touchdown PCR protocol was used in all amplifications and Sanger sequencing reactions were prepared using BigDye v3.1 mastermix (Applied Biosystems, Foster City, USA). Genotyping and Sanger sequencing reactions were run on an ABI3730XL automated DNA sequencer at the Georgia Genomics Facility and analyzed using GENEMARKER [69] and Sequencher (Gene Codes Corporation, Ann Arbor, USA) software, respectively.
For whole genome sequencing of bulked segregants, we generated equimolar amounts of DNA from green (N = 26) and white hybrid seedlings (N = 34). Green and white DNA was pooled separately and sent to the Duke Center for Genomic and Computational Biology, where Illumina libraries with unique barcodes were prepared and sequenced using the Illumina Hi-seq platform (100bp single-end reads). Reads from both pools were aligned to the M. guttatus (IM62) reference genome (https://phytozome.jgi.doe.gov), along with previously generated whole genome re-sequence data for DPR104-nas [29]. Reads were aligned using Burrows-Wheeler Aligner (bwa, [70]) with a minimum alignment quality threshold of Q29 (filtering done with samtools, [71]). We identified 235,922 SNPs that differentiated the IM62 reference genome from DPR104-nas using the samtools mpileup function, which provided a list of SNPs that differentiate these two lineages. We used the samtools mpileup function to estimate the frequency of each SNP (‘alternate allele frequency’) within white and green BSA pools. Since SNPs were not based on differences between DPR104-nas and DPR102-gutt, our analysis assumes that M. guttatus lines IM62 and DPR102-gutt (which is not sequenced) share a common set of SNPs.
qPCR and RNA sequencing
To examine the potential for transcriptional misexpression associated with hybrid lethality, we performed quantitative PCR on a subset of strong candidate genes within hl13 and hl14 (9 genes total, S2 Table). We compared gene expression patterns in seedlings from DPR102-gutt, DPR104-nas, green F2s, and white F2s. We extracted RNA from pools of 10 seedlings for each genotypic class using a Zymo MicroRNA Kit (Zymo Research, Irvine, USA) followed by cDNA synthesis with GOscript Reverse Transcriptase (Promega, Madison, USA). We designed exon-specific primers to amplify fragments of each gene (S4 Table), amplified fragments using standard touchdown PCR, and visualized gene fragments on a 1% agarose gel.
We performed an RNAseq experiment to compare genome-wide expression profiles between white and green seedlings. We used lines of DPR102-gutt and DPR104-nas that had been inbred for 5 generations and their green and white F2 progeny, which resulted in three classes of green seedlings (DPR102-gutt, DPR104-nas, and green F2s) and a single class of white seedlings (white F2s). Seedlings with fully expanded cotyledons began to emerge within 3 days and continued to emerge for a week thereafter. We collected pools of 10 seedlings from each biological class directly into 2mL Eppendorf tubes filled with liquid nitrogen. We then extracted RNA from these pools using the Zymo Quick-RNA microprep kit (Zymo Research, Irvine, USA) and estimated RNA concentration using a qubit fluorometer (Life Technologies, Paisley, UK). High quality RNA was subsequently submitted to the Duke Center for Genomic and Computational Biology, where Kapa Stranded mRNA-Seq libraries (Kapa Biosystems, Wilmington, USA) were prepared and samples were sequenced across a single lane of Illumina Hiseq 4000 with single-end 50 bp reads. In total, our analysis involved three replicates each of DPR102-gutt and DPR104 green seedlings, five replicates of green F2 seedlings, and six replicates of white F2 seedlings, where each replicate was a pool of 10 seedlings.
