

ABSTRACT
The RAS/MAPK signaling pathway is one of the most investigated pathways, owing to its established role in numerous cellular processes and implication in cancer. Germline mutations in genes encoding members of the RAS/MAPK pathway also cause severe developmental syndromes collectively known as RASopathies. These syndromes share overlapping characteristics, including craniofacial dysmorphology, cardiac malformations, cutaneous abnormalities and developmental delay. Cardio-facio-cutaneous syndrome (CFC) is a rare RASopathy associated with mutations in BRAF, KRAS, MEK1 (MAP2K1) and MEK2 (MAP2K2). MEK1 and MEK2 mutations are found in ∼25% of the CFC patients and the MEK1Y130C substitution is the most common one. However, little is known about the origins and mechanisms responsible for the development of CFC. To our knowledge, no mouse model carrying RASopathy-linked Mek1 or Mek2 gene mutations has been reported. To investigate the molecular and developmental consequences of the Mek1Y130C mutation, we generated a mouse line carrying this mutation. Analysis of mice from a Mek1 allelic series revealed that the Mek1Y130C allele expresses both wild-type and Y130C mutant forms of MEK1. However, despite reduced levels of MEK1 protein and the lower abundance of MEK1 Y130C protein than wild type, Mek1Y130C mutants showed increased ERK (MAPK) protein activation in response to growth factors, supporting a role for MEK1 Y130C in hyperactivation of the RAS/MAPK pathway, leading to CFC. Mek1Y130C mutant mice exhibited pulmonary artery stenosis, cranial dysmorphia and neurological anomalies, including increased numbers of GFAP+ astrocytes and Olig2+ oligodendrocytes in regions of the cerebral cortex. These data indicate that the Mek1Y130C mutation recapitulates major aspects of CFC, providing a new animal model to investigate the physiopathology of this RASopathy.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Cardio-facio-cutaneous syndrome, MEK1 Y130C mutation, Mouse model, Pulmonary artery stenosis, RAS/MAPK pathway, Neurological defects
Summary: A mouse model for cardio-facio-cutaneous syndrome caused by MEK1 Y130C mutant protein reveals the role of hyperactivation of the RAS/MAPK pathway in the development of the syndrome.
INTRODUCTION
The RAS/MAPK signaling pathway is one of the best-characterized signaling systems, owing to its regulation of various cellular processes, including proliferation, differentiation, survival and cell death (Shaul and Seger, 2007; Sun et al., 2015). Somatic deregulation of the RAS/MAPK pathway is among the primary causes of cancer, leading this pathway to be heavily studied in the context of oncogenesis. Phases II and III studies have tested MEK (MAP2K) inhibitors for treatment of several tumor types, including breast, colon, endometrial, melanoma and non-small cell lung cancers (Catalanotti et al., 2013; Coleman et al., 2015; Dhillon, 2016; Flaherty et al., 2012; Haura et al., 2010; Lugowska et al., 2015; Rinehart et al., 2004; Signorelli and Shah Gandhi, 2016).
Germline mutations in genes encoding members of the RAS/MAPK pathway also cause developmental syndromes, such as neurofibromatosis type 1 (NF1), Noonan syndrome (NS), Costello syndrome (CS), cardio-facio-cutaneous syndrome (CFC), LEOPARD syndrome and Legius syndrome, all grouped under the appellation of RASopathies (Aoki et al., 2016; Jindal et al., 2015; Rauen, 2013; Rauen et al., 2015). These syndromes share many characteristics, such as craniofacial dysmorphology, cardiac malformations, cutaneous abnormalities and neurocognitive delay. The first RASopathy to be described was NF1, caused by mutations in the gene coding for neurofibromin1, a RAS-GTPase-activating protein (RAS-GAP). The clinical diagnosis of NF1 is based on the presence of café-au-lait maculae. NF1 patients have variable expressivity but often show anomalies of the central nervous system (Chen et al., 2015). One of the brain abnormalities associated with NF1 is astrogliosis. Astrogliosis and astrocyte activation are common responses to brain injury, and commonly marked by upregulation of proteins such as cytokines, growth factors, transcription factors and the astrocyte intermediate filament protein glial fibrillary acidic protein (GFAP) (Pekny et al., 2014; Rizvi et al., 1999; Sofroniew and Vinters, 2010). During the past decade, additional genes involved in RASopathies have been identified. For example, NS is associated with mutations in A2ML1, CBL, KRAS, LZTR1, MAP3K8, MYST4, NRAS, PTPN11, RAF1, RASA2, RRAS, RIT1, SHOC2, SOS1, SOS2 and SPRY1, whereas mutations in HRAS and SPRED1 have been described for CS and Legius syndrome, respectively (Jindal et al., 2015; Tidyman and Rauen, 2016a,b).
CFC is a rare syndrome with a prevalence of 1/810,000 in Japan (Orphanet; www.orpha.net/consor/cgi-bin/index.php). To date, a few hundred cases have been reported worldwide (Roberts et al., 2006; Seth et al., 2016). CFC patients have multiple congenital abnormalities that overlap with NS and CS, including craniofacial defects, hypertrophic cardiomyopathy, pulmonary artery stenosis, neurological defects and neurocognitive delay (Roberts et al., 2006). Four genes have been associated with CFC: BRAF, KRAS, MEK1 (MAP2K1) and MEK2 (MAP2K2). Mutations in BRAF correspond to ∼75% of the cases, whereas MEK1 and MEK2 mutations are found in ∼25% of patients (Dentici et al., 2009; Niihori et al., 2006; Rodriguez-Viciana et al., 2006). MEK1 and MEK2 are dual-specificity serine/threonine and tyrosine kinases responsible for ERK1 (MAPK3) and ERK2 (MAPK1) activation. Most MEK mutations in CFC individuals are missense, but cases carrying a Mek2 deletion have also been reported (Dentici et al., 2009; Nowaczyk et al., 2014).
