

ARE-mRNAs are actively degraded with tristetraprolin (TTP) in resting cells while they turn into stable messengers in activated cells. P38 plays a crucial role in stabilizing ARE-mRNA. Here we reveal that P38 activation represses the interaction between TTP and Ago2, thus restraining TTP from being targeted into processing bodies and stabilizing ARE-mRNA.
Abstract
Tristetraprolin (TTP) destabilizes AU-rich element (ARE)-containing mRNA by directly binding with their 3′UTR. P38 stimulation substantially increases ARE-mRNA stability, at least through repressing TTP. However, the mechanism by which P38 keeps TTP inactive has not been fully understood. TTP and ARE-mRNA localize to processing bodies (PBs), the mRNA granules associated with mRNA silencing. Here, we detected the influence of P38 on TTP localization within PBs and found that P38 regulates TTP localization within PBs. Through luciferase-based systems, we demonstrated that PBs depletion significantly increased ARE-mRNA stability inhibited by TTP. Additionally, we provided evidence that the microRNA-induced silencing complex (miRISC) core member Ago2 is required for TTP distribution within PBs. Importantly, the cooperation of TTP and Ago2 is a prerequisite for effective ARE-mRNA degradation. Moreover, Dcp1a and Dcp2 act downstream of Ago2 and TTP engaging in ARE-mRNA decay. Finally, we demonstrated that P38 activation represses the interaction between TTP and Ago2 due to TTP phosphorylation, which impairs TTP localization within PBs and ARE-mRNA degradation. Collectively, our study revealed a novel mechanism through which P38 activation repressed the cooperation of TTP with Ago2, thus ensuring that ARE-mRNA does not associate with PBs and remains stable.
INTRODUCTION
Posttranscriptional regulation of gene expression involves RNA sequences that collaborate with trans-acting factors to regulate mRNA instability and translation. AU-rich elements (AREs) are key posttranscriptional regulation elements that promote translation silencing and rapid turnover, which reside in the 3′ untranslated region (UTR) of many human mRNAs (referred to as ARE-mRNAs) including those encoding interleukins, cytokines, and proto-oncogenes (Chen and Shyu, 1995; Shim and Karin, 2002; Zhang et al., 2002; Mazan-Mamczarz et al., 2006). AREs have been well characterized as regulators of ARE-mRNA instability and translation repression (Mukherjee et al., 2002; Qi et al., 2012). Targeted deletion of the tumor necrosis factor alpha (TNF-α) mRNA ARE in mice (ΔARE mice) results in TNF-α overproduction and the development of a systemic inflammatory syndrome (Kontoyiannis et al., 1999).
A number of ARE-binding proteins (AUBPs), which activate or suppress target gene decay, have been identified. Tristetraprolin (TTP) (also known as Nup475, TIS11, or Zfp36) is one of the best-characterized ARE-binding proteins containing three tetraproline (PPPP) and two CCCH zinc finger domains required for interaction with ARE. It mediates ARE-mRNA instability and translational repression (Qi et al., 2012; Tao and Gao, 2015; Fu et al., 2016) by directly binding to ARE. The importance of TTP as a physiological regulator of gene expression is manifested in TTP knockout mice, in which chronic inflammation has been directly attributed to elevated levels of TNF-α and granulocyte-macrophage colony-stimulating factor (GM-CSF) protein in TTP−/− macrophages (Carballo et al., 1997, 2000). TTP promotes ARE-mRNA degradation through recruiting a number of mRNA decay enzymes responsible for deadenylation, decapping, and exonucleolytic decay (Fenger-Gron et al., 2005; Lykke-Andersen and Wagner, 2005). TTP could also be involved in a microRNA16-mediated ARE mRNA degradation, through interaction with Argonaute 2 (Ago2) (Jing et al., 2005). Ago2 is the essential functional effector of the microRNP–the slicer in microRNA-mediated decay or the repressor in microRNA-mediated translational repression (Meister et al., 2004; Kiriakidou et al., 2007).
On the other hand, ARE also confers its transcript stability in response to rapidly changing cellular environment. Several cell signaling pathways are known to modulate mRNA stabilization or translation (Lee et al., 2011; Tiedje et al., 2012; Rataj et al., 2016). The P38 mitogen-activated protein kinase (MAPK) pathway plays a very important role in regulating ARE-mRNA stability (Mahtani et al., 2001; Dean et al., 2004; Brook et al., 2006; Hitti et al., 2006; Tudor et al., 2009; McGuire et al., 2017). P38 is activated by proinflammatory stimuli such as bacterial lipopolysaccharide (LPS), TNF-α, and then phosphorylates its own substrates, including the kinase MAPK-activated protein kinase 2 (MAPKAPK2, MK2). P38 activation has been shown to impair ARE-containing mRNA degradation, leading to mRNA stabilization. Macrophages from MK2−/− mice showed significantly decreased levels of TNF-α, IL-1, IL-6, and IFN γ due to reduced cytokine mRNA stability (Kotlyarov et al., 1999; Neininger et al., 2002). MK2 can deliver signals from P38 and then directly phosphorylate TTP at Ser-52 and Ser-178. Phosphorylation of TTP by MK2 creates a substrate for the adaptor protein 14-3-3 (Stoecklin et al., 2004), which protects TTP from dephosphorylation by protein phosphatase 2A (PP2A) (Sun et al., 2007). It has been reported that phosphorylated TTP lose the ability to combine with ARE-mRNA, thus stabilizing mRNA (Carballo et al., 2001; Cao, 2004; Hitti et al., 2006). However, a growing body of evidence indicate that TTP effectively retains its affinity with target genes under P38 activation condition (Mahtani et al., 2001; Stoecklin et al., 2004; Sun et al., 2007; Clement et al., 2011). Moreover, extant studies show that TTP phosphorylation prevents recruitment of cytoplasm deadenylase complex (Marchese et al., 2010; Clement et al., 2011), thereby promoting ARE-mRNA stability. Although it is presently understood that ARE-mRNA stabilization regulation is mediated by P38 and TTP, the detailed underlying mechanisms require further investigation.
TTP and ARE-mRNA were observed to reside in discrete cytoplasm foci called processing bodies (PBs) (Franks and Lykke-Andersen, 2007; Balagopal and Parker, 2009), in which a number of proteins involved in translational silencing and mRNA decay (Eulalio et al., 2007; Balagopal and Parker, 2009). These proteins include the decapping enzyme Dcp2, the 5′-to-3′exonuclease Xrn1, and factors that stimulate decapping or inhibit general translation (Sheth and Parker, 2003). As PBs are very dynamic RNA granules, they can rapidly exchange components with the surrounding cytoplasm and can thus quickly emerge and disappear (Buchan, 2014). In addition, the assembly and formation of PBs are precisely controlled by certain signal pathways (Rzeczkowski et al., 2011; Conforto et al., 2012; Blanco et al., 2014), implying that PBs may play a critical role in mRNA posttranscriptional regulation.
The goal of this study was to verify the function of PBs in P38-mediated ARE-mRNA posttranscriptional modulation. We provided the preliminary evidence that PB integrity is important for ARE-mRNA destabilization induced by TTP. Ago2 is responsible for delivering TTP into PBs for effective degradation of ARE-mRNA. Dcp1a and Dcp2 within PBs act downstream of Ago2 participating in ARE-mRNA decay. P38 activation represses the interaction of TTP with Ago2 by phosphorylating TTP at Ser-52 and Ser-178, thereby blocking the main mechanism by which TTP is directed into PBs, promoting ARE-mRNA stabilization. P38 activity regulates ARE-mRNA turnover by dynamically modifying the cooperation between TTP and Ago2.
RESULTS
P38 activation represses the accumulations of TTP and ARE-mRNA within PBs
We first detected the distribution of TTP in HeLa cells. Owing to the lack of a TTP antibody suitable for immunofluorescence imaging, a construct coding for TTP-EGFP (enhanced green fluorescent protein) was transiently delivered into HeLa cells. Dcp1a, a well-characterized PB component for mRNA decapping in eukaryotes, was used as a marker to visualize PBs by fluorescence microscopy. The endogenous Dcp1a was stained with its polyclonal antibody and looked distributed in a focal pattern (Supplemental Figure S1A, top panel), which was expected as PBs. The endogenous Dcp1a distribution pattern was consistent with that of ectopically expressed Dcp1a fused with red fluorescent protein (RFP-Dcp1a) (Supplemental Figure S1A, bottom panel), thus we used the anti-Dcp1a antibody in the subsequent experiments. TTP-EGFP appeared to cluster in cytoplasmic foci that overlapped PBs either marked by an anti-Dcp1a antibody or by RFP- Dcp1a (Supplemental Figure S1A). We conclude that TTP mainly accumulated within PBs in HeLa cells.
It is well known that in mouse macrophages, such as RAW 264.7 cells, TTP is transiently induced and extensively phosphorylated on LPS stimulation. We tested whether TTP localization would produce any change in this scenario. TNF-α can simultaneously activate several MAPKs family members (P38 MAPK, ERK, and JNK) and was used to treat HeLa cells at the concentration of 10 ng/ml for 2 h. TTP became extensively diffused in the cytoplasm and did not localize to PBs (Figure 1, A and B), while the focal pattern of PBs was preserved under TNF-α stimulation (Figure 1A). The diffused localization of TTP was confirmed by transient expression of HA-TTP followed by anti-HA staining (unpublished data). These results suggest that TTP localization within PBs could be regulated by the MAPK signal cascade.
FIGURE 1:

P38 activation promotes the dissociations of TTP and ARE-mRNA from PBs. (A) HeLa cells transiently transfected with TTP-EGFP were pretreated with DMSO (10 μM) as a control or SB (10 μM), PD (30 μM), and SP (50 μM) for 1 h before TNF-α addition (10 ng/ml) for 2 h. PBs were visualized using the anti-Dcp1a antibody. (B) Graph showing the P-body numbers per cell that is positive for TTP as in A. Error bars present SD (n = 30 cells per group). **p ≤ 0.01. (C) HeLa cells were transfected with TTP-EGFP plasmid, together with plasmid expressing MKK6b(E), MKK6b(A), or its empty vector. PBs were displayed with anti-Dcp1a staining. (D) Graph showing the P-body numbers per cell which is positive for TTP as in C. Error bars present SD (n = 30 cells per group). *p ≤ 0.05; **p ≤ 0.01. (E) Schematic of the MS2 system for fluorescence-based mRNA visualization. The mRNA containing MS2 sites present downstream of the ARE-containing GM-CSF 3′ UTR is bound by GFP-tagged MS2-binding protein, allowing fluorescent visualization of the mRNA. (F) HeLa cells were transfected with the MS2 dual plasmid system using the luciferase-GM-CSF 3′ UTR mRNA expression constructs, along with MS2-GFP. MS2-GFP was used to visualize mRNA. Cells were cultured in media alone (Control), treated for 2 h with TNF-α (10 ng/ml), and pretreated with SB (10 μM) for 1 h before TNF-α addition (10 ng/ml). PBs were visualized using anti-Dcp1a antibody. Results shown are representative of those from three experiments. (G) Graph showing the P-body numbers per cell that is positive for MS2-GFP. Error bars present SD (n = 30 cells per group). *p ≤ 0.05; **p ≤ 0.01.
