

Summary
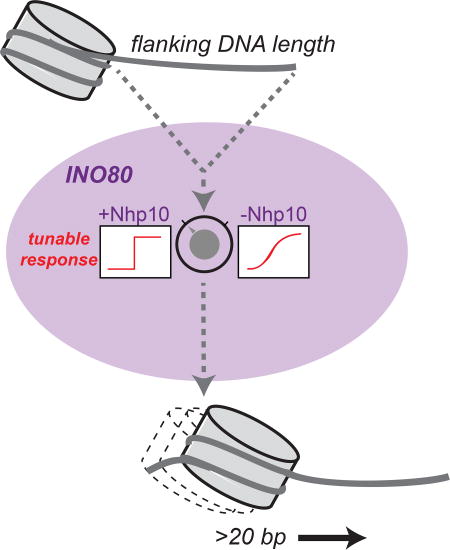
The yeast INO80 chromatin remodeling complex plays essential roles in regulating DNA damage repair, replication and promoter architecture. INO80’s role in these processes is likely related to its ability to slide nucleosomes, but the underlying mechanism is poorly understood. Here we use ensemble and single-molecule enzymology to study INO80-catalyzed nucleosome sliding. We find that the rate of nucleosome sliding by INO80 increases ~100-fold when the flanking DNA length is increased from 40 bp to 60 bp. Furthermore, once sliding is initiated, INO80 moves the nucleosome rapidly at least 20 bp without pausing to re-assess flanking DNA length, and can change the direction of nucleosome sliding without dissociation. Finally, we show that the Nhp10 module of INO80 plays an auto-inhibitory role, tuning INO80’s switch-like response to flanking DNA. Our results indicate that INO80 is a highly processive remodeling motor that is tightly regulated by both substrate cues and non-catalytic subunits.
Keywords: chromatin remodeling, INO80, DNA length sensing, single molecule FRET
eTOC blurb
The INO80 chromatin remodeling complex has essential roles in DNA damage repair, DNA replication, and transcription, but remains relatively poorly characterized. Here we use quantitative enzymology and single molecule biophysics to describe a kinetic mechanism for nucleosome sliding by INO80, which likely has implications for INO80’s specialized in vivo roles.
Introduction
Dynamic accessibility of a cell’s genome is key to the regulation of DNA-based processes. In eukaryotes, these dynamics are ultimately gated by the nucleosome, the fundamental unit of chromatin, consisting of 147 base pairs (bp) of DNA wrapped around a core of eight histone proteins. ATP-dependent chromatin remodeling enzymes convert the chemical energy of ATP into mechanical forces that break and re-form histone-DNA contacts. These small-scale changes to nucleosome structure, coupled with the action of other chromatin factors, translate into large-scale changes to the chromatin landscape, and ultimately regulate every aspect of genome biology from transcription to DNA damage repair (Clapier and Cairns, 2009; Zhou et al., 2016). Of the four main families of remodelers (ISWI, SWI/SNF, CHD1, and INO80), the INO80 family is the most recently discovered and its biochemical mechanisms remain the most elusive (Morrison and Shen, 2009; Shen et al., 2000; Zhou et al., 2016).
The INO80 remodeler from S. cerevisiae is a multi-subunit complex consisting of a core remodeling ATPase (Ino80) and 14 other subunits (Morrison and Shen, 2009; Shen et al., 2000). In vivo, INO80 has roles in regulating transcription, DNA damage repair, replication, and metabolic regulation (Morrison and Shen, 2009; Yao et al., 2016). In vitro, INO80 slides mononucleosomes towards the center of short DNAs and spaces a tri-nucleosomal array (Udugama et al., 2011). Genome-wide studies have shown that ATP-dependent remodeling activity is required for establishing the specific nucleosome architecture at yeast promoters (Yao et al., 2016; Zhang et al., 2011). Using in vitro reconstituted yeast chromatin, it was recently shown that INO80 is the only remodeler that is sufficient for positioning the +1 nucleosome at these promoters (Krietenstein et al., 2016), consistent with earlier data showing that several INO80 subunits bind specifically to the “+1” nucleosome adjacent to the transcription start site (Yen et al., 2013; 2012). In combination with the known biochemical activities of INO80, these data suggest that the nucleosome sliding activity of INO80 observed on single nucleosomes may play a critical role in positioning nucleosomes at the TSS, particularly the +1 nucleosome.
However, much is still unclear about the mechanism of nucleosome sliding by INO80, including how substrate cues are “read” by the enzyme and which motifs in the enzyme itself regulate this activity. Here we describe a combined approach using a variety of ensemble nucleosome remodeling assays as well as single-molecule FRET to probe the underlying mechanism of how yeast INO80 uses flanking DNA length as a substrate cue for nucleosome movement. We find that INO80 exhibits a switch-like response to flanking DNA length, consistent with and extending previous studies (Udugama et al., 2011). We further demonstrate that regulation of nucleosome sliding by flanking DNA length in INO80 is not directly coupled to ATPase activity, unlike in the ISWI family (Whitehouse et al., 2003; Yang et al., 2006). Once sliding is initiated, INO80 rapidly slides a nucleosome at least 20 bp, and is capable of sliding a nucleosome on a long stretch of DNA continuously and bi-directionally without dissociating. Finally, we describe an auto-inhibitory activity of INO80 that is specific for slowing sliding of nucleosomes with short flanking DNA. Taken together, our results suggest that INO80 has a distinct mechanism from other remodeling families, which likely has implications for INO80’s specialized in vivo roles at the TSS and in double-strand break repair.
Results
Flanking DNA modulates the overall remodeling rate of INO80, but not its ATPase activity
Previous work using end-point sliding assays has shown that yeast INO80 can slide mononucleosomes toward the center of a short DNA and evenly space a tri-nucleosomal array, but both activities require at least 50 base pairs (bp) of extranucleosomal DNA (Udugama et al., 2011). We used the strong nucleosome positioning sequence 601 to construct initially end-positioned nucleosomes with varying amounts of extranucleosomal DNA, and found that by native gel remodeling, efficient nucleosome sliding requires >40 bp of flanking DNA (Figure 1A), in agreement with this previous work (Udugama et al., 2011). We also noticed that while nucleosomes with 60 bp of flanking DNA (“0/60”) are moved to a more centered position that runs as a single discrete band on a native gel, nucleosomes with 80 bp of flanking DNA (“0/80”) are slid toward the center, but result in a final distribution of products that runs as a doublet on a native gel (Figure 1A). The appearance of this doublet is sequence-specific (Figure S1A), suggesting that DNA sequence can affect the final product distribution. However, DNA sequence does not affect the rate of remodeling (Figure S1B–D), similar to what was reported for ISWI remodelers (Partensky and Narlikar, 2009) and for INO80 in a reconstituted yeast chromatin system (Krietenstein et al., 2016).
Figure 1. Flanking DNA length modulates nucleosome sliding by INO80, but not ATPase activity.

(A) Left, schematic of the native gel assay. Right, native gel showing the remodeling of nucleosomes over time with 40 bp, 60 bp, or 80 bp of flanking DNA (“0/40”, “0/60”, and “0/80” nucleosomes), under single turnover conditions and with saturating enzyme and ATP.
(B) Left, schematic of the ensemble FRET remodeling assay and example raw data for 0/40, 0/60, and 0/80 nucleosomes.
(C) Rate constants for remodeling of 0/40, 0/60, 0/80, and 0/100 nucleosomes by ensemble FRET. These assays were performed under single turnover conditions and with saturating enzyme and ATP.
(D) Rate constants for ATP hydrolysis for 0/40, 0/60, and 0/80 nucleosomes derived from initial rates. These assays were performed under multiple turnover conditions, with IN080 in excess and saturating over nucleosomes, and ATP in excess of INO80.
(E) Schematic of the REA assay. Remodeling by INO80 (DNA unpeeling, a conformational change, or nucleosome sliding) exposes a Pst1 site located 18 bp from the short end of the DNA. After digestion of the histones with Proteinase K, uncut and cut products are separated by native gel.
(F) Quantification of the fraction of the DNA cut by the Pst1 restriction enzyme as a function of time for 0/40, 0/60 and 0/80 nucleosomes.
(G) Quantification of the rate constant for cutting by Pst1 for 0/40, 0/60, and 0/80 nucleosomes. These assays were performed under single turnover conditions and with saturating enzyme and ATP.
Data in (C), (D), and (G) represent the mean ± the S.E.M. for three replicates.
Thus as shown previously, yeast INO80 exhibits a switch-like response to flanking DNA, in terms of sliding outcome, requiring at least ~50 bp of flanking DNA for efficient sliding (Figure 1A, (Udugama et al., 2011)). To gain mechanistic insight into this behavior, we used a FRET-based assay to understand how the maximal rates of nucleosome sliding (kobs) change as a function of flanking DNA length. We found that varying the flanking DNA from 40 bp to 80 bp increases the kobs for remodeling by 300-fold (Figure 1B–C). However, the largest increase in rate constant (~100-fold) occurs when flanking DNA length increases from 40 bp to 60 bp. As these experiments were performed under saturating concentrations of INO80, the differences in rate constant arise from differences in a step that occurs post-binding.
Our observation that INO80’s nucleosome sliding activity is modulated by flanking DNA length is reminiscent of the well-studied ISWI remodeling enzymes, which move nucleosomes faster toward longer flanking DNA by coupling length sensing to the ATPase activity of the motor (Whitehouse et al., 2003; Yang et al., 2006). To test whether this paradigm holds true for INO80, we measured the ATPase activity of INO80 when stimulated by nucleosomes with varying lengths of flanking DNA, using the same INO80 and nucleosome concentrations as in the FRET-based assay. Despite the 300-fold difference in the remodeling rate constant by FRET between 0/40 and 0/80 nucleosomes, INO80’s ATPase activity stimulated by these two substrates is comparable (Figure 1D). This result is consistent with a previous study showing that increasing flanking DNA length results in a less than 2-fold effect on ATPase activity (Udugama et al., 2011), and demonstrates that DNA length sensing by INO80 is not directly coupled to ATPase activity. Taken together, these findings stand in contrast to what has been shown for ISWI-family motors, raising the possibility that the INO80 family uses a different mechanism to slide nucleosomes. These results also indicate that the substantially slower rate of sliding with 0/40 nucleosomes is not due to an inability of INO80 to bind these nucleosomes, because the rate of ATP hydrolysis with 0/40 nucleosomes is comparable to that with 0/80nucleosomes.