We utilized the cufflinks pipeline [72] to assess patterns of differential expression among the four genotypic classes (DPR102-gutt, DPR104-nas, green F2s, and white F2s). We aligned trimmed and filtered reads (Q>20) to the M. guttatus IM62 reference genome in TopHat2 [73], which resulted in an average of 19 million reads aligned per biological replicate. We then assembled transcriptomes in cufflinks using default settings, the reference IM62 transcriptome to guide the assembly (-g), and allowing for both fragment bias (-b) and multi-read (-u) corrections. We used ‘cuffnorm’ to normalize transcript abundance for each genotypic class and ‘cuffdiff’ to calculate differential expression for all pairwise comparisons using default parameters for both program. For data management and sorting, we used Microsoft excel, the R statistical package [74], and the R package CummeRbund [72]. Gene ontology (GO) enrichment analyses were carried out for particular subsets of data that exhibited patterns of differential expression between white and green seedlings. To perform these analyses, we used GOstat [75] and GO::TermFinder [76] implemented in the Phytomine user interface (https://phytozome.jgi.doe.gov). For GO term analyses, we used all annotated genes in the v2.0 IM62 M. guttatus reference assembly to serve as the background population and used a Bonferroni cutoff value of 0.05 to test for significant GO term enrichment in our subset of differentially expressed genes. For our analysis of chloroplast-encoded genes, we generated a list of putative chloroplast genes, since no chloroplast genome assembly is currently available for M. guttatus. We generated this set by first downloading a list of 135 genes present in the chloroplast genome in A. thaliana from the TAIR database (www.arabidopsis.org). We used this list to identify M. guttatus homologs in the Phytomine database (https://phytozome.jgi.doe.gov). This approach yielded a set of 52 putative Mimulus chloroplast genes that are currently included (and, presumably, misassembled) in the nuclear genome (no homologs were identified for the other 83 genes used in our search). A substantial fraction of these genes (69%) occur along chromosome 4 from positions 6,719,000–7,985,375, which contains 183 genes total.
Gene sequencing and phylogenetic analyses
To obtain full-length pTAC14 sequences from DPR102-gutt and DPR104-nas, we amplified both genomic and cDNA using primers designed within conserved exonic sequence. PCR fragments were amplified using Phusion High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, USA) and either directly sequenced or sequenced after cloning into the TOPO TA Vector (Thermo Fisher, Carlsbad, USA). Additionally, we extracted full-length transcript sequences from RNAseq data by performing de novo assemblies on reads that mapped to candidate genes using the Geneious Assembler (Biomatters, Newark, USA). When reads mapped to duplicated genes, they were combined into a single de novo assembly and 95% confidence consensus sequences were constructed. Using PHYML [77], we constructed a neighbor-joining tree for pTAC14 using 4,319 bp of genomic sequence (excluding 5’ and 3’ UTRs and insertions/deletions coded as single variants) with branch support determined with 1000 bootstraps. For the tree presented in Fig 4B, we used the general time-reversible model with four substitution rate categories and allowed the program to estimate the proportion of variable sites and the gamma distribution parameter (varying these parameters produced identical consensus trees).
Supporting information
White seedlings segregate in 1:15 in reciprocal F2 hybrids of M. guttatus (DPR102-gutt) and M. nasutus (DPR104-nas). Photos kindly provided by Adam J Bewick.
(TIF)
Difference in average allele frequency between green and white pools (plotted along the fourteen Mimulus chromosomes) was calculated in 200-SNP windows with 100-SNP overlap. The 0.5% most divergent windows are highlighted as black dots, which are all located at the distal end of chromosome 13 in contiguous windows and represent the candidate hl13 region. Red dots, which overlap with the previously mapped interval for hl14, show the 0.5% least divergent windows (calculated as absolute difference in allele frequency).
(TIF)
Exons are highlighted blue. Black boxes show ten SNPs that were used to positionally map copies Mg.pTAC14_1, Mg.pTAC14_2, and Mn.pTAC14_1 in DPR104-nas x DPR102-gutt F2 seedlings. Red box indicates site of frameshift mutation in Mg.pTAC14_1.
