

Summary
15 years ago, the fundamental biology of an inflammatory signaling complex eventually dubbed “the inflammasome” began to unravel in chronologic parallel with the discovery that many inflammatory diseases were associated with its hyperactivity. Though the genetic origins of Familial Mediterranean Fever (FMF, caused my mutations in MEFV) were discovered first, it would take nearly two decades before the mechanistic connections to a Pyrin inflammasome were made1. In the interim, the intensive study of the NLRP3 inflammasome, and the diseases associated with its hyperactivation, have largely dictated the paradigm of inflammasome composition and function. Despite impressive gains, focusing on NLRP3 left gaps in our understanding of inflammasome biology. Foremost among these gaps were how inflammasomes become activated and the connections between inflammasome structure and function. Fortunately, work in another inflammasome inducer, NLRC4, grew to fill those gaps. The current understanding of the NLRC4 inflammasome is perhaps the most comprehensive illustration of the inflammasome paradigm: trigger (e.g. cytosolic flagellin), sensor (NAIP), nucleator (NLRC4), adaptor (ASC), and effector (CASP1). Detailed work has also identified observations that challenge this paradigm. Simultaneously, the features unique to each inflammasome offer a lesson in contrast, providing perspectives on inflammasome activation, regulation, and function. In this review, we endeavor to highlight recent breakthroughs related to NLRC4 inflammasome structure and activation, important in vivo work in infection and systemic inflammation, and the characterization of a spectrum of human NLRC4-associated autoinflammatory diseases.
ORIGINS
NLRC4 was originally described by Poyet and colleagues as a pro-apoptotic protein. Searching for structural homologues of APAF1: a protein known to activate apoptotic caspases in response to cytosolic cytochrome c, and containing a Caspase-Activation and Recruitment Domain (CARD) and ATP-binding site2, they identified a protein capable of activating Caspase-1 (CASP1, a.k.a. IL-1 converting enzyme, ICE) and dubbed it ICE-protease Activating Factor (IPAF). When its domain structure clearly placed it in the family of Nucleotide binding/Leucine-Rich Repeat (NLR) proteins, and by virtue of its amino-terminal CARD domain, it was renamed NLRC4.
In the 15 years since, the known interaction with CASP1 has resulted in its inclusion in the pantheon of proteins known to oligomerize and induce inflammasome-activation. We now appreciate that this oligomerization results in an exponential cascade of adapter recruitment and CASP1 autoproteolysis known as inflammasome activation, with resultant cleavage of the inflammatory substrates proIL-1β, pro-IL18, and Gasdermin D. Herein, we examine the unique features of NLRC4 as a both an NLR protein and inflammasome activator. We will focus predominantly on the many recent advances in 1) the structural biology that helps define NLRC4 function, 2) the sensor proteins, known as NAIPs (NLR family Apoptosis Inhibitory Proteins), that integrate cytosolic pathogen sensing and NLRC4 inflammasome activation, 3) the importance of NLRC4 activation in host defense, particularly against enteric pathogens, and 4) the special association of NLRC4 with IL-18 in human autoinflammatory disease.
TRIGGERING THE NLRC4 INFLAMMASOME
The initial discovery of IPAF (later NLRC4), was accompanied by the observation that the full-length protein was unable to activate CASP1 in transfected cells, while expression of truncated versions of the protein lacking the C terminal series of leucine rich repeats (LRRs) did induce CASP1 activation2. Since this observation, it has become clear that the activation of NLRC4 and other inflammasome activating proteins is tightly regulated by transcriptional and post-transcriptional mechanisms in order to prevent unregulated inflammatory signaling by these complexes. NLRC4 expression is upregulated by pro-inflammatory stimuli such as TNFα as well as through genotoxic stress-mediated P53 activation3, 4. However, consistent with its role in danger sensing, numerous studies have demonstrated that basal levels of NLRC4 expression are sufficient to support NLRC4 inflammasome activation in both epithelial and immune cells. NLRC4 inflammasome activation is primarily regulated at the post-transcriptional level. The two best-described regulatory mechanisms are ligand binding and phosphorylation.
The first demonstration that activation of NLRC4 was likely ligand regulated was the demonstration that murine macrophages lacking NLRC4 failed to activate CASP1 after exposure to Salmonella typhimurium5. Two groups subsequently demonstrated that intra-cytoplasmic flagellin from salmonella was capable of inducing NLRC4-mediated CASP1 activation6, 7. The common presumption in the field held that the LRR domain of NLRC4 acted as a direct flagellin receptor, as had been the case for the LRR domain of TLR58. Miao and colleagues went on to demonstrate that components of the Salmonella type III secretion system (T3SS) could induce murine NLRC4–dependent CASP1 activation in the absence of flagellin, suggesting that activation of NLRC4 by bacterial ligands might be more complex9, 10. Building upon the subsequent observation that NAIP5 also appeared to be required for flagellin induced CASP1 activation, Kofoed and Vance and Zhao et al. nearly simultaneously showed that mouse NAIP5 and NAIP2 formed complexes with NLRC4 and either flagellin or T3SS rod protein (Salmonella PrgJ) respectively11, 12. Additional studies later revealed that Naip6 could substitute for Naip5 in the recognition of flagellin and that Naip1 demonstrated a similar ability to induce NLRC4 inflammasome activation in response to T3SS needle protein PrgI13, 14. Thus, different Naip genes in mice encode proteins capable of conferring specific ligand-mediated activation of NLRC4 to a number of bacterial ligands. However, humans lack the duplication of the NAIP gene seen in mice. Nevertheless, the single human NAIP was shown to permit T3SS rod protein PrgJ mediated NLRC4 activation. Recently, a splice variant transcribed from human NAIP was demonstrated to promote flagellin-induced NLRC4 inflammasome activation in human cells15.