Based on these observations, we hypothesized that the ATPase activity of INO80 is used to generate another type of remodeled intermediate that is not detected by native gel or FRET. To test this hypothesis, we used a restriction enzyme accessibility (REA) assay to measure rates of remodeling by INO80 (Figure 1E) (Narlikar et al., 2001; Polach and Widom, 1995). In principle, the REA assay can report on changes to the accessibility of the nucleosomal DNA arising from changes that do not require nucleosome sliding, such as DNA unpeeling. By REA, we found that nucleosomes with 40 bp of flanking DNA show an increase in DNA accessibility at a maximal rate constant that is 10-fold faster than that observed by FRET (0.05 ± 0.02 min−1 vs 0.005 ± 0.006 min−1, respectively) (Figure 1F,G). In the absence of ATP, changes in accessibility were substantially lower (Figure 1F). These observations suggest that INO80 can alter histone-DNA contacts of a nucleosome with shorter flanking DNA, but cannot efficiently convert this altered nucleosome into a slid product unless the nucleosome has greater than 40 bp of flanking DNA. Interestingly, the 300-fold difference in kobs between nucleosomes with 40 bp vs. 80 bp of flanking DNA as measured by FRET decreases to 12-fold when measured by REA, suggesting that creation of the REA-sensitive intermediate versus a slid product depend differently on flanking DNA length. Importantly, this difference in kobs as measured by REA does not appear to be the result of a difference in the stoichiometry of INO80 on the respective nucleosomes, or artifactual differences between the REA and FRET sliding assays (Figure S2D–H). Interestingly, we found that moving the restriction site to other positions along the nucleosomal DNA resulted in substantially decreased changes in accessibility (Figure S2A–C), suggesting that the DNA accessibility created by INO80 is restricted to the H2A/H2B surface. We note that we do not see evidence for dimer loss being essential for nucleosome sliding (Figure S1E–G).
Remodeling by INO80 is dominated by a long initial pause, followed by rapid nucleosome mobilization
Single molecule FRET (smFRET) has proven to be a powerful tool for dissecting the individual kinetic steps in the remodeling reactions of other families, most notably the ISWI family (Blosser et al., 2009; Deindl et al., 2013; Harada et al., 2016; Hwang et al., 2014). We therefore adapted the smFRET assay to study INO80 to gain additional mechanistic insight. Figure 2B shows several examples of smFRET measurements of INO80 remodeling individual 3/78 nucleosomes. As expected, the overall FRET efficiency decreases over time, consistent with INO80 sliding the nucleosome away from the DNA end . Although we cannot rule out the formal possibility that part or all of the observed change in FRET is due to a process other than sliding—for example, unpeeling of the DNA away from the octamer surface, which would also result in a change in inter-dye distance—we note that the behavior shown in Figure 2B is ATP-dependent, with changes in FRET that are not instantaneous (Figure S3H).
Figure 2. Remodeling by INO80 at the single nucleosome level is preceded by a long pause, followed by rapid nucleosome translocation.

(A) Schematic of the smFRET assay. Nucleosomes are immobilized on the surface of microscope slides and imaged with a prism-based TIRF microscope (see Methods). For the 3/78 nucleosome shown here, the nucleosome starts 3 bp from the Cy5-labeled DNA end, with a 78 bp linker attaching the nucleosome to the surface, resulting in an initial high FRET efficiency (Figure S3A). As remodeling proceeds, the nucleosome is moved towards the 78 bp flanking DNA and the FRET efficiency is reduced.
(B) Example timecourses of remodeling from individual, surface-attached, 3/78 nucleosomes in the presence of saturating INO80 and ATP (15 nM and 1 mM respectively), imaged at 7.4 Hz and smoothed (for visualization only) with a median filter with a 1-second window. Vertical yellow line indicates the time at which ATP is introduced into the sample chamber to start the remodeling reaction. We refer to the long initial pause exhibited by all trajectories as pinitial, and to a secondary pause, exhibited in about three quarters of remodeling events (Figure S3B,C), as psecond. Additional example timecourses are shown in Figure S4.
(C) Kernel density estimation (KDE) plot of the change in nucleosome position between pinitial and psecond for the 78 3/78 nucleosomes (out of 100 3/78 nucleosomes total with 1 mM ATP) that exhibit a secondary pause. KDEs are conceptually similar to histograms, but have a smoothing parameter rather than a bin size (see also Figure S3). The peak in the density around 10 bp indicates that the majority of the trajectories that show secondary pauses on 3/78 nucleosomes have translocated the nucleosome about 8–12 bp before the secondary pause is encountered. KDE has a Gaussian kernel with bandwidth 0.025.
(D) Quantification of the average durations of the initial and secondary pauses as a function of entry DNA length, for 3/60, 3/70, and 3/78 nucleosomes. Some traces exhibit more than one secondary pause; however these traces are too few to gain accurate statistics beyond the psecond pause.
(E) Quantification of the average durations of the initial and secondary pauses as a function of ATP concentration, for 3/78 nucleosomes. 1 mM ATP is saturating, while 0.1 mM and 0.2 mM are both sub-saturating, though still well above the Km (Figure S3D,E; (Udugama et al., 2011)). Dark blue data are the same as in C. The increase in pinitial duration with decreasing ATP concentration is greater than the error on the measurements; the slight decrease in psecond, however, is not. Data in (D) and (E) represent means±S.E.M. obtained via a bootstrapping approach (see Methods).
(F) Overall remodeling rates for 3/60 and 3/78 nucleosomes, as measured by ensemble FRET, at 20°C in imaging buffer (see Methods), under single turnover conditions and with saturating enzyme and ATP. Each curve is the average of two independent replicates as described in the Methods. After 5 minutes (vertical dashed line), the ensemble remodeling reaction on 3/78 nucleosomes is nearly 80% complete, while the reaction on 3/60 nucleosomes is only about 50% complete.
(G) Example timecourses of remodeling of 3/60 nucleosomes in the presence of 1 mM ATP, with the laser that excites the Cy3 dye turned off for 5 minutes as indicated. While the laser is off, all dye intensities go to zero. Three kinds of behavior are observed: some nucleosomes are remodeled before the laser is turned off (Class 1, top); some nucleosomes are remodeled after the laser is back on (Class 3, bottom); and some nucleosomes have high Cy5 intensity before the laser is turned off, but high Cy3 intensity when the laser is turned back on (Class 2, middle). These Class 2 nucleosomes must have remodeled during the 5 minutes that the laser was off, because no change in Cy5 signal is observed with the laser off in the absence of ATP.
(H) Quantification of how many nucleosomes are remodeled in the first 5 minutes of the smFRET reaction when the laser is turned off as in the examples in G. Data represent means±S.E.M. for three replicates, where the fraction remodeled was calculated for each replicate according to the expression below the bar graph.
At a simple level, the example timecourses in Figure 2B suggest INO80 shares some features previously described for ISWI remodelers. ISWI remodelers exhibit a stereotyped behavior when observed at the single nucleosome level: remodeling proceeds through an alternating series of “pauses”, during which the FRET is constant and no change in nucleosome position is detected, and translocation events, during which the nucleosome is slid (Blosser et al., 2009; Deindl et al., 2013; Hwang et al., 2014). An emerging model is that the pauses in the ISWI reaction represent a rate-limiting regulatory step, during which substrate cues are periodically assessed and used to gate translocation activity (Zhou et al., 2016). Nucleosome sliding by INO80 similarly appears to contain pause and translocation phases: the reaction begins with an extremely long pause (about a minute long on average; Figure 2D), and translocation is often interrupted (about 75% of the time; Figure S3D,E) by at least one additional pause that appears to occur at roughly half-maximal FRET (Figure 2C and Figure S3D,E). These pauses are candidates for regulatory steps as in the ISWI paradigm and are discussed further below. Translocation itself is fast, even at 20°C (~7 bp/s at saturating ATP; Figure S3H), comparable to the translocation rate of the ISWI family member ACF at 30°C (~7–9 bp/s (Blosser et al., 2009)). As discussed in the next section, however, these similarities between the INO80 and the ISWI do not extend beyond a qualitative level.
The initial pause is ATP dependent, but not DNA length sensitive
Analogous to INO80, many ISWI remodelers are sensitive to flanking DNA length (He et al., 2006; Whitehouse et al., 2003; Yang et al., 2006; Zofall et al., 2004). smFRET studies with ISWI remodelers have shown that DNA length sensing occurs during the pause phases of remodeling: increasing flanking DNA length decreases the durations of the pauses, but does not affect translocation rates (Blosser et al., 2009; Hwang et al., 2014). Furthermore, a reduction in ATP concentration results in longer pauses and slower translocation for ISWI complexes, implying that ATP is required for both the pause and translocation phases (Blosser et al., 2009; Deindl et al., 2013; Hwang et al., 2014).
We performed similar experiments with INO80 to ask which step(s) in its remodeling reaction are sensitive to flanking DNA length or ATP concentration. For these experiments we used nucleosomes containing flanking DNA lengths of either 60, 70 or 78 bp, and varied ATP concentration from 0.1 mM to 1 mM. As with ISWI remodelers, the translocation phases of INO80 are sensitive to ATP concentration and not to flanking DNA length (Figure S3H). The initial pause (pinitial in Figure 2B) is sensitive to ATP concentration, but, surprisingly, is not sensitive to flanking DNA length (Figure 2D,E). The ATP dependence of pinitial quantitatively recapitulates the ATP dependence of the overall remodeling rate constant measured by ensemble FRET, suggesting that pinitial contains the main ATP-dependent step observed in ensemble assays (Figure S3G).
