

ABSTRACT
Dysregulated secretion and extracellular activation of TGF-β1 stimulates myofibroblasts to accumulate disordered and stiff extracellular matrix (ECM) leading to fibrosis. Fibronectin immobilizes latent TGF-β-binding protein-1 (LTBP-1) and thus stores TGF-β1 in the ECM. Because the ED-A fibronectin splice variant is prominently expressed during fibrosis and supports myofibroblast activation, we investigated whether ED-A promotes LTBP-1–fibronectin interactions. Using stiffness-tuneable substrates for human dermal fibroblast cultures, we showed that high ECM stiffness promotes expression and colocalization of LTBP-1 and ED-A-containing fibronectin. When rescuing fibronectin-depleted fibroblasts with specific fibronectin splice variants, LTBP-1 bound more efficiently to ED-A-containing fibronectin than to ED-B-containing fibronectin and fibronectin lacking splice domains. Function blocking of the ED-A domain using antibodies and competitive peptides resulted in reduced LTBP-1 binding to ED-A-containing fibronectin, reduced LTBP-1 incorporation into the fibroblast ECM and reduced TGF-β1 activation. Similar results were obtained by blocking the heparin-binding stretch FNIII12-13-14 (HepII), adjacent to the ED-A domain in fibronectin. Collectively, our results suggest that the ED-A domain enhances association of the latent TGF-β1 by promoting weak direct binding to LTBP-1 and by enhancing heparin-mediated protein interactions through HepII in fibronectin.
KEY WORDS: Myofibroblast, Fibrosis, Wound healing, Transforming growth factor β1, TGF-β1, Growth factor activation
Highlighted Article: The presence of the ED-A splice domain enhances the ability of fibronectin to bind latent pro-fibrotic TGF-β1 and thus supports activation of myofibroblasts, which are main drivers of fibrosis.
INTRODUCTION
Tissue fibrosis manifests as severe deformities in the skin and leads to reduced function and/or failure of vital organs like lung, heart, liver and kidney (Wynn and Ramalingam, 2012). Myofibroblasts are responsible for the irreversible accumulation and excessive remodeling of collagenous extracellular matrix (ECM) that characterizes fibrosis (Hinz, 2016; Klingberg et al., 2013; Tomasek et al., 2002). Three key conditions coordinate myofibroblast activation from a variety of different precursor cells: (1) the presence of a mechanically resistant ECM (Arora et al., 1999; Li et al., 2017), (2) biologically active TGF-β1 (Desmoulière et al., 1993) and (3) extradomain-A (ED-A)-containing fibronectin (FN; also known as FN1) (hereafter denoted ED-A FN) (Serini et al., 1998). However, it is still unclear how TGF-β1 and mechanical stress collaborate with ED-A FN to promote myofibroblast activation.
Fibroblasts cultured from different organs and species have been shown to activate latent TGF-β1 from stores in the ECM by a process that requires cell contraction and a sufficiently stressed ECM (Hinz, 2015; Sarrazy et al., 2014; Wipff et al., 2007). In the soluble latent form, TGF-β1 is non-covalently bound to its pro-peptide form, known as latency-associated peptide (LAP) (Robertson et al., 2015). Covalent binding of the LAP portion of latent TGF-β1 to the latent TGF-β binding protein-1 (LTBP-1) intracellularly forms a large latent complex that is incorporated into the ECM upon secretion. LTBP-1 has been shown to mainly interact with two ECM proteins, FN and fibrillin-1 (Hynes, 2009; Ramirez and Rifkin, 2009; Zilberberg et al., 2012) with FN acting as master template for the initial LTBP-1 incorporation into the maturating ECM (Dallas et al., 2005; Klingberg et al., 2014; Koli et al., 2005). FN exists in two principal forms that are generated by alternative splicing from one single gene: (1) plasma FN that is secreted by hepatocytes into the circulation, and (2) insoluble cellular FN that is secreted by a variety of different cells, including fibroblasts (Klingberg et al., 2013; Pankov and Yamada, 2002; Singh et al., 2010; White et al., 2008; Zollinger and Smith, 2017). Cellular, but not plasma FN, contains the alternatively spliced FN type III (FNIII) extradomains ED-A (also known as EIIIA or FNIII EDA) and/or ED-B, which are transiently expressed during embryogenesis (Astrof and Hynes, 2009; Peters and Hynes, 1996). Under normal conditions, ED-A FN, ED-B FN and ED-A/B FN (i.e. containing both ED-A and ED-B) are typically not expressed in adult connective tissue, but become re-expressed as ‘oncofetal FNs’ during the ECM remodeling associated with wound repair, fibrosis and tumor development. The presence of ED-A FN is characteristic for tissue repair/healing and fibrosis, whereas ED-B FN is most frequently associated with tumor development and angiogenesis (Astrof et al., 2004; Bhattacharyya et al., 2014; Jarnagin et al., 1994; Kelsh et al., 2015; Kumra and Reinhardt, 2015; Sackey-Aboagye et al., 2016; Serini and Gabbiani, 1999; White et al., 2008).
Expression of ED-A FN precedes and is necessary for myofibroblast activation from various different precursor cells and in different fibrotic conditions (Arslan et al., 2011; Booth et al., 2012; Hirshoren et al., 2013; Kohan et al., 2011, 2010). The myofibroblast-permissive action of ED-A FN is inhibited by the ED-A FN function-blocking antibody IST-9 and recombinant ED-A peptides (Hinz et al., 2001; Serini et al., 1998). Consistent with this, ED-A FN-null (ED-A FN−/−) mice display abnormal healing of skin wounds (Muro et al., 2003) and are protected against bleomycin-induced lung fibrosis (Muro et al., 2008). ED-A FN−/− fibroblasts exhibit reduced responsiveness to active TGF-β1, and fail to activate latent TGF-β1 for yet unknown reasons (Muro et al., 2008). Collectively, these findings led us to test the hypothesis that ED-A FN is controlling the storage and/or activation of latent TGF-β1 in the ECM. Our data show that: (1) ED-A FN and LTBP-1 expression are co-upregulated during myofibroblast activation by ECM stiffness and colocalize in the myofibroblast ECM; (2) LTBP-1 incorporation into fibroblast ECM is enhanced by ED-A FN compared to what is seen with ED-B or plasma FN; (3) binding of purified LTBP-1 to recombinant ED-A domain peptides and full-length FNs without the ED-A domain is lower than with full-length ED-A FN; (4) LTBP-1 association with ED-A FN is inhibited by antibodies directed against either ED-A or the adjacent heparin-binding domains FNIII12-13-14 (denoted HepII). Incorporation of LTBP-1 into ED-A FN-containing ECM is blocked by heparan sulfate (HS). These findings suggest that ED-A plays a dual role in guiding LTBP-1 to FN by promoting specific, but low-affinity, interactions and enhancing the availability of the HepII heparin-binding sites in FN. Because reducing incorporation of LTBP-1 into the ECM by means of ED-A blocking antibodies also results in reduced TGF-β1 activation by fibroblast cultures, we propose that ED-A FN presents a potential target for anti-fibrosis strategies.
RESULTS
ECM stiffness regulates expression of ED-A FN and LTBP-1
Mechanical stress arising from ECM stiffening during remodeling is pivotal for myofibroblast activation (Hinz, 2010, 2015). To test whether expression of ED-A FN and LTBP-1 and secretion into the myofibroblast ECM are controlled by mechanical factors, we cultured primary human dermal fibroblasts (hDfs) on differently compliant silicone substrates for 7 days. A Young's modulus of 3 kPa was chosen to simulate the mechanical conditions of normal skin (Achterberg et al., 2014), 100 kPa substrates were used to simulate fibrotic tissue stiffness (Li et al., 2017) and intermediate stiffnesses (10 kPa and 25 kPa) for ECM in tissue under remodeling. Substrates of 3000 kPa provided mechanical growth conditions comparable to those on non-physiologically stiff conventional tissue plastic culture. Immunofluorescence staining demonstrated increasing expression of both ED-A FN and LTBP-1 with increasing substrate stiffness. Both proteins colocalized in the ECM, which was most pronounced in hDf cultures on 100 kPa and 3000 kPa stiff substrates (Fig. 1A). The pattern was similar when co-staining for LTBP-1 and total FN. Because corresponding expression levels of ED-B FN were overall low (Fig. 1A, insets), ED-A FN appears to be the predominant FN splice variant in hDf cultures. Western blotting of lysates of cells plus ECM confirmed increasing ED-A FN and LTBP-1 expression with increasing ECM stiffness, as was the case for expression of the myofibroblast marker α-smooth muscle actin (α-SMA) (Fig. 1B). To support the finding that LTBP-1 and ED-A FN colocalize in the ECM, we performed western blotting in non-reducing conditions (Fig. 1C). hDf cultures were differentially fractionated into conditioned medium supernatants (SN, 10× concentrated) and NH4OH-extraced ECM (ECM) after 7 days growth on plastic. Cell lysates were used as controls; these were obtained from a dish of cells grown in parallel by performing gentle trypsinization, followed by resuspension in lysis buffer (Fig. 1C, ‘cells’, controlled by GAPDH and α-tubulin). ED-A FN and total FN were predominantly present in the conditioned supernatants and ECM fractions and migrated at an apparent molecular mass of ∼220 kDa whereas LTBP-1 (∼180 kDa) mainly associated with the ECM and cell fractions (Fig. 1C).