Mouse models have been generated that recapitulate genetic defects observed in CFC, but all focused on BRAF mutations. The first CFC mouse model expresses low levels of the oncogenic BRAF V600E protein (B-Raf+/LSLV600E mutants), a constitutively BRAF active form linked to cancer, but not reported in CFC patients (Urosevic et al., 2011). It recapitulated three of the major CFC symptoms: facial dysmorphia, cardiomegaly and epileptic seizures. In parallel, mice carrying a BRAF L597V mutation detected in CFC patients also showed CFC characteristics: short stature, facial dysmorphia and cardiac enlargement (Andreadi et al., 2012). A third mouse model carrying the most prevalent CFC mutation, BRAF Q241R, showed embryonic skeletal abnormalities, lymphatic defects, cardiac defects and liver necrosis (Inoue et al., 2014). Despite these existing models, little is known about when and how CFC phenotypes develop and progress (Rodriguez-Viciana and Rauen, 2008).
No mouse model carrying CFC mutations in the Mek1 or Mek2 genes has been reported. Of MEK1 and MEK2 mutations in humans, the MEK1Y130C mutation is the most common (Nava et al., 2007; Tidyman and Rauen, 2016a,b). To investigate the molecular and developmental effects of this mutation, we targeted the MEK1 Y130C point mutation to the third exon of the endogenous Mek1 gene. Heterozygous Mek1+/Y130C mice were found viable and fertile. While characterizing the Mek1Y130C mutation, we identified an intragenic duplication produced by unequal crossing over. The Mek1Y130C mutant allele produced both endogenous wild-type (wt) MEK1 and MEK1 Y130C proteins. The MEK1 Y130C mutant protein was more active than endogenous MEK1 and produced augmented levels of phosphorylated ERK protein in response to growth factors. Moreover, all Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mice presented cranial, neurological and cardiac phenotypes similar to those observed in CFC individuals. This supports a role for deregulation of the RAS/MAPK pathway in development of CFC. Our study is the first to report a CFC mouse model carrying a Mek1 mutation.
RESULTS
Viability of Mek1Y130C mice
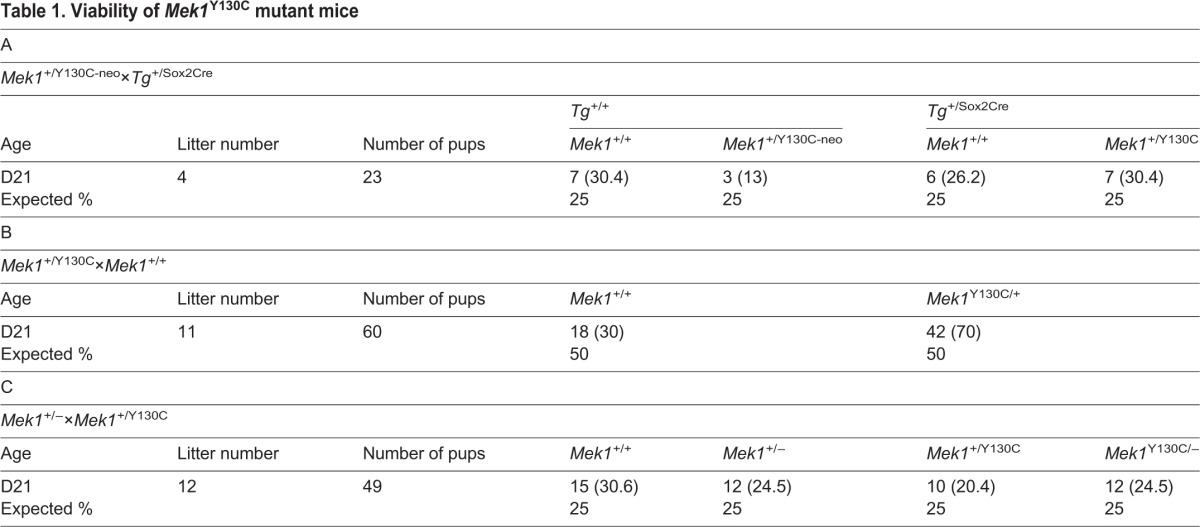
We designed a targeting vector carrying an A to G substitution in the Mek1 third exon to introduce the conditional Y130C mutation into the MEK1 protein (Fig. 1A). The neo selection cassette flanked by loxP sites was inserted in the second intron of Mek1, generating a null allele, owing to the presence of a polyadenylation signal after the neo sequences that interrupts the transcription of the Mek1 gene. Cre-mediated deletion of the neo sequences allowed the production of the Mek1Y130C allele. An XbaI site was inserted into the third intron to facilitate the identification of the targeted allele by Southern blot analysis (Fig. 1A). The XbaI digestion, followed by hybridization with a 3′ probe (probe c; Fig. 1A), generated 8.3 and 6.6 kb fragments for the endogenous and targeted alleles, respectively (Fig. 1B,C). Germline transmission of the Mek1Y130C-neo allele was verified by Southern blot analysis using an EcoRI digestion that distinguished the wt allele (Mek1+; 6.6 kb) and the Mek1Y130C-neo allele (5.6 kb) with a 5′ probe (probe a; Fig. 1B,C). One germline transmitter was obtained and used to establish the Mek1+/Y130C-neo line.
