

COG is a multisubunit vesicle tethering complex in the Golgi. We demonstrate that both COG subcomplexes can be permanently attached to Golgi membranes and that major COG functions do not require cycling between the membrane and cytosol.
Abstract
The conserved oligomeric Golgi (COG) complex is a vesicle tether of the “complexes associated with tethering containing helical rods” family, which functions on the cytoplasmic side of Golgi. It is currently unknown whether COG function, or function of any multisubunit vesicular tether, depends on cycling between the membrane and cytosol. Therefore, we permanently anchored key subunits of COG subcomplexes (COG4, COG7, and COG8) to Golgi membranes using transmembrane protein TMEM115 (TMEM-COG). All TMEM-COG subunits tested were Golgi localized, integrated into the COG complex, and stabilized membrane association of endogenous subunits. Interestingly, TMEM-COG4 and TMEM-COG7 equally rescued COG function in organization of Golgi markers, glycosylation, and abundance of COG-sensitive proteins. In contrast, TMEM-COG8 was not as effective, indicating that N-terminal attachment of COG8 interfered with overall COG structure and function, and none of the TMEM-COG subunits rescued the abnormal Golgi architecture caused by COG knockout. Collectively, these data indicate that both subcomplexes of the COG complex can perform most of COG function when permanently attached to membranes and that the cytosolic pool of COG is not completely essential to COG function.
INTRODUCTION
The conserved oligomeric Golgi (COG) complex is an evolutionarily conserved protein complex whose major function is to recycle resident Golgi proteins in a retrograde manner along the Golgi stack (Whyte and Munro 2001; Suvorova et al., 2002; Ungar et al., 2002; Shestakova et al., 2007). COG is a multisubunit vesicle tethering complex composed of eight subunits functionally and structurally subdivided into two subcomplexes: lobe A (COG1-4) and lobe B (COG5-8) (Ungar et al., 2002; Fotso et al., 2005; Cavanaugh et al., 2007; Richardson et al., 2009; Lees et al., 2010; Ha et al., 2014, 2016). Investigation of COG knockout (KO) cell lines and COG mutant patient samples showed that each COG subunit is equally important to COG function (Foulquier et al., 2006; Bailey Blackburn et al., 2016). Loss of one subunit can destabilize the other COG subunits and decrease functionality of the entire COG complex. Although decreased COG function does not affect viability in 293T cells and CHO cells (Kingsley and Krieger 1984; Bailey Blackburn et al., 2016), it can be lethal in yeast and humans (VanRheenen et al., 1998, 1999; Kim et al., 1999; Ram et al., 2002; Suvorova et al., 2002; Morava et al., 2007; Zeevaert et al., 2009). The COG complex interacts with core vesicle trafficking components such as small GTPases, coiled-coil tethers, Sec1/Munc18 proteins, SNAREs, and vesicle coat proteins (Bonifacino and Glick 2004; Willett et al., 2013b).
It was proposed that these protein–protein interactions aid COG in tethering intra-Golgi vesicles to the Golgi stack. During Golgi trafficking, resident proteins, like glycosylation enzymes, require recycling from the late Golgi to the early Golgi cisternae to maintain the integrity of the nontemplate driven glycosylation system (Glick et al., 1997; Glick and Nakano 2009; Mironov et al., 1997; Pelham 2001). Glycosylation enzymes MAN2A1, MGAT1, B4GALT1, and ST6GAL1 are mislocalized in COG-deficient cells (Shestakova et al., 2006; Pokrovskaya et al., 2011), and all COG subunits, with the exception of COG3, have mutations described as contributing to human congenital disorders of glycosylation (CDG) (Climer et al., 2015; Haijes et al., 2018).
Structurally, each COG subunit is predicted to have flexible coiled coil regions, placing the COG complex in the “complexes associated with tethering containing helical rods” (CATCHR) family with other multisubunit tethering complexes like DSL1, Golgi-associated retrograde protein (GARP), endosome-associated recycling protein (EARP), and Exocyst (Whyte and Munro 2001; Whyte and Munro 2002; Yu and Hughson 2010; Chou et al., 2016). The long flexible structure of COG subunits is visible by electron microscopy (EM) (Ungar et al., 2002; Ha et al., 2016). These flexible extensions likely mediate sequential or simultaneous interactions with the various components of the vesicle trafficking machinery. Using a cleavable fusion construct that leashes COG1 (lobe A) and COG8 (lobe B) together, we demonstrated that the functional COG complex cycles between separate subcomplexes and the complete eight-subunit complex suggesting that COG function requires this cycling (Willett et al 2016).
Endogenous COG subunits are found in association with Golgi membranes and vesicles. The precise molecular and functional description of the interaction between COG and cellular membranes is unknown (Willett et al., 2014). Currently, no inherent transmembrane or lipid binding domains have been discovered in the COG complex, which suggests that the COG–membrane interaction must be mediated through association with other transmembrane and peripheral membrane proteins. The most likely mediators of the COG–membrane interaction are the core components of the trafficking machinery.
Gentle mechanical disruption of cellular membranes with subsequent subcellular fractionation demonstrates that COG is distributed roughly 60:40 between the membrane and cytosolic fractions. Interestingly, it has been observed that this subcellular distribution can be altered by a single COG subunit deficiency, leading to an increase in the cytosolic pool of other COG subunits (Willett et al., 2014). Thus, COG function may require dynamic movement between the Golgi and vesicle membranes. Consistent with this hypothesis, rapid on/off membrane cycling was observed for other peripheral membrane components of vesicle trafficking machinery such as coat protein complex I (COPI) vesicle coat (Presley et al., 2002; Elsner et al., 2003) and coiled-coil tether p115 (Brandon et al., 2006) but this phenomenon has not been studied for the CATCHR tethers. YFP-COG3 recovers from photobleaching no faster than GFP-tagged SNARE GS15 (Willett et al., 2014), indicating that COG could recycle on the membranes similarly to transmembrane proteins and the kinetics of COG recycling process are likely slow. It is possible that in intact cells COG moves down the stack with the transport vesicle while constantly maintaining an interaction with the membrane, and the soluble pool is an artifact created by mechanical disruption or the result of de novo synthesis. Therefore, to determine whether COG function depends on the cytosolic pool of COG subunits, we permanently anchored COG to Golgi membranes by creating a fusion construct with a known medial Golgi localized protein, TMEM115. One subunit from each COG subcomplex, COG4 for lobe A and COG7 for lobe B, was chosen as an anchor. COG8 was also anchored since several COG models have predicted the existence of COG1–COG8 (inter-A-B lobe) association (Ungar et al., 2005). Using a set of previously generated HEK 293T cell lines deficient in individual COG subunits through CRISPR/Cas9 targeted deletion (Blackburn and Lupashin, 2016), we generated lentivirally transduced stable cell lines expressing one COG subunit permanently tethered to the membrane. Detailed analysis of these cells, using superresolution and electron microscopy, subcellular fractionation, coimmunoprecipitation (IP), and Western blot (WB), revealed that that both subcomplexes of the COG complex are mostly functional when permanently attached to membranes and that the cytosolic pool of COG is likely not essential to COG function.
RESULTS
TMEM115-tethered COG subunits are Golgi localized
To determine whether the cytosolic pool is required for COG function, we permanently anchored COG subunits using either N- or C-terminal transmembrane domain (TMD) of Golgi-localized, COG interacting proteins (Supplemental Figure 1A). Initially, we added only the C-terminal TMD of SNAREs STX5, GS15, and GS27 to create COG8-TMD constructs. All constructs were expressed in HeLa cells and show a membrane pattern but failed to deliver COG8 to the Golgi. COG8-TMD constructs appeared to be ER localized instead (Supplemental Figure 1B). This result indicated that neither Golgi SNARE TMDs nor COG8 possess secretory signals that would direct them into COPII vesicles for ER exit. Alternatively, the TMDs could anchor the COG complex too close to the membrane to allow efficient folding and exit from ER. To provide the missing secretory signals, we anchored COG subunits by C-terminal fusion to trans-Golgi SNARE STX16. In this construct, the SNARE domain of STX16 was deleted (STX16-ΔSNARE) to avoid any potential functional competition with the endogenous STX16. Transiently expressed mCherry-tagged STX16-ΔSNARE showed Golgi localization by colocalizing with Golgi marker GalNacT2-GFP but did not localize COG4, COG7, or COG8 to Golgi membranes (Supplemental Figure 1C). In all cases, the hybrid protein showed punctate staining likely indicating endosomal localization that was previously observed for the endogenous STX16 (Willett et al., 2013a).