The question remained how bacterial ligand binding connected NAIP, NLRC4, and inflammasome activation. As an early clue, initial studies of NLRC4 revealed heterotypic interactions with several NLR proteins, namely NOD1, NOD2, NLRP3, and NAIP, through their Nucleotide Binding Domains using overexpression and co-immunoprecipitation16. Later structural work suggested that ligand binding alters the structure of NAIP proteins such that they can interact with NLRC4 and induce its oligomerization, and is discussed in detail below. Nonetheless, NLRC4 may have heterotypic interactions with other NLR proteins, potentially via a similar structural mechanism. Super-resolution microscopy showed that NLRC4 and NLRP3 co-localize to inflammasome structures in infected cells, though these studies lack the resolution to demonstrate direct protein:protein interactions between NLRC4 and NLRP317, 18. It remains unknown whether the formation of hetero-oligomeric structures with other NLR proteins is a common feature of NLR signaling or whether the ability of NLRC4 to respond to bacterial ligand-bound NAIP represents a mechanism of NLR signaling unique to NLRC4.
Because the consequences of unregulated inflammatory signaling can be quite deleterious to the health of an organism, it is not surprising that additional regulatory mechanisms have been identified in NLRC4 activation. These include post-translational modifications, particularly phosphorylation and possibly ubiquitination. Murine NLRC4 expressed in macrophages was found to be phosphorylated on Serine 533 (S533) in response to Salmonella exposure. Further, an NLRC4 mutant unable to be phosphorylated at this site was expressed in macrophages lacking wild type NLRC4 (Nlrc4−/−), and exhibited blunted activation of caspase-1, induction of cell death, and secretion of IL-1β after Salmonella infection19. The δ isoform of Protein Kinase C (PKCδ) was found to phosphorylate NLRC4 in vitro and both inhibitor administration and genetic depletion of PKCδ was also found to impair NLRC4-inflammasome dependent downstream signaling in this system19. Recently, another kinase, Leucine Rich Repeat-containing Kinase-2 (LRRK2), was also found to associate with NLRC4. Association of NLRC4 with LRRK2 was accompanied by phosphorylation at S53320. As previously reported for PKCδ, genetic reduction of LRRK2 expression also resulted in diminished NLRC4 inflammasome activation. Surprisingly, X-ray crystallographic studies found S533 was phosphorylated in purified recombinant NLRC4 expressed in insect cells. This protein was crystalized in its inactive monomeric state and the phosphorylation of this serine residue seemed to stabilize interactions within the inactive molecule. It is likely that these phosphorylation-dependent contacts that would stabilize the inactive state are artifacts induced during NLRC4 crystal formation. However, the finding suggests that the phosphorylation alone is insufficient to induce activation and oligomerization of NLRC4 on its own. This is further supported by the finding that H. pylori flagellin is able to induce NLRC4 phosphorylation but does not trigger NAIP5-induced events suggestive of NLRC4 inflammasome activation including caspase-1 activation or IL-1β secretion. These data support the possibility that phosphorylation of NLRC4 may be involved in a preactivation step, possibly causing the release of NLRC4 from a basal inactive protein complex, allowing the protein to respond to liganded NAIP proteins, and/or enabling NLRC4 to homo-oligomerize more readily once the association with ligand-bound NAIP is initiated21. Though key questions about the identity of the kinases responsible for NLRC4 phosphorylation and whether they play a predominant or redundant role remain open, the substantial functional data suggest this is likely a critical component in the regulatory machinery responsible for ensuring NLRC4 is activated appropriately in response to infection.
STRUCTURAL FEATURES OF NLRC4
Relative to other NLR proteins, the existence of several detailed structural studies of NLRC4 has been a boon to understanding the potential molecular mechanisms of NLRC4 activation. Like all NLR family members, NLRC4 shares a common three-domain structure: an N-terminal homotypic interaction domain, a central nucleotide binding domain, and a series of C-terminal LRRs. The homotypic interaction domain of NLRC4 consists of the first 94 amino acids of the protein, which is predicted to fold into a common structural pattern known as the CARD (Caspase-Activation and Recruitment Domain). The CARD typically folds into 6 anti-parallel α-helices packed around a hydrophobic core. The central Nucleotide Binding Domain falls into the superfamily of AAA+ ATPases and shares structural features with other NLR proteins, including a core NTPase domain and two helical domains (HD1 and HD2) that are interrupted by a region termed a winged helical domain (WD). The NLRC4 C-terminal 440 amino acids consist of 15 repeating structural units each a leucine rich beta strand linked to a short 8 to 15 amino acid alpha helix, so called leucine rich repeats.
Recombinant mouse NLRC4 lacking the N-terminal CARD domain was isolated in sufficient quantity and quality to permit X-ray crystallographic analysis of the structure. This monomeric protein was found to have extensive intramolecular interactions between the C-terminal LRR domain and the Nucleotide Binding Domain. The monomeric structure is stabilized by these interactions along with interactions with ADP occupying the nucleotide binding site22. This group further demonstrated that mutation of a key residue in proximity to ADP, H443, led to spontaneous formation of oligomeric NLRC4, and ostensibly inflammasome activation. Many of the human NLRC4 gain-of-function mutations (including H443, described in detail below) cluster near the ADP binding site, suggesting that nucleotide binding or ATP hydrolysis are fundamental regulators of the conformational changes necessary for oligomerization and inflammasome activation. Correspondingly, mutation of the key lysine in the walker A motif of the NBD results in an NLRC4 protein that fails to bind ATP and is unable to induce caspase-1 activation when over expressed23.
Two studies of NLRC4 co-expressed with NAIP2 and the T3SS needle PrgJ revealed that ligand-bound NAIP associated with NLRC4 and appeared to form disk-like structures consisting of (now opened) NLRC4 monomers. The NLRC4 monomers in this disk make extensive intermolecular contacts with opposite faces of neighboring NLRC4 molecules24, 25. These two studies demonstrated a single NAIP was present in these oligomeric structures that otherwise contained 11-fold symmetry; suggesting a single NAIP could initiate the assembly of 10 NLRC4 molecules into a wheel shaped inflammasome. A third cryo-EM based study on NLRC4 complexes began with coexpression of NAIP5 and Flagellin (FliC-D0L) in HEK293 cells. In contrast to the previous cryo-EM work, it revealed NLRC4-containing structures that formed rod shaped oligomers consisting of helically arranged monomers rather than closed discs26. It is unclear whether NLRC4 may form either complex when activated, whether there is a true difference in the oligomeric structure based on the activating ligand-NAIP protein complex, or the extent to which either or both may represent overexpression artifact.