Even more surprisingly, the secondary pauses are not sensitive to either flanking DNA length or ATP concentration (Figure 2D,E). If either pinitial or psecond were sensitive to flanking DNA length, we would have expected these pause durations to decrease with increasing flanking DNA. However, we do not observe a statistically significant trend in either pinitial or psecond as a function of increasing flanking DNA length from 60 to 80 bp (Figure 1C). These results suggest that the secondary pauses do not play a regulatory role in nucleosome sliding. Rather, we favor an interpretation of our results in which the secondary pauses of the INO80 remodeling reaction are simply a consequence of how the nucleosomal DNA wraps around the histone octamer. The regulatory pauses in ISWI are non-random, with the first pause occurring before an initial large (~7 bp) translocation event, and the subsequent pauses occurring before smaller (~3 bp) translocation events, independent of DNA sequence (Blosser et al., 2009; Deindl et al., 2013; Hwang et al., 2014). In contrast, the secondary pauses in the INO80 reaction occur predominantly after 10 bp of movement, and less commonly after 5 or 15 bp of movement (Figure 2C, Figure S8B,C). These values are intriguingly close to the rotational periodicity of nucleosomal DNA (Luger et al., 1997). Furthermore, the distribution of the locations at which psecond occurs, but not the pause durations, are sensitive to DNA sequence (Figure S4F–G), consistent with INO80 pausing at sites where the DNA forms favorable contacts with the histone core (Hall et al., 2009).
These results have important implications for the mechanism of remodeling by INO80: it appears that once translocation has begun, it is not interrupted by regular pauses that allow re-interrogation of flanking DNA length after the nucleosomes has been moved a short distance, at least not within the 20–25 bp for which the nucleosome is in FRET range (Figure S3B). For comparison, the ISWI-family remodeler ACF, which has a similar length-sensing regime to INO80 in terms of the modulation of its overall remodeling rate, has about three length-sensitive pauses within the first 20 bp of nucleosome movement (Blosser et al., 2009).
Comparing our smFRET and ensemble FRET data raises a paradox, as flanking DNA length modulates the overall rate of INO80 remodeling as measured by ensemble FRET (Figure 1C), but does not appear to modulate any step as observed by smFRET (Figure 2D, Figure S3). In reconciling these differences, we noticed that the overall remodeling rate by ensemble FRET is slower than the remodeling we observe at the single nucleosome level (Figure S4E), which is a significant departure from previous smFRET results with, for example, ISWI family remodelers, for which ensemble and smFRET remodeling rates correspond well (Blosser et al., 2009). We therefore hypothesized that photobleaching, which affects smFRET but not ensemble FRET measurements, may be masking a slow phase of the overall remodeling reaction, and that this slow phase might be DNA length-sensitive.
To test this possibility, we reduced the photobleaching effect by acquiring smFRET movies during which the imaging laser was turned off for 5 minutes early in the reaction (Figure 2G). Photobleaching of the fluorescent dyes occurs only when the laser is on. If the photobleaching process were the cause of the discrepancy we observe between ensemble FRET and smFRET, then the fraction of 3/60 versus 3/78 nucleosomes remodeled during these 5 minutes in the dark should mirror the fraction of the corresponding ensemble FRET reactions that have gone to completion in 5 minutes. Under smFRET buffer conditions, the ensemble FRET reaction goes to ~50% and ~80% completion by 5 minutes for 3/60 and 3/78 nucleosomes respectively, consistent with a flanking DNA length dependence (Figure 2F). Similarly, by smFRET we find that ~40% and ~70% of 3/60 and 3/78 nucleosomes, respectively, are remodeled after 5 minutes (Figure 2H). Thus without photobleaching, we now observe a flanking DNA length dependence at the single nucleosome level that mirrors what we observe by ensemble FRET. We therefore conclude that the INO80 remodeling reaction consists of a fast, DNA length-insensitive population that we observe under continuous laser illumination, and a slower, DNA length-sensitive population that we observe only when the masking effect of photobleaching is removed (see Discussion).
INO80 processively and continuously moves a centered nucleosome significant distances back and forth along the DNA
The results of Figure 2 suggest a potentially important feature of INO80’s mechanism: processivity, which we define as a measure of how many basepairs the nucleosome can be translocated before dissociation of INO80. The experiments in Figure 2 were performed under chase conditions, and INO80 was observed under these conditions to slide end-positioned nucleosomes at least 20 bp, as discussed above. To more directly test for processivity, we generated FRET-labeled “78/78” nucleosomes, with the 601 positioning sequence flanked on both sides by 78 bp, and the Cy5 dye moved to an internal position 9 bp from the nucleosome edge, which increases the distance for which the nucleosome is in FRET range on both sides of the nucleosome’s initial starting point, so that back-and-forth motion of the nucleosome can be measured (Figure 3A, Figure S3A).
Figure 3. INO80 processively and continuously moves a nucleosome bi-directionally on long DNA.

(A) Schematic of the centered 78/78 construct. These nucleosomes have an internal Cy5 9 bp from one edge of the 601 sequence that initially positions the nucleosome, such that they start in mid-high FRET (Figure S3A). If INO80 slides the nucleosome towards the surface, the FRET will decrease according to our calibration curve; if the nucleosome is moved towards the dye, the FRET will increase, and then decrease as the dye-labeled base-pair enters the nucleosome.
(B) Native gel showing the remodeling of the 78/78 nucleosomes in A, under single turnover conditions and with saturating enzyme and ATP. Remodeling was performed at 20°C for consistency with smFRET experiments.
(C) Example traces of remodeling of 78/78 nucleosomes with saturating INO80 and ATP (15 nM and 1 mM respectively). See Methods for a description of the three classes of behavior. The two Class III examples illustrate “double chase” experiments. The percent of the ~100 total traces (from the single-chase and double-chase experiments combined) that we classified into each type of behavior are given in parenthesis. One of the Class I traces has not been assigned a bp scale on the right axis because it represents movement of the Cy5 into the nucleosome, where our calibration curve is not valid (Figure S3B). The percentage of trajectories showing direction reversals with these initially centered constructs is double the percentage with end-positioned constructs (40% for the 78/78 construct, versus 20% of all end-positioned constructs in Figure 2).
(D) No remodeling was observed after a second chase into buffer only (no ATP).
Consistent with recent work with human INO80 (Willhoft et al., 2017), we find by gel remodeling that yeast INO80 mobilizes this 78/78 construct, yielding a distribution of nucleosome positions, some of which are off-center (Figure 3B). Similarly, we find by smFRET that INO80 quickly slides these nucleosomes out of FRET range, as with the end-positioned constructs (Figure 3C). INO80 is also capable of sliding a nucleosome back and forth along the DNA without releasing it, as indicated by the alternating gains and losses of FRET in the observed bi-directional and multi-directional remodeling behaviors (Figure 3C). These results indicate that INO80 can catalyze many rounds of translocation without dissociating.
To confirm that the bi-directional, processive behavior we observe is due to the same INO80 complex that initially binds the nucleosome, we performed “double chase” experiments, in which a second buffer exchange into 1 mM ATP was performed roughly a minute after the addition of ATP that started the remodeling reaction (Figure 3C, bottom panels). As a control, we also chased with buffer only, which stops remodeling, indicating that we are fully exchanging the buffer in the sample chamber during these washes (Figure 3D). Thus INO80 is capable of moving a nucleosome long distances bi-directionally along a DNA strand without dissociating. Although it is currently not clear how INO80 switches its direction of translocation under chase conditions, one possibility is that it can bind a nucleosome as a dimer. Cooperative binding and sliding of nucleosomes by two INO80’s has recently been suggested for a human INO80 core complex (Willhoft et al., 2017).
Deletion of nhp10 results in impaired length sensing by INO80
We next investigated what motif(s) in INO80 might be involved in regulating the switch-like increase in nucleosome sliding upon increasing flanking DNA length beyond 40 bp. Studies of both yeast INO80 (Tosi et al., 2013) and human INO80 (Chen et al., 2013) have shown that the complex is organized into modules, each composed of 3–4 subunits that make distinct contributions to nucleosome remodeling. For example, the Arp8 module, composed of Arp8, Arp4, Act1, Taf14 and Ies4, is marginally involved in nucleosome and DNA binding, but is critical for the catalysis of nucleosome remodeling (Chen et al., 2013; Tosi et al., 2013). In contrast, the yeast-specific Nhp10 module, composed of Nhp10, Ies1, Ies3 and Ies5, is not critical for nucleosome movement or ATP hydrolysis but exhibits strong DNA and nucleosome binding properties on its own (Shen et al., 2003b; Tosi et al., 2013). Based on this observation, we hypothesized that the Nhp10 module may contribute to INO80’s ability to discriminate between nucleosome substrates based on flanking DNA length.
To test this possibility, we purified INO80 complexes from yeast containing a deletion of nhp10. As seen previously, when compared to wild-type (WT) INO80 on an SDS-PAGE gel, the INO80(Δnhp10) complex shows the absence of bands corresponding to components of the Nhp10 module, such as Ies3 (purple star, Figure 4A) (Shen et al., 2003a; Tosi et al., 2013). We also noticed that in our purifications of INO80(Δnhp10), the majority of the Ino80 band migrated faster on an SDS-PAGE gel than in WT INO80 (green star, Figure 4A), which we reasoned may correspond to a truncation of Ino80 that occurs as a result of deleting nhp10. Based on mass spectrometry data, we indeed found that the first ~200 amino acids of the N-terminus of Ino80 is missing in INO80(Δnhp10) but remains intact in WT INO80 (Figure S5A). Previous work has shown that the Nhp10 module interacts with these first 200 amino acids of Ino80 (Tosi et al., 2013), suggesting that deletion of the nhp10 causes both the loss of the Nhp10 module from the INO80 complex as well as the destabilization and degradation of the N-terminal region of the Ino80 ATPase. To further test this possibility, we purified INO80 complexes from yeast containing an N-terminal deletion of Ino80. The INO80(Δ2-200-ino80) complexes lacked the same subunits of the Nhp10 module (Figure 4A), further supporting the idea that the N-terminus of Ino80 and the Nhp10 module are structurally inter-dependent.
Figure 4. Deletion of nhp10 results in impaired DNA length sensing by INO80.

(A) Left, denaturing gels showing purifications of Ino80-FLAG(WT), Ino80-FLAG(Δ2-200-ino80) and Ino80-FLAG(Δnhp10). Green and purple stars represent where the Ino80 ATPase and Ies3, a major component of the Nhp10 module run on the gel, respectively. Right, schematic illustrating the compositions of mutant INO80 complexes purified from Δnhp10 and Δ2-200-ino80 strains.