(JPEG)
{kind=link}
PCR products run on 1% agarose gel with 2-log ladder. Control transcript is Migut.M00195 (ACYL-COENZYME A OXIDASE-LIKE PROTEIN). Note that DNA and RNA was extracted from pools of 10 seedlings for each genotype.
(TIF)
Differential expression among chloroplast genes transcribed by PEP (psaA, psaB, psbA, psbB, petB, petD), PEP and NEP (atpA and ndhB), and NEP (rpoA, rpoB, and rpoC1). Green: average log2 fold-change in all pairwise comparisons among green seedlings (DPR102-gutt, DPR104-nas, Green F2). White: average log2 fold-change in pairwise comparisons between white F2 seedlings and green seedlings. *Significantly down-regulated in white seedlings (all pairwise comparisons, p<5.0−5). **Significantly up-regulated in white seedlings (all pairwise comparisons, p<5.0−5).
(TIF)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Acknowledgments
We thank Noland Martin and John Willis for first identifying white seedlings at Don Pedro Reservoir and encouraging us to study them. We thank Amanda Kenney for her help with BSA experiments and intellectual contributions. Our discussions with Dave Hall, Mike Arnold, John Burke, Kelly Dyer, and CJ Tsai were always enlightening and played an important role in shaping the design and interpretation of this study. John Willis, Dave Hall, Nick Arthur, Nick Batora, Rachel Kerwin, Sam Mantel, and two anonymous reviewers provided thoughtful comments, which greatly improved the quality of our manuscript.
Data Availability
The genomic and transcript sequences used in our bulked segregant and RNAseq experiments are available under the GenBank accession SRP127135.
Funding Statement
This work was supported by an NIH Training Grant Fellowship T32 GM007103 to MPZ and a National Science Foundation (NSF) grant DEB-1350935 to ALS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bateson W. Heredity and variation in modern lights. Darwin and Modern Science. 1909;85:101. [Google Scholar]
- 2.Dobzhansky T, Dobzhansky TG. Genetics and the Origin of Species: Columbia University Press; 1937. [Google Scholar]
- 3.Muller HJ, editor Isolating mechanisms, evolution and temperature. Biol Symp; 1942. [Google Scholar]
- 4.Fishman L, Sweigart AL. When two rights make a wrong: evolutionary genetics of plant hybrid incompatibilities. Annual Review of Plant Biology. in press. [DOI] [PubMed] [Google Scholar]
- 5.Maheshwari S, Barbash DA. The Genetics of Hybrid Incompatibilities. Annual Review of Genetics. 2011;45(1):331–55. [DOI] [PubMed] [Google Scholar]
- 6.Presgraves DC. The molecular evolutionary basis of species formation. Nature Reviews Genetics. 2010;11(3):175–80. doi: 10.1038/nrg2718 [DOI] [PubMed] [Google Scholar]
- 7.Sweigart AL, Willis JH. Molecular evolution and genetics of postzygotic reproductive isolation in plants. F1000 Biology Reports. 2012;4:23 doi: 10.3410/B4-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brideau NJ, Flores HA, Wang J, Maheshwari S, Wang X, Barbash DA. Two Dobzhansky-Muller Genes Interact to Cause Hybrid Lethality in Drosophila. Science. 2006;314(5803):1292–5. doi: 10.1126/science.1133953 [DOI] [PubMed] [Google Scholar]
- 9.Maheshwari S, Wang J, Barbash DA. Recurrent Positive Selection of the Drosophila Hybrid Incompatibility Gene Hmr. Molecular Biology and Evolution. 2008;25(11):2421–30. doi: 10.1093/molbev/msn190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genetics. 2009;5(12):e1000753 doi: 10.1371/journal.pgen.1000753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phadnis N, Orr HA. A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science. 2009;323(5912):376–9. doi: 10.1126/science.1163934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423(6941):715–9. doi: 10.1038/nature01679 [DOI] [PubMed] [Google Scholar]