To identify the potential signal pathways responsible for redistribution of TTP, a number of specific MAPK inhibitors was used to individually pretreat HeLa cells for 1 h before TNF-α incubation, including P38 inhibitor SB 203580 (SB), ERK inhibitor PD 98059 (PD), and JNK inhibitor SP 600125 (SP) with DMSO as negative control. Among these inhibitors only SB prevented diffusion of TTP from PBs to the cytoplasm (Figure 1, A and B), indicating that TTP dispersion from PBs in presence of TNF-α is dependent on P38 activation. P38 activity affects neither the number nor the apparent size of PBs, as was demonstrated in RAW264.7, P38α+/+ or P38α–/– MEF cells, treated with SB, TNF-α or LPS (Supplemental Figure S1, B and C), suggesting that the causation that promotes TTP release from PBs comes from TTP itself rather than of PBs. We further determine whether the increased foci structure under SB treatment is stress granules (SGs), which do not appear in the cytoplasm in general condition. Our results showed that SB treatment did not promote SGs generation as visualized by anti-HuR staining, although SG was greatly induced by FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) treatment (Supplemental Figure S1D). In contrast, PD and SP had no effect on TTP localization within PBs on TNF-α stimulation (Figure 1, A and B). Collectively, these results suggest that P38, but not ERK or JNK, specifically regulates TTP localization in PBs.
MKK6b is a specific activator kinase of P38. Its constitutively active mutant MKK6b (E) constantly keeps P38 phosphorylated and active, while catalytically dead mutant MKK6b (A) is incapable of P38 activation (Han et al., 1996; Raingeaud et al., 1996; Stein et al., 1996; Huang et al., 1997). MKK6b (E) or MKK6b (A) was ectopically expressed in HeLa cells to modulate P38 activity. Like TNF-α stimulation, P38 activation by MKK6b (E) reduced TTP localization within PBs (Figure 1, C and D), while P38 inhibition by MKK6b (A) yielded the opposite effect (Figure 1, C and D). Together, these results confirmed that P38 activity directly affects TTP targeting within PBs.
P38 activation also represses ARE-mRNA localization within PBs
Since TTP is an ARE-binding protein and P38 modifies its localization within PBs, we reasoned that P38 activity could also regulate ARE-mRNA accumulation in PBs. To test this possibility, a chimera reporter was constructed that fuses luciferase coding sequence with ARE-containing human GM-CSF mRNA 3′ UTR and 24 binding sites for the MS2 coat protein (Figure 1E). These MS2-binding sites allowed us to follow the localization of ARE-mRNAs by coexpression of an MS2-GFP fusion protein (Liu et al., 2005). In HeLa cells, the reporter with GM-CSF 3′ UTR exhibited the distribution pattern of discrete cytoplasmic foci marked by Dcp1a, indicating localization of ARE-mRNA in PBs (Figure 1F). TNF-α stimulation rendered GM-CSF reporter mRNA substantially diffused into cytoplasm while SB treatment abolished the diffusion of the reporter mRNA from PBs (Figure 1, F and G). Therefore, ARE-mRNA and TTP demonstrated an identical localization response to TNF-α stimulation. The aforementioned data indicate that TTP and ARE-mRNA can be localized to PBs, depending on P38 activity.
P-body integrity is essential for ARE-mRNA degradation triggered by TTP
Since P38 activity modulates the TTP and ARE-mRNA localization in PBs, we speculate that PBs may be responsible for ARE-mRNA degradation, and P38 stabilizes ARE-mRNA through inhibition of TTP targeting into PBs. To test our hypothesis, we set out to detect the effect of PB depletion on ARE-mRNA abundance. A construct described previously was adopted (Qi et al., 2012), comprising a firefly luciferase reporter followed by the ARE-containing 3′ UTR of GM-CSF mRNA. Renilla luciferase (RL) was chosen as an internal control for all firefly luciferase assays. As shown in Supplemental Figure S2A, TTP overexpression decreased GM-CSF reporter mRNA level by ∼50% compared with Septin1, a cell mitosis-related cytoskeletal protein that served as a control. Similar results were obtained with a different set of FL- or RL-specific primers (unpublished data).
Several factors have been reported to be essential for P-body integrity, including the decapping enhancer Hedls or Lsm1. To disrupt the PB structure, we silenced Hedls with two small interfering RNAs (siRNAs) in 293T cells. The siHedls-1 and siHedls-2 decreased the abundance of Hedls protein to ∼50 and 20% of control siRNA, respectively (Supplemental Figure S2B). As shown in Figure 2A, no PBs could be observed, as evidenced by the uniform cytoplasm distribution of Dcp1a after Hedls silencing. To determine whether PB integrity affects ARE-mRNA turnover, GM-CSF reporter mRNA was assayed with RNAi of Hedls. In this case, the FL mRNA level in the presence of HA-TTP was normalized to that with HA-Septin1 to minimize the potential nonspecific effects of RNA silencing. As expected, overexpression of TTP reduced FL-GM-CSF reporter mRNA level by ∼40% of that measured in the control. Silencing Hedls by siHedls-1 recovered the mRNA level from 40% to 80% in the presence of TTP, and similar change was obtained for siHedls-2 (Figure 2B). An additional reporter FL-TNF hosting the entire human TNF-α 3′UTR was used to further examine our observation. Indeed, Hedls knockdown by either siHedls-1 or siHedls-2 alleviated the diminishing of the FL-TNF reporter mRNA induced by TTP (Figure 2C). Thus, these experiments imply that PB integrity may be involved in ARE-mRNA turnover mediated by TTP.
FIGURE 2:

PB integrity is indispensable for ARE-mRNA degradation induced by TTP. (A) Hedls knockdown completely destroyed the PB integrity. HeLa cells transfected with control siRNA (siNC) or siHedls-1 or siHedls-2 were stained with anti-Dcp1a antibody to visualize PBs. (B) PB depletion by Hedls knockdown increases FL-GM-CSF reporter mRNA level mediated by TTP. 293T cells transfected with control siRNA (siNC) or siHedls-1 or siHedls-2 were also transfected with FL-GM-CSF reporter plasmid and overexpression plasmids HA-tagged TTP or Septin1. The normalized values of FL mRNA level were set to 1 for cells transfected with Septin1 in each knockdown condition. Means and SD from three independent experiments are shown. *p < 0.05; **p < 0.01. (C) Results of an experiment similar to that for B, except that the reporter FL-GM-CSF was replaced with FL-TNF. *p < 0.05. (D) Lsm1 knockdown destroyed the PB integrity. HeLa cells transfected with control siRNA (siNC) or siLsm1 were stained with anti-Dcp1a antibody to visualize PBs. (E) PB depletion by Lsm1 knockdown increases FL-GM-CSF reporter mRNA level induced by TTP. 293T cells transfected with control siRNA (siNC) or siLsm1 were also transfected with FL-GM-CSF reporter plasmid and overexpression plasmids. The normalized values of FL mRNA level were set to 1 for cells transfected with Septin1 in each knockdown condition. *p < 0.05. (F) Results of an experiment similar to that for E, except that the reporter FL-GM-CSF was replaced with FL-TNF. *p < 0.05. (G) RCK silencing diminished the cytoplasm PB foci as determined by anti-Dcp1a staining. (H) The experiment was the same as for E, except that the siLsm1 was replaced with siRCK. *p < 0.05. (I) EIF4E-T knockdown destroyed the PB integrity as shown by anti-Dcp1a staining. (J) The experiment was the same as for B, except that the siHedls-1 and siHedls-2 was replaced with sieIF4E-T-1 and sieIF4E-T-2. *p < 0.05. (K) Hedls knockdown increased ARE-mRNA stability induced by TTP. HeLa cells were transfected with control siRNA (siNC) or siHedls-1 twice. In the second transfection, β-GM-CSF reporter plasmid, HA-TTP or HA-Septin1, and the internal control plasmid FL-GAPDH were also included in the transfection mixture in the presence of tetracycline (50 ng/ml). Pulse-chase experiments were performed as described under Material and Methods. The normalized values of β-globin mRNA level were set to 100% for cells transfected with TTP compared with cells transfected with Septin1, at the 0 time point, in each knockdown condition. Means and SD from three independent experiments are shown. (L) The experiment was the same as for K, except that the siHedls-1 was replaced with sieIF4E-T-1.
Next, PBs were disassembled by depression of the decapping activator Lsm1, which is essential for their formation. Knockdown of Lsm1 was analyzed with real-time PCR (Supplemental Figure S2C) and its effect on PB depletion was monitored by anti-Dcp1a staining (Figure 2D). As expected, Lsm1 silencing reversed the decrease of GM-CSF reporter mRNA level triggered by TTP (Figure 2E). Importantly, introduction of the siRNA-resistant form of Myc-Lsm1 (Myc-Lsm1r) restored the decreased mRNA level of GM-CSF reporter in the presence of TTP (Figure 2E). Consistently, a similar effect was also found with FL-TNF reporter mRNA (Figure 2F).
DEAD-box helicase 6 (DDX6, also called RCK/P54), a conserved helicase family member, has been shown to be essential for PB integrity (Ayache et al., 2015). SiRNAs against RCK eliminated the endogenous RCK protein expression in 293T cells (Qi et al., 2012) and subsequently the visible PB foci (Figure 2G). RCK knockdown also restored GM-CSF reporter mRNA level reduced by TTP (Figure 2H), although its effect was smaller than that of Hedls or Lsm1 silencing. Also, the derepression was diminished with the introduction of a siRNA-resistant form of Myc-RCK (Myc-RCKr) (Figure 2H), indicating that the effect is specific to RCK.