Fig. 1.

Generation of the Mek1Y130C allele. (A) Mek1 gene-targeting strategy for the generation of the Y130C point mutation. Exons are represented as white boxes. The targeting vector contains a PGKneo selection cassette flanked by loxP sites (black triangles), a PGK-DTA selection cassette (gray box), an A to G substitution in exon 3 (asterisk), and a new XbaI site (in bold) inserted into the third intron to facilitate the identification of the targeted allele by Southern blot analysis. Insertion of the targeting vector by homologous recombination will generate the Mek1Y130C-neo-targeted allele. Cre-mediated recombination of the Mek1Y130C-neo allele will generate the Mek1Y130C allele. Location of the 5′, internal and 3′ probes used for Southern analyses is indicated as gray boxes (a,b,c, respectively). (B) Schematic representation of DNA fragments obtained after EcoRI, XbaI and NheI digestions detected by Southern analyses with the 5′, 3′ and internal probes, respectively. (C) Southern blot analyses for ES cell screening (left panel; XbaI digestion with probe c), germline transmission (right panel; EcoRI digestion with probe a) and Cre-mediated deletion (lower panel; NheI digestion with probe b). The position of the different alleles is indicated on the side of the gels. (D) Sequence analysis of amplified exon 3 fragment from Mek1+/+ (left panel) and Mek1+/Y130C (right panel) specimens, confirming the presence of the A to G transition mutation, leading to the substitution of a tyrosine for a cysteine.
To assess the viability of Mek1+/Y130C mutants, Mek1+/Y130C-neo mice were bred to Sox2Cre mice to remove the PGKneopA cassette and generate the Mek1Y130C allele. Southern blot analysis, using NheI digestion and the internal probe b, identified the Mek1Y130C allele, which corresponded to a 2.8 kb fragment, compared to the 16.3 and 4.5 kb fragments associated with the wt and Mek1Y130C-neo alleles, respectively (Fig. 1C). The Mendelian ratio of Mek1+/Y130C mice was obtained at weaning (Table 1A). Moreover, normal transmission of the Mek1Y130C allele was observed when Mek1+/Y130C mice were crossed to Mek1+/+ animals, indicating that Mek1+/Y130C mice were viable and fertile (Table 1B). Genomic DNA from Mek1+/Y130C mice was used to amplify the third exon of Mek1 for sequence analysis. This confirmed that both Mek1+ and Mek1Y130C alleles, the latter with the A to G transition in Mek1 third exon, were present in Mek1+/Y130C mutants (Fig. 1D).
Table 1.
Viability of Mek1Y130C mutant mice
The Mek1Y130C allele contains a partial duplication of the Mek1 gene
To investigate the biochemical properties of the Mek1Y130C allele in vitro, we intercrossed Mek1+/− mice with Mek1+/Y130C mice to generate mouse embryonic fibroblasts (MEFs) from embryonic day (E) 13.5 Mek1Y130C/− embryos to circumvent the absence of a MEK1 Y130C-specific antibody. Mek1+, Mek1Y130C and Mek1− null alleles were resolved by Southern blot analysis as StuI fragments of 2.0, 2.2 and 4.2 kb, respectively (Fig. 2A). Unexpectedly, this breeding produced animals carrying the three Mek1 alleles (Fig. 2B). Moreover, Mek1+/Y130C specimens showed a stronger signal for the Mek1+ allele than for the Mek1Y130C allele. In Mek1+/Y130C mutants, the Mek1+ allele signal was similar to that observed in Mek1+/+ specimens, suggesting that duplication of the Mek1 gene might have happened when generating the Mek1Y130C allele. We thus examined whether duplication occurred on mouse chromosome 9, and whether it encompassed the entire Mek1 gene. First, we performed a whole-genome sequencing analysis of DNA from Mek1Y130C/Y130C MEFs. This revealed a 61.4 kb duplication that initiates 5.6 kb upstream of the Mek1 transcription start site (TSS) of Mek1 gene and finishes in the fifth intron, with equal representation of wt and Y130C sequences (Fig. 2C). A single break junction was detected, located between the fifth intron in the 5′ copy and the upstream sequence of Mek1 locus in the 3′ copy (Fig. 2C; Table S1). These data established that an intragenic duplication occurred due to unequal crossing over.
Fig. 2.

The Mek1Y130C allele contains a partial duplication of the Mek1 gene. (A) Schematic of the endogenous Mek1 gene, the Mek1Y130C allele and the Mek1 null allele with the expected DNA fragments after StuI digestion and detection with probe b. (B) Southern blot analysis of tail DNA from a litter obtained following Mek1+/−×Mek1+/Y130C breeding. DNA was digested with StuI, blotted and hybridized with probe b. Mice positive for the Mek1Y130C allele always carried a wt allele even when a null allele was detected. The wt allele showed a stronger signal equivalent to the one of wt mice, suggesting Mek1 gene duplication in the Mek1Y130C allele. (C) Whole-genome sequencing alignment reveals a 61.5 kb Mek1 duplication starting 6.5 kb upstream of the Mek1 transcription start site and ending in the fifth intron. Only one break point was observed between the fifth intron in 5′ and Mek1 promoter sequences in 3′, suggesting intragenic duplication occurring by unequal crossing over.