(B) Remodeling rate constants measured by ensemble FRET for 0/40, 0/60 and 0/80 nucleosomes and the various INO80 mutants described in A. These assays were performed under single turnover conditions and with saturating enzyme and ATP.
(C) Native gel remodeling of 0/40 nucleosomes by INO80(Δnhp10) and INO80(Δ2-200-ino80).
These assays were performed under single turnover conditions and with saturating enzyme and ATP. Gel remodeling of 0/60 and 0/80 nucleosomes are shown in Figure S5.
(D) Quantification of fraction cut by REA on 0/80 nucleosomes, with either INO80(WT) or INO80(Δnhp10).
(E) Same as D but with 0/40 nucleosomes. For 0/60 nucleosomes, see Figure S5. Note difference in x-axis from D.
(F) Rate constants measured by REA for 0/40, 0/60 and 0/80 nucleosomes with INO80(WT) or INO80(Δnhp10). These assays were performed under single turnover conditions and with saturating enzyme and ATP. Data in (B) and (F) represent means±S.E.M. for three replicates.
We next compared the remodeling rates of these different mutant INO80 complexes on nucleosomes with 40, 60 or 80 bp flanking DNA, using ensemble FRET. We found that neither INO80(Δ2-200-ino80) nor INO80(Δnhp10) has a defect in remodeling nucleosomes with 80 bp flanking DNA, consistent with published data (Tosi et al., 2013). However, to our surprise, we observed a 100-fold increase in remodeling activity on nucleosomes with 40 bp of flanking DNA with the mutant complexes compared to WT INO80 (Figure 4B). In contrast remodeling of 0/60 and 0/80 nucleosomes increases by only ~ 3-fold and ~1.3-fold, respectively, with the mutant complexes (Figure 4B). Similarly, by native gel, we observe the generation of a more centered product with 0/40 nucleosomes and INO80(Δ2-200-ino80) or INO80(Δnhp10), suggesting that the low-FRET product formed by INO80(Δ2-200-ino80) and INO80(Δnhp10) is indeed a slid nucleosome (Figure 4C). In addition, the final distribution of products generated from 0/60 and 0/80 nucleosomes by the mutant INO80 complexes is similar to that generated by WT INO80, consistent with the results of the FRET-based assay (Figure 4C and Figure S5). Taken together, these results suggest that either the Nhp10 module or the N-terminus of Ino80 is auto-inhibitory for remodeling nucleosomes with 40 bp of flanking DNA.
Comparing the remodeling rates measured by FRET vs. REA with WT INO80 reveals a 25-fold difference in flanking length discrimination. This comparsion leads us to hypothesize that overall nucleosome sliding is more sensitive to flanking DNA length than generation and maintenance of the REA-sensitive intermediate. To further test this hypothesis, we measured remodeling rate constants by REA for INO80(Δnhp10) on 0/40, 0/60 and 0/80 nucleosomes. Surprisingly, we found no significant difference in remodeling rates by REA between WT and INO80(Δnhp10), for any of the nucleosome substrates (Figure 4D–F, Figure S5). This result is in contrast to the 100-fold increase observed by FRET for sliding 0/40 nucleosomes, and is consistent with our hypothesis that generation of the REA-sensitive intermediate and nucleosome sliding happen in distinct kinetic steps. These data also suggest that inhibition of sliding of nucleosomes with ≤40 bp of flanking DNA occurs at a step after generation of the REA intermediate (see Discussion).
Discussion
Compared to chromatin remodeling complexes from other families such as ISWI and SWI/SNF, the biophysical mechanism of INO80 is less well understood. In particular, while flanking DNA length has been identified as a key substrate cue in the nucleosome sliding reaction by INO80, the mechanism behind coupling of this cue to nucleosome movement has been unclear (Udugama et al., 2011, Willhoft et al., 2017). Our study uses a complementary set of single molecule and ensemble assays, provideing a starting point for building a kinetic model for how INO80 preferentially slides nucleosomes with longer flanking DNA lengths.
Toward assembling this model, we first summarize three key findings: (i) by ensemble FRET, increasing the flanking DNA length from 40 bp to 80 bp results in a switch-like increase in remodeling rates, with the greatest increase (~100 fold) occurring between 40 bp and 60 bp, and a smaller (~3-fold) but reproducible increase occurring between 60 bp and 80 bp (Figure 1C); (ii) by a restriction enzyme accessibility (REA) assay, nucleosomes with 40 bp flanking DNA are remodeled 10-fold faster than by ensemble FRET, and flanking DNA length regulates DNA accessibility by only 12-fold compared to the 300-fold effect observed for sliding (Figure 1G), and (iii) once translocation has been initiated, flanking DNA length does not affect either the translocation rate measured by smFRET, or any pause durations within at least the first 20 bp that the nucleosome is moved (Figure 2D,E, Figure S3).
Based on these data, we propose the following model for how INO80 couples flanking DNA length sensing and nucleosome movement (Figure 5). After the addition of ATP, an intermediate that is detectable by REA is formed, a process to which we assign a rate constant kconf. This intermediate can either collapse (kcollapse) back to its original structure, or be be slid (kslide). Two steps in this model are regulated by flanking DNA length: kcollapse and kslide. We propose that kcollapse represents the primary DNA length sensitive step of INO80, such that kcollapse increases with decreasing flanking DNA length. Thus nucleosomes with 40 bp flanking DNA will more often collapse back to the original structure than nucleosomes with 80 bp flanking DNA. We propose further that the Nhp10 module imposes a secondary DNA length sensing mechanism by modulating kslide for substrates with flanking DNA between 40 bp and 60 bp. We discuss below how this branching pathway explains the two populations we observe by smFRET.
Figure 5. Model for the length-dependent regulation of sliding by INO80.

Dashed purple box indicates the steps in the reaction detected by smFRET under continuous laser illumination (pinitialobs). Solid purple box indicates pinitialactual, which encompasses all of the events that preceed translocation in our model, including the length-sensitive kcollapse pathway that is too slow to be detected by smFRET under continuous laser illumination. For simplicity, we have shown the Nhp10 module to only inhibit kslide (see also Discussion).
A prediction of our model in Figure 5 is that the generation of an REA-sensitive nucleosome (kconf) occurs independently of sliding (kslide). Consistent with this prediction, INO80(Δnhp10) increases the sliding rate of nucleosomes with 40 bp of flanking DNA by 100-fold, but increases the rate of cutting observed by REA on 0/40 nucleosomes less than 2-fold compared to WT (Figure 4). This result most simply suggests that the Nhp10 module or the N-terminus of Ino80 primarily regulates length-sensitive sliding by altering kslide. However, we cannot rule out the possibility that this module has small effects on kcollapse. We note in particular that the difference in REA between 0/40 and 0/80 nucleosomes is comparable between the INO80(Δnhp10) mutant and WT (Figure 4F), consistent with our model that Nhp10 primarily affects kslide.
In our model, kconf and kcollapse both contribute to the duration of the long initial pause observed by smFRET that preceeds translocation. We find that under continuous laser illumination, the duration of this initial pause, which we will call pinitialobs, is ATP-dependent, but not sensitive to flanking DNA lengths between 60 and 80 bp (Figure 2D,E). According to our model, the ATP dependence of pinitialobs comes from the ATP-dependent generation of the REA-accessible intermediate. Our model further predicts that kcollapse should contribute a sensitivity to flanking DNA lengths between 60 bp and 80 bp. However, we argue that the collapse pathway is relatively slow compared to the photobleaching process in smFRET, so pinitialobs appears length insensitive because it captures primarily those nucleosomes that go directly to the kslide pathway. Our experiment with the imaging laser off for 5 minutes (Figure 2H) suggests that if we could watch nucleosomes remodel in the absence of photobleaching, the duration of this “real” initial pause, pinitialactual, would be both ATP- and DNA length-dependent, because it would capture nucleosomes that go through the length-sensitive collapse pathway and re-enter the kconf pathway, as well as those that go directly to kslide.
This model for flanking DNA length sensing represents a significant departure from the paradigm of the better studied, DNA-length-sensitive ISWI family remodelers. Our smFRET results also highlight another significant difference from the ISWI paradigm: the lack of a regulatory, substrate-cue-sensitive pause that interrupts translocation after the nucleosome has moved only a short distance (~3–7 bp) (Blosser et al., 2009; Deindl et al., 2013; Hwang et al., 2014). In contrast, once translocation is initiated, INO80 moves a nucleosome quickly (~7 bp/s) by at least 20 bp without a re-assessment of flanking DNA length (Figure 2, Figure S3). This ability to rapidly slide a nucleosome a significant distance could be important for INO80’s roles in DNA damage repair, perhaps by allowing INO80 to clear nucleosomes quickly from sites of DNA damage. This rapid sliding could also be critical for maintaining the +1 position of the nucleosome at transcription start sites, where INO80 appears to have a specialized role (Krietenstein et al., 2016).
Given how quickly and how far INO80 is capable of moving a nucleosome once sliding is initiated, it is perhaps unsurprising that the initiation of translocation is gated by two flanking DNA length sensitive steps (Figure 5). We still do not know, however, how the translocation phase itself is regulated. Does translocation continue until the nucleosome has been moved a certain distance, greater than 20 bp? Or is the end of translocation, like its initiation, regulated by the length of the DNA flanking the nucleosome? A model was recently proposed for human INO80 in which ATPase activity becomes gradually uncoupled from sliding as the nucleosome reaches the center of a DNA, such that the nucleosome “slows to a stop” as the midpoint is reached (Willhoft et al., 2017). Our single molecule data are inconsistent with a gradual uncoupling model, as we do not detect any changes in translocation rate that are dependent on nucleosome position, regardless of the initial position. We look forward to future work identifying the regulatory cues that end INO80’s rapid translocation phase, and that lead to the processive, bi-directional translocation we observe on sufficiently long DNAs like the 78/78 construct.