In all three cryo-EM studies, the NLRC4 structure is opened relative to the X-ray crystallography structure of monomeric NLRC4. This opening is accomplished by roughly a 90-degree rotation of the LRR domain at a hinge region between the LRR and the central Nucleotide Binding Domain. This rotation results in the surface localization of a patch of basic amino acids in the NBD that can then interact with an acidic surface of the next NLRC4 monomer added to the structure25, 27. The rotation also positions an acidic region of the LRR to interact with LRR of the previously added NLRC4 monomer, stabilizing the opened structure. In this way, the opened NLRC4 can catalyze the addition of new NLRC4 monomers to the oligomeric structure. The open structure of NAIP is predicted to have similarly positioned basic residues as found in the open NLRC4 unit but it lacks the reciprocal acidic surface. Thus, NAIP is able to engage a single NLRC4 to initiate inflammasome assembly but cannot be further incorporated into an assembling inflammasome, resulting in one NAIP per oligomer. These data also support the ability of hyperactive NLRC4 to induce oligomerization and inflammasome activation without NAIP, with relevance to the pathophysiology of human autoinflammatory disease (as discussed below). These studies offer an unprecedented level of structural detail that is not currently available for NLRP3, NOD2, or other NLR proteins. It is unknown whether the deduced structural changes that occur between these states for NAIP/NLRC4 oligomers will hold true in other NLR proteins.
NLRC4 EFFECTOR MECHANISMS
Activation of the NLRC4 inflammasome appears to activate several different host signaling events. NLRC4 was first identified as potential nucleotide binding protein that was capable of activating CASP1, the primary protease responsible for converting pro-IL-1β to active, secreted IL-1β2, 28. CASP1 is also known to carry out similar proteolytic maturation of IL-1829, 30. Thus, the effects of NLRC4 activation on cytokine signaling are dependent on 1) the presence/abundance of pro-cytokine substrates, 2) the expression of IL-1 and IL-18 receptors in both immune and non-immune cells, and 3) the site of receptor expression (paracrine versus endocrine), all covered in depth in other sections of this special issue [REFs]. However, CASP1 has other intracellular targets that can regulate cellular function. CASP1 can induce proteolytic cleavage of the transcription factor, sterol response element binding protein-1 (SREBP1), leading to alteration in sterol metabolism in the target cells31. CASP1 also proteolytically activates a cytosolic protein known as Gasdermin-D, and the amino-terminal fragment resulting from this cleavage subsequently assembles into cytoplasmic membrane pores32. It is now known that activation of Gasdermin induces a loss of cell membrane integrity and ultimately induces the cell death process known as pyroptosis. Unlike apoptosis (classic programmed cell death), pyroptosis result in spillage of proinflammatory intracellular components like the protein HMGB1 that also cause further inflammatory signaling in nearby host cells. This may be the mechanism by which activated cytokines are released from macrophages. In addition to inducing inflammatory responses in the host, pyroptosis also contributes to host defense by disrupting the intracellular environment conducive to the pathogens that activate NLRC433.
In addition to processing pro-cytokines and inducing pyroptosis, CASP1 activation downstream of NLRC4 results in the rapid production of pro-inflammatory prostaglandin and leukotrienes including PGE2 and LTB4. Moltke and colleagues showed that a flagellin-derived NAIP-NLRC4 activator induced calcium influx in peritoneal macrophages that liberated arachidonic acid by activating calcium dependent phospholipase A234. Arachidonic acid mobilized by CASP1 activation then fed into prostaglandin biosynthesis through cyclooxygenase-1 and leukotriene biosynthesis by the action of lipoxygenases. It remains unclear by what mechanism CASP1 drives arachidonic acid liberation, and whether this mechanism is somehow specific to NLRC4 activation. Proteomic studies have identified as many as 1000 potential protein targets of CASP1, suggesting that additional downstream signaling events may occur in response to NLRC4-inflammasome mediated CASP1 activation beyond the three downstream signaling pathways discussed above35.
Another unique aspect of NLRC4 activation is its ability to interact directly with CASP1. The CARD domain of NLRC4 can interact with the CARD domain of pro-CASP1, allowing NLRC4 to directly activate the protease. However, NLRC4-induced CASP1 activation is enhanced in cells by the presence of the adapter ASC (apoptotic speck protein containing a CARD). ASC consists of a pyrin domain and CARD domain. Both inflammasome activation and initiation of apoptosis can cause the condensation of a cell’s cytoplasmic ASC into a potentially massive insoluble oligomer known as the apoptotic speck36. In vitro assembly of ASC fibrils seems to result from homopolymeric helices consisting of pyrin domains37. This brings the CARD domains of ASC in proximity allowing them to undergo a similar polymerization, which can lead to polymerization of pro-CASP1 CARD domains and result in activation of many pro-CASP1 molecules. The other major inflammasome-forming proteins, including NLRP3, NLRP1, PYRIN, and AIM2, all require ASC to activate caspase-1. Activated NLRC4 also associates with ASC and colocalizes with the ASC-containing Speck after activation by Salmonella18, 38. These findings are consistent with the ability of ASC to enhance NLRC4-induced CASP1 activation.
While CASP1 activation is a shared downstream effector mechanism between NLRC4 and other inflammasome forming proteins, at least one other effector protease has been described for NLRC4. NLRC4 was noted to recruit pro-CASP8 to the inflammasome complex38. CASP8 recruitment and activation in response to Salmonella infection requires both NLRC4 and ASC. CASP8 activation appears to be able to substitute for CASP1 in the processing of pro-IL-1β38. However, unlike CASP1, CASP8 is a pro-apoptotic caspase. In cells in which CASP1 or Gasdermin are depleted, preventing pyroptosis, Salmonella-induced activation of NLRC4 will result in CASP8-dependent apoptosis39.