The next generation whole-genome sequencing did not distinguish whether the Y130C mutation was in the 5′ or 3′ duplication of the Mek1Y130C allele. To define this, we took advantage of the SbfI site located in the loxP sequence introduced in the second intron of the Y130C copy. If the Y130C mutation is present in the 5′ duplication, the SbfI digestion using a probe located in the fifth intron will generate two fragments of 70 and 26 kb in Southern blot analysis. Conversely, if the mutation is located in the 3′ duplication, two DNA bands of 61 and 35 kb will be produced. The wt allele will generate a 70 kb band (Fig. S1A). As expected, the 70 kb DNA fragment was detected in the wt specimen, whereas fragments of 70 and 26 kb were obtained in the Mek1Y130C/Y130C sample (Fig. S1B). Thus, the Mek1Y130C allele contained a duplication generated by unequal crossing over and the Y130C point mutation was located in the 5′ duplication.
The Mek1Y130C allele produces the MEK1 Y130C protein in vivo
The genomic organization of the duplication should allow the production of an unspliced heteronuclear transcript containing the duplicated exons because the sole polyadenylation signal was present after Mek1 exon 11 (Fig. S1A). By differential splicing, both Mek1+ and Mek1Y130C transcripts could be generated from the Mek1Y130C allele. Alternatively, only the 3′ duplication, which encodes the consecutive 11 Mek1 exons initiated from the interrupted Mek1 promoter, could generate a functional transcript and the MEK1 protein. To determine whether both transcripts were produced, quantitative RT-PCR (qRT-PCR) using oligo (dT) for reverse transcription and primers located in the third and fourth exons was performed to establish Mek1 expression levels in Mek1+/+ and Mek1Y130C/− kidneys, lungs and thymus (Fig. 3A). Cumulative (wt and Y130C) Mek1 transcript levels in Mek1Y130C/− tissues were roughly half those found in Mek1+/+ specimens (Fig. 3B; data not shown). Moreover, western blot analyses showed that MEK1 protein levels in Mek1Y130C/− and Mek1+/− mutants were reduced by half when compared to wt specimens, indicating a correlation between transcript and protein levels (Fig. 3C). qRT-PCR products were sequenced and both wt and mutant transcripts were detected in Mek1Y130C/− specimens (Fig. 3D; data not shown). Overall, wt levels of RNA and protein were produced from the Y130C allele. This suggested that the duplication did not lead to a RNA product that is degraded.
Fig. 3.

MEK1 Y130C expression levels. (A) Schematic representation of Mek1 mRNA with the eleven exons. Blue and red boxes represent the translated region (amino acids 1 to 393) and the kinase domain, respectively. The position of the Y130C point mutation is indicated, as well as that of primers C and D used for qRT-PCR. (B) Mek1 mRNA expression levels were assessed by qRT-PCR analysis on RNA isolated from Mek1+/+ (n=4), Mek1+/− (n=4) and Mek1Y130C/− (n=6) kidneys. (C) MEK1 protein levels were evaluated by western blot analysis of total protein extracts from Mek1+/+ (n=3), Mek1+/− (n=3) and Mek1Y130C/− (n=3) kidneys. Vinculin was used as a loading control. Quantification showed a significant diminution of Mek1 mRNA and MEK1 protein levels in Mek1+/− and Mek1Y130C/− mutants compared to Mek1+/+ specimens. Values are reported as mean±s.e.m. (D) qRT-PCR products obtained from Mek1+/+ and Mek1Y130C/− samples analyzed in B were sequenced. Sequence from Mek1Y130C/− samples showed equal representation of A and G at the mutation site, indicating the presence of transcript encoding the Y130C mutation. (E) Relative wt MEK1 and MEK1 Y130C protein levels were quantified by PRM-targeted mass spectrometry using the CNSPYIVGFYGAFYSDGE wt and CNSPYIVGFCGAFYSDGE Y130C peptides (mean±s.d.; n=2 biological replicates).
To quantify the relative abundance of wt and MEK1 Y130C proteins, MEK1 proteins were immunoprecipitated from Mek1Y130C/− MEF protein extracts and analyzed by parallel reaction monitoring (PRM)-targeted proteomics (Gallien et al., 2012; Peterson et al., 2012). Using isotope-labeled synthetic peptides to discriminate between MEK1 and MEK1 Y130C proteins, the analysis confirmed that both proteins were indeed present in Mek1Y130C/− MEFs (Fig. S2). In addition, the PRM analysis clearly showed that MEK1 Y130C protein was present at lower levels when compared to wt MEK1 protein (Fig. 3E; Table S2). Altogether, these data revealed that transcription of the duplicated Mek1Y130C allele results in transcripts that can generate both MEK1 and MEK1 Y130C proteins.