- 13.Tang S, Presgraves DC. Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science. 2009;323(5915):779–82. doi: 10.1126/science.1169123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, Chen H, Lin Y-S, Shen J-B, Shan J-X, Qi P, et al. A two-locus interaction causes interspecific hybrid weakness in rice. Nature Communications. 2014;5:3357 doi: 10.1038/ncomms4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sicard A, Kappel C, Josephs EB, Lee YW, Marona C, Stinchcombe JR, et al. Divergent sorting of a balanced ancestral polymorphism underlies the establishment of gene-flow barriers in Capsella. Nature Communications. 2015;6:7960 doi: 10.1038/ncomms8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wright KM, Lloyd D, Lowry DB, Macnair MR, Willis JH. Indirect evolution of hybrid lethality due to linkage with selected locus in Mimulus guttatus. PLoS Biology. 2013;11(2):e1001497 doi: 10.1371/journal.pbio.1001497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Case AL, Finseth FR, Barr CM, Fishman L. Selfish evolution of cytonuclear hybrid incompatibility in Mimulus. Proceedings of the Royal Society B: Biological Sciences. 2016;283(1838). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao Y, Hartl DL, Laurie CC. Sex-ratio segregation distortion associated with reproductive isolation in Drosophila. Proceedings of the National Academy of Sciences. 2001;98(23):13183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Sun T, Woldesellassie F, Xiao H, Tao Y. Sex Ratio Meiotic Drive as a Plausible Evolutionary Mechanism for Hybrid Male Sterility. PLOS Genetics. 2015;11(3):e1005073 doi: 10.1371/journal.pgen.1005073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizuta Y, Harushima Y, Kurata N. Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proceedings of the National Academy of Sciences. 2010;107(47):20417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen GN, Yamagata Y, Shigematsu Y, Watanabe M, Miyazaki Y, Doi K, et al. Duplication and Loss of Function of Genes Encoding RNA Polymerase III Subunit C4 Causes Hybrid Incompatibility in Rice. G3: Genes|Genomes|Genetics. 2017;7(8):2565 doi: 10.1534/g3.117.043943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamagata Y, Yamamoto E, Aya K, Win KT, Doi K, Sobrizal, et al. Mitochondrial gene in the nuclear genome induces reproductive barrier in rice. Proceedings of the National Academy of Sciences. 2010;107(4):1494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oka H-I. Analysis of Genes Controlling F(1) Sterility in Rice by the Use of Isogenic Lines. Genetics. 1974;77(3):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch M, Force AG. The Origin of Interspecific Genomic Incompatibility via Gene Duplication. The American Naturalist. 2000;156(6):590–605. [DOI] [PubMed] [Google Scholar]
- 25.Werth CR, Windham MD. A Model for Divergent, Allopatric Speciation of Polyploid Pteridophytes Resulting from Silencing of Duplicate-Gene Expression. The American Naturalist. 1991;137(4):515–26. [Google Scholar]
- 26.Masly JP, Jones CD, Noor MA, Locke J, Orr HA. Gene transposition as a cause of hybrid sterility in Drosophila. Science. 2006;313(5792):1448–50. doi: 10.1126/science.1128721 [DOI] [PubMed] [Google Scholar]
- 27.Bikard D, Patel D, Le Metté C, Giorgi V, Camilleri C, Bennett MJ, et al. Divergent Evolution of Duplicate Genes Leads to Genetic Incompatibilities Within A. thaliana. Science. 2009;323(5914):623–6. doi: 10.1126/science.1165917 [DOI] [PubMed] [Google Scholar]
- 28.Durand S, Bouché N, Strand EP, Loudet O, Camilleri C. Rapid establishment of genetic incompatibility through natural epigenetic variation. Current Biology. 2012;22(4):326–31. doi: 10.1016/j.cub.2011.12.054 [DOI] [PubMed] [Google Scholar]