NLRC4 IN HOST DEFENSE
NLRC4 is a critical component of defense against enteric pathogens
NLRC4 activation in response to Salmonella suggested the protein may play an important role in defense against enteric pathogens. Further supporting this hypothesis, Salmonella typhimurium, but not commensal intestinal microbes, were found to induce CASP1 activation and IL-1β secretion through NLRC4 in intestinal macrophages40. After Salmonella PrgJ was found to activate the NAIP:NLRC4 inflammasome, homologs of this T3SS Needle protein from other enteric pathogens including Shigella flexneri (MxiH) and enterohemorrhagic E. coli (EprI) were also found to activate NAIP1 – NLRC414. Interestingly, ligands from commensal organisms, like flagellin from non-pathogenic E. coli, were found to poorly activate NLRC4 when compared to Salmonella-derived flagellin41. A role for NLRC4 in defense against Salmonella infection was clearly demonstrated when Nlrc4−/− mice were found to exhibit increased susceptibility to orogastric challenge with Salmonella, showing decreased survival and increased systemic bacterial loads40, 42. Interestingly, this difference was not seen when mice were challenged intraperitoneally, suggesting NLRC4-mediated signaling was redundant to other pathogen sensing mechanisms for Salmonella outside of the GI tract. Similar to the impact of Nlrc4 deletion on Salmonella infection outcomes, infection with the rodent GI pathogen Citrobacter rodentium was more severe in NLRC4 deficient animals. Nlrc4−/− mice infected by orogastric inoculation with C. rodentium were found to have increased Citrobacter bacterial loads, persistent weight loss, and worse colonic pathology than similarly treated WT mice43. Combined, these studies all suggest that the NLRC4 inflammasome plays an important role in facilitating the clearance of bacterial enteric pathogens.
While many initial studies focused on recognition of intracellular Salmonella by macrophages or phagocytic cells, recent studies have demonstrated that multiple defensive mechanisms initiated by NLRC4 in both epithelial and immune cells are responsible for restricting Salmonella and likely other enteric pathogens. Although many early studies focused on the NLRC4’s ability to drive IL-1β secretion, a transgenic mouse that overexpressed a secreted IL-18 decoy receptor was found to partially recapitulate the increased susceptibility to orogastric S. typhimurium seen with Nlrc4 deletion42. Likewise, Nlrc4-dependent IL-18 secretion activated Natural Killer cells, which were essential for early colonic inflammation in Salmonella infection44.
Increasingly, we can also appreciate that non-hematopoietic NLRC4 inflammasome signaling, primarily in gut epithelium, plays a role in controlling enteric bacterial infections. Norlander and colleagues showed that reconstituting Nlrc4−/− mice with wild-type bone marrow did not restore control of Citrobacter rodentium infection45. These observations using bone marrow transplantation have subsequently been supported by tissue-specific gene knock out experiments. Sellin and colleagues demonstrated that tissue-specific deletion of Naip1-6 in mouse gut epithelium led to increased Salmonella bacterial burdens early in infection. Specifically, expulsion of Salmonella-infected enterocytes into the gut lumen was dependent on intestinal expression of NAIP proteins46. Later studies with gut epithelium specific knock-out of Nlrc4 also showed that expulsion of infected enterocytes relied on NLRC4 using both in-vivo animal studies and using an elegant primary cell organoid system47. As with the peritoneal macrophages34, NLRC4-dependent enterocyte expulsion was accompanied by eicosanoid production. Remarkably, the enterocyte death and expulsion did not entirely require CASP1, demonstrating that an NLRC4-induced, CASP8-mediated cell death program also contributed to pathogen-infected enterocyte expulsion. Combined, these studies demonstrate that the NAIP-NLRC4 inflammasome plays an important role in controlling enteric infection by bacterial pathogens in both the epithelium and immune compartments.
NLRC4 is a critical component of defense against systemic pathogens
Though the preponderance of murine in vivo investigation of NLRC4 has focused on enteric infections or cellular responses to enteric pathogens, a great deal of work has been done examining its role combatting non-enteric pathogens. Additionally, the in-vivo utilization of non-infectious NLRC4 stimuli has resulted in some startling findings with relevance to the physiology of acute systemic inflammation and the Systemic Inflammatory Response Syndrome (SIRS).
Given the robust response of NLRC4 to NAIP proteins activated by cytosolic flagellin or components the T3SS, a number of non-enteric bacteria require NLRC4 for full recognition and clearance. Listeria monocytogenes triggers multiple inflammasomes in macrophages, but requires NLRC4 for inflammasome activation unless cells are first primed with LPS48. Similarly, Pseudomonas aeruginosa peritonitis triggers the NLRC4 inflammasome in a flagellin-independent manner49, and NLRC4 can protect from lung infection with non-flagellated bacteria such as Klebsiella pneumonia50. As with enteric pathogens like Salmonella, pulmonary pathogens like Burkholderia pseudomallei can trigger both the NLRC4 and NLRP3 inflammasomes and both are required for full protection51. These non-enteric pathogens nevertheless trigger NLRC4 via mechanisms similar to classical NLRC4-activating enteric pathogens like Salmonella and Shigella: through detection of flagellin or components of bacterial T3SS by NAIP proteins. However, some bacteria, namely the intracellular rickettsial pathogen Anaplasma phagoctyophilum, may induce macrophage NLRC4 activation without expression of these PAMPs, via a mechanism dependent on Cyclo-oxygenase 2 (COX2)-mediated prostaglandin synthesis24, 52.
Maltez and colleagues recently dissected complementary roles for the outcomes of NLRC4 inflammasome activation after infection with the ubiquitous intracellular bacterium C. violaceum. Clearance of C. violaceum from the spleen required only pyroptosis, whereas bacterial clearance from the liver required NLRC4-driven IL-18 to induce appropriate cytotoxicity in NK cells53. This group was intrigued about the mechanism by which c. violaceum and similar intracellular bacteria were cleared from the spleen in the absence of cytokine production. They identified that inflammasome activation within infected macrophages induced CASP1-dependent but IL-1β and IL-18 independent Pyroptosis-associated Intracellular Traps (PITs)33. PITs consisted of swollen macrophages with disrupted cellular and nuclear membranes and trapped intracellular bacteria within their borders. These PITs were readily taken up by activated neutrophils, and the trapped bacteria were subsequently cleared through neutrophil NADPH-oxidase activity33. This mechanism may be particularly important for clearance of the fastidious intracellular bacteria that trigger NLRC4 inflammasomes.