Increased activation of the RAS/MAPK pathway in MEFs carrying the Mek1Y130C allele
In vitro studies suggested that CFC mutations are gain-of-function mutations that hyperactivate the RAS/MAPK pathway (Rodriguez-Viciana and Rauen, 2008; Rodriguez-Viciana et al., 2006). Conversely, the CFC mouse model with a mutation in Braf (BRAF Q241R) developed severe phenotypes without any major impact on the activation of the RAS/MAPK pathway in whole embryo (Inoue et al., 2014). However, a slight increase in ERK phosphorylation was observed in brain samples, and treatment of these mice with MEK inhibitors partially rescued the phenotype, suggesting that the brain defects were due to hyperactivated RAS/MAPK pathway. Moreover, overexpression of a Mek1Y130C transgene in HEK293T cells indicated that the MEK1 Y130C protein is more active than the wt form (Rodriguez-Viciana et al., 2006). To investigate the biochemical properties of the MEK1 Y130C protein, we looked at its ability to phosphorylate ERK in quiescent cells and in response to stimulation by growth factors and serum. ERK phosphorylation was quantified in wt-, Mek1+/Y130C- and Mek1Y130C/−-established MEFs stimulated with fetal bovine serum (FBS), FGF2 or EGF (Fig. 4A-C). Following serum deprivation, ERK activation in Mek1+/Y130C and Mek1Y130C/− MEFs showed a more elevated trend than in Mek1+/+ cells. In response to FGF2 or EGF, both Mek1+/Y130C and Mek1Y130C/− MEFs presented similar kinetics of ERK activation, with higher levels of ERK phosphorylation than Mek1+/+ cells (P<0.001 and P<0.00001 by ANOVA, respectively). To confirm that the increased phosphorylation activity was due to the MEK1 Y130C protein, we compared ERK phosphorylation in primary MEFs derived from wt, Mek1Y130C/− and Mek1Y130C/Y130C embryos (Fig. 4D). Mek1Y130C/− and Mek1Y130C/Y130C MEFs produced lower levels of MEK1 protein (wt and Y130C) when compared to wt MEFs (Fig. S3). Despite that, in response to EGF, both Mek1Y130C/− and Mek1Y130C/Y130C MEFs showed similar kinetics of ERK activation with higher levels of ERK phosphorylation than Mek1+/+ MEFs (Fig. 4D; P<0.0001 by ANOVA). Thus, these results are in agreement with the notion that the MEK1 Y130C mutation generated a more active form of MEK1.
Fig. 4.

RAS/MAPK pathway hyperactivation in MEFs carrying Mek1Y130C allele. (A-C) Mek1+/+-, Mek1+/Y130C- and Mek1Y130C/−-established MEF lines were stimulated with 20% FBS (in A, Mek1Y130C/− MEFs were not tested), 2 ng/ml of FGF2 (B) or 2 ng/ml of EGF (C), and phosphorylation of ERK1/2 was assessed by quantitative immunoblotting along with total vinculin as a loading control. (D) Three Mek1+/+, Mek1Y130C/− and Mek1Y130C/Y130C primary MEF cultures were treated with 2 ng/ml EGF and phosphorylation of ERK1/2 was assessed. Values are reported as mean±s.e.m. in arbitrary units (n=3).
Heart and cranial defects in Mek1Y130C mutant mice
All Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mice were viable and fertile. To assess whether the MEK1 Y130C mutation reproduced CFC phenotypes, these three lines were compared with Mek1+/+ and Mek1+/− animals. All mouse lines presented comparable length and weight at adult ages. The heart weight/body weight ratio in adult mutants was examined to determine whether they developed hypertrophic cardiomyopathy. No differences were detected. Similarly, no variation in the weight ratio was observed for the liver, kidney and spleen (data not shown). No major anomalies were observed in the gross morphology of embryos at E13.5 for the different Mek1Y130C genotypes (Fig. 5A). Another heart defect frequently found in CFC individuals is pulmonary artery stenosis (Araki et al., 2004; Chen et al., 2010; Moriya et al., 2015). At E13.5, pulmonary artery stenosis was seen in all Mek1Y130C mutants (Fig. 5B). The measurement of the lumen area of the pulmonary artery showed no statistical difference between Mek1+/+ and Mek1+/− specimens (Fig. 5C). However, the left and right pulmonary artery lumen areas were significantly reduced in all genotypic combinations carrying a Mek1Y130C allele when compared to controls, indicating that one Mek1Y130C mutant allele was sufficient to cause pulmonary stenosis. To determine whether this phenotype was associated with the hyperactivation of the RAS/MAPK pathway, ERK phosphorylation was monitored by immunofluorescence staining in pulmonary arteries from E13.5 Mek1+/+, Mek1+/−, Mek1Y130C/− and Mek1Y130C/Y130C embryos. No variation in immunostaining was detected between the different genotypes (Fig. 5D).
Fig. 5.

Mek1Y130C mutant mice present cardiac and cranial anomalies. (A) E13.5 control (Mek1+/+ and Mek+/−) and Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mutant embryos did not present overt morphological anomalies. (B) H&E staining of transverse sections from pulmonary arteries of E13.5 control (Mek1+/+ and Mek1+/−) and Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mutant embryos. Pulmonary stenosis was observed in all specimens carrying a Mek1Y130C allele (arrowhead). e, esophagus; lb, left bronchia; lpa, left pulmonary artery (arrow); rb, right bronchia; rpa, right pulmonary artery. (C) Measurement of arterial lumen area confirmed the pulmonary stenosis for the left and right arteries in Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mutants. (D) Immunostaining for phospho-ERK (pERK) was performed on sections of pulmonary arteries from E13.5 Mek1+/+, Mek1+/− and Mek1Y130C/− embryos. No difference was observed. (E) Morphometric characteristics of mice skulls. Length and width of the skull as well as inner canthal width were measured in cm on Alcian Blue/Alizarin Red-stained skulls. Mek1Y130C/− mice presented facial dysmorphia with reduced skull width and increased inner canthal width. Mek1Y130C/Y130C mutants presented reduced skull length. *P<0.05; **P<0.01; ***P<0.005. Scale bars: 25 µm (A); 100 µm (D); 200 µm (B).