Recently, Golgi transmembrane protein, TMEM115 was shown to interact with the COG complex (Ong et al., 2014). TMEM115 putatively contains 4 N-terminal TMDs and a Golgi localization sequence. Initially, we attempted to use only a fragment of TMEM115 by using truncations that contain a published Golgi localization sequence. However, these truncations failed to reach the Golgi, and only full-length TMEM115-COG-mCherry (TMEM-COG) fusion proteins were Golgi localized in HEK293T cells (Figure 1A and Supplemental Figure 2). These data indicate that there is no information innate to any tested COG subunit that determines Golgi localization. COG must be Golgi localized due to interactions with Golgi trafficking machinery and/or Golgi resident proteins. Transient expression of TMEM-COG resulted in variable expression of fusion constructs (unpublished data). Therefore, we used lentiviral transduction to stably rescue our COG KO HEK 293T cells with the TMEM-COG fusion proteins permanently anchored on Golgi membranes (Figure 1). From here onward, TMEM-COG is referencing stable expression of an anchored COG subunit in an HEK293T COG KO cell line. Except for Figure 2, expression of TMEM-COG subunits was controlled by FACS sorting cell populations expressing low levels of TMEM-COG. As controls, the 293T parental cell line and/or 293T COG4 and COG7 KO cell lines stably expressing unrestrained COG4-GFP and COG7-GFP were tested in all experiments (Table 1).
FIGURE 1:
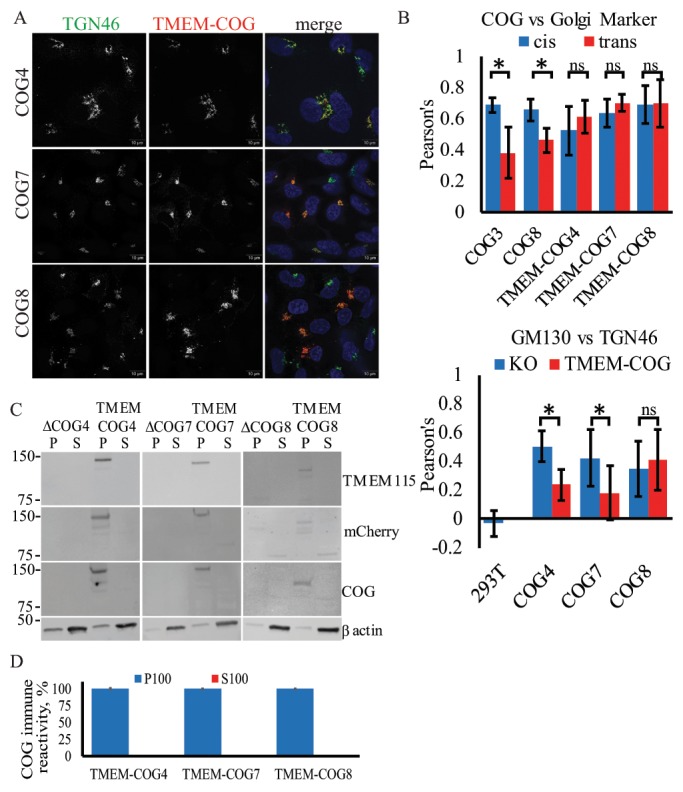
Stable TMEM-anchored COG subunits are Golgi localized and enriched in TGN. HEK293T cells stably expressing TMEM-COG4 in COG4 KO cells, TMEM-COG7 in COG7 KO cells, and TMEM-COG8 in COG8 KO cells. (A) Elyra 3D-SIM superresolution images of TMEM-COG (red), endogenous TGN46 (green), and 4’,6-diamidino-2-phenylindole (DAPI, blue). Maximum intensity projections. (B) Colocalization of COG with Golgi markers. Images were collected and analyzed with LSM880 Airyscan using ZEN colocalization software. At least 20 Golgi were quantified per comparison. Bottom: Colocalization of GM130 and TGN46 in WT, COG KO cells, and stable TMEM-COG cells. Top: TMEM-COG4, TMEM-COG7, and TMEM-COG8 show colocalization with both GM130 (blue) and TGN46 (red). * denotes p < 0.05 in a Student’s t test. Error bars represent SD. (C) Distribution of TMEM-COG in P100 membrane and S100 soluble fractions. Left panels depict COG4 KO and TMEM-COG4 cells probed with antibodies to TMEM115, mCherry, and COG4. Middle panels depict COG7 KO and TMEM-COG7 cells probed with antibodies to TMEM115, mCherry, and COG7. Right panels depict COG8 KO and TMEM-COG8 cells probed with antibodies to TMEM115, mCherry, and COG8. (D) Quantification of COG antibody detection of P100 and S100 on three blots each. No detectable COG in S100 samples.
FIGURE 2:
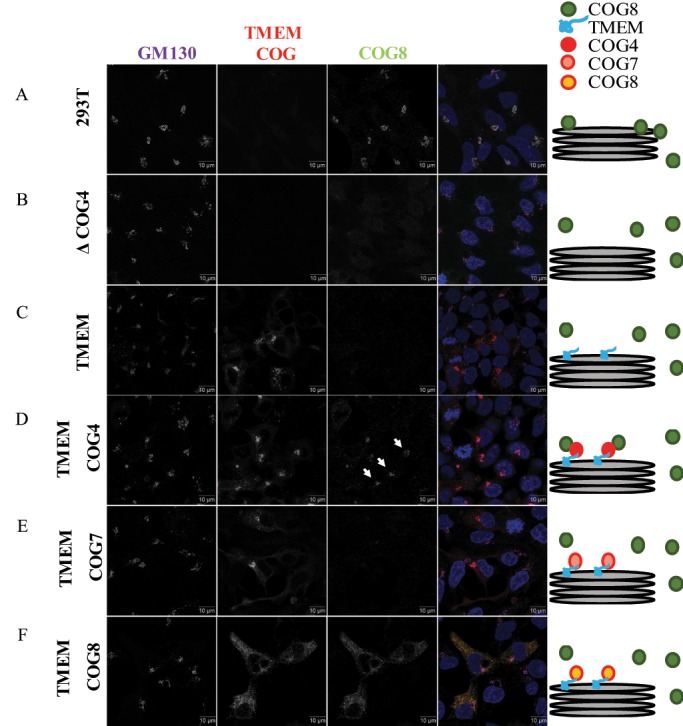
TMEM-COG4 expression rescues COG4 KO-induced mislocalization of endogenous COG8. HEK293T cells stably expressing TMEM-COG4, TMEM-COG7, or TMEM-COG8 in COG4 KO cells. (A) WT HEK293T cells. (B) COG4 KO HEK293T. (C) TMEM-mCherry. (D) TMEM-COG4 (arrows indicate Golgi-localized COG8). (E) TMEM-COG7. (F) TMEM-COG8. Stained for endogenous COG8 (green) and GM130 (purple). Scale bars = 10 microns.