In addition to function in translational repression, RCK has also been demonstrated involved in mRNA decapping (Fenger-Gron et al., 2005). To confirm that the effect of PB depletion as seen above are genuinely resulted from PB disorganization, rather than the impaired decapping process, we dispersed PBs by eIF4E-T knockdown, eIF4E-T is a blocking partner of the translational initiation factor eIF4E (eukaryotic initiation factor 4E, eIF4E) and has been shown to be required for the accumulation of mRNA degrading enzymes Lsm1, RCK/P54 and Ccr4 in PB and mRNA instability (Andrei et al., 2005). RNAi reduced eIF4E-T protein to ∼20 and 25%, respectively (Supplemental Figure S2D), compared with corresponding control. EIF4E-T depletion caused Dcp1a to disperse throughout the cytoplasm and did not concentrate in foci any longer, indicative of disrupted PBs (Figure 2I). Interestingly, depletion of eIF4E-T relieved the mRNA level of GM-CSF reporter from 45% to 75% in the presence of TTP, with either siRNA (Figure 2J). A similar change was also obtained with FL-TNF (unpublished data). Together, the above results suggested the possibility that PB integrity is involved in ARE-mRNA degradation initiated by TTP.
To further determine whether PBs affect ARE-mRNA stability, we adopted a tetracycline-regulatory pulse-chase experiment described previously to trace ARE-mRNA stability (Lykke-Andersen and Wagner, 2005; Franks and Lykke-Andersen, 2007). A mRNA reporter β-GM-CSF was constructed by placing GM-CSF 3′UTR downstream of human β–globin coding region. Transcription of the reporter gene is driven by a tetracycline response promoter, which is activated by a tetracycline-repressible activator protein when tetracycline is absent. A FL mRNA with the 3′UTR of GAPDH (FL-GAPDH) under the control of the CMV promoter served as an internal control. To specifically monitor the β-GM-CSF reporter mRNA stability influenced by TTP, we cotransfected the β-GM-CSF reporter together with Septin1 as a control. We found that the half-life of β-GM-CSF reporter mRNA was ∼80 min in HeLa cells (Figure 2K). PB depletion by Hedls knockdown enhanced β-GM-CSF reporter mRNA stability (Figure 2K), with a half-life ∼135 min, suggesting that the PBs are involved in the decrease of ARE-mRNA stability. Importantly, TTP overexpression caused a twofold reduction in the half-life of the β-GM-CSF reporter mRNA and the accelerated degradation was inhibited with Hedls knockdown (Figure 2K). A similar effect of derepression was also observed on eIF4E-T knockdown (Figure 2L), confirming that PBs modulated ARE-mRNA stability initiated by TTP. We noticed that PB depletion by Hedls or eIF4E-T knockdown restored the β-GM-CSF reporter mRNA level suppressed by TTP comparable with that without TTP overexpression (Figure 2, K and L), implicating that the PB-mediated mRNA decay pathway is the main downstream mechanism responsible for TTP-mediated degradation of ARE-mRNA. Collectively, these ARE-containing reporter mRNA experiments suggest that PB depletion represses ARE-mRNA instability prompted by TTP.
Ago2 mediates TTP targeting into P-body
We proposed that TTP promotes ARE-mRNA turnover by delivering them into PBs in which multiple mRNA decay enzymes reside. Thus, delivery into PBs should be a potential mechanism for TTP to induce target mRNA degradation. TTP, as an RNA-binding protein, interacts with PBs by two possible mechanisms. One possibility is that TTP spontaneously assemble visible PBs by recruiting factors responsible for RNA degradation after it binds to ARE-mRNA. Alternatively, TTP and its ARE-mRNA partner are delivered into well-assembled PBs. TTP protein level is quite small due to low transcription rate and relatively quick mRNA degradation in the general condition. Hence we could not knock down TTP to validate its role in PB organization and then turned to determine whether TTP overexpression could restore PB formation after depleting PBs. Our assay showed that RCK knockdown completely diminished cytoplasm PBs as visualized by anti-Dcp1a staining (Figure 3A). TTP overexpression could not restore PB number, as displayed by diffused distribution of Dcp1a as well as TTP (Figure 3A). Similar results were obtained when Hedls was knockdown (unpublished data). Furthermore, we attempted to determine whether TTP expression would augment PB de novo biogenesis. Our results showed that TTP expression slightly increase PB number but with no statistical significance even at a higher transfection dosage (Figure 3, B and C), indicating that TTP itself could not significantly promote PB production. Instead, the fluorescence intensity and apparent size of PBs marked by Dcp1a were enhanced with TTP expression (Figure 3B), implying that TTP may help PB assembly by interaction with other mRNA decay components. Overall, the above results indicate that TTP itself was not sufficient to promote PB de novo formation, excluding the first possibility. Alternatively, TTP is associated with PBs by an unidentified factor. Ago2 is the component of PBs and has been reported to participate in TTP-mediated ARE-mRNA decay, and therefore we tested whether Ago2 is essential for TTP recruitment into PBs. Ago2 knockdown was achieved by RNAi (Supplemental Figure S3A) and led to the decline of PB localization, as well as to an increase in cytoplasm distribution of TTP (Figure 3, D and E). In contrast, Ago2 knockdown slightly reduced the number of PBs with no statistical significance (Figure 3E and Supplmental Figure S3, B and C), suggesting that Ago2 is specifically required for TTP accumulation in PBs, whereas function in PB formation is only a secondary effect. Additionally, Ago2 overexpression substantially increased the fluorescence intensity of TTP within the PBs and reduced its distribution in the cytoplasm (Figure 3, F and G); Moreover, overexpression of Ago2 also increased the number of PBs that are positive for TTP (Figure 3, F and G). However, the total number of PBs displayed by Dcp1a was marginally increased with Ago2 expression with no statistical significance (Figure 3G and Supplemental Figure S3, D and E). This observation indicates that Ago2 promoted TTP targeting into PBs. Overall, Ago2 knockdown and overexpression experiments demonstrated that Ago2 specifically mediated TTP association with PBs.
FIGURE 3:

Ago2 is responsible for delivering TTP into PBs. (A) HeLa cells were transfected with siRCK to deplete the cytoplasm PBs. Scrambled siRNA (siNC) served as negative control. TTP-EGFP plasmid was also included in the transfection mixture. PBs were visualized with anti-Dcp1a antibody. (B) TTP overexpression did not increase the P-body numbers. HeLa cells were transfected with TTP-EGFP plasmid and the corresponding empty vector served as negative control. PBs were displayed by anti-Dcp1a antibody. (C) Graph showing the numbers of P-body as in B. Error bars represent standard error calculations obtained from averaging the PB number for 50 cells. ns, no significance. (D) Ago2 knockdown significantly reduced TTP localization in PBs. HeLa cells were treated with siAgo2 or siNC twice. TTP-EGFP plasmid was included in the second transfection mixture. PBs were shown by anti-Dcp1a staining. (E) Graph showing the P-body numbers that are positive for TTP, and the total P-body numbers per cell, in siNC and siAgo2 conditions. Error bars represent standard error calculations obtained from averaging the PB number for 50 cells. *p < 0.05; **p < 0.01; ns, no significance. (F) Ago2 overexpression significantly increased the localization of TTP within PBs. HeLa cells were transfected with TTP-EGFP with or without Myc-Ago2 cotransfection. PBs were visualized with anti-Dcp1a antibody. (G) Graph showing the P-body numbers that are positive for TTP and the total P-body numbers per cell. Error bars represent standard error calculations obtained from averaging the PB number for 50 cells. *p ≤ 0.05; ns, no significance. (H) Both TTP and Ago2 could organize microscopically visible granules formation. HeLa cells were treated with siRCK twice to deplete PBs. TTP-EGFP plasmid was included in the second transfection mixture with or without Myc-Ago2. PBs were displayed by anti-Dcp1a antibody. (I) Graph showing the P-body numbers that are positive for TTP per cell. Error bars represent standard error calculations obtained from averaging the PB number for 30 cells. **p ≤ 0.01. (J) Western blots of anti-Myc immunoprecipitates from RNase A–treated HEK 293T cells treated with siRCK or siNC (negative control) twice. Plasmids Flag-TTP and Myc-Ago2 were included in the second transfection mixture. Precipitates (left panels) and total extracts (input, right panels) were probed for the presence of coexpressed Flag-TTP and Myc-Ago2, as indicated.
We next sought to establish whether Ago2 and TTP are sufficient for organizing microscopically visible granules formation. TTP and Ago2 were introduced into HeLa cells in which cytoplasmic PBs had been disassembled by RCK knockdown. Interestingly, on RCK depletion, Ago2 and TTP expression led to the focal pattern of TTP that is positive for PB marker Dcp1a, in spite of decreased numbers compared with those under the undisturbed condition (Figure 3, H and I). This experiment showed that the interaction of TTP and Ago2 could organize the formation of Dcp1a-containing granules that are likely to be PBs or their precursor and implies that the interaction of TTP and Ago2 may occur independently of PBs. Indeed, coimmunoprecipitation assay showed that PB deletion by RCK silencing did not interfere with the interactions of TTP with Ago2 (Figure 3J). Together, these data support the conclusion that Ago2 plays a role in targeting TTP into PBs.
Ago2 is required for TTP-mediated ARE-mRNA degradation that is dependent on PB integrity
The results reported above suggest that Ago2 may be involved in ARE-mRNA degradation through delivering TTP into PBs. To determine whether Ago2 is required for ARE-mRNA decay induced by TTP, we silenced Ago2 expression and detected its effect on the ARE reporter mRNA level. Our findings indicated that TTP overexpression reduced GM-CSF reporter mRNA level to ∼50%, and Ago2 knockdown significantly increased the relative mRNA level to ∼80% of that induced by TTP (Figure 4A). In addition, Ago2 overexpression further reinforced the inhibitory effect of TTP on GM-CSF reporter mRNA (Figure 4B). Similar results were also obtained with FL-TNF reporter mRNA (Figure 4, A and B). The ability of Ago2 to stimulate degradation of the reporter mRNA is thus dependent on TTP binding, as Ago2 neither influences TTP-induced decay of a reporter mRNA with a mutated ARE sequence (Supplemental Figure S4A), nor stimulates GM-CSF reporter mRNA degradation in the presence of a TTP mutant incompetent for ARE binding (Supplemental Figure S4B). Furthermore, in the absence of TTP, either knockdown or overexpression of Ago2 did not affect the relative mRNA levels of either ARE reporter (Figure 4, C and D). All these data imply that Ago2 must act in concert with TTP to promote ARE-mRNA degradation.