CFC patients present craniofacial dysmorphia. Observation of mutant mice did not reveal obvious phenotypes. Therefore, we examined the skulls of Mek1+/Y130C, Mek1Y130C/− and Mek1Y130C/Y130C mutants at 6 weeks of age and compared them to Mek1+/+ and Mek1+/− specimens. Three parameters were analyzed: the length and width of the skull, and the width of the inner canthal distance (Fig. 5E). Mek1Y130C/Y130C and Mek1Y130C/− mutants showed significant changes in one or several of these parameters. Thus, the Mek1Y130C allele can instigate cranial dysmorphia in mice, suggesting that it is also the cause of this phenotype in CFC patients. However, in contrast to the pulmonary artery phenotypes, which are dominantly inherited with the Y130C allele, craniofacial defects showed a lower expressivity.
The Mek1Y130C/Y130C mutation causes brain defects
Individuals with CFC exhibit a broad range of neurological abnormalities, including variable penetrance of developmental and neurocognitive delays, with ∼40% of the patients suffering from seizure disorder (Rauen, 2013; Yoon et al., 2007). Astrocytes are involved in several pathologies of the central nervous system. In general, astrogliosis, characterized by an increased GFAP expression, is known to occur in a range of neuropathological states (Middeldorp and Hol, 2011). Extensive work modeling RASopathy-associated mutations in mice has revealed numerous cellular defects in distinct neuronal and glial subtypes that might contribute to these neurological phenotypes (Anastasaki and Gutmann, 2014; Brown et al., 2012; Gauthier et al., 2007; Lee et al., 2014; Li et al., 2012; Lush et al., 2008; Paquin et al., 2009; Pucilowska et al., 2012; Xing et al., 2016). A cellular abnormality frequently observed in postmortem tissues from NF1 patients and in NF1 mouse models is an increased number of GFAP+ astrocytes in cortical and hippocampal gray matter (Gutmann et al., 1999; Hegedus et al., 2007; Nordlund et al., 1995; Rizvi et al., 1999; Zhu et al., 2005). The relative number of GFAP+ astrocytes in the sensory cortex and hippocampal CA1 regions was assessed in Mek1Y130C/Y130C adults (Fig. 6A). Consistent with other RASopathy-linked mutations, an increased density of GFAP+ astrocytes was observed in Mek1Y130C/Y130C mutant sensory cortices (Fig. 6B,C-F). A similar increase in GFAP+ astrocyte numbers in hippocampal CA1 was detected (Fig. 6B,G-J). Enhanced myelination and oligodendrocyte progenitor numbers were also reported in RASopathy models (Bennett et al., 2003; Ehrman et al., 2014; Ishii et al., 2013). The total cortical oligodendrocyte population was therefore analyzed in Mek1Y130C/Y130C mutants by immunolabeling for Olig2, a master transcription factor essential in oligodendrocyte fate decisions (Mizuguchi et al., 2001; Novitch et al., 2001; Zhou et al., 2001). Mek1Y130C/Y130C mice exhibited increased density of Olig2+ nuclei in the sensory cortex relative to littermate controls (Fig. 7). ERK activation was also assayed by phospho-ERK (pERK) immunostaining on brain sections from Mek1+/+ and Mek1Y130C/Y130C adults. Signal was detected in the pyramidal neuron layer of the hippocampal CA1 region in both Mek1+/+ and Mek1Y130C/Y130C specimens, and Mek1Y130C/Y130C mutants showed an increased density of pERK-positive cells (Fig. S4). Taken together, our results demonstrated that regulation of astrocyte oligodendrocyte and pyramidal neuron populations could be an important feature in RASopathy neuropathogenesis, and further supported the validity of the Mek1Y130C mutation as a model for CFC.
Fig. 6.

Increased number of GFAP+ cells in cortical and hippocampal sections of Mek1Y130C/Y130C mutants. (A) Representative coronal brain sections stained for GFAP. Red dashed line boxes denote an area of sensory cortex shown in C-F; yellow dashed line boxes denote an area of hippocampal CA1 presented in G-J. (B) Quantification of relative density of GFAP+ cell counts is shown as a relative number of positive cells/mm2 (n=3). (C-F) Mek1Y130C/Y130C animals exhibited an increased number of GFAP+ astrocytes in the sensory cortex in comparison to wt animals. (G-J) Analysis of GFAP-labeled astrocytes in hippocampal CA1 revealed a modest but significant increase in the relative density of cells/mm2. *P<0.05; **P<0.01. Scale bars: 50 µm (G); 100 µm (C).
Fig. 7.

Mek1Y130C/Y130C cortices exhibit increased density of Olig2+ cells. (A) Representative coronal brain sections stained for the oligodendrocyte transcription factor (Olig2). Red boxes denote an area of sensory cortex presented in C-F. (B) Quantification of relative densities of Olig2+ cells in adult sensory cortex (n=3). (C-F) The relative density of Olig2+ cells was assessed in radial columns of sensory cortex. Note the increased density of Olig2+ cells in the mutant sensory cortex (E,F) relative to controls (C,D). **P<0.01. Scale bars: 100 µm.