Fig. 1.

Stiff ECM co-stimulates expression of α-SMA, ED-A FN and LTBP-1. (A) Primary hDfs were grown on compliant silicone substrates with elastic moduli of 3 kPa, 10 kPa, 25 kPa, 100 kPa and 3000 kPa for 7 days. The ECM produced on all stiffnesses contains LTBP-1 (green), ED-A FN (red) and low amounts of ED-B FN (red and insets), as shown by immunofluorescence microscopy. Images are orthogonal projections of 5 µm-thick confocal z-stacks, where yellow represents colocalization. Scale bar: 20 µm. (B) Expression of ED-A FN, LTBP-1 and α-SMA was determined by western blotting from lysates containing cell and ECM proteins, and quantified by normalizing to vimentin as a loading control. Shown are mean±s.d. from at least five independent experiments. *P<0.05; ns, not significant (one-way ANOVA followed by a post-hoc Dunnett's multiple comparison test). (C) hDfs were grown to confluency for 7 days on tissue culture plastic; supernatants (SN) were collected and concentrated, and the culture then extracted with NH4OH to produce ECM fractions (ECM). Cell fractions were produced in parallel from trypsinized cells. Western blots for high molecular mass proteins (4% SDS gels) were first processed for LTBP-1 (rbAb), then stripped with 200 mM glycine and 1% SDS (pH 2.5), and re-probed for ED-A FN (mAb) and total FN (rbAb). The same fractions were run in parallel on 10% SDS gels and then immunoblotted for GAPDH and α-tubulin to control for ECM fractionation efficacy.
ED-A FN is more potent in guiding LTBP-1 to fibroblast ECM than other FN splice variants
FN is a master regulator of the fibroblast ECM, and loss of FN expression has been previously shown to abolish LTBP-1 incorporation into the ECM of fibroblasts and osteoblasts (Dallas et al., 2005). Similarly, knockdown of FN expression in the human fibroblast cell line MRC-5 using human-specific small interfering (si)RNA directed against total FN almost completely abolished formation of FN fibrils in the ECM (Fig. 2A). FN knockdown efficiency was ∼90% and resulted in co-downregulation of the expression of LTBP-1 and fibrillin-1, another ECM protein dependent on FN (Sabatier et al., 2009) in lysates of cells plus ECM (Fig. 2B). The remaining low levels of LTBP-1 mainly accumulated in concentrated culture supernatants (SN) whereas LTBP-1 was virtually absent from the ECM fraction after FN knockdown (Fig. 2C). As a result, the ratio between the LTBP-1 level after FN knockdown and the LTBP-1 level in control conditions was higher in supernatant than in ECM (Fig. 2C).
Fig. 2.

FN knockdown reduces LTBP-1 expression and association with fibroblast ECM. (A) Human MRC-5 fibroblasts were transfected with human FN targeting (siFN) and non-targeting control (siCON) siRNAs. Transfected cells were assessed after 7 days by immunostaining and compared with non-transfected (NT) fibroblasts. LTBP-1, green; FN, red and insets, nuclei, DAPI (blue). All images are orthogonal projections of 5 µm-thick confocal z-stacks, where yellow shows colocalization. Scale bars: 50 µm. The graph on the right shows mean±s.d. LTBP-1 intensity signals (relative to FN intensity) from confocal images from three independent experiments, calculated over at least five images per experiment. (B,C) Knockdown efficiency and effect of FN loss were assessed by quantitative western blotting for LTBP-1 (rbAb), total FN (rbAb), ED-A FN (mAb), fibrillin-1 (rbAb) of either (B) lysates of cells plus ECM or (C) fractions of fibroblast ECM and concentrated supernatants (SN). Ratios of LTBP-1 in FN-knockout cells versus those in control cells (siFN/siCON) was calculated to demonstrate the shift of LTBP-1 into the supernatant upon loss of FN. Graphs show mean±s.d. from at least three independent experiments. *P<0.05; **P<0.01; ***P<0.005 (one-way ANOVA followed by a post-hoc Dunnett's multiple comparison test).
To investigate whether different splice variants of FN differentially affect expression and ECM organization of LTBP-1, we used 6xHis-tagged full-length rat FN constructs, containing: (1) only the ED-A extradomain (ED-A FN), (2) only the ED-B extradomain (ED-B FN), (3) both, ED-A and ED-B domains (ED-A/B FN), and (4) no extradomains (FN0) (Fig. 3A,B). Expression and purification from human embryonic kidney-293 cells (HEK293) confirmed correct molecular masses and the presence of ED-A in the ED-A FN splice variant (Fig. 3B). When stably overexpressed in wild-type MRC-5 cells, all full-length FN constructs incorporated into the ECM, colocalizing with endogenous FN and LTBP-1 after 7 days culture (Fig. 3C,D).
Fig. 3.

ED-A presence in FN enhances LTBP-1 incorporation into the fibroblast ECM. (A) Full-length rat FN constructs ED-A FN, ED-B FN, ED-A/B FN and FN0 with a C-terminal 6xHis tag were recombinantly expressed in HEK293 cells, purified and (B) western blotted for total FN, His (rb, rabbit, and mouse, m, antibodies) and ED-A FN. (C) Human MRC-5 fibroblasts were stably transfected with 6xHis-tagged rat FN full-length constructs. After 7 days, cultures were stained for the His tag (red), LTBP-1 (green) and nuclei (blue) and visualized at (C) low and (D) higher magnification. (E) Cultures were processed for western blotting to control for construct expression levels. (F) MRC-5 fibroblasts stably transfected with rat FN full-length constructs were transiently transfected with siRNA directed against human FN (siFN). Cells were then immunostained after 7 days culture for (F) the respective splice variant (green), 6xHis (red), and nuclei (DAPI; blue), or (G) for LTBP-1 (green), 6xHis (red), and nuclei (DAPI; blue). All images are orthogonal projections of 5 µm-thick confocal z-stacks, where yellow represents colocalization. Scale bars: 50 µm. (H) LTBP-1 intensity signals (relative to FN intensity) from confocal images from three independent experiment, calculated over at least five images per experiment. NT, not transfected. (I) The same cell populations were processed for western blotting and quantified for the LTBP-1 signal normalized to 6xHis tag and vimentin. Graphs show means±s.d. from at least three independent experiments. *P<0.05; **P<0.01; ***P<0.005 (one-way ANOVA followed by post-hoc Dunnett's multiple comparison).
MRC-5 cell lines, selected to express equal levels of His-tagged rat FN versions (Fig. 3E), were then knocked down for human FN (Fig. 3F–I). The ECM organization of His-tagged rat FN in human FN-depleted MRC-5 (Fig. 3E) was similar to that of FN in wild-type MRC-5 cells (Fig. 3C,D). Rescue of knocked down endogenous human FN with rat ED-A and ED-A/B FN restored the LTBP-1 to ∼40% of that measured in control (not siRNA treated) MRC-5 cultures as assed by quantifying the LTBP-1 signal intensity from immunofluorescence images (Fig. 3G,H) and western blots of cell plus ECM lysates (Fig. 3I). Notably, rescuing human FN-deficient MRC-5 cells with rat ED-A and ED-A/B FN resulted in ∼2-fold higher LTBP-1 incorporation into the ECM (Fig. 3G,H) and expression of LTBP-1 (Fig. 3I) compared with what was seen with ED-B FN and FN0 at similar expression levels (Fig. 3F–I). LTBP-1 levels were ∼4-fold higher than in non-rescued FN-knockdown cells (Fig. 3I). Hence, although all FN variants were able to recruit LTBP-1, ED-A FN was 2-fold more efficient at doing so.