FIGURE 4:
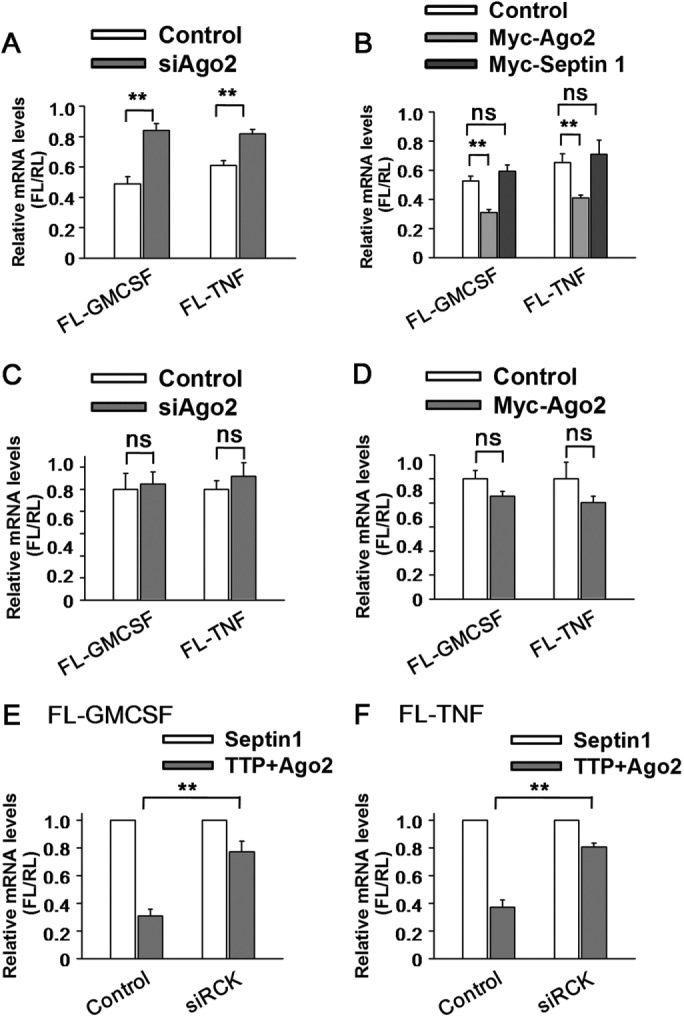
The cooperation of TTP and Ago2 is critical for ARE-mRNA degradation. (A) Ago2, acts together with TTP to promote ARE reporter mRNA decay. 293T cells were transfected with the indicated siRNAs and then transfected with reporter FL-GM-CSF or FL-TNF, RL, and the indicated plasmids expressing HA-tagged TTP or HA-Septin1. Normalized value of FL mRNA level was set to 1 for cells transfected with the HA-Septin1 plasmid and the relative value of FL mRNA was shown for cells transfected with the HA-TTP plasmid in each knockdown condition. Means and SD from three independent experiments are shown. **p < 0.01. (B) 293T cells were transfected with the FL-GM-CSF or FL-TNF reporter and RL plasmids, together with two plasmids, one expressing either HA-tagged TTP or HA-Septin1 and another expressing either Myc-tagged Ago2 or Myc-Septin1 or corresponding empty vector, as indicated. The relative value of FL activity was set to 1 for cells transfected with the plasmid expressing HA-tagged Septin1 and the relative value of FL mRNA was shown for cells transfected with the plasmid expressing HA-tagged TTP in each condition. Means and SD from three independent experiments are shown. **p < 0.01; ns, no significance. (C) Ago2 depletion only did not change ARE reporter mRNA level. 293T cells transfected with control siRNA (siNC) or siAgo2 were also transfected with the indicated reporter plasmids. The normalized value of FL mRNA level was set to 1 for cells transfected with siNC for each reporter mRNA. ns, no significance. (D) 293T cells transfected with Myc-Ago2 or corresponding empty vector (control) were also transfected with the indicated reporter plasmids. The normalized value of FL mRNA level was set to 1 for cells transfected with control plasmid for each reporter mRNA. ns, no significance. (E) 293T cells were transfected with the indicated siRNAs and the reporter FL-GM-CSF and RL plasmids, together with two plasmids, one expressing either HA-tagged TTP or HA-Septin1 and another expressing either Myc-tagged Ago2 or Myc-Septin1. The normalized value of FL mRNA level was set to 1 for cells transfected with plasmids HA-Septin1 and Myc-Septin1 in each knockdown condition. Means and SD from three independent experiments are shown. **p < 0.01. (F) Results of an experiment similar to that for E, except that the reporter FL-GM-CSF was replaced with FL-TNF. **p < 0.01.
To determine whether PB integrity is necessary for ARE-mRNA degradation, we depleted cytoplasm PBs by RCK knockdown in 293T cells. As shown in Figure 4E, TTP and Ago2 expression led to the decline of GM-CSF reporter mRNA level, while the mRNA level was substantially restored by the PB destruction. Similar results were obtained with TNF reporter mRNA (Figure 4F), supporting the denotation that PBs are involved in executing the ARE-mRNA degradation mediated by TTP and Ago2.
Ago2 mediates the interaction between TTP and Dcp1a
Since PBs are engaged in the decapping and degradation of mRNA from the 5′ end, we propose that Ago2 delivers TTP into PBs, after which the decapping machinery proceeds with the subsequent degradation. If this assertion is correct, Ago2 may mediate the interactions of TTP with the decapping activator Dcp1a and decapping enzyme Dcp2. Coimmunoprecipitate (Co-IP) assay showed that TTP copurifies with Dcp1a and Dcp2, while RCK knockdown almost eliminated these complex formations (Figure 5A), implying that the interactions of TTP with Dcp1a and Dcp2 took place inside PBs. In addition, Ago2 knockdown clearly removed either Dcp1a or Dcp2 from TTP complex immunoprecipitated by anti-Flag antibody (Figure 5A). Furthermore, Dcp1a or Dcp2 silencing impaired GM-CSF reporter mRNA decay in the presence of TTP and Ago2 (Figure 5B). Conversely, Dcp1a and Dcp2 expression strengthened the rapid ARE-mRNA turnover (Figure 5C), suggesting that Dcp1a and Dcp2 act downstream of Ago2 in TTP-mediated ARE-mRNA degradation. Moreover, expression of a catalytically inactive mutant form of human Dcp2 (Dcp2-E148Q), which impairs the 5′ cap removal and subsequent 5′-3′ phosphohydrolysis, blocked TTP, and Ago2-induced ARE-mRNA degradation (Figure 5C). In addition, TTP aggregate in PBs was greatly increased in the presence of Dcp2-E148Q compared with that of the wild-type Dcp2 (Figure 5D), implying that ARE-mRNAs are targeted into PBs but are protected from degradation. Thus, the decapping complex Dcp1a-Dcp2 in PBs plays an essential role in the ARE-mRNA decay downstream of TTP and Ago2.
FIGURE 5:

Dcp1a and Dcp2 are responsible for TTP- and Ago2-mediated ARE-mRNA decay. (A) Western blots for the endogenous Dcp1a and Dcp2 proteins that coimmunopurify with Myc-tagged TTP from RNase A–treated HEK 293T cells treated with Scrambled siRNA, siAgo2, or siRCK. Precipitates (left panels) and total extracts (input, right panels) were probed for the presence of Dcp1a, Dcp2, and TTP as indicated. (B) 293T cells were transfected with the FL-GM-CSF reporter and RL plasmids, together with siRNA as indicated. Flag-tagged TTP and Myc-tagged Ago2 were also included in the transfection mixture. Flag-tagged Septin1 and Myc-tagged Septin1 served as control. The relative value of FL mRNA level was set to 1 for cells transfected with the plasmid expressing Septin1 in each knockdown condition. Means and SD from three independent experiments are shown. *p < 0.05. (C) 293T cells were transfected with the FL-GM-CSF reporter and RL plasmids, together with plasmid expressing HA-tagged Dcp1a, Dcp2, Dcp2-E148Q, or Septin1. Plasmid expressing Flag-tagged TTP and Myc-tagged Ago2 were also included in the transfection mixture. Flag-tagged Septin1 and Myc-tagged Septin1 served as control. The relative value of FL mRNA was set to 1 for cells transfected with the Flag-tagged and Myc-tagged Septin1 plasmids in each condition. Means and SD from three independent experiments are shown. *p < 0.05. (D) Dcp2-E148Q mutant significantly retains TTP localization in PBs. HeLa cells were transfected with Myc-tagged wild-type Dcp2 (Dcp2-WT), Dcp2-E148Q mutant, or the corresponding empty expression plasmid (control), together with plasmid TTP-EGFP. PBs were shown by anti-Dcp1a staining.
Phosphorylation of TTP prevents Ago2 recruitment
ARE-mRNA degradation mediated by TTP requires its binding with target mRNA and subsequent recruitment of decay enzymes. This process could be potentially regulated by TTP phosphorylation. A consensus has not been reached yet whether TTP phosphorylation impairs its ability to bind target mRNA (Carballo et al., 2001; Mahtani et al., 2001; Cao, 2004; Stoecklin et al., 2004; Hitti et al., 2006; Sun et al., 2007; Clement et al., 2011). Therefore, we want to determine whether TTP phosphorylation weaken its association with ARE-mRNA in cells. To this end, Myc-tagged TTP was overexpressed in HEK293T in the presence of the constitutively active kinase MKK6b (E) or the inactive kinase MKK6b (A) (Han et al., 1996; Raingeaud et al., 1996; Stein et al., 1996; Huang et al., 1997), after which GM-CSF reporter mRNA was copurified by RNA immunoprecipitation with anti-Myc antibody. As shown in Figure 6A, no significant differences in the abundance of GM-CSF reporter mRNA associated with TTP were observed between the MKK6b (E) or MKK6b (A) expression. Myc-tagged hnRNP A1 served as a positive control for copurification with the GM-CSF reporter mRNA, whereas TTP lacking the RNA-binding domain (TTP1-100) was used as a negative control (Figure 6A). Our data support that TTP phsophorylation by P38 does not impair its ARE-mRNA-binding ability, and thus alternative mechanisms may exist, such as the effect of TTP phosphorylation on the interaction with degradation-related proteins.