The connection between Nlrc4 activation and prostaglandin synthesis was dramatically illustrated by von Moltke and colleagues. This group capitalized on the fact that cytosolic delivery of flagellin monomers could robustly and specifically activate macrophage NAIP5/NLRC4 inflammasome in vitro. They systemically introduced a fusion protein enabling delivery of flagellin intracellularly (FlaTox), and induced an immediate and deadly “eicosanoid storm.” Injected mice developed rapidly fatal vascular leak that was dependent on the NAIP5-NLRC4-CASP1 inflammasome as well as Phospholipase A2 and COX1 activity in resident peritoneal macrophages. Importantly, this response was independent of IL-1β and IL-1834. Subsequently, this group expressed FlaTox specifically in intestinal epithelial cells, with resulting cytokine production and cell death requiring dual effects of both CASP1 and CASP8, as discussed previously47.
Although it is clear that NLRC4 is expressed and active in murine macrophages and intestinal epithelial cells, its expression and role in neutrophils is controversial. In certain intracellular infections (like Burkholderia) or in immune privileged sites (like cornea), pathogens capable of triggering the NLRC4 inflammasome instead drive neutrophil production of IL-1β independently of NLRC451, 54. By contrast, Chen et al. showed a specific function for neutrophil NLRC4 in prolonged IL-1β production, in the absence of neutrophil cell death, in the appropriate clearance of Salmonella55.
NLRC4 AND HUMAN DISEASE
The role of NLRC4 in murine host defense continues to undergo elaboration. Similarly, human NLRC4 is clearly important for in vitro detection and clearance of intracellular bacterial infection, and different isoforms of the single human NAIP appear capable of detecting both T3SS rod proteins and flagellin15. However, the relevance of NLRC4 for the clearance of these pathogens from human hosts in the modern age is less well developed, and immunodeficiency related to NLRC4 loss-of-function (or any inflammasome component, for that matter) has not been described.
Increasingly, investigators are discovering an important role for NLRC4 in driving human autoinflammation. Autoinflammation is the concept of systemic or organ-specific inflammation not attributable to infection, malignancy, or antigen-specific autoimmunity56. The autoinflammatory paradigm was initially molded around monogenic disorders of spontaneous or increased inflammasome activity, and reinforced by the dramatic response of many of these disorders to blockade of IL-1β. The concept has blossomed to include a staggering number of diseases, many with monogenic causes, resulting from dysregulation and hyperactivity of innate immune pathways56. Disorders of inflammasome hyperactivity (inflammasomopathies) typified many of the first-described autoinflammatory diseases, but the genre has expanded far beyond the inflammasome.
Following more than a decade of intense investigation of the PYRIN and NLRP3 inflammasomes, and the related inflammasomopathies FMF and CAPS (Cryopyrin-Associated Periodic Syndromes), respectively, the first description of NLRC4-associated disease came in 2014. Two groups independently and simultaneously described gain-of-function mutations in NLRC4 as the cause of a syndrome of infantile enterocolitis and recurrent Macrophage Activation Syndrome57, 58. Functionally, these mutations induced changes identical to those seen with FMF and CAPS mutations, with spontaneous/increased macrophage inflammasome activity. One NLRC4-MAS patient showed substantial response to IL-1 inhibition.
Three items set the NLRC4 inflammasomopathy apart from the PYRIN and NLRP3 inflammasomopathies, and these features have been largely confirmed by subsequent reports. First, Macrophage Activation Syndrome (MAS) is not a feature of FMF and CAPS. MAS is a life-threatening syndrome of hectic fever, low peripheral cell counts, hepatobiliary dysfunction, coagulopathy, dramatically elevated serum ferritin, and hemophagocytosis59. In one patient, features of MAS began in utero and resulted in placental thrombosis, pre-term birth, and perinatal demise60. As such, MAS is clinically very similar to familial Hemophagocytic Lymphohistiocytosis (fHLH), a disorder caused by impaired granule-mediated cytotoxicity that is typically categorized as an immunodeficiency. MAS typically complicates rheumatic diseases, particularly systemic Juvenile Idiopathic Arthritis (sJIA) and Adult-Onset Still’s Disease (AOSD). Prior to the description of NLRC4-related disease, MAS had very rarely been associated with inflammasomopathy or autoinflammation more generally61. Thus, NLRC4 finally provided a clear link between autoinflammation and the Systemic Inflammatory Response Syndrome (SIRS) via a hyperinflammatory SIRS subtype known as MAS.
Second, the described NLRC4-MAS patients had extraordinary and chronic elevation of peripheral IL-18. Despite both IL-1β and IL-18 being expressed in myeloid cells and requiring inflammasome activity for their maturation, prior to the discovery of NLRC4-MAS neither cytokine had been measured in great abundance in inflammasomopathy patients’ serum. By contrast, extremely high levels of IL-18 were detected in patients with sJIA and AOSD, particularly those who developed MAS62, 63. Thus, even though NLRC4 is both an inflammasomopathy and a cause of MAS, it seems that very high IL-18 levels correlate only with the latter. Interestingly, there is genetic evidence for an association between NLRC4 and IL-18 remote from MAS: small elevations in IL-18, long associated with worse cardiac outcomes, were associated with an NLRC4 polymorphism in a Genome-Wide Association Study (GWAS) of patients with known cardiac disease64.
Despite providing little insight into its sources, IL-18’s compelling role as a biomarker have spurred interest into its role as a therapeutic target. Specifically, an infant with severe NLRC4-MAS refractory to aggressive treatment, including IL-1 blockade, ultimately showed an acute and dramatic response when IL-18 blockade was added65. This was achieved by administration of exogenous recombinant IL-18 binding protein (IL-18BP). Similarly, another infant with NLRC4-MAS may have responded well to blockade of Interferon-γ, a cytokine implicated in fHLH whose production is induced by IL-1866.