Function blocking of the FN ED-A domain inhibits incorporation of LTBP-1 into fibroblast ECM
We next investigated whether function blocking of the ED-A domain in FN with competitive peptides affected the capability of fibroblasts to incorporate LTBP-1 into the ECM in a potential therapeutic setting. We recombinantly produced His-tagged short peptide fragments of rat FN domains in E. coli: (1) ED-A and ED-A with flanking domains (11-ED-A-12) as ‘active’ peptides, and (2) the flanking FNIII domains of ED-A alone (11-12) and only the FNIII11 domain (11) as specificity controls (Fig. 4A). Purity and the correct size of the peptides were confirmed on Coomassie-stained SDS gels (Fig. 4B) and by anti-6xHis western blotting (Fig. 4C). hDfs were then cultured for 7 days in the presence of FN domain peptides to compete with the different endogenous FN domains (Fig. 4D). LTBP-1 incorporation into the fibroblast ECM was reduced in the presence of domain peptides ED-A and 11-ED-A-12 but not by domain peptides FNIII11 or FNIII11-12, shown by performing immunofluorescence staining (Fig. 4E). FN domain peptides did not interfere with the organization and incorporation of endogenous ED-A FN (Fig. 4D). These results were confirmed by western blotting analysis showing an ∼5-fold reduced LTBP-1 level in ECM fractions after treatment with 11-ED-A-12 and ∼3-fold reduced LTBP-1 levels after treatment with ED-A domain peptides compared to the values in controls (Fig. 4E). Addition of FN domain peptides did not affect overall LTBP-1 production but resulted in the accumulation of LTBP-1 in hDf culture supernatants (Fig. 4E). Soluble LTBP-1 was increased by 6-fold after treating hDfs with 11-ED-A-12 and by 3.5-fold after ED-A domain peptide treatment compared to the values in controls (Fig. 4E). These data show that LTBP-1 incorporation into the fibroblast ECM can be competitively inhibited by soluble ED-A-containing peptides.
Fig. 4.

Competitive ED-A domain peptides reduce LTBP-1 incorporation into the ECM. (A) Rat FN peptide constructs, comprising domains 11, 11-12, 11-ED-A-12 and ED-A with a C-terminal 6xHis tag were recombinantly produced in E. coli, purified and characterized on (B) Coomassie blue-stained poly-acrylamide gels and (C) by western blotting for the His tag. MW, molecular mass markers. (D) hDfs were cultured for 7 days with the addition of FNIII domain peptides (10 µg/ml), replenished daily. PBS served as control. Cells were then processed for immunostaining against ED-A FN (red) and LTBP-1 (green). Scale bar: 25 µm. (E) ECM fractions (ECM) from NH4OH-extracted and concentrated conditioned supernatants (SN) produced from the same cultures were immunoblotted for LTBP-1. Solubilized cell fractions were blotted for vimentin as a general loading control. Graphs represent western blotting quantifications normalized to vimentin, showing means±s.d. from at least three independent experiments. *P<0.05; **P<0.01 compared to PBS controls (one-way ANOVA followed by post-hoc Dunnett's multiple comparison test).
Next, we assessed whether the ED-A domain-specific blocking antibody IST-9 (Carnemolla et al., 1987; Serini et al., 1998) had similar effects in inhibiting LTBP-1 anchoring with the ECM and can thus be used to prevent TGF-β activation from the large latent complex. Immunofluorescence analysis showed that treating hDfs for 7 days with IST-9 reduced integration of LTBP-1 into the ECM of hDfs and colocalization with ED-A FN compared to what was seen upon treatment with control antibody BC-1 (binds to FN when ED-B is present) and IgG (Fig. 5A). The amount and organization of ED-A FN, and lower expression of ED-B FN remained unaffected by IST-9 or control antibodies (Fig. 5A). Treatment of hDf cultures with IST-9 but not with control antibodies increased levels of total TGF-β but decreased active TGF-β in the supernatants, as measured by using TGF-β-reporter cells (Fig. 5B). Collectively, these data show that blocking the ED-A domain in FN with specific peptides and antibodies substantially decreases the ability of fibroblasts to immobilize LTBP-1 in the ECM. Loss of LTBP-1 from the ED-A FN ECM reduces the ability of fibroblasts to store, and thus activate, latent TGF-β1.
Fig. 5.
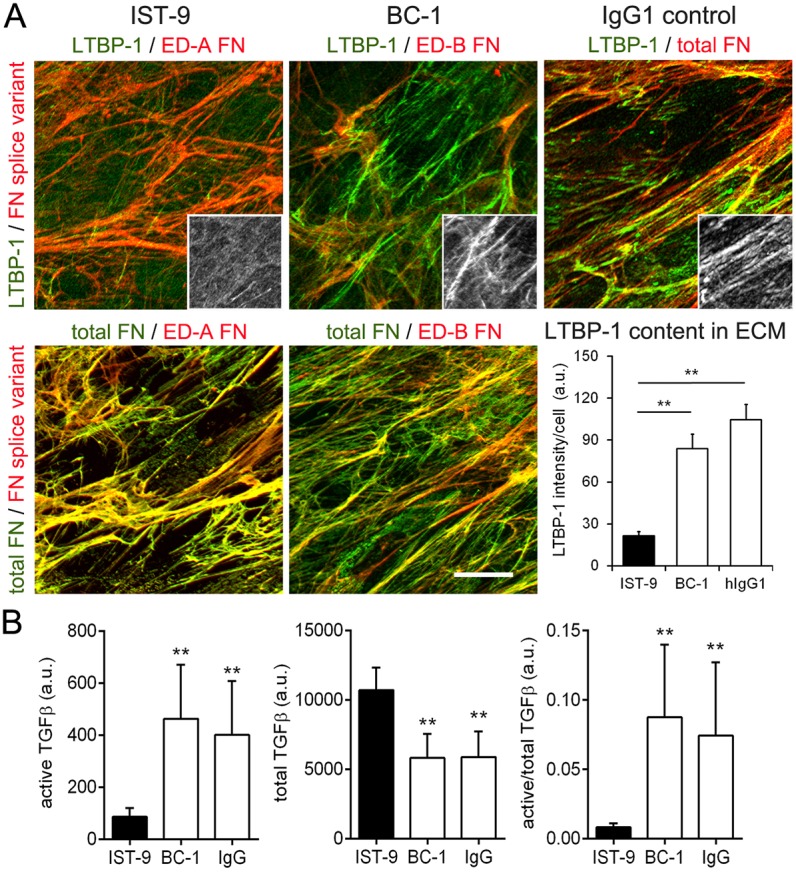
Blocking ED-A FN with IST-9 antibody inhibits incorporation of LTBP-1 into the ECM and reduces latent TGF-β activation. Cultures of hDfs were incubated with IST-9 or control anti-ED-B FN (BC-1) or human IgG1 (all 100 µg/ml) for 7 days. Cultures were assessed by immunofluorescence staining for ED-A FN (mAb), ED-B FN (mAb), total FN (rbAb) and LTBP-1 (rbAb, insets) and LTBP-1 was quantified by measuring the fluorescence signal intensity, normalized to cell numbers (DAPI count) in the respective image field (DAPI not shown). Mean±s.d. values were calculated over at least five images per experiment and three independent experiments. All images are orthogonal projections of 5 µm-thick confocal z-stacks, where yellow represents colocalization. Scale bar: 25 µm. (B) After treatment with ED-A domain-blocking antibodies and controls, supernatants of hDf cultures were collected. To assess TGF-β levels, TGF-β reporter cells were incubated with either native conditioned medium (active TGF-β) or heat-activated medium for 10 min at 80°C (total TGF-β) for 16 h. Reporter cell activity was corrected for the baseline in non-conditioned culture medium. All experiments were performed at least three times. *P<0.05, **P<0.001 compared with IgG control (one-way ANOVA followed by a post-hoc Dunnett's multiple comparison test).