FIGURE 6:

P38 activation represses the interaction of TTP with Ago2 or with Dcps. (A) 293T cells were transfected with empty expression plasmid (control) or plasmids expressing MKK6b(E) or MKK6b(A), together with plasmids FL-GM-CSF and Myc-tagged TTP-WT. Myc-tagged hnRNP-A1 and TTP fragment (TTP1-100) served as positive and negative control for the wild-type TTP, respectively. Cytoplasmic lysates were used for mRNP immunoprecipitation (IP) assays with antibody to Myc followed by qRT-PCR to measure FL-GM-CSF mRNA abundance. GAPDH mRNA served as internal control. Means and SD from three independent experiments are shown. ns, no significance. (B) Western blots of anti-HA immunoprecipitates from RNase A–treated extracts of HEK 293T cells transiently expressing HA-tagged Ago2 and Myc-tagged TTP in the absence or presence of SB 203580 for 2 h. Precipitates and cell lysates were probed for the presence of coexpressed Myc-TTP, HA-Ago2, as indicated. (C) The same cell extracts as for B were immunoprecipitated with anti-Myc antibody. Precipitates and cell lysates were probed for the presence of HA-Ago2, Myc-TTP, as indicated. (D) Western blots of anti-Myc immunoprecipitates from RNase A–treated HEK 293T cells transiently expressing three plasmids: one expressing Flag-tagged TTP, another expressing Myc-tagged Ago2, third expressing either HA-tagged MKK6b(A) or HA-MKK6b(E) or corresponding empty vector. Precipitates (left panels) and total extracts (input, right panels) were probed for the presence of coexpressed Flag-TTP, Myc-Ago2, as indicated. (E) Results of an experiment similar to that for panel D, except that the overexpressed Myc-tagged Ago2 was replaced with endogenous Ago2. (F) Results of an experiment similar to that for panel E, except that the endogenous Ago2 was replaced with the endogenous Dcp1a and Dcp2.
We therefore tested the effect of TTP phosphorylation on the interaction with Ago2 by using the coimmunoprecipitation assay. For this purpose, 293T cells were ectopically expressed with Myc-tagged TTP and HA-tagged Ago2, followed by treatment with SB 203580 for 2 h. Among the immunoprecipitation complexes with anti-HA antibody, Myc-TTP level was elevated in the presence of SB 203580 compared with control, although SB 203580 treatment did not change TTP or Ago2 expression (Figure 6B). Similar results were obtained when the same cell lysate was immnoprecipitated with anti-Myc antibody (Figure 6C). These findings indicate that P38 inhibition promotes complex formation between TTP and Ago2.
Next, MKK6b (A) or MKK6b (E) were introduced together with Myc-Ago2 and Flag-TTP into 293T cells to specifically inhibit or activate P38, while Co-IP by anti-Myc antibody was analyzed 48 h later. As seen in Figure 6D, P38 inhibition significantly increased the presence of TTP in the Ago2 precipitate, whereas P38 activation almost completely abolished Ago2-complexed TTP. Importantly, the interaction of TTP with the endogenous Ago2 was verified under different P38 activity conditions and similar results were obtained (Figure 6E). Furthermore, the interactions of TTP with PB components Dcp1a or Dcp2 were also impaired by P38 activation and were strengthened by P38 inhibition (Figure 6F). Thus, we concluded that P38 activation inhibits the interaction of TTP with its partner Ago2, as well as PB components.
Ser-52 and Ser-178 of TTP are the main phosphorylation sites mediating its interaction with Ago2 and ARE-mRNA degradation
Ser-52 and Ser-178 of TTP are the phosphorylation targets of the P38-MK2 signal pathway. To identify the target site mediating the interaction between TTP and Ago2, the TTP mutant resistant to P38 phosphorylation, TTP-AA was employed. We found that TTP-AA copurified with Myc-tagged Ago2 more than the wild-type TTP did (Figure 7A). Furthermore, the interaction between TTP-AA and Ago2 was not weakened by P38 stimulation (Figure 7B), suggesting that phosphorylation of TTP at the two sites was essential for its dissociation from Ago2. In addition, the interaction between TTP-AA and Ago2 was not further strengthened by P38 inhibition (Figure 7B), implying that Ser-52 and Ser-178 of TTP are the main phosphorylation sites responsible for P38 signaling. Similarly, association between TTP-AA and Dcp1a was neither attenuated by P38 activation nor enhanced by P38 inhibition (Figure 7C).
FIGURE 7:

Ser-52 and Ser-178 of TTP are the main phosphorylation sites mediating its interaction with Ago2 and ARE-mRNA degradation. (A) 293T cells tranfected with Myc-tagged Ago2 and Flag-tagged TTP-WT or TTP-AA were subjected to anti-Myc immunopurification (IP), and precipitates were detected via immunoblotting with anti-Flag antibody. (B) Coimmunoprecipitation assays performed with extracts derived from 293T cells coexpressing Flag-tagged TTP-AA together with MKK6b(E) or MKK6b(A). Immunoprecipitations (IP) were performed with anti-Flag antibody. Pellet and 5% of input fractions were loaded on the left and the right and subjected to immunoblotting with anti-Flag or anti-Ago2 antibodies as indicated. (C) Results of an experiment similar to that for B, except that the anti-Ago2 antibody was replaced with anti-Dcp1a antibody. (D) HeLa cells were transfected with plasmids expressing either TTP-WT-EGFP or TTP-AA-EGFP. PBs were displayed with anti-Dcp1a antibody (left panel). Graph showing the P-body numbers that are positive for TTP, and the total P-body numbers per cell, in TTP-WT-EGFP and TTP-AA-EGFP transfection conditions (right panel). Error bars represent standard error calculations obtained from averaging the PB number for 50 cells. *p < 0.05; ns, no significance. (E) HeLa cells were transfected with two plasmids, one expressing TTP-AA-EGFP and another expressing MKK6b(E), MKK6b(A), or a corresponding empty vector. PBs were visualized by anti-Dcp1a staining. (F) 293T cells were transfected with the FL-GM-CSF reporter and RL plasmids, together with two plasmids, one expressing HA-tagged TTP-WT, TTP-AA, or Septin1 and another expressing either MKK6b(E) or corresponding empty vector, as indicated. The relative value of FL mRNA was set to 1 for cells transfected with the plasmid expressing HA-tagged Septin1 in each condition. Means and SD from three independent experiments are shown. *p < 0.05; **p < 0.01; ns, no significance. (G) 293T cells were transfected with the indicated siRNAs and then transfected with reporter FL-GM-CSF and RL plasmids, together with HA-tagged TTP-AA or HA-Septin1. The normalized value of FL mRNA level was set to 1 for cells transfected with plasmids HA-Septin1 in each knockdown condition. Means and SD from three independent experiments are shown. **p < 0.01.
To determine whether the TTP mutant could respond to P38 to alter its localization within PBs, we expressed TTP-AA-EGFP in HeLa cells and traced its localization. TTP-AA localized to PBs under a natural condition, with an apparent size of PBs larger than that hosting the wild-type TTP (Figure 7D, left panel). TTP-AA did not increase total PB numbers per cell but significantly enhanced the number of PBs that was positive for TTP (Figure 7D, right panel). TTP-AA did not diffuse into the cytoplasm and persisted in clustering within PBs when P38 was activated (Figure 7E). Conversely, its localization to PBs was not further enhanced by P38 inhibition (Figure 7E). These findings suggest that the phosphorylation status of TTP induced by P38 is critical for its PB localization.
Finally, we studied the ARE-mRNA degradation regulated by TTP-AA and found that TTP-AA more effectively promoted GM-CSF reporter mRNA decay than the wild-type TTP did (Figure 7F). The mRNA decay of GM-CSF reporter was clearly blocked by P38 activation in the presence of wild-type TTP (Figure 7F), whereas it persisted in presence of TTP-AA in spite of high P38 activity (Figure 7F). Moreover, Ago2 knockdown alleviated GM-CSF reporter mRNA decay induced by TTP-AA (Figure 7G), and a similar effect was observed in the condition of Dcp1a knockdown or destruction of PBs (Figure 7G). Thus TTP-AA utilized the downstream decay pathway in the same manner as the wild-type TTP did. Together, these results indicated that phosphorylation at Ser-52 and Ser-178 of TTP by P38 mitigates its interaction with Ago2 and relieves the ARE-mRNA instability.
DISCUSSION
Control of mRNA turnover plays a critical role in the regulation of gene expression. Cell signaling pathways can influence mRNA decay rates, yet the mechanism by which cell signaling information is communicated to mRNA decay factors is poorly understood. In this study, we showed that, in the case of the ARE-mRNA decay pathway, TTP activates rapid degradation of ARE-mRNAs by delivering it into PBs, which are dependent on the recruitment of microRNA-induced silencing complex (miRISC) member Ago2. On activation of the P38 MAPK signaling pathway, the phosphorylation of TTP resulted in impaired Ago2 recruitment, whereby ARE-mRNA were removed from PBs and became stabilized. Thus, recruitment of Ago2 was a regulated point in ARE-mRNA decay prompted by TTP.