Third, the infantile enterocolitis associated with NLRC4 hyperactivity is distinct amongst both autoinflammatory diseases and other causes of very early onset Inflammatory Bowel Disease (VEO-IBD). The described cases of NLRC4-associated enterocolitis all occurred early in infancy, although not always in the neonatal period57, 58, 60, 65. Inflammation could involve the entirety of the intestines, from stomach through colon, although more mildly affected patients appeared to have primarily duodenal inflammation. Most strikingly, gastrointestinal disease in those patients who survived infancy largely disappeared, even though bouts of MAS persisted and levels of serum IL-18 remained highly elevated57, 58, 65. In the context of murine NLRC4’s extraordinary role in gut host defense, this observation suggests a complex interaction of NLRC4, IL-18, and the maturation of intestinal microbial and immune homeostasis.
Though MAS and enterocolitis have been the most dramatic clinical manifestations of NLRC4-associated disease, other more typical autoinflammatory symptoms have been described. Kitamura and colleagues described an extended family with a relatively mild phenotype of cold-induced urticaria67. Response to IL-1 inhibition and peripheral IL-18 were not assessed. Subsequently, Volker-Touw et al. described another extended family with a spectrum of autoinflammatory phenotypes, including urticarial and nodular rashes, conjunctivitis, arthritis, IBD, and sensorineural hearing loss (a hallmark symptom of CAPS) that segregated with a missense NLRC4 mutation68. Response to IL-1 inhibition was highly variable in this kindred, and biopsy of the nodular rash demonstrated lymphohistiocytic infiltrate as opposed to the neutrophilic infiltrate of other inflammasomopathy rashes. Serum IL-18 was profoundly elevated in affected members of this kindred. Finally, Kawasaki et al. described a patient with clinical symptoms of severe CAPS whose disease was entirely responsive to IL-1 blockade, and who was eventually found to carry a high-frequency somatic NLRC4 mutation69. Thus, not all patients with NLRC4 inflammasomopathy develop MAS or enterocolitis, but these other presentations are nevertheless consistent with those observed in many other autoinflammatory diseases. The specific proclivity of murine NLRC4 activation for eicosanoid production has not been evaluated in these patients, particularly those with incomplete responses to IL-1β and/or IL-18 blockade, but COX inhibition has been insufficient treatment in all but the most mildly-affected patients.
The sources of phenotypic heterogeneity are difficult to establish in any monogenic disease, although here again NLRC4 offers some interesting clues. Despite small numbers of patients described thusfar, there may be a genotype/phenotype correlation between the amino acid position of NLRC4 mutation and the phenotypes described. MAS associated mutations have been concentrated around amino acid positions 337 to 341, within Helical Domain (HD)-1. Mutations associated with more classical inflammasomopathy symptoms have been identified at positions 443 and 445, within the Winged Helix Domain (WHD). Notably, the H443P mutation, was uniquely associated with increased interaction with SUG1 and ubiquitinated proteins, and ultimately drove a CASP8 dependent apoptosis in a human lung epithelial cell line70. This increased activity was independent of S533 phosphorylation, consistent with in vitro work with other NLRC4 gain-of-function mutations (unpublished observations). Levels of CASP1 as a competing effector of the NLRC4 inflammasome in this cell line were not assessed. Somatic mutations, albeit variable in their presentation, occurred at positions 171 (in utero MAS, placental thrombosis) and 177 (severe CAPS), within the Nucleotide Binding Domain (NBD). All of the mutations published to date have occurred within close proximity of the putative ADP/ATP binding site22.
As translational investigation and clinical observations accumulate, we will refine our understanding of the links between NLRC4 hyperactivity, autoinflammation, IL-18, infantile enterocolitis, and MAS. Simultaneously, we hope these findings will illuminate circumstances in which NLRC4 or associated signaling pathways are important in more common diseases like sJIA and AOSD, and potentially in host defense and beyond.
SUMMARY
Despite a relatively delayed start, research on the NLRC4 inflammasome has provided foundational insights into inflammasome biology, and increasingly autoinflammatory disease (see Figure). The complex interactions between NAIP proteins and NLRC4, rather than defining the limits of inflammasome activation, offer detailed insights into the types of inputs evolution has selected. The recent elaboration of the murine crystal and cryo-EM structures have provided a molecular basis for NLR autoinhibition and activation, as well as a living framework for the inflammasome paradigm. A large body of work on the microbiology and cellular immunology of murine Nlrc4 activity has secured its place in the firmament of innate immune host defense mechanisms, and identified avenues of research on novel sites of action (myeloid versus epithelial), regulatory mechanisms (e.g. phosphorylation), inflammasome interacting partners (e.g. other NLRs, Caspase-8), and effector mechanisms (e.g. eicosanoid induction). Finally, the discovery and study of human NLRC4 inflammasomopathies have provided several clues, most notably the unique associations with IL-18, MAS, and infantile enterocolitis, on which future work will surely build. Overall, the foundations of NLRC4 biology, from monomeric structure through human phenotypes, are uniquely comprehensive for inflammasomes and have provided critical insights and tools to probe this important innate immune mechanism.
Figure.