The ED-A domain plays a potential dual role in mediating the association of LTBP-1 with FN
To test whether the ED-A domain directly promotes LTBP-1 binding, we immobilized purified 6xHis-tagged FN domain peptides and full-length FN splice variants on enzyme-linked immunosorbent assay (ELISA) plates. The coated plates were subsequently incubated with purified LTBP-1 (Buscemi et al., 2011; Klingberg et al., 2014), which was then immunolocalized and quantified (Fig. 6). Plates coated with LTBP-1 at the same concentration served as the standard for maximal signal. LTBP-1 did not bind to FNIII domain peptides 11 and 11-12; binding was significantly higher to ED-A (1.4-fold) and 11-ED-A-12 domain peptides (1.7-fold over controls) (Fig. 6A). LTBP-1 binding to ED-A and 11-ED-A-12 was completely abolished in the presence of the ED-A-blocking antibody IST-9 as an additional binding specificity control (Fig. 6B). Despite being specific, LTBP-1 binding to ED-A-containing peptides was low and reached only 14–17% of the maximal signal (Fig. 6A,B).
Fig. 6.

Binding of LTBP-1 to purified FN domain peptides and full-length FN constructs. (A) ELISAs were performed by (A,B) immobilizing FN domain peptides and (C,D) full-length FN variants on multi-well plates (10 µg) and measuring the binding interaction with added LTBP-1 (10 µg). Control wells were either directly coated with 10 µg LTBP-1 for maximal signal (no additional LTBP-1 added in the incubation period) or PBS-treated for background binding. (B,D) ELISAs were repeated in the presence of anti-ED-A (IST-9) or control IgG antibodies (100 µg/ml). All ELISA quantifications show mean±s.d. values from at least three independent experiments. *P<0.05; **P<0.01; n.s., not significant (one-way ANOVA followed by a post-hoc Dunnett's multiple comparison test).
In contrast, binding of LTBP-1 to full-length ED-A FN and ED-A/B FN was as high as 50% of the maximal LTBP-1-binding signal (Fig. 6C). Binding of LTBP-1 was ∼2-fold higher when full-length FN contained the ED-A domain than for FN0 and ED-B FN, both lacking ED-A (Fig. 6C). Presence of ED-B did not further enhance binding of LTBP-1 to full-length ED-A FN (ED-A/B FN) and ED-B alone had no enhancing effect (ED-B FN) (Fig. 6C). Blocking ED-A with IST-9 reduced LTBP-1 binding to ED-A FN to the level of that seen with FN0 (Fig. 6D). Collectively, the solid-state ELISA binding studies showed that LTBP-1 binds to all full-length FN constructs, which was enhanced by 2-fold when the ED-A domain was present. Binding of LTBP-1 to ED-A domain peptides was specific but low, suggesting an additional mechanism modulates how ED-A enhances LTBP-1 binding in the context of the FN molecule.
Thus, we tested whether the ED-A domain can potentiate LTBP-1 binding by modulating the availability of other binding domains in full-length FN. LTBP-1 binding to FN has been shown to be mediated by heparin (Chen et al., 2007; Massam-Wu et al., 2010), and heparin binding occurs in a stretch directly adjacent to ED-A in the FNIII domains 12-13-14 (HepII) (Clark et al., 2003; Mostafavi-Pour et al., 2001). Consequently, preincubation of LTBP-1 with heparan sulfate (HS) but not control chondroitin sulfate (CS) reduced binding to ED-A FN (and FN0) in ELISA assays with purified FN (Fig. 7A). Adding HS to hDf during the 7 day culture period resulted in almost complete inhibition of LTBP-1 incorporation into the ECM as shown on western blots with ECM fractions (Fig. 7B) and by immunofluorescence studies of hDF cultures (Fig. 7C). In addition, adding the HepII-blocking antibody A32 (Underwood et al., 1992) to hDf cultures reduced the ECM contents of LTBP-1 compared to what was seen with control antibody treatment, with moderate effects on FN organization (Fig. 7D). Blocking HepII also reduced the binding of purified LTBP-1 to full-length FN0 and ED-A FN splice variants (Fig. 7E) but not to ED-A-containing domain peptides lacking the HepII domain stretch (Fig. 7F). Collectively, these data suggest that the ED-A domain may increase the accessibility of the adjacent heparin-binding stretch FNIII12-13-14 (HepII) for LTBP-1 binding, in addition to weakly, but specifically, interacting with LTBP-1 in a direct fashion.
Fig. 7.

Binding of LTBP-1 to ED-A FN depends on heparin and the FN HepII domain. (A) ELISA with immobilized full-length FN constructs (10 µg) performed with soluble LTBP-1 (10 µg) that was pre-incubated with HS (500 µg/ml) to saturate heparin-binding sites or CS as control. HS and CS were added for 7 days to hDf cultures that were then processed for (B) western blotting of NH4OH-extracted ECM fractions and (C) immunofluorescence for LTBP-1 (green) and ED-A FN (red). (D) Cultures of hDfa were incubated with anti-HepII or control IgG antibody (100 µg/ml) for 7 days and then assessed by immunofluorescence co-staining for total FN (rbAb, red) and either HepII (mIgG1, green) or LTBP-1 (mIgG, green and insets). Images are orthogonal projections of 5 µm-thick confocal z-stacks, where yellow represents colocalization. Scale bars: 20 µm. (E) ELISAs were performed by immobilizing (E) full-length FN variants or (F) FN domain peptides and on multi-well plates (10 µg) and measuring the binding interaction with added LTBP-1 (10 µg). Control wells were either directly coated with 10 µg LTBP-1 for maximal signal (no additional LTBP-1 added in the incubation period) or PBS treated for background binding. ELISAs were performed either in PBS (control) or in the presence of anti-HepII or control IgG antibodies (100 µg/ml). All ELISA quantifications show mean±s.d. values from at least four independent experiments *P<0.05; **P<0.01 (one-way ANOVA followed by a post-hoc Dunnett's multiple comparison test).
DISCUSSION
The ED-A splice variant of FN is an important element controlling myofibroblast activation during wound healing and development of fibrosis (Klingberg et al., 2013; Shinde et al., 2015; White et al., 2008). Part of this action appears to be mediated by binding of ED-A FN-specific integrins. Integrins α9β1 and α4β1 recognize the EDGIHEL motif in ED-A FN (Liao et al., 2002; Shinde et al., 2008), and blocking antibodies against α4-integrins reduce the extent of bleomycin-induced lung fibrosis in mice (Gailit et al., 1993; Wang et al., 2000). This effect is likely caused by affecting inflammatory cells that most prominently express α4β1 integrin. The ED-A FN-binding integrin α4β7 has been directly implicated in myofibroblast differentiation of murine lung fibroblasts (Kohan et al., 2011, 2010). ED-A FN−/− mice are protected against bleomycin-induced skin and lung fibrosis (Muro et al., 2003, 2008) but not from experimentally induced liver fibrosis (Olsen et al., 2012). Notably, ED-A FN−/− liver hepatic stellate cells are able to become myofibroblasts in vivo and in vitro, suggesting that lacking ED-A FN can be compensated for by other myofibroblast-inducing factors. Myofibroblast activation further depends on the presence of active TGF-β1 and mechanical stress arising from the stiff ECM of the scar tissue. Although previous studies have shown that ED-A FN−/− mice and fibroblasts exhibit reduced levels of total and active TGF-β1 in conditions of lung fibrosis (Muro et al., 2008), the mechanistic link between ED-A FN and TGF-β1 activation or storage remained elusive. TGF-β1 activation by αv integrins depends on LTBP-1 binding to the ECM, which provides physical resistance against the cell pulling that is required to induce a conformational change in the latent complex (Annes et al., 2004; Buscemi et al., 2011; Shi et al., 2011; Wipff et al., 2007). We hypothesized that ED-A FN stores latent TGF-β1 in the myofibroblast ECM particularly efficiently. Our central finding is that the presence of the ED-A domain enhances the capacity of FN to recruit LTBP-1 to the ECM, which is required for the sequestration and subsequent activation of latent TGF-β1. Consistently, inhibition of the ED-A domain in our fibroblast cultures reduced levels of active TGF-β1 and increased the release of soluble LTBP-1 and latent TGF-β1 into culture medium.