TTP promoted ARE-mRNA degradation requires PB integrity
TTP is a well-known ARE-mRNA posttranscriptional regulator that functions through binding to the ARE domain in the 3′UTR (Clark and Dean, 2016). However, the precise mechanism underlying this role is not well understood. It is thus noteworthy that TTP and ARE-mRNA concentrated in cytoplasm discrete foci called PBs (Franks and Lykke-Andersen, 2007), a site involved in mRNA decay and translation repression. However, it is unclear whether PBs serve as granules where ARE-mRNA is quickly degraded or act as a reservoir for their storage. Increased abundance and size of PBs by inhibiting mRNA turnover after decapping or PB deficiency by inhibiting mRNA decay before decapping, suggested a possible site of PBs for regulation of mRNA degradation (Sheth and Parker, 2003). Yet, contrary results presented that the combined deletion of decapping activator Edc3 and the Q/N-rich domain of Lsm4 led to reduced stability of PGC1 and MFA2 mRNA and concomitant disappearance of PBs under general and stress conditions in Saccharomyces cerevisiae, pointing out PBs implicating in mRNA storage but not decay, at least under some circumstances (Huch et al., 2016; Huch and Nissan, 2017). In contrast to the conflicting discoveries, studies showed that TTP and ARE-mRNA accumulate in PBs following depletion of Dcp2 and Xrn1, both of which are catalytic components of PBs involved in mRNA 5′-to-3′ decay. In addition, a K63-branched ubiquitin mutation (K63R) led to strong stabilization of prototypical IL-1 target gene mRNAs IL-6, IL-8, and CXCL3, along with the disappearance of PBs (Tenekeci et al., 2016), suggesting a coupling of ARE-mRNA degradation and PB integrity. Notably, it was determined that Dcp1a silencing increased ARE-mRNA stability and the PB abundance, although knocking down Lsm1 caused PBs to disappear and increased ARE-mRNA stability, suggesting that PBs may not simply be involved in ARE-mRNA stability regulation (Stoecklin et al., 2006). PBs organize from the complex of mRNA and proteins. To date, several factors required for its integrity have been identified, such as decapping activator Hedls, Lsm1, RCK, or eIF4E-T. Our observations demonstrated that PB depletion by Hedls, Lsm1, or helicase RCK/P54 knockdown greatly relieved GM-CSF reporter mRNA level repressed by TTP (Figure 2, B, E, H, and K). Similar results were obtained in the experiments with TNF-α reporter mRNA (Figure 2, C and F). In addition, PB depletion by eIF4E-T knockdown also rescued decreased GM-CSF reporter mRNA level induced by TTP (Figure 2, J and L), implying that PBs were specifically involved in ARE-mRNA degradation prompted by TTP. Additional evidence further showed that Dcp2-E148Q overexpression not only inhibited ARE-mRNA degradation mediated by TTP and Ago2 (Figure 5C) but also increased ARE-mRNA localization within PBs (Figure 5D). Therefore, our data favor the denotation that PBs likely execute the ARE-mRNA decay. However, eIF4E-T has been verified to complex with TTP and assist in the degradation process of ARE-mRNA induced by TTP (Nishimura et al., 2015). Thus, the increased stability of GM-CSF reporter mRNA from eIF4E-T knockdown (Figure 2, J and L) may arise from the consequence of its non-PB effects. Collectively, our result provides some clues that PBs may be likely the location where ARE-mRNA was actively degraded. However, we could not completely exclude the possibility that ARE-mRNA degradation actually happens in some place other than PB and ARE-mRNA localization in PBs as the consequence of RNA decay. This issue is of great interest for future research.
TTP delivers ARE-mRNA into PBs, thus facilitating its contact with decay-related enzymes and subsequent degradation. TTP could enter preexisting PBs via a certain mechanism or organize PB formation by recruiting obligatory partners. Our data showed TTP expression extended the apparent size of PBs but did not alter their number (Figure 3, B and C), and it could not rescue the PB depletion induced by RCK knockdown (Figure 3H). Therefore our data support that TTP and associated ARE-mRNA join in a mature PBs and implicate that a potential component, such as a mRNA decay-related factor, is engaged in delivering TTP into PBs.
Ago2 delivers TTP into PBs and is involved in TTP-mediated ARE-mRNA degradation
Our studies provided the important evidence that TTP is the prerequisite for Ago2 participation in ARE-mRNA decay. Without cotransfection of TTP, Ago2 has no significant effect on ARE-mRNA expression at the mRNA level (Figure 4, C and D). However, when TTP was cotransfected, Ago2 led to decreased ARE-mRNA expression at the mRNA level (Figure 4B), clearly indicating that Ago2 is an ARE-mRNA decay factor. As Ago2 depends on TTP, without TTP, it could not effectively bind to ARE-mRNA. In other words, Ago2 may preferentially interact with TTP relative to other ARE-binding factors. On the other hand, TTP promotes ARE-mRNA degradation dependent on Ago2. As shown in the experiments where Ago2 was depleted, in the presence of TTP, Ago2 was indispensable for ARE-mRNA degradation (Figure 4A). Consistently, Ago2 overexpression further strengthened ARE-mRNA repression induced by TTP, suggesting that Ago2 acts downstream of TTP (Figure 4B).
As noted above, TTP itself could not organize microscopically visible granule formation after PBs were destroyed, indicating that other factors participated in this process. Ago2 is an interesting candidate because of its closer correlation with TTP and ARE-mRNA degradation (Jing et al., 2005). We cotransfected Ago2 together with TTP into HeLa cells and found that granule formation was somehow recovered, but their number and apparent size were still smaller than those observed under the general conditions (Figure 3H). We posit that Ago2 recruited by TTP will further attract other interaction components, leading to greater aggregation formation, which can be seen under the microscope. Dcp1a, a critical decapping activator, was positive in these foci, indicating that a portion of Dcp1a is recruited into this complex. This is consistent with the finding that Ago2 facilitates the association of TTP with Dcp1a and Dcp1a acts downstream of Ago2 (Figure 5A). However, RCK depletion diminished the interaction between TTP and Dcp1a (Figure 5A), suggesting that this interaction mainly occurs within PBs.
Previous studies showed that P38-MK2 activation impaired the deadenylase recruitment to ARE-mRNA due to TTP phosphorylation (Marchese et al., 2010; Clement et al., 2011). It is thus important to ascertain whether Ago2 mediates the connection between TTP and deadenylase. Given the closer relation of Ago2 and deadenylase complex in a microRNA (miRNA)-mediated pathway, we hypothesize that Ago2 is responsible for deadenylase recruitment. Thus, Ago2 dissociation from TTP by P38 stimulation leads to deadenylase recruitment failure.
Owing to the involvement of Ago2 in TTP-mediated ARE-mRNA decay (Jing et al., 2005), we could not exclude the possibility that miRNAs, such as miR-16, may participate in TTP association with Ago2 targeting to PBs. Further work is needed to elucidate the function and mechanism of miRNAs and their role in the cooperation of TTP and Ago2.
Phosphorylation of TTP by P38 inhibits Ago2 recruitment
It has been shown that phosphorylation of TTP by P38 is associated with impaired decay of ARE-mRNA (Mahtani et al., 2001; Dean et al., 2004; Brook et al., 2006; Hitti et al., 2006; Tudor et al., 2009; McGuire et al., 2017), but the mechanism behind this process remains insufficiently understood. Previous observations based on in vitro RNA gel shift experiments suggested that phosphorylation of TTP decreases its ability to bind ARE-mRNA in vitro (Carballo et al., 2001; Cao, 2004; Hitti et al., 2006), whereas other studies have suggested otherwise (Mahtani et al., 2001; Stoecklin et al., 2004; Sun et al., 2007; Clement et al., 2011). Our results are consistent with the latter. First, by RNA immunoprecipitate assay, we did not observe a significant decrease in the ability of phosphorylated TTP to bind ARE reporter mRNA in 293T cells (Figure 6A). Second, phosphorylation of TTP by P38 impairs its ability to activate the decay of an mRNA substrate to which it is tethered (unpublished data), indicating that phosphorylation of TTP affects a step downstream of mRNA binding. However, it is also possible that the binding of the phosphorylated TTP with its target is finely controlled and is thus beyond the limit that we can presently detect and discriminate.
Rather than substrate binding defect, our observations suggest that the phosphorylation state of TTP affects its association with miRISC member Ago2. First, TTP dephosphorylation by P38 inhibitor SB 203580 increased the copurification of TTP with Ago2 (Figure 6, B and C). Second, P38 inhibition by MKK6b (A) also increased the coprecipitation of TTP with Ago2 (Figure 6, D and E). Third, TTP phosphorylation by P38 upstream constitutively activated kinase MKK6b (E) and clearly diminished the interaction of TTP and Ago2 (Figure 6, D and E). This reduced association defect is, at least in part, due to the phosphorylation of P38 target residues Ser-52 and Ser-178 of TTP, as evidenced by the restoration of TTP-Ago2 interactions during P38 activation when TTP is mutated in these residues (TTP-AA) (Figure 7B). Taken together, these observations suggest that, on activation of the P38 MAPK pathway, the phosphorylation of TTP leaves TTP associated with target mRNAs yet unable to recruit Ago2 complexes to promote mRNA decay. If phosphorylated TTP remains associated with target mRNA, it may be possible that phosphorylated TTP prevents ARE-mRNA from being degraded by other ARE-binding proteins. In addition, phosphorylated TTP may also be involved in contacting with other mRNA stability or translation-related modulators, allowing ARE-mRNA regulation at multiple levels. Future studies should reveal additional functions and mechanisms for phosphorylated TTP on ARE-mRNA expression under P38 activation condition.
How does phosphorylation of TTP impede Ago2 deadenylase recruitment? Previous studies have shown that phosphorylation of TTP by P38-MK2 at Ser-52 or Ser-178 induces binding by the adaptor protein 14-3-3 (Chrestensen et al., 2004; Stoecklin et al., 2004). It is known that the association of TTP with 14-3-3 protects TTP from dephosphorylation by PP2A (Sun et al., 2007), ensuring TNF-α mRNA stabilization. In addition, the association of TTP with 14-3-3 blocks its contact with decay-related enzymes, such as deadenylase complex, preventing ARE-mRNA from being degraded (Clement et al., 2011). Our findings showed that Ago2 dissociation from TTP is dependent on the phosphorylation of TTP at Ser-52 and Ser-178, which is also the target for 14-3-3. Thus, we hypothesized that TTP phosphorylation at Ser-52 and Ser-178 led to 14-3-3 recruitment. Simultaneously, Ago2 contact with TTP was blocked, thus promoting ARE-mRNA stabilization. It is interesting to note that P38 activation induced during neuronal differentiation up-regulates the phosphorylation of Ago2, which promotes the dissociation of miRNA from Ago2 and ensures normal neuronal differentiation (Patranabis and Bhattacharyya, 2016), indicating that Ago2 could also be phosphorylated by P38. Additionally, the amino terminus of human Ago2 was also demonstrated to bind 14-3-3 proteins (Stoica et al., 2006). We proposed that, like TTP, Ago2 could associate with 14-3-3 after it is phosphorylated by P38, which would result in the dissociation of TTP from Ago2. Further experiments are needed to determine the precise role of 14-3-3 in TTP and Ago2 combination and its implication in ARE-mRNA stability under P38 activation condition. Perhaps Ser-52 and Ser-178 are not the only two sites that can be phosphorylated by certain signal pathways. For example, hyperphosphorylation of the TTP-AA mutant protein, which cannot be phosphorylated by P38-MK2, partially impairs deadenylase association (Clement et al., 2011), suggesting that other phosphorylation sites that regulate TTP function exist. Further studies are required to identify signal pathways other than P38 involved in TTP phosphorylation by a similar or different mechanism and its effects on ARE-mRNA stability regulation. Moreover, not only TTP but also other AUBPs could be regulated by phosphorylation, including TTP paralogues BRF-1 and BRF-2, AUF1, and KSRP (Schmidlin et al., 2004; Gherzi et al., 2006; Maitra et al., 2008; Shen and Malter, 2015). The phosphorylation of KSRP by P38 leads to 14-3-3 association and the inhibition of KSRP’s ability to activate mRNA decay (Gherzi et al., 2006). Thus, in future studies, it would be beneficial to explore the mechanism by which phosphorylation regulates the activity of mRNA decay factor.