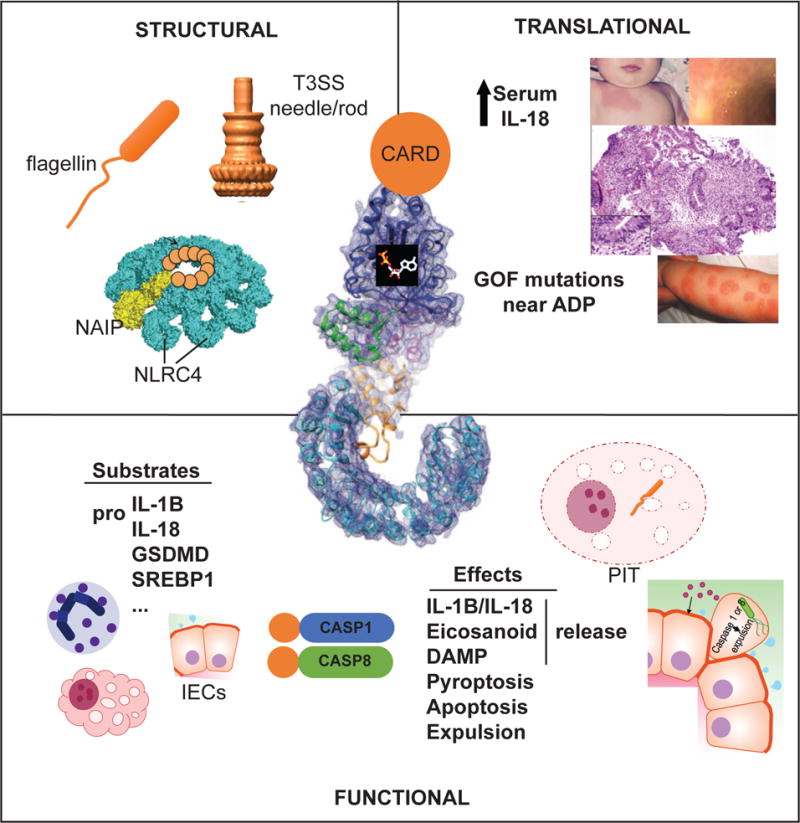
Structural, functional, and translational insights gleaned from the study of NLRC4 inflammasomes. Structural: NAIP proteins detect monomers of flagellin or Type III Secretion System (T3SS) needle or rod proteins and nucleate the oligomerization of NLRC4 to initiate inflammasome activation. Functional: NLRC4 inflammasomes present in macrophages, neutrophils, and Intestinal Epithelial Cells (IECs) mediate substrate cleavage via CASP1 or CASP8 to induce effector programs including release of inflammatory mediators and various cell death programs including Pore-Induced Intracellular Traps (PITs), and IEC expulsion. Translational: Gain-of-function (GOF) mutations clustering near NLRC4’s ADP-binding site have been associated with skin rash, enterocolitis, Macrophage Activation Syndrome, and chronically elevated IL-18. Figures adapted from Galan JE et al.10, Zhang L et al.25, Rauch I et al.47, Canna SW et al.65, and Kitamura A et al.67.
Acknowledgments
JAD was supported by NIH R01 AI088255; SWC was supported by NIH K22 AI123366 and The RK Mellon Foundation Institute for Pediatric Research.
Footnotes
DR SCOTT W CANNA (Orcid ID : 0000-0003-3837-5337)
DISCLOSURES/CONFLICTS OF INTEREST
SWC is an ad hoc consultant for AB2Bio, Ltd. The authors have no relevant financial or other conflicts of interest.
References
- 1.Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–41. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 2.Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276:28309–13. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 3.Gutierrez O, Pipaon C, Fernandez-Luna JL. Ipaf is upregulated by tumor necrosis factor-alpha in human leukemia cells. FEBS Lett. 2004;568:79–82. doi: 10.1016/j.febslet.2004.04.095. [DOI] [PubMed] [Google Scholar]
- 4.Sadasivam S, Gupta S, Radha V, Batta K, Kundu TK, Swarup G. Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene. 2005;24:627–36. doi: 10.1038/sj.onc.1208201. [DOI] [PubMed] [Google Scholar]
- 5.Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 6.Miao EA, Alpuche-Aranda CM, Dors M, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–75. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 7.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–82. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 8.Mizel SB, West AP, Hantgan RR. Identification of a sequence in human toll-like receptor 5 required for the binding of Gram-negative flagellin. J Biol Chem. 2003;278:23624–9. doi: 10.1074/jbc.M303481200. [DOI] [PubMed] [Google Scholar]
- 9.Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–80. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–73. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 12.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–5. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J Immunol. 2013;191:3986–9. doi: 10.4049/jimmunol.1301549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. 2013;110:14408–13. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kortmann J, Brubaker SW, Monack DM. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J Immunol. 2015;195:815–9. doi: 10.4049/jimmunol.1403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damiano JS, Oliveira V, Welsh K, Reed JC. Heterotypic interactions among NACHT domains: implications for regulation of innate immune responses. Biochem J. 2004;381:213–9. doi: 10.1042/BJ20031506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu Y, Misaghi S, Newton K, et al. NLRP3 recruitment by NLRC4 during Salmonella infection. J Exp Med. 2016;213:877–85. doi: 10.1084/jem.20132234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Man SM, Hopkins LJ, Nugent E, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111:7403–8. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qu Y, Misaghi S, Izrael-Tomasevic A, et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature. 2012;490:539–42. doi: 10.1038/nature11429. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Liu X, Li Y, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med. 2017 doi: 10.1084/jem.20170014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, Lamkanfi M. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci U S A. 2015;112:1541–6. doi: 10.1073/pnas.1417945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu Z, Yan C, Liu P, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science. 2013;341:172–5. doi: 10.1126/science.1236381. [DOI] [PubMed] [Google Scholar]
- 23.Lu C, Wang A, Wang L, Dorsch M, Ocain TD, Xu Y. Nucleotide binding to CARD12 and its role in CARD12-mediated caspase-1 activation. Biochem Biophys Res Commun. 2005;331:1114–9. doi: 10.1016/j.bbrc.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Shaw DK, Hammond HL, et al. The Prostaglandin E2-EP3 Receptor Axis Regulates Anaplasma phagocytophilum-Mediated NLRC4 Inflammasome Activation. PLoS pathogens. 2016;12:e1005803. doi: 10.1371/journal.ppat.1005803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Chen S, Ruan J, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science. 2015;350:404–9. doi: 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diebolder CA, Halff EF, Koster AJ, Huizinga EG, Koning RI. Cryoelectron Tomography of the NAIP5/NLRC4 Inflammasome: Implications for NLR Activation. Structure. 2015;23:2349–57. doi: 10.1016/j.str.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Hu Z, Zhou Q, Zhang C, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science. 2015;350:399–404. doi: 10.1126/science.aac5489. [DOI] [PubMed] [Google Scholar]
- 28.Damiano JS, Stehlik C, Pio F, Godzik A, Reed JC. CLAN, a novel human CED-4-like gene. Genomics. 2001;75:77–83. doi: 10.1006/geno.2001.6579. [DOI] [PubMed] [Google Scholar]