Binding to the ECM is mediated through the N-terminus of LTBP-1 (Dallas et al., 2000; Nunes et al., 1997; Unsold et al., 2001), and the minimal ECM-binding sequence that allows integrin-mediated TGF-β1 activation comprises amino acids 402–449 in the N-terminal hinge domain in LTBP-1 (Annes et al., 2004; Fontana et al., 2005). LTBP-1 binds to different proteins of the ECM, including FNs, fibrillins and fibulins, in a carefully orchestrated sequence of changing binding partners (Todorovic and Rifkin, 2012). Initial ECM targeting of LTBP-1 is dependent on FN, but a transfer to fibrillin-1-containing microfibrils was shown to subsequently occur in cell culture models (Chaudhry et al., 2007; Dallas et al., 2000, 2005; Isogai et al., 2003; Ono et al., 2009; Sabatier et al., 2009, 2013). The partner domains in either fibrillin or FN that bind the hinge region of LTBP-1 have not yet been identified.
Our results show that presence of the ED-A domain in FN enhances LTBP-1 incorporation into the ECM of fibroblast cultures over FN lacking the ED-A domain as follows: (1) inhibition of ED-A using specific antibodies and competitive peptides prevents LTBP-1 targeting to the ECM and instead leads to its release into the cell culture supernatant; (2) expression of ED-A FN in FN-depleted fibroblasts rescue LTBP-1 targeting to the ECM more efficiently than ED-B FN or FN without extradomains; and (3) full-length purified FN binds purified LTBP-1 more efficiently when the ED-A domain is present. Although recombinant ED-A domain peptides display a low potential to directly bind purified LTBP-1 in vitro, they potently blocked targeting of LTBP-1 to the fibroblast ECM. We thus consider that ED-A creates a favorable FN conformation for LTBP-1 binding, in addition to being a direct binding partner.
Previous findings support the idea that the presence of ED-A primes the FN structure for LTBP-1 binding. LTBP-1 contains a sensitive proline-rich hinge region with a heparin-binding consensus sequence, and LTBP-1 binding to both fibrillin-1 and FN has been shown to be mediated by heparin (Chen et al., 2007; Massam-Wu et al., 2010). In addition to heparin-binding domains in proximity to the N-terminus of FN, a major heparin-binding domain – HepII – is located in the FNIII domain stretch 12-13-14, adjacent to ED-A (Clark et al., 2003; Mostafavi-Pour et al., 2001). HepII plays a crucial role in growth factor binding to FN (Mitsi et al., 2008; Wan et al., 2013). Moreover, ligation of αv integrins to the RGD binding site in the FNIII domain 10, and α5β1 integrin to the additional consensus sequence in FNIII 9 has been shown to enhance cell responses to various growth factors that promiscuously bind to FNIII domains 12-13-14 (Martino and Hubbell, 2010). Our own results demonstrate that LTBP-1 binding to ED-A FN is abolished by saturating heparin-binding sites with HS and upon treatment with blocking antibodies directed against HepII. It is thus conceivable that the presence of ED-A places the HepII heparin-binding site into a favorable position for FN interaction with LTBP-1. ED-A is a cryptic FN domain subject to regulatory cell processes (Julier et al., 2015; Klein et al., 2003), and mechanical stress has recently been shown to collaborate with heparin binding to FNIII 11-12-13 to determine FN structure (Hubbard et al., 2014; Zollinger and Smith, 2017). It remains to be shown whether mechanical stress can also modulate the function of ED-A in LTBP-1 binding to FN as suggested by increasing colocalization of both proteins in ECM with increasing stiffness in our fibroblast cultures.
In summary, we found that the mechanical conditions that fibroblasts encounter in advanced stages of wound healing and fibrosis stimulate ED-A FN and LTBP-1 co-expression, and their interaction in the ECM. The ED-A domain plays a supporting role in promoting FN interactions with LTBP-1, and the HepII domain stretch adjacent to ED-A in FN appears to enhance this binding. We propose that blocking the interaction of LTBP-1 with ED-A FN by using competitive domain peptides or specific antibodies is a potential strategy to specifically reduce TGF-β1 storage in the myofibroblast-associated ECM and ultimately development of fibrosis.
MATERIALS AND METHODS
Cell culture
Normal fibroblasts were explanted from human dermal tissue samples (n=5), received from Dr Benjamin A. Alman (Sick Kids Hospital, Toronto, ON) as described previously (Klingberg et al., 2014). Written consent for the use of human biopsy material was obtained from patients and procedures approved by the Institutional Review Board of the Hospital for Sick Children (Toronto, Canada). HDfs between passages (P)2–P5 and lineage MRC-5 fibroblasts (ATCC, Manassass, VA) were maintained in standard cell culture (Dulbecco's modified Eagle's medium, DMEM; Life Technologies, Burlington, ON), supplemented with 10% fetal bovine serum (Sigma-Aldrich, Oakville, ON) and penicillin-streptomycin (Life Technologies). HEK293 cells, stably expressing LTBP-1–EGFP or different rat FN constructs were selected and maintained in Zeocin™ and G418 (Life Technologies) (Klingberg et al., 2014). In select experiments, mechanical growth conditions for fibroblasts were controlled by using deformable silicone (polydimethylsiloxane) substrates with a Young's modulus of 3, 10, 25, 100 and 3000 kPa (Excellness Biotech SA, Lausanne, Switzerland) that were coated with 2 µg/cm2 gelatin (Sigma-Aldrich) (Li et al., 2017).
Reagents and antibodies
siRNA constructs directed against human FN were designed and purchased from Thermo Fisher Scientific (Burlington, ON). All cell transfections were performed according to the manufacturer's specifications by using an electroporation device (NEON, Life Technologies), with two pulses at 1150 V and 20 ms duration. Primary antibodies used in this study were directed against: α-SMA (clone SM1, a kind gift of Giulio Gabbiani, University of Geneva, Switzerland, 1:100), (total) FN (rbAb, Sigma-Aldrich, F3648, 1:100), ED-A FN (mAb, clone IST-9, Santa Cruz Biotech, Dallas, TX, sc-59826, 1:100), ED-B FN (mAb, clone BC-1, Abcam, Cambridge, MA, ab154210, 1:100), FN HepII (mIgG1, clone Ab32, Thermo Fisher CSI 005-32-02, 1:50) LTBP-1 (mAb, R&D Systems, Minneapolis, MN, MAB388, 1:100; and rbAb39, a very generous gift from Carl-Hendrik Heldin, Uppsala University, Sweden, 1:200), HIS (mAb A00186 and rbAb A00174, Genscript, Piscataway, NJ, 1:200), fibrillin-1 (rF6H ID 157, a kind gift from D. Reinhardt, McGill University, Montreal, Canada, 1:50), and vimentin (mAb, Dako, Burlington, ON, M0725, 1:400). Secondary antibodies used were: goat anti-mouse-IgG conjugated to Alexa Fluor 568 (Life Technologies, A-11004, 1:250), goat anti-mouse-IgG1 conjugated to FITC (Southern Biotechnology, Birmingham, AL, 1070-02, 1:100), goat anti-mouse-IgG2a conjugated to TRITC (Southern Biotechnology, 1080-03, 1:100), goat anti-mouse-IgG2b conjugated to TRITC (Southern Biotechnology, 1090-03, 1:100), and goat anti-rabbit-IgG conjugated to TRITC and FITC (Sigma-Aldrich, F9887, 1:100). To stain DNA and nuclei, 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich, D9542) was used (1:50). For blocking experiments, hDfs were cultured in standard conditions for 2 days and then incubated with anti-ED-A FN (mAb clone IST-9, sodium azide-free, Abcam, ab6328) or anti-HepII (Ab32; Underwood et al., 1992) at 100 µg/ml (Hinz et al., 2001; Serini et al., 1998) or purified FN peptide fragments (50 µg/ml) for another 5 days with daily replenishments. Controls were 10% FBS, BC-1 (Abcam) and human IgG1 antibodies (Sigma-Aldrich, I4506), all at 100 µg/ml. Heparan sulfate and control chondroitin sulfate were added at 500 µg/ml with the same time course and replenishments.