- 29.Brandvain Y, Kenney AM, Flagel L, Coop G, Sweigart AL. Speciation and introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genetics. 2014;10(6):e1004410 doi: 10.1371/journal.pgen.1004410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz A, Macnair M. Pollen tube competition as a mechanism of prezygotic reproductive isolation between Mimulus nasutus and its presumed progenitor M. guttatus. New Phytologist. 1999;144(3):471–8. [DOI] [PubMed] [Google Scholar]
- 31.Kenney AM, Sweigart AL. Reproductive isolation and introgression between sympatric Mimulus species. Molecular Ecology. 2016;25(11):2499–517. doi: 10.1111/mec.13630 [DOI] [PubMed] [Google Scholar]
- 32.Kiang Y, Hamrick J. Reproductive isolation in the Mimulus guttatus M. nasutus complex. American Midland Naturalist. 1978:269–76. [Google Scholar]
- 33.Martin NH, Willis JH. Ecological divergence associated with mating system causes nearly complete reproductive isolation between sympatric Mimulus species. Evolution. 2007;61(1):68–82. doi: 10.1111/j.1558-5646.2007.00006.x [DOI] [PubMed] [Google Scholar]
- 34.Case AL, Willis JH. Hybrid male sterility in Mimulus (Phrymaceae) is associated with a geographically restricted mitochondrial rearrangement. Evolution. 2008;62(5):1026–39. doi: 10.1111/j.1558-5646.2008.00360.x [DOI] [PubMed] [Google Scholar]
- 35.Fishman L, Willis JH. A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution. 2006;60(7):1372–81. [DOI] [PubMed] [Google Scholar]
- 36.Kiang Y, Libby W. Maintenance of a lethal in a natural population of Mimulus guttatus. The American Naturalist. 1972;106(949):351–67. [Google Scholar]
- 37.Martin NH, Willis JH. Geographical variation in postzygotic isolation and its genetic basis within and between two Mimulus species. Philosophical Transactions of the Royal Society of London B: Biological Sciences. 2010;365(1552):2469–78. doi: 10.1098/rstb.2010.0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sweigart AL, Fishman L, Willis JH. A Simple Genetic Incompatibility Causes Hybrid Male Sterility in Mimulus. Genetics. 2006;172(4):2465–79. doi: 10.1534/genetics.105.053686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweigart AL, Flagel LE. Evidence of natural selection acting on a polymorphic hybrid incompatibility locus in Mimulus. Genetics. 2015;199(2):543–54. doi: 10.1534/genetics.114.171819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vickery RK Jr. Case studies in the evolution of species complexes in Mimulus. Evolutionary Biology: Springer; 1978. p. 405–507. [Google Scholar]
- 41.Sweigart A, Willis J. Patterns of nucleotide diversity are affected by mating system and asymmetric introgression in two species of Mimulus. Evolution. 2003;57:2490–506. [DOI] [PubMed] [Google Scholar]
- 42.Gao Z-P, Yu Q-B, Zhao T-T, Ma Q, Chen G-X, Yang Z-N. A functional component of the transcriptionally active chromosome complex, Arabidopsis pTAC14, interacts with pTAC12/HEMERA and regulates plastid gene expression. Plant Physiology. 2011;157(4):1733–45. doi: 10.1104/pp.111.184762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellsten U, Wright KM, Jenkins J, Shu S, Yuan Y, Wessler SR, et al. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proceedings of the National Academy of Sciences. 2013;110(48):19478–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hajdukiewicz PT, Allison LA, Maliga P. The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. The EMBO Journal. 1997;16(13):4041–8. doi: 10.1093/emboj/16.13.4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swiatecka-Hagenbruch M, Liere K, Börner T. High diversity of plastidial promoters in Arabidopsis thaliana. Molecular Genetics and Genomics. 2007;277(6):725–34. doi: 10.1007/s00438-007-0222-4 [DOI] [PubMed] [Google Scholar]