TABLE 1:
Cell lines
| Cell line name | Description | Citation |
|---|---|---|
| COG4 KO | CRISPR/Cas9-mediated deletion of endogenous COG4 in HEK293T cells | Bailey Blackburn et al., 2016 |
| COG7 KO | CRISPR/Cas9-mediated deletion of endogenous COG7 in HEK293T cells | Bailey Blackburn et al., 2016 |
| COG8 KO | CRISPR/Cas9-mediated deletion of endogenous COG8 in HEK293T cells | Bailey Blackburn et al., 2016 |
| TMEM | TMEM115-mCherry stably expressed in COG4, COG7 or COG8 KO cell lines. | This study |
| TMEM-COG4 | TMEM115-COG4mCherry stably expressed in COG4 KO cells. | This study |
| TMEM-COG7 | TMEM115-COG7mCherry stably expressed in COG7 KO cells. | This study |
| TMEM-COG8 | TMEM115-COG8mCherry stably expressed in COG8 KO cells. | This study |
| COG4-GFP | COG4-GFP stably expressed in COG4 KO cells. | This study |
| COG7-GFP | COG7-2xGFP-3myc stably expressed in COG7 KO cells. | This study |
| COG4 KO TMEM-COG7 | TMEM115-COG7mCherry stably expressed in COG4 KO cells. | This study |
| COG4 KO TMEM-COG8 | TMEM115-COG8mCherry stably expressed in COG4 KO cells. | This study |
Endogenous and TMEM-COG subunits are associated with multiple Golgi cisternae
To determine the exact location of TMEM-COG constructs, we utilized superresolution microscopy by using either 3D-SIM Elyra or the LSM880 laser scanning microscope with Airyscan (Zeiss) and compared the colocalization of unrestrained endogenous COG subunits and membrane-tethered TMEM-COG subunits with Golgi cisternal markers: cis-Golgi GM130 and Giantin, and trans-Golgi Network (TGN) protein TGN46 (Figure 1, A and B). TMEM-COG4, TMEM-COG7, and TMEM-COG8 were all Golgi localized (Figure 1A) and show equal colocalization with both cis and trans markers (Figure 1B, top). Because COG4 and COG7 antibodies are poor reagents for immunofluorescence, we tested endogenous COG3 and COG8 colocalization. Again, components of both lobes were found in all cisternae but preferentially localized to the cis face compared with the trans face of the Golgi in WT 293T cells (Figure 1B, top), indicating that the anchor changed COG distribution across the Golgi stack compared with endogenous counterparts or that Golgi markers themselves are not correctly distributed in Golgi containing TMEM-COG. This was tested by comparing colocalization between the two markers in WT 293T cells and COG KO cells. Superresolution microscopy did not show colocalization of cis and trans Golgi markers in WT cells, but colocalization between GM130 and TGN46 was increased in COG4, COG7, and COG8 KO cells (Figure 1B, bottom). This indicates that COG KO either destabilizes Golgi cis-trans organization or disrupts the Golgi ribbon into small mini-stacks that are below the resolution of fluorescence microscopy used in this study. TMEM-COG4 and TMEM-COG7 expression can significantly reduce the colocalization between cis and trans Golgi markers indicating that these anchored subunits are restoring important Golgi compartmentalization that was severely distorted in COG KO cells. Golgi compartmentalization is not restored in the presence of anchored TMEM-COG8.
In addition to TMEM-COG localization on the Golgi, subcellular fractionation demonstrated that TMEM-COG subunits are proteolytically stable and permanently associated with membranes. We could not detect the presence of cleaved COG in the S100 (cytosolic) fraction of the cells analyzed by antibodies to TMEM115, COG, or mCherry (Figure 1C). Blot quantification revealed that less than 2% of immune-positive material was detected in the S100 fraction (Figure 1D).
Taken together, the colocalization and subcellular fractionation data indicate that TMEM-COG subunits are permanently membrane localized, evenly distributed between the cis and TGN, and COG4/COG7 function in Golgi cisternal organization are not completely dependent on the cytosolic pool of COG. Despite TMEM-COG8 Golgi localization, lobe B function, and/or overall COG complex function in cisternal organization is not rescued in these cells.
TMEM-COG subunits restore endogenous COG localization to the Golgi membranes
Our lab has previously shown that endogenous COG8 is not Golgi localized in HeLa cells transiently depleted by small interfering RNA (siRNA) to COG4 (Willett et al., 2014). Therefore, we used Airyscan superresolution microscopy to determine whether stable expression of TMEM-COG could restore endogenous COG8 to Golgi membranes in COG4 KO cells. Consistent with previously published data, endogenous COG8 was preferentially localized in the Golgi of HEK293T cells (Figure 2A) (Bailey Blackburn et al., 2016). COG4 KO resulted in mislocalization of COG8 away from Golgi membranes (Figure 2B). Stable expression of TMEM-COG4 restored endogenous COG8 to the Golgi (Figure 2D), whereas TMEM alone could not recruit COG8 (Figure 2C). Additionally, stable expression of TMEM-COG7 and TMEM-COG8 could not restore endogenous COG8 to the Golgi of COG4 KO cells (Figure 2, E and F). These data indicated that permanent attachment of COG4 to membranes did not interfere with its physiological interaction with other COG subunits. They also indicated that overexpression of TMEM-COG7 and TMEM-COG8 were also not sufficient to functionally replace endogenous COG4 in cells with a COG4 deficiency.
To verify biochemically that the endogenous COG subunits are recruited to Golgi membranes by TMEM-COG, we used subcellular fractionation to determine whether COG3 and COG6 are enriched in the membrane fraction. In WT 293T cells, COG3 and COG6 showed ∼60:40 distributions between the membrane bound and membrane free, respectively (Figure 3, A and B, 293T). In COG4, COG7, and COG8 KO cells, COG3 (60–75% cytosolic) and COG6 (80–95% cytosolic) are more abundant in the soluble fraction than the WT counterpart (Figure 3, A–C). This is consistent with our previously published data that showed on COG4 depletion by siRNA in HeLa cells, COG3, COG6, and COG8 appear predominantly in the cytosolic fraction (Willett et al., 2014). Interestingly, in the presence of anchored TMEM-COG4, 80% of endogenous COG3 was enriched within the membrane-bound fraction (Figure 3, A and B) and endogenous COG6 distribution was restored to WT levels (Figure 3C). Additionally, TMEM-COG7 and TMEM-COG8 recruited 75% of lobe A and 70–80% of lobe B on membranes as suggested by the presence of COG3 and COG6, respectively. Further, TMEM-COG7 can restore the abundance of COG6 which was degraded in COG7 KO cells (Figure 3, A and C). These data indicated that TMEM-COG4, -COG7, and -COG8 can restore endogenous COG interactions with membranes.
FIGURE 3:

TMEM-COG incorporates into the COG complex and increases membrane localization of endogenous COG subunits. HEK293T cells stably expressing TMEM-COG4 in COG4 KO cells, TMEM-COG7 in COG7 KO cells, and TMEM-COG8 in COG8 KO cells. COG4-GFP and COG7-GFP stable expression in COG4 KO cells and COG7 KO cells, respectively. (A–C) Western blot analysis of proteins isolated from WT, KO, and TMEM-COG membrane (P100, blue) and soluble (S100, red) fractions. Quantification of at least three replicates of (B) endogenous COG3 and (C) endogenous COG6 distribution. *p < 0.05 in a Student’s t test. Error bars represent SD. (D) Pull down of endogenous COG3 and COG6 by GFP-tagged COG4 and COG7, TMEM-COG4, TMEM-COG7, and TMEM-COG8.
TMEM-COG efficiently interacts with endogenous COG subunits
To determine whether TMEM-COG subunits can physically interact with endogenous subunits to form the complete COG complex, we performed pull-down experiments with beads bound by mCherry-binding protein (MBP) or GFP-binding protein (GBP). Cellular lysates from stable cell lines expressing TMEM-COG and COG-GFP were incubated with MBP or GBP beads and probed for endogenous COG3 and COG6. Endogenous COG3 and COG6 were pulled down only by TMEM-COG and COG-GFP and not by GFP, mCh, or TMEM alone (Figure 3D). Importantly, the relative amounts of recovered COG3 and COG6 were similar for both membrane-free and membrane-attached fluorescently tagged-COG constructs, which strongly indicated that permanent membrane attachment of any tested COG subunits did not interfere with their incorporation into the COG complex. Endogenous COG was missing in pull downs using only GFP and mCherry, as well as TMEM alone which supported the conclusion that the interaction is mediated by the COG subunit, not TMEM115 or GFP/mCherry. These data indicate that TMEM-COG subunits can interact with endogenous COG subunits to form the complete COG complex.