As noted above, the Nhp10 module specifically inhibits sliding of 0/40 nucleosomes. This inhibitory mechanism imposes a switch-like response on INO80’s sliding activity for substrates with flanking DNA between 40 bp and 60 bp. Interestingly, Nhp10 is not found in humans, but human INO80 does contain several metazoan-specific subunits that also bind to the N-terminus of the Ino80 ATPase (Chen et al., 2011). Indeed, a core complex of human INO80 lacking these N-terminal subunits does not exhibit the switch-like behavior as a function of DNA length that we observe with yeast INO80 (Willhoft et al., 2017). Rather than a 100-fold increase in overall remodeling rate constant between nucleosomes with 40 bp and 60 bp flanking DNA, the core human INO80 complex exhibits a gradual increase in remodeling rate between 20 bp and 80 bp flanking DNA—in particular, only a 5-fold increase between 40 bp and 60 bp flanking DNA (Willhoft et al., 2017). While the sensitivity to flanking DNA length has not yet been tested for the complete human INO80 complex, this observation raises the possibility that there exist human INO80 subunits that play a similar role to that of the Nhp10 module in yeast.
Auto-inhibitory motifs have been discovered in several chromatin remodeling enzymes. For example, the chromodomains of CHD1 have been shown to inhibit ATPase activity (Hauk et al., 2010) while the AutoN motif of ISWI inhibits both ATPase and remodeling (Clapier and Cairns, 2012). Both of these mechanisms are distinct from that observed for the Nhp10 module, which only inhibits remodeling of nucleosomes with short flanking DNA (Figure 4), and has no effect on ATPase rates (Tosi et al., 2013). More similar to Nhp10 is the NegC module of ISWI, which has been shown to regulate the remodeling activity of the human ISWI ATPase subunit SNF2h in a DNA-length dependent manner, with the strongest inhibitory effect (10-fold) on nucleosomes without flanking DNA (Leonard and Narlikar, 2015). An important distinction between NegC and Nhp10 is that Nhp10 is a separate polypeptide from the remodeling ATPase, suggesting that the presence of this module in any given INO80 complex is a potential source of regulation in vivo. Such differential regulation based on subunit composition has already been demonstrated for a positive regulator of INO80, the Arp5/Ies6 module, which has been shown for yeast INO80 to be required for both ATPase and remodeling activity (Chen et al., 2013; Yao et al., 2016). In vivo, the most highly transcribed genes also contain more Arp5, suggesting that different levels of Arp5/Ies6 may have different effects on promoter architecture (Yao et al., 2016). Our data with INO80(Δnhp10) suggest that the Nhp10 module may act analogously, perhaps providing a means to regulate nucleosome sliding at sites of DNA damage. Our study, in combination with previous work on INO80, demonstrates how remodeling activity can be tuned by subunit composition, and we speculate that such tunability is important to meet the varying chromatin remodeling needs at different genomic contexts.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Geeta J. Narlikar (geeta.narlikar@ucsf.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Yeast strains
All yeast used for protein purification were grown at 30 degrees. For all of the experiments using wild type INO80, yeast were first inoculated in YPD from a frozen glycerol stock and then transferred into SC media. The yeast were then grown until saturation, 50g/liter of YPD powder was added, and cells were harvested six hours later. For experiments involving mutants of INO80, yeast were grown only in YPD and harvested upon saturation. We tested the activity of wild type INO80 purified using either of these growth protocols and found no difference.
METHOD DETAILS
Purification of INO80 complexes from yeast
INO80 was purified by FLAG immunoprecipitation based on previously published methods (Shen, 2004). Briefly, S. cerevisae with endogenously FLAG-tagged INO80 (Shen, 2004) was grown in YEPD at 30°C to saturation. Cells were pelleted by centrifugation for 10 min at 5000 rpm, resuspended with buffer H0.3 (25 mM HEPES, pH 7.5, 1 mM EDTA, pH 8.0, 10% glycerol, 0.02% NP-40, 0.3 M KCl), and pelleted again. Pelleted cells were then extruded through a 60 mL syringe into liquid nitrogen to create “noodles”. Cell “noodles” were then lysed using a Tissue Lyser II (Qiagen, Hilden Germany) or a freezer mill (SPEX 6970 EFM), cooled in liquid nitrogen. Frozen lysate powder was resuspended in equal volume of H0.3 and spun in an SW28 rotor for 2 hr at 25,000 rpm at 4°C. Clarified lysate was mixed with equal volume buffer H0.3 and applied to FLAG M2-affinity resin (1 mL bead slurry per 40 mL of cleared lysate) equilibrated with H0.3 and incubated for 3 hours at 4°C. An additional dose of protease inhibitors was added halfway through the incubation. Resin was washed with 3×50 mL buffer H0.5 (H0.3 buffer except with 0.5 M KCl) followed by 3×10 mL washes with buffer H0.1 (0.1 M KCl) and eluted with H0.1 supplemented with 1mg/mL FLAG peptide. Eluate was concentrated, aliquoted, flash frozen in liquid nitrogen, and stored at −80°C. INO80 concentration was determined by SDS-PAGE with BSA standards, based on the intensity of the Ino80-FLAG band.
To generate the INO80(Δ2-200-ino80) construct, amino acids 2-200 of Ino80 were deleted by knock-in at the endogenous locus in the Ino80-FLAG strain, using a NAT marker −700 bp upstream of the ORF. The mutation was verified by colony PCR and by sequencing. INO80(Δnhp10) was made similarly except that a KanMX marker was knocked into the endogenous Nhp10 locus. Mutant complexes were purified as described above for WT INO80.
Mass spectrometry of INO80 complexes
1.5 µg of WT INO80 or INO80(Δnhp10) were run on a 4–20% SDS gel and stained with Colloidal Blue (ThermoFisher Scientific, Waltham, MA). The INO80 band and the truncated band were cut out and submitted to the UC Davis Mass Spectrometry Facilities (Davis, CA) for analysis. The samples were trypsin digested and run on a Xevo G2 QTof coupled to a nanoAcquity UPLC system (Waters, Milford, MA). RAW MSe files were processed using Protein Lynx Global Server (PLGS) version 2.5.3 (Waters, Milford, MA). Sequences were searched against the S. cerevisiae database form uniprot.org, as well as common contaminants including human keratins, porcine trypsin, bovine serum albumin, and bovine beta-casein. Searches were performed with trypsin specificity and allowed for three missed cleavages.
Nucleosome labeling and reconstitution
Recombinant Xenopus laevis histones were expressed and purified from E. coli as previously described (Luger et al., 1999). Histone octamer was reconstituted as previously described (Luger et al., 1999; Zhou and Narlikar, 2016). FRET-labeled nucleosomes, with the donor Cy3 (for smFRET) or the acceptor Cy5 (for ensemble) on histone H3, were generated via a cysteine introduced at position 33, and were labeled prior to histone octamer assembly via cysteine-maleimide chemistry. Octamer for smFRET was assembled using a 2:1 unlabeled:labeled H3 mixture, to generate nucleosomes with mostly one H3 or neither H3 labeled; labeled octamer for ensemble FRET was assembled with all labeled histone H3. Nucleosomes for REA were not labeled on the histones. Cy3-labeled (for ensemble assays) and Cyanine 5 SE-labeled and biotinylated DNAs (for smFRET) were generated by PCR with HPLC-purified, labeled primers (Cy5 end-labeled primers: TriLink Biotechnologies, San Diego, CA; Cy5 internally labeled primers for centered constructs, IBA Life Sciences, Göttingen, Germany; Cy3 and biotinylated primers: IDT, Coralville, IA) and purified by PAGE. In most cases, nucleosomes were assembled using the 601 nucleosome positioning (Lowary and Widom, 1998). Where indicated, the naturally occurring 5S sequence from X. borealis, with a different arbitrary sequence in the flanking DNA, was used instead (Wolffe et al., 1986). These DNAs were assembled with either labeled or unlabeled octamers by salt gradient dialysis, purified by glycerol gradient centrifugation, and quantified by native gel (Zhou and Narlikar, 2016). DNA sequences used in this work are given in Data S1.
Native gel remodeling assay
All gel remodeling reactions were performed under single turnover conditions (enzyme in excess of nucleosomes), where enzyme concentration is saturating. Saturation was determined by increasing the concentration of enzyme by 3-fold without an observable increase in the rate constant. Reactions were performed at 30°C with 10 nM FRET-labeled nucleosomes, saturating enzyme (15 nM for WT INO80, 30 nM for INO80(Δnhp10) and INO80(Δ2-200-ino80)), 40 mM Tris pH 7.5, 60 mM KCl, 2 mM ATP•MgCl2, 1.1 mM MgCl2, 0.02% NP40, 1%(v/v) glycerol, and 0.5 mg/mL FLAG peptide. Reactions were assembled without ATP-Mg2+ and incubated at 30 °C for 10 minutes prior to the addition of ATP. At the times indicated, a small portion of the reaction was removed and quenched with excess ADP and plasmid DNA. Time points were then resolved by native PAGE (6% acrylamide, 0.5XTBE) and scanned on a Typhoon variable mode imager (GE Life Sciences, Pittsburgh, PA) by scanning for Cy3. Gels were then quantified by densitometry using ImageJ.
ATPase assay
ATPase reactions were performed under multiple turnover conditions (ATP in excess of enzyme). Reactions were performed at 30°C with 5 nM nucleosomes, 15 nM INO80, 40 mM Tris pH 7.5, 50 mM KCl, 100 mM ATP•MgCl2, 0.5 mM MgCl2, and trace amounts of γ-32P-ATP. Reactions were assembled without ATP-Mg2+ and incubated at 30 °C for 10 minutes prior to the addition of ATP. 2.5 µL time points were quenched with an equal volume of 50 mM Tris pH 7.5, 3% SDS, and 100 mM EDTA. Inorganic phosphate was resolved from ATP on a PEI-cellulose TLC plate (Select Scientific) with 0.5 M LiCl/1M formic acid mobile phase. Plates were dried, exposed to a phosphorscreen overnight, and scanned on a Typhoon variable mode imager. Rate constants were determined by fitting a line through the first 10% of inorganic phosphate generated using Prism.