- 46.Kremnev D, Strand Å. Plastid encoded RNA polymerase activity and expression of photosynthesis genes required for embryo and seed development in Arabidopsis. Frontiers in Plant Science. 2014;5 doi: 10.3389/fpls.2014.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demarsy E, Buhr F, Lambert E, Lerbs-Mache S. Characterization of the plastid-specific germination and seedling establishment transcriptional programme. Journal of Experimental botany. 2012;63(2):925–39. doi: 10.1093/jxb/err322 [DOI] [PubMed] [Google Scholar]
- 48.Ishizaki Y, Tsunoyama Y, Hatano K, Ando K, Kato K, Shinmyo A, et al. A nuclear‐encoded sigma factor, Arabidopsis SIG6, recognizes sigma‐70 type chloroplast promoters and regulates early chloroplast development in cotyledons. The Plant Journal. 2005;42(2):133–44. doi: 10.1111/j.1365-313X.2005.02362.x [DOI] [PubMed] [Google Scholar]
- 49.Liere K, Börner T. Transcription and transcriptional regulation in plastids. Cell and Molecular Biology of Plastids: Springer; 2007. p. 121–74. [Google Scholar]
- 50.Pfannschmidt T, Blanvillain R, Merendino L, Courtois F, Chevalier F, Liebers M, et al. Plastid RNA polymerases: orchestration of enzymes with different evolutionary origins controls chloroplast biogenesis during the plant life cycle. Journal of Experimental Botany. 2015;66(22):6957–73. doi: 10.1093/jxb/erv415 [DOI] [PubMed] [Google Scholar]
- 51.Blevins T, Wang J, Pflieger D, Pontvianne F, Pikaard CS. Hybrid incompatibility caused by an epiallele. Proceedings of the National Academy of Sciences. 2017:201700368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch M, Conery JS. The Evolutionary Fate and Consequences of Duplicate Genes. Science. 2000;290(5494):1151–5. [DOI] [PubMed] [Google Scholar]
- 53.Flagel LE, Wendel JF. Gene duplication and evolutionary novelty in plants. New Phytologist. 2009;183(3):557–64. doi: 10.1111/j.1469-8137.2009.02923.x [DOI] [PubMed] [Google Scholar]
- 54.Flagel LE, Willis JH, Vision TJ. The standing pool of genomic structural variation in a natural population of Mimulus guttatus. Genome Biology and Evolution. 2013;6(1):53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly JK, Koseva B, Mojica JP. The genomic signal of partial sweeps in Mimulus guttatus. Genome Biology and Evolution. 2013;5(8):1457–69. doi: 10.1093/gbe/evt100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mojica JP, Lee YW, Willis JH, Kelly JK. Spatially and temporally varying selection on intrapopulation quantitative trait loci for a life history trade‐off in Mimulus guttatus. Molecular Ecology. 2012;21(15):3718–28. doi: 10.1111/j.1365-294X.2012.05662.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puzey JR, Willis JH, Kelly JK. Population structure and local selection yield high genomic variation in Mimulus guttatus. Molecular Ecology. 2017;26(2):519–35. doi: 10.1111/mec.13922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macnair M. A polymorphism for a lethal phenotype governed by two duplicate genes in Mimulus. Heredity. 1993;70:362–9. [Google Scholar]
- 59.Willis JH. Genetic analysis of inbreeding depression caused by chlorophyll-deficient lethals in Mimulus guttatus. Heredity. 1992;69:562–. [Google Scholar]
- 60.Bank C, Bürger R, Hermisson J. The Limits to Parapatric Speciation: Dobzhansky–Muller Incompatibilities in a Continent–Island Model. Genetics. 2012;191(3):845–63. doi: 10.1534/genetics.111.137513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barton N, Bengtsson BO. The barrier to genetic exchange between hybridising populations. Heredity. 1986;57(3):357–76. [DOI] [PubMed] [Google Scholar]
- 62.Gavrilets S. Hybrid Zones With Dobzhansky-Type Epistatic Selection. Evolution. 1997;51(4):1027–35. doi: 10.1111/j.1558-5646.1997.tb03949.x [DOI] [PubMed] [Google Scholar]