Cell fractionation and western blotting
hDfs were cultured to confluency for 7 days in all experiments. For regular western blots (‘whole lysates’), culture medium was removed, and cultures were washed with PBS and scraped into standard lysis buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 0.1% Bromophenol Blue and 10% glycerol). To blot proteins from conditioned medium, culture supernatants were harvested, concentrated 10-fold with size exclusion centrifugation filters, and dialyzed against RIPA buffer (150 mM NaCl, 1 mM EDTA, 25 mM Tris-HCl pH 7.4 and 1% Triton X-100), supplemented with protease inhibitor cocktail (1:50 dilution, Sigma P8340) and 1 mM sodium orthovanadate and thoroughly sonicated. After removing supernatants, the same cultures were then used to either prepare cell fractions or ECM fractions. To obtain ECM fractions (‘ECM’), cells were removed using decellularization buffer (20 mM NH4OH and 0.5% Triton X-100 in PBS), rinsed twice in the same buffer, washed three times with PBS, and then scraped into RIPA buffer. To obtain cell fractions (‘cells’), cells were gently trypsinized (0.25% trypsin, 5 min), centrifuged (800 g for 5 min) and the pellet lysed in RIPA buffer. Remaining material from these dishes was not used to produce ECM fractions. Western blotting was performed in reducing conditions (α-SMA and vimentin) or non-reducing conditions (all ECM proteins) on 8% and 10% SDS-PAGE gels. Proteins were transferred onto nitrocellulose membranes by using a wet transfer technique. Protein membranes were blocked with 5% skim milk, and primary antibodies were detected with fluorescently labeled anti-mouse-IgG or anti-rabbit-IgG conjugated to 680 nm 800 nm IRDye®, respectively, secondary antibodies (1:10,000, LICOR Biosciences, LIC-926-68020 and LIC-926-32211). Signals were detected and quantified with a LICOR Fx imaging system (LI-COR Biosciences, Lincoln, NE).
Protein purification
Rat FN domain peptides were expressed in E. coli, purified, and characterized as published in detail previously (Kohan et al., 2010). Full-length FN constructs were produced by cloning the entire sequences of the rat FN splice variants (Schwarzbauer et al., 1987) into pcDNA3.1 using the Invitrogen TOPO TA cloning method (Invitrogen) (Sackey-Aboagye et al., 2016). All 6xHis-tagged proteins were purified from serum-free conditioned medium from transfected HEK293 cells. In brief, conditioned medium was collected and dialyzed against phosphate-buffered saline (PBS, Life Technologies) before it was run through an ion metal affinity chromatography column with HIS-Select® Nickel Affinity Gel (Sigma-Aldrich). Columns were washed with PBS buffer containing 0, 10 or 15 mM imidazole (Sigma-Aldrich). Fractions containing LTBP-1 were eluted with 250 mM imidazole. Full-length protein constructs were detected by western blotting using anti-His antibodies.
Immunofluorescence, microscopy and quantitative image analysis
Samples were treated in sequence for immunostaining: fixation with 3% paraformaldehyde for 10 min, permeabilization with 0.2% Triton X-100 (Sigma-Aldrich), incubation with primary antibodies for 1 h, and labeling with secondary antibodies for 1 h, all at room temperature. Fluorescence microscopy images were acquired with an Axio Imager upright microscope equipped with an AxioCam HRm camera, Apotome 2 structured illumination and ZEN software (Zeiss, Oberkochem, Germany). Plan-Apochromat objectives were used (Zeiss, 40×, NA 1.2, and Zeiss, 63×, NA 1.4, Oil-DIC) in addition to a Fluar objective (Zeiss, 20×, NA 0.75). Confocal images were acquired at the Centre for Microfluidics Systems, University of Toronto, using a Nikon Eclipse Ti microscope system and Apo 60× objective. Quantitative image analysis was performed using ImageJ (http://imagej.nih.gov/ij/) using customized macros (available upon request). Figures were assembled in Adobe Photoshop CS5 (Adobe Systems, San Jose, CA).
ELISA
To study protein–protein interactions, ELISA with fluorescent detection was established by coating black clear bottom 96-well plates with 10 µg of full-length FN splice variants, domain peptides of FN or 10 µg LTBP-1 (control) overnight at 4°C. Wells were then incubated with 0.5% BSA and 10 µg/ml heparin in PBS for 1 h. After three washes with PBS, 10 µg of LTBP-1 was added to the wells for 2 h at 4°C, with the exception of LTBP-1 control wells. In select experiments, LTBP-1 was pre-incubated with 0.5 mg/ml BSA or chondroitin sulfate (controls) or HS, to block heparin-binding sites, before adding the whole solution to full-length FNs for binding assays. Subsequently, wells were washed with PBS, stained for LTBP-1 and signals detected with fluorescent antibodies in a LICOR Fx imaging system (LI-COR Biosciences).
TGF-β1 bioassay
Active and total TGF-β were quantified by using transformed reporter mink lung epithelial cells (TMLCs), producing luciferase under the control of the PAI-1 promoter in response to TGF-β (Abe et al., 1994). After treatment with ED-A domain blocking antibodies and controls, supernatants of hDf cultures were collected. To assess TGF-β levels, TMLCs (60,000 cells/cm2) were adhered for 4 h before being subjected to either native conditioned medium (active TGF-β) or heat-activated medium for 10 min at 80°C (total TGF-β) for an additional 16 h. All results were corrected for TMLC baseline luciferase production in non-conditioned culture medium.
Statistical analysis
When applicable, data are presented as means±s.d. Differences between groups were assessed with a one-way analysis of variance (ANOVA) followed by a post-hoc Dunnett's multiple comparison test and the significance level set at P≤0.05. Statistical analyses and data plots were performed using Prism software (GraphPad, San Diego, CA). *P≤0.05; **P≤0.01; ***P≤0.005.
Acknowledgements
We thank Dr Pierre Jurdic (Université de Lyon, France) for providing Flp-In™ 293 cells, Drs Christine Chaponnier and Giulio Gabbiani (University of Geneva, Switzerland) for providing antibodies directed against α-SMA, Dr Carl-Hendrik Heldin (Uppsala University, Sweden) for anti-LTBP-1 (Ab39) and Dr Dieter Reinhardt for anti-fibrillin-1 (McGill University, Canada). Dr Christopher A. G. McCulloch and Dr Craig Simmons (University of Toronto, Canada) are acknowledged for technical support and advice.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: F.K., R.W., E.W., B.H.; Methodology: F.K., G.C., S.B., M.C., A.K., A.O., M.I., R.W., E.W.; Validation: F.K., G.C., M.C., A.K., M.L., B.H.; Investigation: F.K., G.C., M.W., S.B., M.C., A.K., M.I., B.H.; Resources: B.H.; Data curation: F.K., G.C., M.W., S.B., M.C., A.K., A.O., M.I., M.L.; Writing - original draft: F.K., G.C., M.C., A.K., B.H.; Writing - review & editing: R.W., E.W., B.H.; Supervision: B.H.; Project administration: B.H.; Funding acquisition: B.H.
Funding
This research was supported by the Canadian Institutes of Health Research (CIHR; grants #375597, #210820, #286920 and #286720), the Collaborative Health Research Programme (CIHR and Natural Sciences and Engineering Research Council of Canada grant #413783), the Canada Foundation for Innovation and Ontario Research Fund (CFI/ORF grants #26653 and 36050), the E-Rare Joint Transnational Program ‘Development of Innovative Therapeutic Approaches for Rare Diseases’ (Grant # ERL-138395) (all to B.H.), and from the US National Institutes of Health (PO1 grants DK-058123 to R.G.W., and HL085083 to E.S.W.). Data presented herein was further receiving support from the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement #237946, the CIHR Cell Signals Training program (F.K.) and an NIH training grant to A.L.O. (F30-DK081265-01). M.W. was supported by post-doc fellowships from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research) and the CIHR. Deposited in PMC for release after 12 months.