Restriction enzyme accessibility (REA) assays
With the exception of the titration experiments in Figure S2G–H, all REA reactions were performed under single turnover conditions (enzyme in excess of nucleosomes) and at 30°C. Enzyme concentration was also determined to be saturating using methods described previously. Final conditions were: saturating enzyme (20 nM for WT INO80, 60 nM for INO80(Δnhp10)), 15 nM nucleosomes, 40 mM Tris pH 7.5, 60 mM KCl, 1 mM ATP•MgCl2, 5 mM MgCl2, 0.01% NP40, 0.5 mg/mL FLAG peptide, and 3 U/uL Pst1 (NEB). Final conditions for the titration experiments in Figure S2G–H were the same, except that 20 nM nucleosomes and 10 nM, 20 nM, or 40 nM WT INO80 were used. Reactions were assembled without ATP•MgCl2 and incubated at 30°C for 15 minutes before addition of ATP. Time points were quenched with an equal volume of stop solution (70 mM EDTA, 20 mM Tris, pH 7.5–7.7, 2% SDS, 20% glycerol, 0.2 mg/ml xylene cyanole and 0.2 bromophenol blue). After all time points were completed, Proteinase K was added to each sample to a final concentration of 4 mg/mL and incubated for 20 minutes at 50°C. Time points were then resolved by native PAGE (6% acrylamide, 0.5XTBE) and scanned on a Typhoon variable mode imager (GE Life Sciences, Pittsburgh, PA). All gels were imaged by scanning for Cy3, except for the gels in Figure S2C, which were stained with SYBR Gold (S11494, ThermoFisher Scientific, Waltham, MA). Fraction of DNA cut was quantified by densitometry using ImageJ. The data were fit to a single exponential decay using Prism 6 (GraphPad, La Jolla, CA) (Equation 1),
| (1) |
where y0 is the initial fraction un-cut, kobs is the observed rate constant (min−1), and p is the fraction DNA un-cut at plateau.
For the REA assays with the Pst18 site in the linker, the same equation was used, except that y0 represented the initial fraction DNA cut and p is the fraction DNA cut at the plateau.
Ensemble FRET remodeling assay
For all ensemble FRET remodeling experiments except those in Figure 2F and Figure S4E, assays were performed under the same conditions as gel remodeling assays, except that 5 nM nucleosomes were used. For the reactions in Figure 2F and Figure S4E, 7.5 nM nucleosomes were used. Reactions were initiated by addition of ATP, and then the Cy5 emission intensity was measured every second in a K2 fluorometer (ISS) equipped with a 550 nm short pass excitation filter and a 535 nm long pass emission filter. Reactions were excited at 515 nm and emission was measured at 665 nm. The resulting curves were fit to a two-phase exponential decay (Equation 2),
| (2) |
where ffast is the fraction in the fast phase and kfast and kslow are the remodeling rates of the fast and slow phase respectively. All rate constants reported for ensemble FRET assays (“kobs” on figure axes) are the kfast value obtained from the fit, with the exception of the ensemble FRET rate constants reported in Figures S3G and S4E. Data in Figures S3G and S4E were obtained at 20°C rather than 30°C, and so these slower data were fit to a single exponential decay, with the reported rate constant being the decay rate of the single exponential.
The data in Figure 2F represent the average of two independent remodeling reactions. Each dataset (i.e. independent replicate of Cy5 intensity over time) was down-sampled by taking the average Cy5 intensity over non-overlapping 3-second windows. Two such down-sampled replicates were then averaged to obtain each of the curves in Figure 2F.
Single molecule FRET measurements
Cleaning and PEGylation of quartz slides
GE 124 quartz slides (G. Finkenbeiner, Waltham, MA) with laser-drilled holes for tubing attachment were cleaned by sonication in a bath sonicator in 2.5% Alconox for 10 minutes, rinsed thoroughly with water, then sonicated in acetone (HPLC grade, Sigma-Aldrich) for 15 minutes followed by methanol (HPLC grade, Sigma-Aldrich) for 15 minutes. Slides were rinsed with water, and then slides and coverslips (24 mm × 50 mm, No. 1.5, VWR, Radnor, PA) were sonicated in 1 M KOH (Sigma-Aldrich) for not more than 20 minutes, followed by sonication in water for 20 minutes. A propane torch was used to burn any remaining epoxy from previous experiments off of the slides as the final cleaning step.
Slides and coverslips were silanized with a mixture of methanol (Sigma-Aldrich), glacial acetic acid (Sigma-Aldrich), and N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (APTMES, United Chemicals A0700, Bristol, PA) at a volumetric ratio of 200 methanol:1 acetic acid:2 APTMES. Slides and coverslips were sonicated in this mixture for 30 seconds, incubated in the dark for 10 minutes, then sonicated for another 30 seconds and incubated for another 10 minutes. Slides and coverslips were rinsed with methanol and dried with compressed air.
A PEG mixture consisting of 32 mPEG-SVA:1 biotin-PEG-SVA (Laysan Bio, Arab, AL) by weight was diluted to 0.25 mg/µL in 0.1 M sodium bicarbonate and then applied to the slides and coverslips and incubated for an hour, followed by an additional incubation with 0.33 mg/µL mPEG alone for an additional hour. Slides and coverslips were rinsed with water, dried with compressed air, and stored in the dark under vacuum at −20°C. No deterioration in surface quality was observed even after several months under these storage conditions.
Sample preparation
PEGylated slides were brought to room temperature, and then chambers were constructed between a slide and a coverslip with double-sided tape. Thin-walled tubing (ETT-28 or ETT-26, Weico Wire and Cable, Edgewood, NY) was affixed to each chamber's input and output holes for buffer exchange. Chambers were first washed with buffer SPB (50 mM HEPES-KOH, pH 7.5 at 22°C, and 60 mM KCl), and then passivated with 0 .2 mg/mL acetylated BSA (R3691, Promega, Madison, WI) in SPB. After five minutes, chambers were washed with Wash Buffer (12 mM HEPES-KOH, pH 7.5 at 22°C, 50 mM KCl, 0.52 m M MgCl2, 10% (v/v) glycerol, 0.6 mM EDTA, 0.02% Igepal [Sigma-Aldrich I8896], 1% [w/v] glucose, and 0.1 mg/mL acetylated BSA). Neutravidin (A2666, ThermoFisher Scientific, Waltham, MA) at 0.2 mg/mL in Wash Buffer was then added and incubated for 5 minutes. After washing with Wash Buffer, nucleosomes at 12.5 pM in Wash Buffer were added and incubated for 10 minutes. (If necessary, serial dilutions of nucleosomes were performed in 20% glycerol gradient buffer [20 mM HEPES, pH 7.5 at 4°C, 1 mM EDTA, 20% glycerol, 0.1% Igepal]. Nucleosomes were imaged within 90 minutes of dilution into Wash Buffer.) Unbound nucleosomes were removed by washing with Wash Buffer. All incubations were at 20°C. All flow-throughs were of sufficient volume (150 µL) to ensure complete buffer exchange in the ~30 µL channels.
Data acquisition
Nucleosomes were imaged on a custom-built prism-based TIRF setup, consisting of an inverted Olympus (Burlingame, CA) 1×71 microscope with a PL APO 60× water immersion objective and an Andor (Belfast, Ireland) iXon X3 897 EMCCD camera, illuminated by a 532 nm diode-pumped solid-state laser (R531001GX, Laserglow, Toronto, ON, Canada). Light from the sample was filtered with a 577 nm/690 nm dual band-pass filter (FF01-577/690-25, Semrock, Rochester, NY), passed through an adjustable slit (VA100, Thor Labs, Newton, NJ), and split into donor and acceptor images by a 638 nm longpass dichroic mirror (Thor Labs DMLP638). The two images were focused by separate lenses (Thor Labs AC508-100-A) onto the two halves of the camera.
All smFRET experiments were performed at 20°C to ex tend dye lifetimes. Immediately prior to data acquisition, the sample chamber was flushed with 300 µL of 15 nM INO80 in imaging buffer (53 mM HEPES-KOH, pH 7.5 at 22°C, 9.1 mM Tris-aceta te, pH 7.5 at 22°C [contributed by the Trolox], 53 mM KCl, 0.52 mM MgCl2, 10% glycerol, 0.6 mM EDTA, 0.02% Igepal (Sigma CA-630), 1% [w/v] glucose, 0.1 mg/mL acetylated BSA, 2 mM Trolox [Sigma 238813, made as an 11 mM stock in Tris-acetate, pH'd to 7.5 with 1 M NaOH, and stored at 4°C], 0.03 mM β-mercaptoethanol, 2 U/µL catalase [=0.2 mg/mL; Sigma E3289], and 0.08 U/µL glucose oxidase [0.8 mg/mL, Sigma G2133; made with the catalase as a 100× stock in SPB, and stored at 4°C for not more than one week]). INO80 was allowed to bind nucleosomes for 10 min prior to the addition of ATP. Images were collected using Micro-Manager (www.micro-manager.org, San Francisco, CA) (Edelstein et al., 2010) at 7.4 Hz, with an exposure time of 100 ms. To start each reaction, saturating ATP (1 mM) in 600 µL imaging buffer were added via an automated syringe pump (J-KEM Scientific, St. Louis, MO). Injection times were calibrated using a flash from a 638 nm laser (Vortran Laser Technology 10567, Sacramento, CA) as a fiducial mark, combined with a measurement of the syringe pump delay (2.6±0.3 s). For double-chase experiments, an additional 600 µL of imaging buffer alone, or saturating ATP in imaging buffer, were added via syringe pump, using a flash from a 638 nm laser as a fiducial mark.
Obtaining intensity-versus-time traces from raw image data
To streamline the data analysis process for large data sets, as well as to improve the overall quality of the data, we developed custom in-house software called Traces, available for download at https://github.com/stephlj/Traces, to analyze the raw camera images obtained by Micro-Manager. The Traces analysis pipeline is summarized in Data S2. Traces facilitates the analysis of large data sets by automating many of the analysis steps, offering computational and manual checks at each of these steps to ensure data quality, and allowing the user to save and rerun various parts of the analysis without needing to rerun all of it. Moreover, Traces includes a simple graphical user interface that allows the user to examine the original camera images alongside the calculated dye intensities and FRET values, which is instrumental in excluding spurious events. More details can be found in the manual that accompanies the Traces software.