- 63.Kondrashov AS, Morgan M. Accumulation of Dobzhansky-Muller incompatibilities within a spatially structured population. Evolution. 2003;57(1):151–3. [DOI] [PubMed] [Google Scholar]
- 64.Lemmon AR, Kirkpatrick M. Reinforcement and the genetics of hybrid incompatibilities. Genetics. 2006;173(2):1145–55. doi: 10.1534/genetics.105.048199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424(6945):194 doi: 10.1038/nature01771 [DOI] [PubMed] [Google Scholar]
- 66.Doyle J, Doyle J. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull. 1987;19(11):11–5. [Google Scholar]
- 67.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, et al. The Genome Portal of the Department of Energy Joint Genome Institute. Nucleic Acids Research. 2012;40(D1):D26–D32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Research. 2014;42(Database issue):D26–D31. doi: 10.1093/nar/gkt1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hulce D, Li X, Snyder-Leiby T, Liu CJ. GeneMarker® genotyping software: tools to Increase the statistical power of DNA fragment analysis. Journal of biomolecular techniques: JBT. 2011;22(Suppl):S35. [Google Scholar]
- 70.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–95. doi: 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14(4):R36 doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Team RC. R language definition Vienna, Austria: R foundation for statistical computing; 2000. [Google Scholar]
- 75.Beißbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20(9):1464–5. doi: 10.1093/bioinformatics/bth088 [DOI] [PubMed] [Google Scholar]
- 76.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, et al. GO:: TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–5. doi: 10.1093/bioinformatics/bth456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
White seedlings segregate in 1:15 in reciprocal F2 hybrids of M. guttatus (DPR102-gutt) and M. nasutus (DPR104-nas). Photos kindly provided by Adam J Bewick.
(TIF)
Difference in average allele frequency between green and white pools (plotted along the fourteen Mimulus chromosomes) was calculated in 200-SNP windows with 100-SNP overlap. The 0.5% most divergent windows are highlighted as black dots, which are all located at the distal end of chromosome 13 in contiguous windows and represent the candidate hl13 region. Red dots, which overlap with the previously mapped interval for hl14, show the 0.5% least divergent windows (calculated as absolute difference in allele frequency).
(TIF)
Exons are highlighted blue. Black boxes show ten SNPs that were used to positionally map copies Mg.pTAC14_1, Mg.pTAC14_2, and Mn.pTAC14_1 in DPR104-nas x DPR102-gutt F2 seedlings. Red box indicates site of frameshift mutation in Mg.pTAC14_1.
(JPEG)
PCR products run on 1% agarose gel with 2-log ladder. Control transcript is Migut.M00195 (ACYL-COENZYME A OXIDASE-LIKE PROTEIN). Note that DNA and RNA was extracted from pools of 10 seedlings for each genotype.
(TIF)
Differential expression among chloroplast genes transcribed by PEP (psaA, psaB, psbA, psbB, petB, petD), PEP and NEP (atpA and ndhB), and NEP (rpoA, rpoB, and rpoC1). Green: average log2 fold-change in all pairwise comparisons among green seedlings (DPR102-gutt, DPR104-nas, Green F2). White: average log2 fold-change in pairwise comparisons between white F2 seedlings and green seedlings. *Significantly down-regulated in white seedlings (all pairwise comparisons, p<5.0−5). **Significantly up-regulated in white seedlings (all pairwise comparisons, p<5.0−5).
(TIF)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Data Availability Statement
The genomic and transcript sequences used in our bulked segregant and RNAseq experiments are available under the GenBank accession SRP127135.