TMEM-COG4 and -COG7 subunits are mostly functional for stabilization of COG-sensitive Golgi proteins and glycosylation
To determine what effect the permanent membrane anchor imparts on COG function, we tested whether TMEM-COG could rescue trafficking and glycosylation defects specific to COG deficiency. First, we analyzed the protein expression of several proteins that are sensitive to COG deficiency: GPP130, B4GalT1, and Lamp2 (Figure 4, A–C) (Oka et al., 2004; Pokrovskaya et al., 2011; Bailey Blackburn et al., 2016). By WB, these proteins showed reduced abundance in COG4, COG7, and COG8 KO cells that are mostly restored in TMEM-COG4 and -COG7 cells but not in TMEM-COG8 cells. This would indicate that COG-sensitive Golgi proteins are stabilized by expression of TMEM-COG4 and TMEM-COG7 but not by TMEM-COG8. Interestingly, GPP130, B4GalT1, and Lamp2 also show altered SDS–PAGE migration in COG deficient cells, which indicated altered posttranslational modifications. Since COG deficiency is known to affect glycosylation, and it is likely that these proteins are misglycosylated. The dramatic change in Lamp2 migration, whose predicted polypeptide molecular weight is estimated at 45 kDa, can be attributed to ∼24 N- and O-glycosylation sites. Fully modified Lamp2 appears at a molecular weight above 100 kDa, and is decreased to ∼60 kDa in COG KO cells (Figure 4, A and C). Most of the Lamp2 in COG-GFP stable cell lines is fully glycosylated, whereas Lamp2 in TMEM-COG cell lines demonstrated partial restoration of glycosylation (Figure 4, B and C). These data demonstrated that TMEM-COG4 and -COG7 can partially restore glycosylation and prevent degradation of these COG-sensitive proteins. However, TMEM-COG8 is less effective at restoring COG function and the COG-sensitive proteins are only slightly restored.
FIGURE 4:
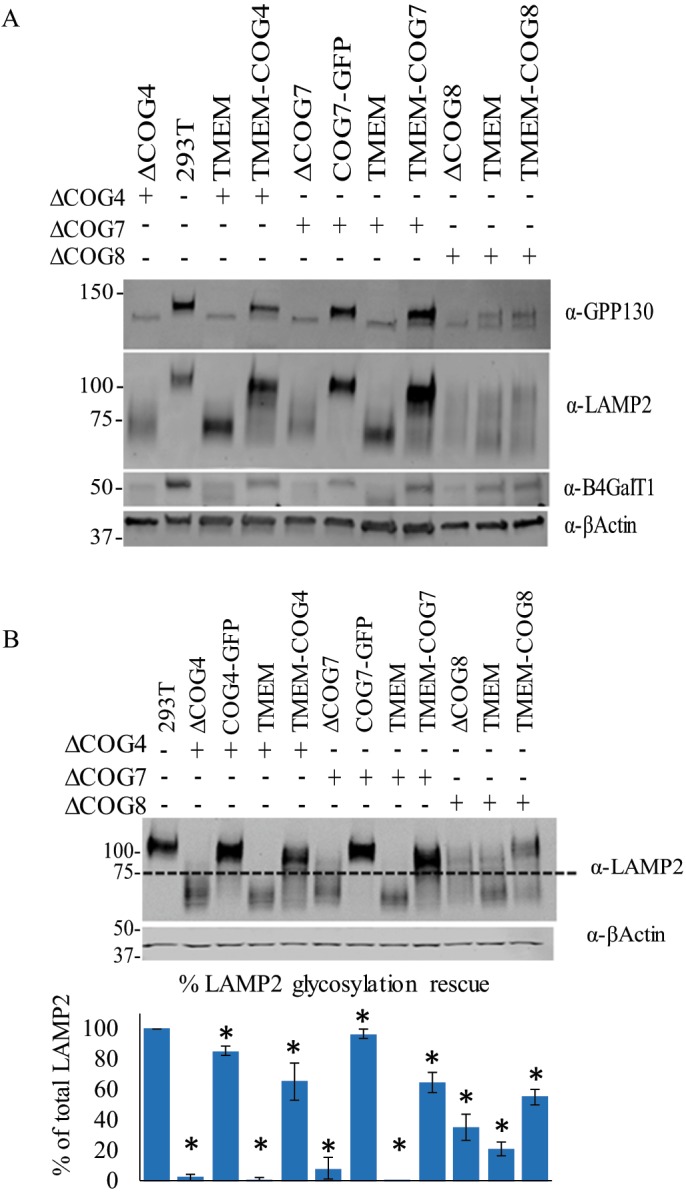
TMEM-COG4 and TMEM-COG7 support Golgi glycosylation and stability of COG-sensitive proteins. HEK293T cells stably expressing TMEM-COG4 in COG4 KO cells, TMEM-COG7 in COG7 KO cells, and TMEM-COG8 in COG8 KO cells. COG4-GFP and COG7-GFP stable expression in COG4 KO cells and COG7 KO cells, respectively. (A) TMEM-COG4 and TMEM-COG7, but not TMEM-COG8, rescue abundance of COG-sensitive proteins: GPP130, LAMP2, and B4GalT1. (B) TMEM-COG mostly restores Lamp2 glycosylation. Western blot and quantification of LAMP2 fully glycosylated species. Dotted line is an arbitrary separation of fully glycosylated and underglycosylated LAMP2. Quantification of three replicates. *p < 0.05 in a Student’s t test compared with 293T. Error bars represent SD.
TMEM-COG restores Cathepsin D sorting
Cathepsin D is cleaved several times throughout the secretory pathway and sorted at the TGN to the endo-lysosomal compartment (Gieselmann et al., 1985; Masson et al., 2010). WT cells do not secrete Cathepsin D into the media, but all COG KO cells abnormally secrete pro-/intermediate Cathepsin D indicating a post-Golgi sorting defect and/or lysosomal defect (Figure 5) (Bailey Blackburn et al., 2016). COG4-GFP and COG7-GFP can rescue the sorting defect of COG4 and COG7 KO, respectively. Importantly, TMEM-COG4, TMEM-COG7, and TMEM-COG8 significantly rescue Cathepsin D sorting (Figure 5B), which indicated that post-Golgi sorting is not dependent on transient COG–membrane interactions.
FIGURE 5:
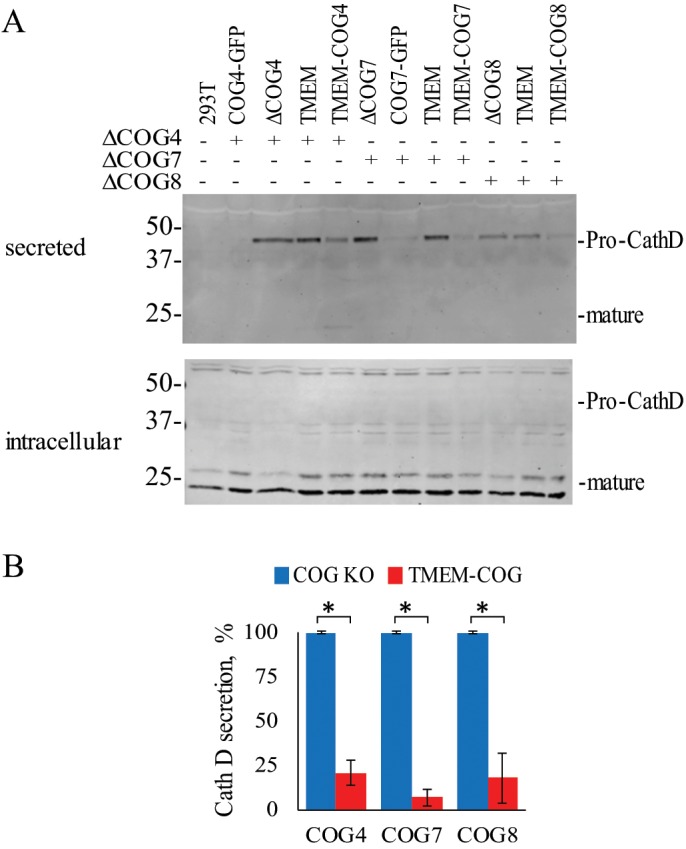
TMEM-COG expression restores Cathepsin D sorting defect of COG KO cells. HEK293T cells stably expressing TMEM-COG4 in COG4 KO cells, TMEM-COG7 in COG7 KO cells, and TMEM-COG8 in COG8 KO cells. COG4-GFP and COG7-GFP stable expression in COG4 KO cells and COG7 KO cells, respectively. (A) Cathepsin D secretion in control, KO, and rescue cells. (B) Comparison of three replicates of pro-cathepsin D secretion in COG4, COG7, and COG8 KO cells rescued with TMEM-COG4, TMEM-COG7, and TMEM-COG8, respectively. *p < 0.05 in a Student’s t test. Error bars represent SD.