DISCUSSION
In this study, we described the generation of a hypermorphic Mek1 allele carrying the Y130C mutation found in a significant subset of CFC patients. The Mek1Y130C allele contained a partial duplication of the Mek1 gene, leading to the production from the same allele of both an endogenous MEK1 protein and the MEK1 Y130C protein at lower levels. Despite this, mice heterozygous and homozygous for the Mek1Y130C allele present several characteristics of CFC, including one of the most frequent cardiac defects in CFC; namely, pulmonary stenosis, cranial dysmorphogenesis and neurological abnormalities (Table S3) (Roberts et al., 2006). Characterization of MEFs carrying the Mek1Y130C allele also revealed higher ERK phosphorylation levels in response to growth factors. This increased activation of the RAS/MAPK pathway is similar to that observed in the Raf1+/L613V and Sos1+/E846K mouse models of NS. It supports the notion that CFC, like other RASopathies, results from abnormal hyperactivation of the RAS/MAPK pathway (Chen et al., 2010; Wu et al., 2011).
In contrast to other mouse models of RASopathies, neither heterozygous nor homozygous mice carrying the Mek1Y130C mutation exhibit embryonic or premature lethality. Indeed, it was shown that Braf+/Q241R and B-Raf+/LSLV600E mutant embryos die during gestation or early after birth and present several phenotypes reminiscent of CFC (Inoue et al., 2014; Urosevic et al., 2011). In the case of B-Raf+/LSLV600E mutants, perinatal survival varies depending on the genetic background. Similarly, Raf1+/L613V and Ptpn11+/D61G mice, as well as K-RasV14I/V14I homozygous mutants, which all model Noonan syndrome, display reduced (or no) survival in the C57BL/6J genetic background, whereas they are viable in a mixed background (Araki et al., 2004; Hernández-Porras et al., 2014, 2015; Wu et al., 2011). These data suggested that modifier loci might contribute to the phenotypic variation observed between RASopathy patients carrying the same allele. At least two factors might explain the milder phenotype observed in Mek1Y130C mutant mice. First, the low levels of MEK1 Y130C protein produced by the Mek1Y130C allele could minimize the phenotypic manifestation of CFC. Second, the 129S6 genetic background, in which the mutation was maintained, might contain modifier loci that could mask manifestations of the CFC phenotype.
Hyperactivation of the RAS/MAPK pathway in response to growth factors in cells carrying the Mek1Y130C allele supports a role for this signaling pathway in the development of CFC. MEK1 Y130C expression was evaluated in Mek1Y130C/− MEFs by comparing the levels of wt MEK1 and MEK1 Y130C proteins produced by the Mek1Y130C allele using PRM. An endogenous peptide corresponding to MEK1 Y130C co-eluted with its synthetic counterpart and was clearly detected in MEK1 immuno-precipitates (Fig. S2D). The precise quantification of endogenous MEK1 Y130C levels was complicated by the interference in the quantification of the MEK1 Y130C synthetic peptide when added to total MEK1 immuno-precipitates. Tandem mass spectrometry (MS/MS) analyses confirmed the identification of the MEK1 Y130C peptide in Mek1Y130C mutant, and evidence indicated that MEK1 Y130C protein levels are lower than those of the wt MEK1 protein. Nonetheless, in all genotypes tested, ERK phosphorylation was increased in the presence of low levels of MEK1 Y130C protein, strongly suggesting that the MEK1 Y130C is several times more active than MEK1. This is consistent with previous reported overexpression experiments (Rodriguez-Viciana and Rauen, 2008).
The genomic organization of the Mek1Y130C allele could account for the differences in MEK1 and MEK1 Y130C protein levels. The Y130C point mutation is inserted in the 5′ duplication, implying that production of MEK1 Y130C protein requires transcription of the entire Mek1Y130C locus to produce by alternative splicing the Mek1Y130C full-length transcript (Fig. S1A). The production of the endogenous Mek1 full-length transcript could proceed via the transcription from the truncated Mek1 promoter in the 3′ duplication. qRT-PCR experiments showed that the Mek1Y130C allele was transcribed as efficiently as the Mek1+ allele. Moreover, sequence analysis of the qRT-PCR products revealed an equivalent representation of the wt transcript and the one carrying the Y130C mutation in Mek1Y130C/− specimens. As the sequence encompassing the third and fourth exons was the only one to be analyzed in depth, we cannot rule out the possibility that incorrect alternative splicing of the Mek1 Y130C full-length transcript can contribute to reduced MEK1 Y130C protein levels.
The sequence duplicated in the Mek1Y130C allele includes the Mek1 first intron of 39 kb in which the Uchl4 gene is located (Osawa et al., 2001). Uchl4 is part of a gene family that encodes ubiquitin carboxyl-terminal hydrolases implicated in proteolytic processing of polymeric ubiquitin. This activity is important for cytoplasmic protein degradation and for recycling free ubiquitin via cleavage of ubiquitylated peptides produced by the proteasomal degradation of polyubiquitylated proteins (Larsen et al., 1998). Perturbation of this process may have a major impact on cytoplasmic protein degradation pathway in neurodegenerative disorders, such as Parkinson's disease. For instance, a dominant mutation in Uchl1, which decreases the in vitro hydrolytic activity of UCHL1, has been linked to higher risk of developing Parkinson's disease (Liu et al., 2002). Moreover, inactivation of Uchl3, the closest member of Uchl4, causes learning deficits in mice owing to significantly increased working memory errors (Wood et al., 2005). In both cases, the phenotype is due to a loss of function. The duplication of the Uchl4 gene in the Mek1Y130C allele generates a gain of function (Fig. S5). However, gain in ubiquitin carboxyl-terminal hydrolase activity might also be detrimental for protein homeostasis. More experiments are required to determine whether Uchl4 gene duplication can contribute to the phenotypes observed in Mek1Y130C mice.