References
- Abe M., Harpel J. G., Metz C. N., Nunes I., Loskutoff D. J. and Rifkin D. B. (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276-284. 10.1006/abio.1994.1042 [DOI] [PubMed] [Google Scholar]
- Achterberg V. F., Buscemi L., Diekmann H., Smith-Clerc J., Schwengler H., Meister J.-J., Wenck H., Gallinat S. and Hinz B. (2014). The nano-scale mechanical properties of the extracellular matrix regulate dermal fibroblast function. J. Invest. Dermatol. 134, 1862-1872. 10.1038/jid.2014.90 [DOI] [PubMed] [Google Scholar]
- Annes J. P., Chen Y., Munger J. S. and Rifkin D. B. (2004). Integrin α V β 6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 165, 723-734. 10.1083/jcb.200312172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora P. D., Narani N. and McCulloch C. A. (1999). The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am. J. Pathol. 154, 871-882. 10.1016/S0002-9440(10)65334-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan F., Smeets M. B., Riem Vis P. W., Karper J. C., Quax P. H., Bongartz L. G., Peters J. H., Hoefer I. E., Doevendans P. A., Pasterkamp G. et al. (2011). Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ. Res. 108, 582-592. 10.1161/CIRCRESAHA.110.224428 [DOI] [PubMed] [Google Scholar]
- Astrof S. and Hynes R. O. (2009). Fibronectins in vascular morphogenesis. Angiogenesis 12, 165-175. 10.1007/s10456-009-9136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrof S., Crowley D., George E. L., Fukuda T., Sekiguchi K., Hanahan D. and Hynes R. O. (2004). Direct test of potential roles of EIIIA and EIIIB alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol. Cell. Biol. 24, 8662-8670. 10.1128/MCB.24.19.8662-8670.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S., Tamaki Z., Wang W., Hinchcliff M., Hoover P., Getsios S., White E. S. and Varga J. (2014). FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med. 6, 232ra50 10.1126/scitranslmed.3008264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth A. J., Wood S. C., Cornett A. M., Dreffs A. A., Lu G., Muro A. F., White E. S. and Bishop D. K. (2012). Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J. Pathol. 226, 609-618. 10.1002/path.3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi L., Ramonet D., Klingberg F., Formey A., Smith-Clerc J., Meister J.-J. and Hinz B. (2011). The single-molecule mechanics of the latent TGF-beta1 complex. Curr. Biol. 21, 2046-2054. 10.1016/j.cub.2011.11.037 [DOI] [PubMed] [Google Scholar]
- Carnemolla B., Borsi L., Zardi L., Owens R. J. and Baralle F. E. (1987). Localization of the cellular-fibronectin-specific epitope recognized by the monoclonal antibody IST-9 using fusion proteins expressed in E. coli. FEBS Lett. 215, 269-273. 10.1016/0014-5793(87)80160-6 [DOI] [PubMed] [Google Scholar]
- Chaudhry S. S., Cain S. A., Morgan A., Dallas S. L., Shuttleworth C. A. and Kielty C. M. (2007). Fibrillin-1 regulates the bioavailability of TGFbeta1. J. Cell Biol. 176, 355-367. 10.1083/jcb.200608167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Sivakumar P., Barley C., Peters D. M., Gomes R. R., Farach-Carson M. C. and Dallas S. L. (2007). Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-beta) by modulating assembly of latent TGF-beta-binding protein-1. J. Biol. Chem. 282, 26418-26430. 10.1074/jbc.M703341200 [DOI] [PubMed] [Google Scholar]
- Clark R. A. F., An J.-Q., Greiling D., Khan A. and Schwarzbauer J. E. (2003). Fibroblast migration on fibronectin requires three distinct functional domains. J. Invest. Dermatol. 121, 695-705. 10.1046/j.1523-1747.2003.12484.x [DOI] [PubMed] [Google Scholar]
- Dallas S. L., Keene D. R., Bruder S. P., Saharinen J., Sakai L. Y., Mundy G. R. and Bonewald L. F. (2000). Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J. Bone Miner. Res. 15, 68-81. 10.1359/jbmr.2000.15.1.68 [DOI] [PubMed] [Google Scholar]
- Dallas S. L., Sivakumar P., Jones C. J. P., Chen Q., Peters D. M., Mosher D. F., Humphries M. J. and Kielty C. M. (2005). Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J. Biol. Chem. 280, 18871-18880. 10.1074/jbc.M410762200 [DOI] [PubMed] [Google Scholar]
- Desmoulière A., Geinoz A., Gabbiani F. and Gabbiani G. (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103-111. 10.1083/jcb.122.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Chen Y., Prijatelj P., Sakai T., Fassler R., Sakai L. Y. and Rifkin D. B. (2005). Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 19, 1798-1808. 10.1096/fj.05-4134com [DOI] [PubMed] [Google Scholar]
- Gailit J., Pierschbacher M. and Clark R. A. F. (1993). Expression of functional alpha 4 beta 1 integrin by human dermal fibroblasts. J. Invest. Dermatol. 100, 323-328. 10.1111/1523-1747.ep12470011 [DOI] [PubMed] [Google Scholar]
- Hinz B. (2010). The myofibroblast: paradigm for a mechanically active cell. J. Biomech. 43, 146-155. 10.1016/j.jbiomech.2009.09.020 [DOI] [PubMed] [Google Scholar]
- Hinz B. (2015). The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 47, 54-65. 10.1016/j.matbio.2015.05.006 [DOI] [PubMed] [Google Scholar]
- Hinz B. (2016). Myofibroblasts. Exp. Eye Res. 142, 56-70. 10.1016/j.exer.2015.07.009 [DOI] [PubMed] [Google Scholar]
- Hinz B., Celetta G., Tomasek J. J., Gabbiani G. and Chaponnier C. (2001). Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 12, 2730-2741. 10.1091/mbc.12.9.2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshoren N., Kohan M., Assayag M., Neuman T., Vernea F., Muro A., Eliashar R. and Berkman N. (2013). Extra domain-A fibronectin is necessary for the development of nasal remodeling in chronic allergen-induced rhinitis. Ann. Allergy Asthma Immunol. 110, 322-327. 10.1016/j.anai.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Hubbard B., Buczek-Thomas J. A., Nugent M. A. and Smith M. L. (2014). Heparin-dependent regulation of fibronectin matrix conformation. Matrix Biol. 34, 124-131. 10.1016/j.matbio.2013.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. (2009). The extracellular matrix: not just pretty fibrils. Science 326, 1216-1219. 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai Z., Ono R. N., Ushiro S., Keene D. R., Chen Y., Mazzieri R., Charbonneau N. L., Reinhardt D. P., Rifkin D. B. and Sakai L. Y. (2003). Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278, 2750-2757. 10.1074/jbc.M209256200 [DOI] [PubMed] [Google Scholar]
- Jarnagin W. R., Rockey D. C., Koteliansky V. E., Wang S. S. and Bissell D. M. (1994). Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J. Cell Biol. 127, 2037-2048. 10.1083/jcb.127.6.2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julier Z., Martino M. M., de Titta A., Jeanbart L. and Hubbell J. A. (2015). The TLR4 agonist fibronectin extra domain A is cryptic, exposed by elastase-2; use in a fibrin matrix cancer vaccine. Sci. Rep. 5, 8569 10.1038/srep08569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsh R. M., McKeown-Longo P. J. and Clark R. A. F. (2015). EDA fibronectin in keloids create a vicious cycle of fibrotic tumor formation. J. Invest. Dermatol. 135, 1714-1718. 10.1038/jid.2015.155 [DOI] [PubMed] [Google Scholar]
- Klein R. M., Zheng M., Ambesi A., Van De Water L. and McKeown-Longo P. J. (2003). Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J. Cell Sci. 116, 4663-4674. 10.1242/jcs.00778 [DOI] [PubMed] [Google Scholar]
- Klingberg F., Hinz B. and White E. S. (2013). The myofibroblast matrix: implications for tissue repair and fibrosis. J. Pathol. 229, 298-309. 10.1002/path.4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingberg F., Chow M. L., Koehler A., Boo S., Buscemi L., Quinn T. M., Costell M., Alman B. A., Genot E. and Hinz B. (2014). Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J. Cell Biol. 207, 283-297. 10.1083/jcb.201402006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan M., Muro A. F., White E. S. and Berkman N. (2010). EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 24, 4503-4512. 10.1096/fj.10-154435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan M., Muro A. F., Bader R. and Berkman N. (2011). The extra domain A of fibronectin is essential for allergen-induced airway fibrosis and hyperresponsiveness in mice. J. Allergy Clin. Immunol. 127, 439-446 e1-5. 10.1016/j.jaci.2010.10.021 [DOI] [PubMed] [Google Scholar]
- Koli K., Hyytiäinen M., Ryynänen M. J. and Keski-Oja J. (2005). Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp. Cell Res. 310, 370-382. 10.1016/j.yexcr.2005.08.008 [DOI] [PubMed] [Google Scholar]
- Kumra H. and Reinhardt D. P. (2015). Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 97, 101-110. 10.1016/j.addr.2015.11.014 [DOI] [PubMed] [Google Scholar]