TMEM-COG cannot rescue fine Golgi structure
Electron microscopy of high-pressure-frozen cells growing on sapphire disks indicates that mammalian Golgi cisternae in HEK293T cells are thin and tightly stacked (Figure 6A, WT, and Supplemental Figures 3 and 4 WT). On KO of COG4, COG7, or COG8 the cisternae become swollen, irregular structures (Figure 6, A–D, and Supplemental Figure 3). Stable rescue with TMEM-COG4, TMEM-COG7, or TMEM-COG8, in respective COG KO cell lines, does not restore the organized and tightly stacked cisternal structure (Figure 6, A–D, and Supplemental Figure 4). WT cells processed at the same time as COG4, COG7, and COG8 KO cells demonstrated a Golgi stack that showed a fourfold increase in length compared with width (Figure 6B, top). The width of the Golgi stacks in COG4, COG7, and COG8 KO cells were twofold shorter compare to WT (Figure 6B, top), and the expression of TMEM-COG4, -COG7, and -COG8 did not ameliorate this change in Golgi dimensions (Figure 6B, bottom). Additionally, COG4, COG7, and COG8 KO increased the number of spherical, noncisternal elements (NCGE) in the Golgi vicinity compared with the WT Golgi (Figure 6D). TMEM-COG4, -COG7, and -COG8 did not reduce the number of NCGE to the level of WT. However, COG4 KO caused less NCGE (Figure 6C), indicating that COG4 may be less critical for this aspect of Golgi architecture. TMEM-COG4 showed the closest resemblance to WT Golgi structure, which could indicate that the functional COG complex is oriented so that the N-terminus of COG4 is toward the membrane. COG7 and COG8 KO resulted in the greatest destruction to Golgi architecture and increased the number of NCGEs. TMEM-COG7 and TMEM-COG8 did not rescue this phenotype, which indicated that lobe B interactions could have the greatest impact on Golgi morphology and anchored lobe B subunits are predominantly inhibitory to COG function related to fine Golgi architecture. This result is consistent with a COG1–COG8 bridge, which serves as the functional interaction between the two COG lobes. Taken together, these data demonstrate that COG functions partially in the maintenance of Golgi structure and that TMEM-COG cannot fully perform this role.
FIGURE 6:

TMEM-COG does not restore fine Golgi structure. HEK293T cells stably expressing TMEM-COG4 in COG4 KO cells, TMEM-COG7 in COG7 KO cells, and TMEM-COG8 in COG8 KO cells. (A–D) 293T, COG KO, and TMEM-COG transmission EM analysis of Golgi structure. (A) Golgi structure. * Noncisternal Golgi Elements (NCGE). (B) Quantification of Golgi length (blue) and width (red) in 10 cells per condition. (C) Quantification of NCGE. Error bars represent SD. (D) Description of all NCGE in 10 cells per condition. Colored bars represent the diameter (in nm) of individual NCGEs.
DISCUSSION
The COG complex is found in a roughly 60:40 distribution between membrane-bound and the cytosol within cells (Willett et al., 2014). Therefore, COG may need to briefly dissociate from cellular membranes to traffic between the Golgi cisternae, or the soluble pool of COG could be the result of de novo synthesis. To address the functional aspect of the COG-membrane association, we generated cell lines that stably express a COG subunit that is permanently tethered to membranes. Anchored COG subunits are Golgi localized, can integrate into the COG complex, and (with respect to TMEM-COG4 and TMEM-COG7) can restore most of COG function in COG KO cells. These data indicated that neither lobe A (COG4) nor lobe B (COG7) needs to freely associate with or dissociate from Golgi and vesicle membranes.
TMEM-COG4 recruits 80% of its lobe A partner COG3 and restores wild-type distribution of lobe B subunit COG6, indicating that permanently anchored lobe A does not interfere with the normal COG complex assembly–disassembly cycle (Willett et al., 2016). TMEM-COG constructs were surprisingly stable (Figure 1C); therefore, the appearance of a small amount (∼20%) of partner COG subunit in the detached form was not caused by potential proteolytic instability of TMEM-COGs but was likely a result of cell mechanical disruption procedure.
TMEM-COG7 recruits most of lobe B (COG6) and lobe A (COG3) to the membranes. Importantly, cells expressing TMEM-COG4 and TMEM-COG7 show very similar phenotypes in all functional tests, indicating that the increase in lobe A membrane association can be tolerated and that COG function is maintained when most of COG subunits are membrane associated.
Another possibility is that the remaining 20–25% of soluble COG subunits (Figure 3) are capable of forming an incomplete, “crippled” complex and this “crippled” complex is performing the function perceived in the rescued TMEM-COG cells. Indeed, another CATCHR complex, exocyst, has been found to be partially functional in the absence of one subunit (Finger and Novick 1997; Wiederkehr et al., 2004). However, our previously published data (Bailey Blackburn et al., 2016) demonstrate that in the case of human cells, the remaining “crippled” COG arrangements that may form in the absence of any one COG subunit are completely defective in core COG functions in Golgi trafficking, glycosylation, and sorting. Quantification of the membrane (P100) and soluble (S100) fraction (Figure 1D) in TMEM-COG cells demonstrates that there is no detectable soluble form of the eighth subunit contributed by the TMEM-COG construct. Nor is there any detectable amount of the missing COG subunit present in the COG KO cells (Figure 1D) (Bailey Blackburn et al., 2016). Therefore, the rescue phenotype must be attributed to the readdition of the missing eighth subunit in the anchored (TMEM-COG) form.
Additionally, anchored COG8 did not restore COG function in glycosylation and protein sorting to the level of TMEM-COG4 and TMEM-COG7. This indicates that N-terminal attachment inhibits COG8 function nearly to the extent of the complete removal of COG8. COG subcomplexes are thought to be joined by a COG1-COG8 interaction bridge via the N-terminal half of COG8 (Willett et al., 2016). It is possible that TMEM-COG8 inhibits the dynamic interaction with COG1, but TMEM-COG8 can still physically interact with lobe A (COG3) and recruit lobe A to the membrane. This indicates that although TMEM-COG8 still forms the COG complex, the placement of COG8 N-terminus toward the membrane likely results in a conformation of COG that destabilizes the structure and function of the entire complex. TMEM-COG4 and TMEM-COG7 must assume orientations such that the COG complex is functionally productive or is close enough that the flexibility of the subunits can compensate for the altered orientation.
Collectively, these data allow us to clarify our current COG trafficking model. The COG complex mediates transport of vesicles from trans-Golgi cisternae to earlier Golgi cisternae (Figure 7A). Here we show that COG does not need to completely dissociate from Golgi membranes to accomplish this task. Therefore, COG must move with the membranes as they traverse the stack in both anterograde and retrograde directions. COG deficiency has no apparent effect on anterograde transport kinetics. However, COG deficiency destabilizes Golgi structure (Figure 7B) likely through altered protein sorting at the TGN, which not only mislocalizes Golgi markers (Figure 1) but also disorganizes the endolysosomal compartment (Bailey Blackburn et al., 2016). Unlike the cis-oriented endogenous COG, TMEM-COG subunits colocalize equally with both cis- and trans-Golgi markers. Even though TMEM-COG subunits are capable of restoring most of COG function, including segregation of cis and trans-Golgi markers and sorting of Cathepsin D, they are unable to restore a highly organized, flattened Golgi structure (Figure 7C). COG may not completely dissociate from the membrane during trafficking, but COG function in Golgi organization likely relies on more rapid recycling between the compartments than is possible by the recycling route used by TMEM115. Since TMEM-COG is embedded in the membrane it must move with the membrane and be packaged into vesicles for redistribution to earlier cisternae (Figure 7C). The altered recycling dynamic could account for the enrichment of TMEM-COG in the TGN and be fundamentally less productive to the maintenance of an elegant Golgi structure.