In summary, we have discovered a novel function for P38 in inducing ARE-mRNA stabilization by hindering the collaboration between TTP and Ago2. These results provide new insight into the mechanism by which P38 activation promotes ARE-mRNA stabilization. Future work is required to understand these mechanisms in more detail.
MATERIALS AND METHODS
Plasmid constructs
Plasmid TTP-EGFP is from our lab preserved. To construct plasmids encoding HA-tagged or Myc-tagged TTP, full-length TTP was amplified from TTP-EGFP and inserted between the EcoRI and XhoI sites of pCMV-HA or pCMV-Myc.
To construct HA-tagged or Myc-tagged Ago2, Ago2 cDNA was amplified from 293T cells and inserted between EcoRI and NotI sites of pCMV-HA or pCMV-Myc to generate HA-tagged Ago2 or Myc-tagged Ago2.
The mRNA localization reporters were as described previously (Liu et al., 2005). We did some modification through replacing the let-7 target sequence with human GM-CSF 3′UTR.
To generate FL-GM-CSF, GM-CSF 3′UTR was inserted between the KpnI and BglII sites of pGL3 basic luciferase report vector. The two sites were also used in the construction of FL-TNF and FL-GAPDH.
Cell culture and luciferase reporter assay
For analysis of ARE reporter mRNA, transfections of 293T cells were performed in 24-well plates with Lipofectamine 2000 (Invitrogen). Transfection mixtures contained 50 ng of FL reporter plasmid. Plasmids expressing various proteins were cotransfected in specific experiments as follows: HA-TTP, HA-TTP-F126N, or HA-Septin1 (10 ng); Myc-RCK or its siRNA resistant version Myc-RCKr (50 ng); Myc-Lsm1 or its siRNA-resistant version, Myc-Lsm1r; Myc-Ago2, Myc-Dcp1a, Myc-Dcp2 or its catalytically inactive mutant Myc-Dcp2-E148Q, or Myc-Septin1; and MKK6b(E) or MKK6b(A) (75 ng) (Han et al., 1996; Raingeaud et al., 1996; Stein et al., 1996; Huang et al., 1997). PcDNA3.0 was added to make a total of 200 ng of plasmid in each experiment; 2 ng of RL was included in all transfection mixes. In all experiments, FL and RL mRNA levels were measured 48 h after transfection. The FL mRNA level was normalized to that of RL.
Quantitative real-time RT-PCR for FL, RL, and GAPDH mRNA
Total RNA was isolated by TRIzol (Invitrogen), and DNA was removed from RNA samples with RNase-free DNase I (TaKaRa). Total RNA was reverse transcribed into cDNA with Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega) and oligo(dT18) according to the manufacturer’s instructions. Fluorescence real-time PCR was performed with the double-stranded DNA dye SYBR green real-time PCR Master Mix (ToYoBo) with the ABI Prism 7900 system (Perkin-Elmer, Torrance, CA). The detailed experimental procedure and analyses were as described previously (Zhao et al., 2004). The following primers were used: for FL, 5′-TGAGTACTTCGAAATGTCCGTTC-3′ (forward) and 5′-GTATTCAGCCCATATCGTTTCAT-3′ (reverse); for RL, 5′-GCAGCATATCTTGAACCATTC-3′ (forward) and 5′-TTGTACAACGTCAGGTTTACC-3′ (reverse); and for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-AGCCACATCGCTCAGACAC-3′ (forward) and 5′-GCCCAATACGACCAAATCC-3′ (reverse). The quantitative real-time RT-PCR (qRT-PCR) products were visualized by fractionation in 2% agarose gels to ensure correct product size. The FL mRNA level was normalized to that of RL.
Transfection of siRNAs
293T cells were split to a density of 3 × 105/well in six-well plates 24 h before transfection. The following siRNAs were used: control siRNA, 5′-CUCGUAAUGCAAUGGGUCCTT-3′; siHedls-1, 5′-GCAACGGAAGGUCCUCUAUTT-3′; siHedls-2, 5′-GCAACUCAGUGGCUACCAATT-3′; Lsm1 siRNA, 5′-GUGACAUCCUGGCCACCUCACUU-3′; siRCK, 5′-GCAGAAACCCUAUGAGAUUUU-3′; siAgo2, 5′-GCACGGAAGUCCAUCUGAATT-3′; siDcp1a, 5′-GCUAGGAAUGGAUAUGAUATT-3′; and siDcp2, 5′-GCUGUAUAUGACUUGCCUATT-3′. On the following day, each cell sample was incubated for 6 h in a 1-ml transfection mixture containing 2 μl of Lipofectamine 2000 reagent (Invitrogen) and siRNAs at a concentration of 50 nM according to the manufacturer’s protocols. Twenty-four hours later, cells were split into 24-well plates (0.9 × 105/well) and further incubated for 24 h. Seventy-five nanomolar siRNA, 200 ng of total plasmids as indicated, and 1 μl of Lipofectamine 2000 were mixed in 500 μl Opti-MEM medium in the second transfection. Forty-eight hours later, cells were harvested for mRNA analysis.
Pulse-chase assay
HeLa cells (Clontech) at 50% confluency in six-well plates were transfected in the presence of 50 ng/ml tetracycline using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer protocols. Transfection mixtures contained 50 ng of internal control FL-GAPDH plasmid, 150 ng of β-GM-CSF, 300 ng of HA-TTP or HA-Septin 1, 500 ng of pTet-TTA, and 100 pmol of siHedls, sieIF4E-T, or siNC in the presence of tetracycline (50 ng/ml). Forty hours after transfection, a transcriptional pulse was initiated by washing cells with phosphate-buffered saline (PBS) and feeding with 2 ml DMEM/10% fetal bovine serum containing no tetracycline. Eight hours later, tetracycline was added to 1.0 μg/ml to stop transcription. Cells were harvested in 1 ml Trizol (Invitrogen) starting at the 0 time point and then every 30 min as indicated in Figure 2, K and L. Total RNA was prepared, and the steady levels of the β-GM-CSF reporter mRNA were determined by real-time PCR.
Coimmunoprecipitation and Western blotting
For co-IP experiments, 293T cells expressing the plasmids as indicated in the figure legends were lysed at 48 h after transfection by incubating for 10 min at 4°C in 600 μl of lysis buffer (20 mM Tris-HCl buffer, pH 7.4, containing 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate, and 1 mM NaF supplemented with a protease inhibitor cocktail [Roche]). Cell lysate (50 μl) was preserved as input, and the remainder was preincubated with protein A/G agarose for 1 h, and then the supernatant was incubated with a monoclonal anti-HA (or anti-Myc) agarose conjugate (Santa Cruz) in the presence of 0.1 μg/ml RNase A. Immunoprecipitations were performed at 4°C for 4 h. The beads were washed five times with the lysis buffer. Coprecipitated proteins were detected by Western blotting using the indicated antibodies as following dilution: horseradish peroxidase (HRP)-conjugated monoclonal anti-Myc antibody at 1:2000 (Santa Cruz); mouse monoclonal anti-HA antibody at 1:5000 (Sigma-Aldrich); mouse monoclonal anti-Flag antibody at 1:5000 (Roche); rabbit polyclonal anti-Dcp1a antibody at 1:10,000 (gift from Jens Lykke-Andersen); and monoclonal anti-GAPDH or anti-β-actin antibody at 1:5000 (Abmart). HRP-conjugated goat anti-mouse IgG antibodies at 1:5000 (Abmart) or goat anti-rabbit at 1:5000 (Abmart) were used as a secondary antibody for detection with a chemiluminescence reagent (peroxide/luminol enhancer; Millipore).
RNA immunoprecipitation
293T cells were transfected with plasmids FL-GM-CSF and Myc-tagged TTP-WT (wild type), together with plasmids expressing MKK6b(E) or MKK6b(A). Myc-tagged hnRNP-A1 and TTP fragment (TTP1-100) served as positive and negative control for the wild-type TTP, respectively (Clement et al., 2011). At 48 h posttransfection, cells were harvested. For Myc immunoprecipitation, cells were lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) for 30 min at 4°C, centrifuged at 12,000 × g for 10 min, and then incubated with a monoclonal anti-Myc agarose conjugate (Santa Cruz) for 16 h at 4°C. The protein complex was washed five times with wash buffer (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl). The immunoprecipitation complex was pelleted by centrifugation and resuspended in 400 μl proteinase K buffer (100 mM Tris-HCl, pH 7.4, 150 mM NaCl, 12.5 mM EDTA, and 1% SDS) with 100 μg proteinase K (Sigma) for 30 min at 37°C. The RNA from the supernatant was extracted by TRIzol LS Reagent (Invitrogen) according to the manufacturer’s instructions. The detailed procedures for DNA removal, reverse transcription, and fluorescence real-time PCR were performed as described above.
Indirect immunofluorescence
HeLa cells were split to a density of 1 × 105/well in six-well plates 24 h before transfection. On the following day, each cell sample was incubated for 6 h in a 1-ml transfection mixture containing 2 μl of Lipofectamine 2000 reagent (Invitrogen) and siRNAs at a concentration of 50 nM according to the manufacturer’s protocols. Twenty-four hours later, cells were divided and applied to chamber slides (1 × 105/well) and further incubated for 24 h. Seventy-five nanomolar siRNA, 500 ng of total plasmids, and 2 μl of Lipofectamine 2000 were mixed in 1 ml Opti-MEM medium in the second transfection. Plasmids included 50 ng of TTP-WT-EGFP or TTP-AA-EGFP, 100 ng of Myc-Ago2 or Myc-Septin1, and 150 ng of HA-tagged-MKK6b(E) or 6b(A). PcDNA3.0 was added to make a total of 500 ng of plasmid in each experiment. Forty-eight hours after transfection, cells were fixed in 4% paraformaldehyde for 15 min and permeabilized and blocked with PBS–1% goat serum–0.1% Triton X-100 for 30 min. For indirect immunofluorescence, all the primary and secondary antibodies were diluted 1:500 with 1% (wt/vol) bovine serum albumin (BSA) in PBS. PBs were detected using rabbit polyclonal anti-Dcp1a antibody and Alexa Fluor 555-labeled goat anti-rabbit IgG (Molecular Probes). Fluorescence was captured with a laser-scanning confocal microscope (Leica TCS SP5, Germary).