- Li C. X., Talele N. P., Boo S., Koehler A., Knee-Walden E., Balestrini J. L., Speight P., Kapus A. and Hinz B. (2017). MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater. 16, 379-389. 10.1038/nmat4780 [DOI] [PubMed] [Google Scholar]
- Liao Y.-F., Gotwals P. J., Koteliansky V. E., Sheppard D. and Van De Water L. (2002). The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J. Biol. Chem. 277, 14467-14474. 10.1074/jbc.M201100200 [DOI] [PubMed] [Google Scholar]
- Martino M. M. and Hubbell J. A. (2010). The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 24, 4711-4721. 10.1096/fj.09-151282 [DOI] [PubMed] [Google Scholar]
- Massam-Wu T., Chiu M., Choudhury R., Chaudhry S. S., Baldwin A. K., McGovern A., Baldock C., Shuttleworth C. A. and Kielty C. M. (2010). Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J. Cell Sci. 123, 3006-3018. 10.1242/jcs.073437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsi M., Forsten-Williams K., Gopalakrishnan M. and Nugent M. A. (2008). A catalytic role of heparin within the extracellular matrix. J. Biol. Chem. 283, 34796-34807. 10.1074/jbc.M806692200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi-Pour Z., Askari J. A., Whittard J. D. and Humphries M. J. (2001). Identification of a novel heparin-binding site in the alternatively spliced IIICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 20, 63-73. 10.1016/S0945-053X(00)00131-1 [DOI] [PubMed] [Google Scholar]
- Muro A. F., Chauhan A. K., Gajovic S., Iaconcig A., Porro F., Stanta G. and Baralle F. E. (2003). Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J. Cell Biol. 162, 149-160. 10.1083/jcb.200212079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro A. F., Moretti F. A., Moore B. B., Yan M., Atrasz R. G., Wilke C. A., Flaherty K. R., Martinez F. J., Tsui J. L., Sheppard D. et al. (2008). An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care. Med. 177, 638-645. 10.1164/rccm.200708-1291OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes I., Gleizes P.-E., Metz C. N. and Rifkin D. B. (1997). Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J. Cell Biol. 136, 1151-1163. 10.1083/jcb.136.5.1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen A. L., Sackey B. K., Marcinkiewicz C., Boettiger D. and Wells R. G. (2012). Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology 142, 928-937 e3. 10.1053/j.gastro.2011.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono R. N., Sengle G., Charbonneau N. L., Carlberg V., Bachinger H. P., Sasaki T., Lee-Arteaga S., Zilberberg L., Rifkin D. B., Ramirez F. et al. (2009). Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J. Biol. Chem. 284, 16872-16881. 10.1074/jbc.M809348200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R. and Yamada K. M. (2002). Fibronectin at a glance. J. Cell Sci. 115, 3861-3863. 10.1242/jcs.00059 [DOI] [PubMed] [Google Scholar]
- Peters J. H. and Hynes R. O. (1996). Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes. Commun. 4, 103-125. 10.3109/15419069609010766 [DOI] [PubMed] [Google Scholar]
- Ramirez F. and Rifkin D. B. (2009). Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr. Opin. Cell Biol. 21, 616-622. 10.1016/j.ceb.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson I. B., Horiguchi M., Zilberberg L., Dabovic B., Hadjiolova K. and Rifkin D. B. (2015). Latent TGF-beta-binding proteins. Matrix Biol. 47, 44-53. 10.1016/j.matbio.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier L., Chen D., Fagotto-Kaufmann C., Hubmacher D., McKee M. D., Annis D. S., Mosher D. F. and Reinhardt D. P. (2009). Fibrillin assembly requires fibronectin. Mol. Biol. Cell 20, 846-858. 10.1091/mbc.E08-08-0830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier L., Djokic J., Fagotto-Kaufmann C., Chen M., Annis D. S., Mosher D. F. and Reinhardt D. P. (2013). Complex contributions of fibronectin to initiation and maturation of microfibrils. Biochem. J. 456, 283-295. 10.1042/BJ20130699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackey-Aboagye B., Olsen A. L., Mukherjee S. M., Ventriglia A., Yokosaki Y., Greenbaum L. E., Lee G. Y., Naga H. and Wells R. G. (2016). Fibronectin extra domain a promotes liver sinusoid repair following hepatectomy. PLoS ONE 11, e0163737 10.1371/journal.pone.0163737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazy V., Koehler A., Chow M. L., Zimina E., Li C. X., Kato H., Caldarone C. A. and Hinz B. (2014). Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 102, 407-417. 10.1093/cvr/cvu053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer J. E., Patel R. S., Fonda D. and Hynes R. O. (1987). Multiple sites of alternative splicing of the rat fibronectin gene transcript. EMBO J. 6, 2573-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serini G. and Gabbiani G. (1999). Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 250, 273-283. 10.1006/excr.1999.4543 [DOI] [PubMed] [Google Scholar]
- Serini G., Bochaton-Piallat M.-L., Ropraz P., Geinoz A., Borsi L., Zardi L. and Gabbiani G. (1998). The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 142, 873-881. 10.1083/jcb.142.3.873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T. and Springer T. A. (2011). Latent TGF-beta structure and activation. Nature 474, 343-349. 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde A. V., Bystroff C., Wang C., Vogelezang M. G., Vincent P. A., Hynes R. O. and Van De Water L. (2008). Identification of the peptide sequences within the EIIIA (EDA) segment of fibronectin that mediate integrin alpha9beta1-dependent cellular activities. J. Biol. Chem. 283, 2858-2870. 10.1074/jbc.M708306200 [DOI] [PubMed] [Google Scholar]
- Shinde A. V., Kelsh R., Peters J. H., Sekiguchi K., Van De Water L. and McKeown-Longo P. J. (2015). The alpha4beta1 integrin and the EDA domain of fibronectin regulate a profibrotic phenotype in dermal fibroblasts. Matrix Biol. 41, 26-35. 10.1016/j.matbio.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Carraher C. and Schwarzbauer J. E. (2010). Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26, 397-419. 10.1146/annurev-cellbio-100109-104020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic V. and Rifkin D. B. (2012). LTBPs, more than just an escort service. J. Cell. Biochem. 113, 410-418. 10.1002/jcb.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C. and Brown R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349-363. 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- Underwood P. A., Dalton B. A., Steele J. G., Bennett F. A. and Strike P. (1992). Anti-fibronectin antibodies that modify heparin binding and cell adhesion: evidence for a new cell binding site in the heparin binding region. J. Cell Sci. 102, 833-845. [DOI] [PubMed] [Google Scholar]
- Unsold C., Hyytiainen M., Bruckner-Tuderman L. and Keski-Oja J. (2001). Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J. Cell Sci. 114, 187-197. [DOI] [PubMed] [Google Scholar]
- Wan A. M. D., Chandler E. M., Madhavan M., Infanger D. W., Ober C. K., Gourdon D., Malliaras G. G. and Fischbach C. (2013). Fibronectin conformation regulates the proangiogenic capability of tumor-associated adipogenic stromal cells. Biochim. Biophys. Acta 1830, 4314-4320. 10.1016/j.bbagen.2013.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Wang Y., Hyde D. M., Gotwals P. J., Lobb R. R., Ryan S. T. and Giri S. N. (2000). Effect of antibody against integrin alpha4 on bleomycin-induced pulmonary fibrosis in mice. Biochem. Pharmacol. 60, 1949-1958. 10.1016/S0006-2952(00)00491-3 [DOI] [PubMed] [Google Scholar]
- White E. S., Baralle F. E. and Muro A. F. (2008). New insights into form and function of fibronectin splice variants. J. Pathol. 216, 1-14. 10.1002/path.2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff P.-J., Rifkin D. B., Meister J.-J. and Hinz B. (2007). Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 179, 1311-1323. 10.1083/jcb.200704042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn T. A. and Ramalingam T. R. (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028-1040. 10.1038/nm.2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg L., Todorovic V., Dabovic B., Horiguchi M., Couroussé T., Sakai L. Y. and Rifkin D. B. (2012). Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J. Cell. Physiol. 227, 3828-3836. 10.1002/jcp.24094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollinger A. J. and Smith M. L. (2017). Fibronectin, the extracellular glue. Matrix Biol. 60-61, 27-37. 10.1016/j.matbio.2016.07.011 [DOI] [PubMed] [Google Scholar]