FIGURE 7:
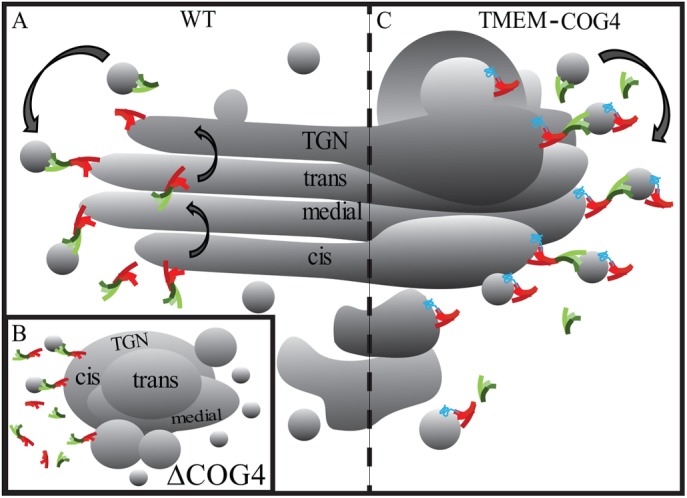
Model depicting COG-dependent intra-Golgi membrane dynamic. (A) The COG complex coordinates retrograde Golgi recycling in WT cells. (B) On COG4 depletion, the COG complex is destabilized destroying Golgi architecture and dysregulating retrograde trafficking and trafficking components. (C) Permanent attachment of COG4 to the membrane with a transmembrane fusion construct restores COG structure and function but only partially restores Golgi organization. Therefore, the trafficking route of TMEM-COG subunits contributed by integral membrane protein TMEM115 likely alters the trafficking dynamic of the COG complex. Trafficking of COG subunits by this method is not amenable to a well-organized Golgi structure and balance between anterograde and retrograde trafficking.
We propose that vesicle tethering complexes could perform the majority of their function in the membrane-associated form. Yeast Dsl1 (Reilly et al., 2001) and mammalian GARP (Liewen et al., 2005) complexes were actually described as preferentially membrane-bound molecules. All vesicle tethers demonstrate multivalent interactions with membrane-bound components of vesicle docking/fusion machinery (Cheung and Pfeffer, 2016). While it is not currently clear what is the exact sequence of protein–protein interactions during vesicle tethering and fusion, our model predicts that these interactions always keep tethering complexes in close proximity to the membrane, making them readily available to interact, proofread, hold, and assist fusion of transport vesicle to their acceptor compartment at every step of secretory and endocytic pathways.
MATERIALS AND METHODS
Antibodies
Antibodies for immunofluorescence microscopy (IF) or WB were purchased through commercial sources, gifts from generous investigators, or generated by this lab. Primary antibodies: mouse anti-βactin (WB 1:500, Sigma), goat anti-B4GalT1 (WB 1:1000, R&D Systems), mouse anti-Cathepsin D (WB 1:500, Sigma), mouse anti-COG3 (WB 1:1000; IF 1:500, this lab), rabbit anti-COG4 (WB 1:1000, this lab), rabbit anti-COG6 (WB 1:1000, this lab), rabbit anti-COG7 (WB 1:500, Dani Ungar), rabbit anti-COG8 (WB 1:500, IF 1:1000, Sigma), rabbit anti-Giantin (1:500, Covance), mouse anti-GM130 (WB 1:1000, BD), rabbit anti-GPP130 (WB 1:1000, Covance), mouse anti-LAMP2 (WB 1:200, DSHB), sheep anti-TGN46 (IF 1:300, Bio-Rad), rabbit anti-mCherry (WB 1:500, this lab), and rabbit anti-TMEM115 (WB 1:500, Sigma). Secondary antibodies: fluorescent dye conjugated AffiniPure Donkey anti-mouse, anti-rabbit, or anti-sheep (IF 1:1000, Jackson Laboratories) and infrared dye IRDye 680 or IRDye 800 anti-mouse and anti-rabbit (WB 1:2000, LI-COR).
Plasmid preparation and transfection
Mammalian expression constructs were generated using standard molecular biology techniques or obtained as generous gifts. See Table 2 for a complete list of plasmids. Plasmids were isolated from bacteria using the QIAprep Spin Miniprep Kits (Qiagen). Plasmid transfections were performed using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s instructions.
TABLE 2:
Plasmids.
| Plasmid name | Source | Citation |
|---|---|---|
| pENTR1A no ccDB (w48-1) | Eric Campeau and Paul Kaufman,a Addgene #17398 | Campeau et al., 2009 |
| pLenti CMV Blast DEST (706-1) | Eric Campeau and Paul Kaufman,a Addgene #17451 | Campeau et al., 2009 |
| pMD2.G | Didier Trono,b Addgene #12259 | Dull et al., 1998 |
| pRSV-Rev | Didier Trono,b Addgene #12253 | Dull et al., 1998 |
| pMDLg/pRRE | Didier Trono,b Addgene #12251 | Dull et al., 1998 |
| TMEM115-mCh pLenti CMV Blast DEST | This study | This study |
| TMEM115-COG4-mCh pLenti CMV Blast DEST | This study | This study |
| TMEM115-COG7-mCh pLenti CMV Blast DEST | This study | This study |
| TMEM115-COG8-mCh pLenti CMV Blast DEST | This study | This study |
| COG4-GFP pLenti CMV Blast DEST | This study | This study |
| COG7-2xGFP-3myc pLenti CMV Blast DEST | This study | This study |
| mCh-STX16∆SD | This study | This study |
| COG4-mCh-STX16∆SD | This study | This study |
| COG7-mCh-STX16∆SD | This study | This study |
| COG8-mCh-STX16∆SD | This study | This study |
| COG8-mCh-GS15TMD | This study | This study |
| COG8-mCh-STX5TMD | This study | This study |
| COG8-mCh-GS27TMD | This study | This study |
aUniversity of Massachusetts Medical School.
bCell Genesys.
Generation of lentiviral particles and stable cell lines
TMEM115-COG-mCh and COG-GFP plasmids were subcloned into the pENTR1A no ccDB (w48-1) (Addgene #17398) entry vector and recombined into pLenti CMV Blast DEST (706-1) (Addgene #17451) destination vector using Gateway LR Clonase II Enzyme Mix (Thermo Fisher) according to the manufacturer’s instructions. Lentiviral particles were generated using the 293FT cell line. In brief, 293FT cells were plated in a six-well plate coated with poly-l-lysine. Cells (100% confluent) were equilibrated in serum-free Opti-MEM with GlutaMAX and 25 µM chloroquine. Equal amounts of the three lentiviral packaging plasmids pMD2.G (Addgene #12259), pRSV-Rev (Addgene #12253), and pMDLg/pRRE (Addgene #12251) plus destination plasmid containing COG or control sequence were transfected using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s instructions. Six hours after transfection, media was supplemented with sodium butyrate to a final concentration of 10 µM. Twenty-four hours after transfection, the media was replaced with serum-free Opti-MEM with GlutaMAX. Forty-eight hours after transfection, the media was collected and cell debris removed by centrifugation at 2000 × g for 10 min and lentiviral media was stored at 4°C overnight. Seventy-two hours after transfection, the media was collected, debris removed, and lentiviral media added to the original collection.