Statistical analysis
Data are reported as means and SD from three independent experiments. For luciferase assays, transfection control plasmid RL was included and the FL mRNA level was normalized to that of RL in all experiments. The statistical significance of the differences between the two groups was determined using Student’s t test; p values of <0.05 were considered significant.
Supplementary Material
Acknowledgments
We are grateful to Parker Roy (University of Colorado Boulder) for kindly providing ARE-mRNA localization plasmids and Jens Lykke-Andersen (University of California, San Diego) for kindly providing anti-Dcp1a antibody, Myc-RCK, β-GM-CSF, and pTet-TTA plasmid. We thank Chen Wang for critically reading the manuscript. We thank all members of the Jing laboratory for helpful discussions and comments on the manuscript. This work was supported in part by the National Key Research & Development Program of China (2017YFA0103700), the Strategic Priority Research Program of the Chinese Academy of Science (XDA16020903), and the National Natural Science Foundation of China (91739301, 31229002, 81301855, and 91339205).
Abbreviations used:
- Ago2
argonaute 2
- PB
processing body
- SG
stress granule
- TTP
tristetraprolin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-02-0105) on February 12, 2018.
REFERENCE
- Andrei MA, Ingelfinger D, Heintzmann R, Achsel T, Rivera-Pomar R, Luhrmann R. (2005). A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA , 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayache J, Benard M, Ernoult-Lange M, Minshall N, Standart N, Kress M, Weil D. (2015). P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol Biol Cell , 2579–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopal V, Parker R. (2009). Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol , 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FF, Sanduja S, Deane NG, Blackshear PJ, Dixon DA. (2014). Transforming growth factor beta regulates P-body formation through induction of the mRNA decay factor tristetraprolin. Mol Cell Biol , 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, Clark AR. (2006). Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol Cell Biol , 2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR. (2014). mRNP granules. Assembly, function, and connections with disease. RNA Biol , 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H. (2004). Expression, purification, and biochemical characterization of the antiinflammatory tristetraprolin: a zinc-dependent mRNA binding protein affected by posttranslational modifications. Biochemistry , 13724–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. (2001). Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem , 42580–42587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Gilkeson GS, Blackshear PJ. (1997). Bone marrow transplantation reproduces the tristetraprolin-deficiency syndrome in recombination activating gene-2 (-/-) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha overproduction. J Clin Invest , 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. (2000). Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood , 1891–1899. [PubMed] [Google Scholar]
- Chen CY, Shyu AB. (1995). AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci , 465–470. [DOI] [PubMed] [Google Scholar]
- Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. (2004). MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14–3-3 binding. J Biol Chem , 10176–10184. [DOI] [PubMed] [Google Scholar]
- Clark AR, Dean JL. (2016). The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: a tale of two phosphatases. Biochem Soc Trans , 1321–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J. (2011). Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol , 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforto TL, Zhang Y, Sherman J, Waxman DJ. (2012). Impact of CUX2 on the female mouse liver transcriptome: activation of female-biased genes and repression of male-biased genes. Mol Cell Biol , 4611–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Sully G, Clark AR, Saklatvala J. (2004). The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal , 1113–1121. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. (2007). P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol , 9–22. [DOI] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. (2005). Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell , 905–915. [DOI] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. (2007). TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev , 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Olsen MT, Webb K, Bennett EJ, Lykke-Andersen J. (2016). Recruitment of the 4EHP-GYF2 cap-binding complex to tetraproline motifs of tristetraprolin promotes repression and degradation of mRNAs with AU-rich elements. RNA , 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherzi R, Trabucchi M, Ponassi M, Ruggiero T, Corte G, Moroni C, Chen CY, Khabar KS, Andersen JS, Briata P. (2006). The RNA-binding protein KSRP promotes decay of beta-catenin mRNA and is inactivated by PI3K-AKT signaling. PLoS Biol , e5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. (1996). Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J Biol Chem , 2886–2891. [DOI] [PubMed] [Google Scholar]
- Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. (2006). Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol , 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J. (1997). Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity , 739–749. [DOI] [PubMed] [Google Scholar]
- Huch S, Muller M, Muppavarapu M, Gommlich J, Balagopal V, Nissan T. (2016). The decapping activator Edc3 and the Q/N-rich domain of Lsm4 function together to enhance mRNA stability and alter mRNA decay pathway dependence in Saccharomyces cerevisiae. Biol Open , 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch S, Nissan T. (2017). An mRNA decapping mutant deficient in P body assembly limits mRNA stabilization in response to osmotic stress. Sci Rep , 44395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. (2005). Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell , 623–634. [DOI] [PubMed] [Google Scholar]
- Kiriakidou M, Tan GS, Lamprinaki S, De Planell-Saguer M, Nelson PT, Mourelatos Z. (2007). An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell , 1141–1151. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. (1999). Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity , 387–398. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. (1999). MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol , 94–97. [DOI] [PubMed] [Google Scholar]
- Lee WH, Lee HH, Vo MT, Kim HJ, Ko MS, Im YC, Min YJ, Lee BJ, Cho WJ, Park JW. (2011). Casein kinase 2 regulates the mRNA-destabilizing activity of tristetraprolin. J Biol Chem , 21577–21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. (2005). MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol , 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Wagner E. (2005). Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev , 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. (2001). Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol , 6461–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra S, Chou CF, Luber CA, Lee KY, Mann M, Chen CY. (2008). The AU-rich element mRNA decay-promoting activity of BRF1 is regulated by mitogen-activated protein kinase-activated protein kinase 2. RNA , 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese FP, Aubareda A, Tudor C, Saklatvala J, Clark AR, Dean JL. (2010). MAPKAP kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J Biol Chem , 27590–27600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Lal A, Martindale JL, Kawai T, Gorospe M. (2006). Translational repression by RNA-binding protein TIAR. Mol Cell Biol , 2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire VA, Rosner D, Ananieva O, Ross EA, Elcombe SE, Naqvi S, van den Bosch MM, Monk CE, Ruiz-Zorrilla Diez T, Clark AR, Arthur JS. (2017). Beta interferon production is regulated by p38 mitogen-activated protein kinase in macrophages via both MSK1/2- and tristetraprolin-dependent pathways. Mol Cell Biol , e00454-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. (2004). Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell , 185–197. [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Gao M, O’Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J. (2002). The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J , 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. (2002). MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem , 3065–3068. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Padamsi Z, Fakim H, Milette S, Dunham WH, Gingras AC, Fabian MR. (2015). The eIF4E-binding protein 4E-T is a component of the mRNA decay machinery that bridges the 5’ and 3’ termini of target mRNAs. Cell Rep , 1425–1436. [DOI] [PubMed] [Google Scholar]
- Patranabis S, Bhattacharyya SN. (2016). Phosphorylation of Ago2 and subsequent inactivation of let-7a RNP-specific microRNAs control differentiation of mammalian sympathetic neurons. Mol Cell Biol , 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi MY, Wang ZZ, Zhang Z, Shao Q, Zeng A, Li XQ, Li WQ, Wang C, Tian FJ, Li Q. et al (2012). AU-rich-element-dependent translation repression requires the cooperation of tristetraprolin and RCK/P54. Mol Cell Biol , 913–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. (1996). MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol , 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rataj F, Planel S, Desroches-Castan A, Le Douce J, Lamribet K, Denis J, Feige JJ, Cherradi N. (2016). The cAMP pathway regulates mRNA decay through phosphorylation of the RNA-binding protein TIS11b/BRF1. Mol Biol Cell , 3841–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzeczkowski K, Beuerlein K, Muller H, Dittrich-Breiholz O, Schneider H, Kettner-Buhrow D, Holtmann H, Kracht M. (2011). c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J Cell Biol , 581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin M, Lu M, Leuenberger SA, Stoecklin G, Mallaun M, Gross B, Gherzi R, Hess D, Hemmings BA, Moroni C. (2004). The ARE-dependent mRNA-destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J , 4760–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen ZJ, Malter JS. (2015). Regulation of AU-rich element RNA binding proteins by phosphorylation and the prolyl isomerase Pin1. Biomolecules , 412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science , 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Karin M. (2002). The control of mRNA stability in response to extracellular stimuli. Mol Cells , 323–331. [PubMed] [Google Scholar]
- Stein B, Brady H, Yang MX, Young DB, Barbosa MS. (1996). Cloning and characterization of MEK6, a novel member of the mitogen-activated protein kinase kinase cascade. J Biol Chem , 11427–11433. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Mayo T, Anderson P. (2006). ARE-mRNA degradation requires the 5’-3’ decay pathway. EMBO Rep , 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. (2004). MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J , 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica C, Carmichael JB, Parker H, Pare J, Hobman TC. (2006). Interactions between the RNA interference effector protein Ago1 and 14-3-3 proteins: consequences for cell cycle progression. J Biol Chem , 37646–37651. [DOI] [PubMed] [Google Scholar]
- Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo RF, Anderson P, Shanley TP. (2007). Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem , 3766–3777. [DOI] [PubMed] [Google Scholar]
- Tao X, Gao G. (2015). Tristetraprolin recruits eukaryotic initiation factor 4E2 to repress translation of AU-rich element-containing mRNAs. Mol Cell Biol , 3921–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenekeci U, Poppe M, Beuerlein K, Buro C, Muller H, Weiser H, Kettner-Buhrow D, Porada K, Newel D, Xu M. et al (2016). K63-Ubiquitylation and TRAF6 pathways regulate mammalian P-body formation and mRNA decapping. Mol Cell , 943–957. [DOI] [PubMed] [Google Scholar]
- Tiedje C, Ronkina N, Tehrani M, Dhamija S, Laass K, Holtmann H, Kotlyarov A, Gaestel M. (2012). The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet , e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor C, Marchese FP, Hitti E, Aubareda A, Rawlinson L, Gaestel M, Blackshear PJ, Clark AR, Saklatvala J, Dean JL. (2009). The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett , 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kruys V, Huez G, Gueydan C. (2002). AU-rich element-mediated translational control: complexity and multiple activities of trans-activating factors. Biochem Soc Trans , 952–958. [DOI] [PubMed] [Google Scholar]
- Zhao KW, Li X, Zhao Q, Huang Y, Li D, Peng ZG, Shen WZ, Zhao J, Zhou Q, Chen Z. et al (2004). Protein kinase Cdelta mediates retinoic acid and phorbol myristate acetate-induced phospholipid scramblase 1 gene expression: its role in leukemic cell differentiation. Blood , 3731–3738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.