In the example traces in Figures 2B, 3C–D and S4A–C, traces were cropped where one of the dyes photobleached.
Microscope calibration for conversion from FRET to base pairs of DNA
Following previous smFRET studies with mononucleosomes (Deindl et al., 2013), we generated a calibration curve of FRET signal as a function of exit DNA linker length, by constructing a series of nucleosomes with 78 bp between the nucleosome and the biotinylated DNA end on one side, and varying DNA lengths between the other side of the nucleosome and the Cy5-labeled DNA end (Gamarra, Johnson, Trnka, Burlingame, and Narlikar, in revision). In contrast to previous work, we observe a nonlinear relationship between FRET and flanking DNA length. We propose that, due to technical differences between our work and others’ (e.g. choice of filter sets and other optical components), previous smFRET studies with mononucleosomes occupied the pseudo-linear range of the FRET-versus-bp relationship, which in our hands is the range from about 5 to 20 bp. We therefore derived a non-linear expression for the relationship between FRET and the length n of DNA flanking the nucleosome (Gamarra, Johnson, Trnka, Burlingame, and Narlikar, in revision). We found we can write the relationship between FRET and n as
| (3) |
where R0 is the Förster radius for Cy3-Cy5 in nm, d0 is the average distance in nm between the dyes when n = 0, and θ is the angle between d0 and the DNA vector. We find a best fit of Equation 3 to our calibration curve to have the parameters R0 = 10.9 nm, d0 = 5.8 nm, and θ = 153.8°. To convert FRET values to base-pairs of DNA translocated out of the nucleosome, we invert Eq. 3 to obtain
| (4) |
Classification of bi-directional movement in Figure 3
We observe three kinds of behavior with the centered 78/78 constructs in Figure 3: "unidirectional" remodeling, in which the nucleosome is moved out of FRET range and does not come back into FRET range before one of the dyes photobleaches (Class I); bidirectional movement, in which the nucleosome initially moves out of FRET range but then returns, and can persist in mid to high FRET even for very long durations, as in the right panel (Class II); and multi-directional movement, in which the nucleosome is continuously moved in and out of FRET range before photobleaching occurs (Class III). All of these traces were acquired under chase conditions, in that, as in Figure 2, unbound INO80 is washed out as ATP is washed in. Double chase experiments are indicated by two vertical yellow lines, where an additional wash with either buffer or ATP was performed subsequent to the wash that added ATP to start the reaction (and concurrently washed out unbound INO80).
QUANTIFICATION AND STATISTICAL ANALYSIS
Error estimation for ensemble measurements
All ensemble measurements of rate constants are reported as the mean of three or more experimental replicates (n=3), along with standard error of the mean (S.E.M.). Significance was determined using unpaired, two-tailed T-tests (shown as asterisks in some bar graphs). These values are reported in the figure legends. None of the data were excluded. Graphing and statistical analyses were done using GraphPad Prism 5.
Hidden Markov model (HMM) analysis for extracting pause durations and FRET values from smFRET data
As has become standard in the field, with the exception of the translocation rates in Figure S3H, we quantified our smFRET data using a hidden Markov model (HMM) approach; that is, remodeling data were approximated as step functions, with the pause phases modeled as states with Gaussian emissions, and the translocation phases as instantaneous transitions between pauses. However, long smFRET trajectories, like those observed for INO80, are subject to an increased number of artifacts, such as dye blinking or slight fluctuations in the noise, which can complicate quantification of smFRET trajectories. Rather than using an HMM software package specifically designed for smFRET (e.g. the vbFRET package (Bronson et al., 2009)), in order to reduce the likelihood of the HMM identifying artifacts as real transitions, and to reduce analysis time, we adapted a computationally fast, versatile, open-source, python-based HMM library called pyhsmm (https://github.com/mattjj/pyhsmm). The HMM code fits a discrete state HMM to each trajectory generated by Traces, operating on the raw (unsmoothed) acceptor and donor intensity signals.
We fit the models using Gibbs sampling. The posterior distributions on states were very concentrated, so we used a single sample from the end of each Gibbs sampling chain as an estimate of the hidden state sequence. We used a hierarchical Dirichlet process (HDP) prior to automatically select the number of states (pause phases) in each trace. Further, we made use of the “sticky” regularizer available in the pyhsmm library to penalize short-lived states and hence prevent the model from assigning new states to spurious noise events. We developed a combination of Matlab and python code to run pyhsmm by shelling out of Matlab's IDE; this code is included in the Traces GitHub repository (https://github.com/stephlj/Traces).
As shown in Figure S3A, we observe two predominant clusters of FRET values, at 0.57 and 0.95 FRET, in the absence of remodeler. These FRET states correspond to two of the four populations of nucleosomes that result from mixing unlabeled H3 with Cy3-labeled H3 during octamer formation: some nucleosomes will have a Cy3 label on the H3 proximal to the Cy5-labeled DNA end, resulting in the higher FRET state, and some will have a Cy3 label on the H3 distal to the Cy5-labeled DNA end, resulting in the mid-FRET state. There will also be a population of nucleosomes with both H3 histones unlabeled, which show no FRET; and a population with both copies of H3 labeled, which are distinguishable by two-step photobleaching of the Cy3 dye, and are excluded. We also excluded any trajectories to which pyhsmm fit an initial FRET value lower than 0.775 FRET, since nucleosomes with distally labeled H3's do not provide as great a dynamic range for monitoring nucleosome remodeling. Each data set (e.g., INO80 with 3/78 nucleosomes and saturating ATP) consists of about 100 trajectories collated from at least 5 different experiments. Errors are derived from a bootstrapping procedure performed over trajectories: for each data set, the ≥100 trajectories were resampled with replacement, and reported values, such as the means of each pause duration, were recalculated for each bootstrapped sample. Reported errors are standard deviations of the bootstrapped values.
Quantification of translocation rates observed by smFRET
The HMM analysis we use to quantify pause durations is based on a common assumption in the field, that states are well-described as having Gaussian-distributed emissions and are separated by instantaneous transitions. This analysis, particularly using pyhsmm, has the advantage of being computationally fast, and accurate for modeling the pauses we observe. However, the translocation phases that separate the pauses are not instantaneous transitions, and so cannot be quantified by this approach.
We therefore developed a continuous-time Bayesian model for analyzing FRET data with non-instantaneous but still fast transitions. Our analysis tool, called Slopey (available at https://github.com/stephlj/slopey), pairs a continuous-time prior with an explicit model of the camera used to acquire discrete-time images, enabling sub-frame inference over translocation durations. Posterior inference in the model is performed using a Metropolis-Hastings Markov chain Monte Carlo (MCMC) algorithm. A schematic of the Slopey algorithm is shown in Data S3.
The continuous-time prior is on the red- and green-channel intensity processes, which we model as non-Markovian continuous-time jump processes. We parameterize this jump process prior in terms of four distributions: (1) a distribution on intensities of the red channel in pause phases, from which red intensity values are sampled independently, (2) a distribution on pause duration times, from which pause durations are sampled independently, (3) a distribution on translocation times, from which translocation times are sampled independently, and (4) a distribution on affine transformations to produce a corresponding green-channel process from the red-channel process. In symbols, we denote the continuous-time red-channel intensity as VR(t) for t ≥ 0 (Data S3A). A sample path of VR(t) is piecewise linear in time, and is constructed by first sampling a sequence of durations , where K is the number of translocation phases, independently according to
and setting a corresponding sequence of times according to , for k = 1,2,…,2K + 1 (e.g. the t1 through t4 in Data S3A). Note that odd indices k correspond to pause phases while even indices k correspond to translocation phases. We then sample a sequence of red-channel pause-phase intensities independently according to
With these values we can write the red-channel intensity process as
for k = 0,2,…,2K, taking t0 = 0. Finally, we write the green-channel intensity process as
where α~ Gamma(g,h) and β~Gamma(i,j) independently.
We use a uniform prior on the translocation phase durations, with prior hyperparameters of minimum duration c = 0.14 seconds (1 frame) and maximum duration d = 4.5 seconds. We use an exponential prior on pause durations, with a prior hyperparameter of average duration 50 seconds (taking a = 1 and b = 1/50). The prior hyperparameters for red-channel (acceptor) intensities are e = 1, f = 2.5. The prior hyperparameters for the affine transformation from red-channel intensities to green-channel intensities are hyperparameters g = 3, h = 3, i = 1, j = 53.
These continuous-time red- and green-channel intensity processes are measured by the camera, which produces a discrete-time sequence of red- and green-channel frames with additive Gaussian noise (Data S3B). For each frame and each channel, the measured value is computed by integrating over a shutter period determined by the camera parameters Tcycle and Tblank (Data S3C). A random phase of u~Uniform(0,Tcycle) is included (Data S3B). Finally, additive Gaussian noise is added with standard deviation σ = 0.075 (Data S3D).
We initialized the Metropolis-Hastings MCMC trajectories using the HMM fit from pyhsmm, and ran each Markov chain for 105 iterations, collecting 200 evenly spaced iterates from the second half of each trajectory to form Monte Carlo estimates of pause and translocation durations. For each estimate we report standard errors (not posterior standard deviations) obtained via a bootstrapping procedure over trajectories (as with the pyhsmm algorithm described above).
Translocation by INO80 is fast, even at 0.1 mM ATP, making it difficult to identify with confidence the times at which translocation phases begin and end when the distance translocated is short (less than ~5 bp). Therefore for each trajectory, we measured the duration of the longest translocation, and averaged these durations to obtain the mean values reported in Figure S3H. In most cases, the translocation duration included in the reported average was the first translocation, except in those cases where distance translocated between pinitial and psecond was short (again, less than ~5 bp).
DATA AND SOFTWARE AVAILABILITY
Traces
Software for obtaining intensity-versus-time information from microscope images, available at https://github.com/stephlj/Traces. Includes code for extracting pause durations using the pyhsmm HMM package.
Slopey
Software for quantifying non-instantaneous transitions between states with Gaussian emissions in smFRET data, available at https://github.com/stephlj.
Supplementary Material
Data File S1. Schematic of nucleosomal DNA constructs used in this work, related to STAR Methods.
Data File S2. Summary of the Traces smFRET analysis software, related to STAR Methods.