Lentiviral media (1 ml) was used to transduce COG4, COG7, or COG8 KO cell lines in a six-well dish. Twenty-four hours after transduction, media was replaced with 10% fetal bovine serum (FBS), without antibiotic/antimicotic in DMEM/F-12. Forty-eight hours after transduction, media was replaced with 10 µg/ml blasticidin selection media. A 10-cm dish of blasticidin-selected cell populations were sorted by FACS Aria (BD Biosciences) for mCherry fluorescence. All data were generated after sorting for low levels of mCherry fluorescence signal, except for Figure 2 which was generated from presort mixed fluorescence cell populations.
Immunofluorescence
Cells were grown on 12-mm glass coverslips (#1, 0.17 mm thickness) so that they would be 70–90% confluent at harvest. In short, cells were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (16% stock solution; Electron Microscopy Sciences). Cells were permeabilized with 0.1% Triton X-100 for 1 min and then treated with 50 mM ammonium chloride for 5 min. Cells were washed with PBS and blocked twice for 10 min with 1% BSA, 0.1% saponin in PBS. Cells were incubated for 1 h with primary antibody diluted in 1% fish gelatin, 0.1% saponin in PBS at room temperature. Cells were washed four times with PBS and incubated for 30 min with fluorescently tagged secondary antibody in fish gelatin, saponin at room temperature. Coverslips were washed four times with PBS, rinsed with double-distilled H2O, and mounted on glass microscope slides using Prolong Gold antifade reagent (Thermo Fisher).
Microscopy: Airyscan, 3D-SIM, and colocalization analysis
Cells were imaged with the 63× oil 1.4 numerical aperture objective of a LSM880 Zeiss Laser inverted microscope outfitted with confocal optics and four lines of excitation lasers (405, 488, 561, and 642 nm) under the control of Zen 2012 software (Zeiss). Cells were also imaged using Zeiss ELYRA S1 Super-Resolution Microscope with the 100 × 1.46 numerical aperture oil objective under the control of the Zen 2012 software (Zeiss). Colocalization was obtained using the colocalization module built into the Zen software.
Subcellular fractionation
Stable cells were harvested at 100% confluence from a 10-cm dish. Cells were washed twice with PBS and then lysed in 20 mM HEPES-KOH, pH 7.2, 150 mM KCl, 2 mM EDTA, 1 mM DTT, 5 μl/ml Halt protease inhibitor cocktail (Thermo Fisher), and 1 mM PMSF at room temperature. Cells were passaged 15 times through a 27g needle to achieve complete lysis. Unlysed cells and debris were removed by centrifugation at 500 × g for 5 min. Supernatant was collected and centrifuged at 100,000 × g in the Beckman Optima ultracentrifuge using TLA 55 rotor for 1 h at 18°C. Supernatant was collected as the soluble fraction (S100). The pellet was solubilized in lysis buffer with 1% Triton X-100. Solubilized pellet was centrifuged at 20,000 × g for 10 min, and the supernatant was collected as the membrane fraction (P100). Quantification is a percentage of the total amount of signal in the P100 and S100 combined and reproduced in three separate cellular passage numbers.
GFP and mCherry pull down
Stable cells were washed twice in PBS and lysed in 50 mM Tris, 150 mM NaCl, pH 7.4, containing 1% Triton, 5 μl/ml Halt protease inhibitor cocktail (Thermo Fisher), and 1 mM PMSF at 4°C for 30 min. Cell lysate was spun at 20,000 × g and supernatant was added to 25 μl of 50% GFP-binding protein-bead (GBP-bead) or mCherry-binding protein (MBP-bead) suspension. Bead and lysates were mixed by rotation for 2 h at room temperature. Unbound material was removed and the beads were washed three times in 0.05% Triton X-100 in PBS and eluted in 2X sample buffer to a total volume of 50 μl.
Western blot
Total lysates, IPs, and subcellular fractions were analyzed by SDS–PAGE and then transferred to nitrocellulose. Blots were incubated with primary antibodies, followed by incubation with appropriate secondary antibodies conjugated with IRDye 680 or IRDye 800 dyes (LI-COR). Blots were scanned and analyzed with an Odyssey IR imaging system (LI-COR).
High-pressure freezing, freeze substitution, and EM
Sapphire disks were coated in carbon followed by collagen (Corning) per the manufacturer’s instructions. Coated disks were sterilized under UV light and transferred to new sterile 3-cm dishes (one dish with three disks per sample) and plated with cells. After reaching 80–100% confluence, cells were equilibrated in fresh media for 2–3 h at 37°C followed by high-pressure freezing. High-pressure freezing was performed in cryo-protectant (PBS with 2% low-melt agarose, 100 m MD-mannitol, and 2% FBS) using a Leica EMPACT2 high-pressure freezer with rapid transfer system, and then disks were transferred under liquid nitrogen into tubes containing a prefrozen staining cocktail (acetone with 2% osmium tetroxide, 0.1% glutaraldehyde, and 1% ddH2O). Next, the tubes were transferred to a freeze substitution chamber at –90°C programmed with the following schedule: –90°C for 22 h, warm 3°C/h to –60°C, –60°C for 8 h, warm 3°C/h to –30°C, –30°C for 8 h, warm 3°C/h to 0°C. Afterward, sample tubes were then placed on ice and washed three times with acetone. Samples were than stained with a 1% tannic acid, 1% double-distilled H2O solution in acetone on ice for 1 h followed by three acetone washes. Next, samples were stained with a 1% OsO4, 1% ddH2O solution in acetone on ice for 1 h followed by three acetone washes. Samples were embedded in Araldite 502/Embed 812 resin with DMP-30 activator added in a Biowave at 70°C under vacuum for 3 min for each embedding step. Samples were then baked at 60°C for 48 h before sectioning. Samples were stained postsectioning with uranylacetate and lead citrate. Ultrathin sections were imaged at 80 kV on a FEI Technai G2 TF20 transmission electron microscope and images were acquired with a FEI Eagle 4k XUSB Digital Camera.
For quantification, 293T COG KO cells were compared with WT cells processed at the same time. Similarly, 293T TMEM-COG stable cell lines were compared with WT cells processed at the same time. Golgi of 10 cells were counted per condition. Golgi dimensions were quantified using the Line Marker tool of TEM Imaging and Analysis software (FEI Company).
Supplementary Material
Acknowledgments
We are thankful to Dani Ungar and Brian Storrie, as well as others who provided reagents. We thank Tetyana Kudlyk for excellent technical support. We also thank the DNA Sequencing and Flow Cytometry cores (National Institutes of Health [NIH] grant P20GM103625), and the Digital Microscopy core facilities for the use of their equipment and expertise. This work was supported by the NIH grants GM083144 and U54 GM105814.
Abbreviations used:
- 3D-SIM
3-dimensional structured illumination microscopy
- CATCHR
complexes associated with tethering containing helical rods
- CDG
congenital disorders of glycosylation
- COG
conserved oligomeric Golgi
- COPII
coat protein complex II
- CRISPR
clustered regularly interspaced short palindromic repeats
- EARP
endosome-associated recycling protein
- EM
electron microscopy
- ER
endoplasmic reticulum
- FACS
fluorescence-activated cell sorting
- GARP
Golgi-associated retrograde protein
- GBP
GFP-binding protein
- GFP
green fluorescent protein
- IP
immunoprecipitation
- KO
knockout
- MBP
mCherry-binding protein
- NCGE
noncisternal Golgi elements
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- TGN
trans-Golgi network
- TMD
transmembrane domain
- TMEM115
transmembrane protein 115
- TMEM-COG
TMEM115-COG-mCherry
- WB
Western blot
- WT
wild type
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-11-0694) on February 19, 2018.