- 29.Gu Y, Kuida K, Tsutsui H, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–9. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 30.Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 31.Im SS, Yousef L, Blaschitz C, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13:540–9. doi: 10.1016/j.cmet.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sborgi L, Ruhl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–28. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Moltke J, Trinidad NJ, Moayeri M, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–11. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamkanfi M, Kanneganti TD, Van Damme P, et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics. 2008;7:2350–63. doi: 10.1074/mcp.M800132-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masumoto J, Taniguchi S, Ayukawa K, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 37.Lu A, Magupalli VG, Ruan J, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell. 2014;156:1193–206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol. 2013;191:5239–46. doi: 10.4049/jimmunol.1301581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mascarenhas DPA, Cerqueira DM, Pereira MSF, et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS pathogens. 2017;13:e1006502. doi: 10.1371/journal.ppat.1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franchi L, Kamada N, Nakamura Y, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13:449–56. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Zhang E, Liu F, et al. Flagellins of Salmonella Typhi and nonpathogenic Escherichia coli are differentially recognized through the NLRC4 pathway in macrophages. J Innate Immun. 2014;6:47–57. doi: 10.1159/000351476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho FA, Nalbantoglu I, Aitken JD, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal immunology. 2012;5:288–98. doi: 10.1038/mi.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, Zaki MH, Vogel P, et al. Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem. 2012;287:16955–64. doi: 10.1074/jbc.M112.358705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller AA, Dolowschiak T, Sellin ME, et al. An NK Cell Perforin Response Elicited via IL-18 Controls Mucosal Inflammation Kinetics during Salmonella Gut Infection. PLoS pathogens. 2016;12:e1005723. doi: 10.1371/journal.ppat.1005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal immunology. 2014;7:775–85. doi: 10.1038/mi.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sellin ME, Muller AA, Felmy B, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell host & microbe. 2014;16:237–48. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Rauch I, Deets KA, Ji DX, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 2017;46:649–59. doi: 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–45. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol. 2012;188:5623–35. doi: 10.4049/jimmunol.1200195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS pathogens. 2011;7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedra JH, Sutterwala FS, Sukumaran B, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J Immunol. 2007;179:4783–91. doi: 10.4049/jimmunol.179.7.4783. [DOI] [PubMed] [Google Scholar]
- 53.Maltez VI, Tubbs AL, Cook KD, et al. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity. 2015;43:987–97. doi: 10.1016/j.immuni.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J Immunol. 2012;189:4231–5. doi: 10.4049/jimmunol.1201447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen KW, Gross CJ, Sotomayor FV, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell reports. 2014;8:570–82. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 56.de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. 2015;33:823–74. doi: 10.1146/annurev-immunol-032414-112227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46:1140–6. doi: 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romberg N, Al Moussawi K, Nelson-Williams C, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46:1135–9. doi: 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weaver LK, Behrens EM. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol. 2014;26:562–9. doi: 10.1097/BOR.0000000000000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang J, Alfano DN, Squires JE, et al. Novel NLRC4 Mutation Causes a Syndrome of Perinatal Autoinflammation With Hemophagocytic Lymphohistiocytosis, Hepatosplenomegaly, Fetal Thrombotic Vasculopathy, and Congenital Anemia and Ascites. Pediatric and Developmental Pathology. 2017 doi: 10.1177/1093526616686890. [DOI] [PubMed] [Google Scholar]
- 61.Rigante D, Emmi G, Fastiggi M, Silvestri E, Cantarini L. Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clinical rheumatology. 2015;34:1333–9. doi: 10.1007/s10067-015-2923-0. [DOI] [PubMed] [Google Scholar]
- 62.Shimizu M, Nakagishi Y, Inoue N, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160:277–81. doi: 10.1016/j.clim.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 63.Girard C, Rech J, Brown M, et al. Elevated serum levels of free interleukin-18 in adult-onset Still’s disease. Rheumatology (Oxford) 2016;55:2237–47. doi: 10.1093/rheumatology/kew300. [DOI] [PubMed] [Google Scholar]
- 64.Johansson A, Eriksson N, Becker RC, et al. The NLRC4 Inflammasome Is an Important Regulator of Interleukin-18 Levels in Patients with Acute Coronary Syndromes: A Genome-Wide Association Study in the PLATO Trial. Circulation Cardiovascular genetics. 2015 doi: 10.1161/CIRCGENETICS.114.000724. [DOI] [PubMed] [Google Scholar]
- 65.Canna SW, Girard C, Malle L, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with Interleukin-18 inhibition. The Journal of allergy and clinical immunology. 2016;139:1698–701. doi: 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bracaglia C, Prencipe G, Gatto A, et al. ANTI INTERFERON-GAMMA (IFN gamma) MONOCLONAL ANTIBODY TREATMENT IN A CHILD WITH NLRC4-RELATED DISEASE AND SEVERE HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS (HLH) Pediatric Blood & Cancer. 2015;62:S123–S. [Google Scholar]
- 67.Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014;211:2385–96. doi: 10.1084/jem.20141091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Volker-Touw CM, de Koning HD, Giltay J, et al. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. The British journal of dermatology. 2016;176:244–8. doi: 10.1111/bjd.14757. [DOI] [PubMed] [Google Scholar]
- 69.Kawasaki Y, Oda H, Ito J, et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell-Based Phenotype Dissection. Arthritis & rheumatology. 2017;69:447–59. doi: 10.1002/art.39960. [DOI] [PubMed] [Google Scholar]
- 70.Raghawan AK, Sripada A, Gopinath G, et al. A Disease-associated Mutant of NLRC4 Shows Enhanced Interaction with SUG1 Leading to Constitutive FADD-dependent Caspase-8 Activation and Cell Death. J Biol Chem. 2017;292:1218–30. doi: 10.1074/jbc.M116.763979. [DOI] [PMC free article] [PubMed] [Google Scholar]