Data File S3. Schematic summary of the Slopey algorithm for quantifying translocation events.
Highlights.
Nucleosome sliding by INO80 exhibits a switch-like response to flanking DNA length
The Nhp10 module tunes the sliding response on nucleosomes with short flanking DNA
INO80 rapidly slides a nucleosome >20 bp without re-assessing DNA length
INO80 is highly processive and can move a nucleosome bi-directionally
Acknowledgments
We thank Julia Tretyakova for histone purifications, and all of the Narlikar lab for helpful discussions. We thank Toshio Tsukiyama for the yeast plasmids to make the Δnhp10 strain, and Hua-Ying Fan for plasmids containing PstI sites at 55, 94 and 118 bp in the nucleosome core. This work was supported by a grant from the NIH to G.J.N. (R01GM073767), from the NIH to A.M. (5R35GM119580), by a Ruth L. Kirschstein National Research Service Award (5F31CA180651-02) to C.Y.Z., and a Leukemia and Lymphoma Society Career Development Program Fellow award to S.L.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Conceptualization: C.Y.Z., S.L.J., L.J.L., and G.J.N.; Methodology: C.Y.Z., S.L.J., L.J.L., and G.J.N.; Software: S.L.J. and M.J.J.; Investigation: C.Y.Z., S.L.J. L.J.L., and A.L.; Resources: S.B. and A.M.; Writing–Original Draft: C.Y.Z., S.L.J., and G.J.N.; Writing–Reviewing & Editing: C.Y.Z., S.L.J., L.J.L., A.L., S.B., A.M., and G.J.N.; Supervision: G.J.N.
Declaration of Interests
The authors declare no competing interests.
References
- Blosser TR, Yang JG, Stone MD, Narlikar GJ, Zhuang X. Dynamics of nucleosome remodelling by individual ACF complexes. Nature. 2009;462:1022–1027. doi: 10.1038/nature08627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko VA, Steele LM, Újvári A, Gaykalova DA, Kulaeva OI, Polikanov YS, Luse DS, Studitsky VM. Nucleosomes Can Form a Polar Barrier to Transcript Elongation by RNA Polymerase II. Mol Cell. 2006;24:469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Brahma S, Udugama MI, Kim J, Hada A, Bhardwaj SK, Hailu SG, Lee T-H, Bartholomew B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nature Communications. 2017;8:15616–12. doi: 10.1038/ncomms15616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson JE, Fei J, Hofman JM, Gonzalez RL, Jr, Wiggins CH. Learning Rates and States from Biophysical Time Series: A Bayesian Approach to Model Selection and Single-Molecule FRET Data. Biophys J. 2009;97:3196–3205. doi: 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Cai Y, Jin J, Florens L, Swanson SK, Washburn MP, Conaway JW, Conaway RC. Subunit organization of the human ino80 chromatin remodeling complex: An evolutionarily conserved core complex catalyzes ATP-dependent nucleosome remodeling. J Biol Chem. 2011;286:11283–11289. doi: 10.1074/jbc.M111.222505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Conaway RC, Conaway JW. Multiple modes of regulation of the human Ino80 SNF2 ATPase by subunits of the INO80 chromatin-remodeling complex. Proc Natl Acad Sci USA. 2013;110:20497–20502. doi: 10.1073/pnas.1317092110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The Biology of Chromatin Remodeling Complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature. 2012;492:280–284. doi: 10.1038/nature11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deindl S, Hwang WL, Hota SK, Blosser TR, Prasad P, Bartholomew B, Zhuang X. ISWI Remodelers Slide Nucleosomes with Coordinated Multi-Base-Pair Entry Steps and Single-Base-Pair Exit Steps. Cell. 2013;152:442–452. doi: 10.1016/j.cell.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H-Y, He X, Kingston RE, Narlikar GJ. Distinct strategies to make nucleosomal DNA accessible. Mol Cell. 2003;11:1311–1322. doi: 10.1016/s1097-2765(03)00192-8. [DOI] [PubMed] [Google Scholar]
- Gaykalova DA, Kulaeva OI, Volokh O, Shaytan AK, Hsieh F-K, Kirpichnikov MP, Sokolova OS, Studitsky VM. Structural analysis of nucleosomal barrier to transcription. Proc Natl Acad Sci USA. 2015;112:E5787–E5795. doi: 10.1073/pnas.1508371112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nuc Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat Struct Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada BT, Hwang WL, Deindl S, Chatterjee N, Bartholomew B, Zhuang X. Stepwise nucleosome translocation by RSC remodeling complexes. eLIFE. 2016;5:3653. doi: 10.7554/eLife.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G, McKnight JN, Nodelman IM, Bowman GD. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell. 2010;39:711–723. doi: 10.1016/j.molcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Fan H-Y, Narlikar GJ, Kingston RE. Human ACF1 alters the remodeling strategy of SNF2h. J Biol Chem. 2006;281:28636–28647. doi: 10.1074/jbc.M603008200. [DOI] [PubMed] [Google Scholar]
- Hwang WL, Deindl S, Harada BT, Zhuang X. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature. 2014;512:213–217. doi: 10.1038/nature13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N, Wal M, Watanabe S, Park B, Peterson CL, Pugh BF, Korber P. Genomic Nucleosome Organization Reconstituted with Pure Proteins. Cell. 2016;167:709–721. doi: 10.1016/j.cell.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JD, Narlikar GJ. A Nucleotide-Driven Switch Regulates Flanking DNA Length Sensing by a Dimeric Chromatin Remodeler. Mol Cell. 2015;57:850–859. doi: 10.1016/j.molcel.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch Y, Cairns BR, Zhang M, Kornberg RD. Activated RSC-nucleosome complex and persistently altered form of the nucleosome. Cell. 1998;94:29–34. doi: 10.1016/s0092-8674(00)81218-0. [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol. 2009;10:373–384. doi: 10.1038/nrm2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Phelan ML, Kingston RE. Generation and interconversion of multiple distinct nucleosomal states as a mechanism for catalyzing chromatin fluidity. Mol Cell. 2001;8:1219–1230. doi: 10.1016/s1097-2765(01)00412-9. [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. Global Regulation of H2A.Z Localization by the INO80 Chromatin-Remodeling Enzyme Is Essential for Genome Integrity. Cell. 2011;144:200–213. doi: 10.1016/j.cell.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partensky PD, Narlikar GJ. Chromatin remodelers act globally, sequence positions nucleosomes locally. J Mol Biol. 2009;391:12–25. doi: 10.1016/j.jmb.2009.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–271. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzler G, Sif S, Kingston RE. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406:541–544. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- Shen X. Preparation and analysis of the INO80 complex. Meth Enzymol. 2004;377:401–412. doi: 10.1016/S0076-6879(03)77026-8. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, Wu C. Involvement of actin-related proteins in ATPdependent chromatin remodeling. Mol Cell. 2003a;12:147–155. doi: 10.1016/s1097-2765(03)00264-8. [DOI] [PubMed] [Google Scholar]
- Shen X, Xiao H, Ranallo R, Wu W-H, Wu C. Modulation of ATP-dependent chromatinremodeling complexes by inositol polyphosphates. Science. 2003b;299:112–114. doi: 10.1126/science.1078068. [DOI] [PubMed] [Google Scholar]
- Shukla MS, Syed SH, Montel F, Faivre-Moskalenko C, Bednar J, Travers A, Angelov D, Dimitrov S. Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc Natl Acad Sci USA. 2010;107:1936–1941. doi: 10.1073/pnas.0904497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi A, Haas C, Herzog F, Gilmozzi A, Berninghausen O, Ungewickell C, Gerhold CB, Lakomek K, Aebersold R, Beckmann R, et al. Structure and Subunit Topology of the INO80 Chromatin Remodeler and Its Nucleosome Complex. Cell. 2013;154:1207–1219. doi: 10.1016/j.cell.2013.08.016. [DOI] [PubMed] [Google Scholar]
- Udugama M, Sabri A, Bartholomew B. The INO80 ATP-dependent chromatin remodeling complex is a nucleosome spacing factor. Mol Cell Biol. 2011;31:662–673. doi: 10.1128/MCB.01035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science. 2013;340:195–199. doi: 10.1126/science.1229758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I, Stockdale C, Flaus A, Szczelkun MD, Owen-Hughes T. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol Cell Biol. 2003;23:1935–1945. doi: 10.1128/MCB.23.6.1935-1945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willhoft O, McCormack EA, Aramayo RJ, Bythell-Douglas R, Ocloo L, Zhang X, Wigley DB. Cross-talk within a functional INO80 complex dimer regulates nucleosome sliding. eLIFE. 2017:1–70. doi: 10.7554/eLife.25782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;13:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- Yao W, King DA, Beckwith SL, Gowans GJ, Yen K, Zhou C, Morrison AJ. The INO80 Complex Requires the Arp5-Ies6 Subcomplex for Chromatin Remodeling and Metabolic Regulation. Mol Cell Biol. 2016;36:979–991. doi: 10.1128/MCB.00801-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Pugh BF. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013;154:1246–1256. doi: 10.1016/j.cell.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G-C, Liu Y-J, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-Scale Identification of Nucleosome Positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CY, Narlikar GJ. Analysis of Nucleosome Sliding by ATP-Dependent Chromatin Remodeling Enzymes. Meth Enzymol. 2016;573:119–135. doi: 10.1016/bs.mie.2016.01.015. [DOI] [PubMed] [Google Scholar]
- Zhou CY, Johnson SL, Gamarra NI, Narlikar GJ. Mechanisms of ATP-Dependent Chromatin Remodeling Motors. Annu. Rev. Biophys. 2016;45:153–181. doi: 10.1146/annurev-biophys-051013-022819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Bartholomew B. Functional Role of Extranucleosomal DNA and the Entry Site of the Nucleosome in Chromatin Remodeling by ISW2. Mol Cell Biol. 2004;24:10047–10057. doi: 10.1128/MCB.24.22.10047-10057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data File S1. Schematic of nucleosomal DNA constructs used in this work, related to STAR Methods.
Data File S2. Summary of the Traces smFRET analysis software, related to STAR Methods.
Data File S3. Schematic summary of the Slopey algorithm for quantifying translocation events.