REFERENCES
- Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin VV. (2016). COG complex complexities: detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front Cell Dev Biol , 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn JB, Lupashin VV. (2016). Creating knockouts of conserved oligomeric Golgi complex subunits using CRISPR-mediated gene editing paired with a selection strategy based on glycosylation defects associated with impaired COG complex function. Methods Mol Biol , 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. (2004). The mechanisms of vesicle budding and fusion. Cell , 153–166. [DOI] [PubMed] [Google Scholar]
- Brandon E, Szul T, Alvarez C, Grabski R, Benjamin R, Kawai R, Sztul E. (2006). On and off membrane dynamics of the endoplasmic reticulum-golgi tethering factor p115 in vivo. Mol Biol Cell , 2996–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One , e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh LF, Chen X, Richardson BC, Ungar D, Pelczer I, Rizo J, Hughson FM. (2007). Structural analysis of conserved oligomeric Golgi complex subunit 2. J Biol Chem , 23418–23426. [DOI] [PubMed] [Google Scholar]
- Cheung PY, Pfeffer SR. (2016). Transport vesicle tethering at the trans Golgi network: coiled coil proteins in action. Front Cell Dev Biol , 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HT, Dukovski D, Chambers MG, Reinisch KM, Walz T. (2016). CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat Struct Mol Biol , 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climer LK, Dobretsov M, Lupashin V. (2015). Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. Front Neurosci , 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol , 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner M, Hashimoto H, Simpson JC, Cassel D, Nilsson T, Weiss M. (2003). Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep , 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger FP, Novick P. (1997). Sec3p is involved in secretion and morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell , 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotso P, Koryakina Y, Pavliv O, Tsiomenko AB, Lupashin VV. (2005). Cog1p plays a central role in the organization of the yeast conserved oligomeric golgi complex. J Biol Chem , 27613–27623. [DOI] [PubMed] [Google Scholar]
- Foulquier F, Vasile E, Schollen E, Callewaert N, Raemaekers T, Quelhas D, Jaeken J, Mills P, Winchester B, Krieger M. (2006). Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA , 3764–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann V, Hasilik A, von Figura K. (1985). Processing of human cathepsin D in lysosomes in vitro. J Biol Chem , 3215–3220. [PubMed] [Google Scholar]
- Glick BS, Elston T, Oster G. (1997). A cisternal maturation mechanism can explain the asymmetry of the Golgi stack. FEBS Lett , 177–181. [DOI] [PubMed] [Google Scholar]
- Glick BS, Nakano A. (2009). Membrane traffic within the Golgi apparatus. Annu Rev Cell Dev Biol , 113–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha JY, Chou HT, Ungar D, Yip CK, Walz T, Hughson FM. (2016). Molecular architecture of the complete COG tethering complex. Nat Struct Mol Biol , 758–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha JY, Pokrovskaya ID, Climer LK, Shimamura GR, Kudlyk T, Jeffrey PD, Lupashin VV, Hughson FM. (2014). Cog5-Cog7 crystal structure reveals interactions essential for the function of a multisubunit tethering complex. Proc Natl Acad Sci USA , 15762–15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijes HA, Jaeken J, Foulquier F, van Hasselt PM. (2018). Hypothesis: lobe A (COG1–4)-CDG causes a more severe phenotype than lobe B (COG5–8)-CDG. J Med Genet , 137–142. [DOI] [PubMed] [Google Scholar]
- Kim DW, Sacher M, Scarpa A, Quinn AM, Ferro-Novick S. (1999). High-copy suppressor analysis reveals a physical interaction between Sec34p and Sec35p, a protein implicated in vesicle docking. Mol Biol Cell , 3317–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley DM, Krieger M. (1984). Receptor-mediated endocytosis of low density lipoprotein: somatic cell mutants define multiple genes required for expression of surface- receptor activity. Proc Natl Acad Sci USA , 5454–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Yip CK, Walz T, Hughson FM. (2010). Molecular organization of the COG vesicle tethering complex. Nat Struct Mol Biol , 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liewen H, Meinhold-Heerlein I, Oliveira V, Schwarzenbacher R, Luo G, Wadle A, Jung M, Pfreundschuh M, Stenner-Liewen F. (2005). Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp Cell Res , 24–34. [DOI] [PubMed] [Google Scholar]
- Masson O, Bach AS, Derocq D, Prebois C, Laurent-Matha V, Pattingre S, Liaudet-Coopman E. (2010). Pathophysiological functions of cathepsin D: Targeting its catalytic activity versus its protein binding activity?. Biochimie , 1635–1643. [DOI] [PubMed] [Google Scholar]
- Mironov AA, Weidman P, Luini A. (1997). Variations on the intracellular transport theme: maturing cisternae and trafficking tubules. J Cell Biol , 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E, Zeevaert R, Korsch E, Huijben K, Wopereis S, Matthijs G, Keymolen K, Lefeber DJ, De Meirleir L, Wevers RA. (2007). A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet , 638–645. [DOI] [PubMed] [Google Scholar]
- Oka T, Ungar D, Hughson FM, Krieger M. (2004). The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell , 2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong YS, Tran TH, Gounko NV, Hong W. (2014). TMEM115 is an integral membrane protein of the Golgi complex involved in retrograde transport. J Cell Sci , 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HR. (2001). Traffic through the Golgi apparatus. J Cell Biol , 1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. (2011). Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology , 1554–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Ward TH, Pfeifer AC, Siggia ED, Phair RD, Lippincott-Schwartz J. (2002). Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature , 187–193. [DOI] [PubMed] [Google Scholar]
- Ram RJ, Li BJ, Kaiser CA. (2002). Identification of Sec36p, Sec37p, and Sec38p: Components of yeast complex that contains Sec34p and Sec35p. Mol Biol Cell , 1484–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly BA, Kraynack BA, VanRheenen SM, Waters MG. (2001). Golgi-to-endoplasmic reticulum (ER) retrograde traffic in yeast requires Dsl1p, a component of the ER target site that interacts with a COPI coat subunit. Mol Biol Cell , 3783–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BC, Smith RD, Ungar D, Nakamura A, Jeffrey PD, Lupashin VV, Hughson FM. (2009). Structural basis for a human glycosylation disorder caused by mutation of the COG4 gene. Proc Natl Acad Sci USA , 13329–13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova A, Smith RD, Pavliv O, Khaidakova G, Lupashin V. (2007). The COG complex interacts with golgi t-SNARE Syntaxin5 to direct trafficking of recycling intra-Golgi vesicles. J Gen Physiol , 11a–11a.17591984 [Google Scholar]
- Shestakova A, Zolov S, Lupashin V. (2006). COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic , 191–204. [DOI] [PubMed] [Google Scholar]
- Suvorova ES, Duden R, Lupashin VV. (2002). The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J Cell Biol , 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, Heuser JE, Krieger M, Waters MG. (2002). Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol , 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar D, Oka T, Vasile E, Krieger M, Hughson FM. (2005). Subunit architecture of the conserved oligomeric Golgi complex. J Biol Chem , 32729–32735. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. (1998). Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol , 1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Sapperstein SK, Chiang EC, Lupashin VV, Barlowe C, Waters MG. (1999). Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J Cell Biol , 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JR, Munro S. (2002). Vesicle tethering complexes in membrane traffic. J Cell Sci , 2627–2637. [DOI] [PubMed] [Google Scholar]
- Whyte JRC, Munro S. (2001). The SeC34/35 golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev Cell , 527–537. [DOI] [PubMed] [Google Scholar]
- Wiederkehr A, De Craene JO, Ferro-Novick S, Novick P. (2004). Functional specialization within a vesicle tethering complex: bypass of a subset of exocyst deletion mutants by Sec1p or Sec4p. J Cell Biol , 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Blackburn JB, Climer L, Pokrovskaya I, Kudlyk T, Wang W, Lupashin V. (2016). COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci Rep , 29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V. (2013a). COG complexes form spatial landmarks for distinct SNARE complexes. Nat Commun , 1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Pokrovskaya I, Kudlyk T, Lupashin V. (2014). Multipronged interaction of the COG complex with intracellular membranes. Cell Logist , e27888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Ungar D, Lupashin V. (2013b). The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem Cell Biol , 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Hughson FM. (2010). Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol , 137–156. [DOI] [PubMed] [Google Scholar]
- Zeevaert R, Foulquier F, Cheillan D, Cloix I, Guffon N, Sturiale L, Garozzo D, Matthijs G, Jaeken J. (2009). A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur J Med Genet , 303–305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.