Hyperactivation of the RAS/MAPK pathway by the MEK1 Y130C protein also supports the notion that the mutation contributes to the neurological anomalies observed in Mek1Y130C/Y130C mutants. We have previously shown that the RAS/MAPK pathway plays a key role in gliogenesis (Li et al., 2012). Deletion of Mek1 and Mek2 genes in radial progenitors prevents gliogenesis, affecting astrocyte and oligodendrocyte differentiation, whereas the expression of a dominant active form of MEK1 in the same lineage leads to increased numbers of astrocytes with coincident reduction in neuron numbers. Reduced neurogenesis is consistent with the idea that hyperactive MEK accelerates radial progenitor progression into a gliogenic mode and prematurely terminates neurogenesis. Data from the Mek1Y130C/Y130C mutants might reflect developmental defects, but they could also indicate astrogliosis in response to neurological lesion, as revealed by the increase in GFAP-positive cells in the cortex and hippocampus. Astrogliosis with accumulation of GFAP-positive cells is a defensive reaction aimed at limiting tissue damage (Stringer, 1996). Patients with CFC present with mild to severe cognitive delay, developmental delay and seizure disorder (Yoon et al., 2007). It will be interesting to test whether Mek1Y130C mutants exhibit defects in learning ability and memory.
The Mek1Y130C mutant mouse reported here is the first mouse model of a CFC-linked Mek1 mutation. It provides a powerful tool to gain further understanding of the physiopathology of at least some CFC clinical features, such as pulmonary stenosis, facial defects and astrogliosis, and to investigate the associated brain defects that are more difficult to address in other CFC mouse models due to the embryonic or neonatal lethality observed.
MATERIALS AND METHODS
Generation of the Mek1Y130C allele
The targeting vector was made using a 5.5 kb genomic fragment that encompasses exons 2 and 3 of the Mek1 gene, containing 2.2 kb of 5′- and 3.3 kb of 3′-sequence homology for homologous recombination to occur. The PGK-neo selection cassette flanked by loxP sites was inserted in the BamHI site located in Mek1 intron 2, while the PGK-DTA negative selection cassette was located in intron 3 at the 3′-end of Mek1 homology. An A to G transition was inserted in exon 3 to generate the MEK1 Y130C mutation (Fig. 1A). Correctly targeted ES clones were injected into MF1 blastocysts to generate chimeras as described (Bélanger et al., 2003). Chimeras were bred with 129S6 mice for transmission of the targeted Mek1Y130C-neo allele and all subsequent breedings were done in this genetic background. Mek1+/Y130C-neo heterozygous mice were bred with Sox2Cre deleter mice to generate the Mek1Y130C allele (Hayashi et al., 2002). The Mek1+/− mouse line was described previously (Bissonauth et al., 2006).
Mice, genotype and tissue collection
The age of the embryos was estimated by considering the morning of the day of the vaginal plug as E0.5. Control and mutant embryos were collected at E13.5. Adult skeletons and organs were collected at 6 weeks of age. For RNA and protein extraction, organs were snap frozen in liquid N2. Genomic DNA from embryonic stem (ES) cells and mouse tail biopsies was extracted, purified and genotyped by Southern analysis using restriction digestions described in the text and Mek1 genomic probes (Fig. 1B). All experiments were performed according to the guidelines of the Canadian Council on Animal Care and approved by the institutional animal care committee.
Pulse field gel electrophoresis
MEF DNA was prepared as described (Aoidi et al., 2016). Purified DNA was digested with the NheI and SbfI restriction endonucleases, fractionated by pulse field gel electrophoresis through 1% agarose gel, which was conducted in 0.5× TBE buffer (45 mM Tris-base, 1 mM EDTA, 45 mM boric acid) with a CHEF DR-II apparatus (Bio-Rad, Hercules, CA), with an initial switch time at 1 s and final switch time at 10 s for 20 h at 14°C and a voltage of 10 V/cm (Herschleb et al., 2007; Jo et al., 2013). The gel was blotted onto N-Hybond membrane (GE Healthcare Life Sciences, Mississauga, ON, Canada), and hybridized to probe d described in Fig. S1A by following procedures recommended by the supplier.
RNA isolation and qRT-PCR
Organs were collected from wt, Mek1Y130C/−, Mek1Y130C/Y130C and Mek1−/− mice as described (Nadeau and Charron, 2014). Total RNA was isolated using TRIzol reagent according to the manufacturer's procedure (Life Technologies Inc., Burlington, ON). cDNA was synthesized with the Superscript II Reverse Transcriptase (Life Technologies) with 1 µg total RNA and oligo (dT). qRT-PCR experiments were performed as described using Rpl19 gene as a control (Boucherat et al., 2012). Mek1 primer sequences were: Forward 5′-CTGATCCACCTGGAGATCAAACC-3′ and Reverse 5′-CTCCCGAAGATAGGTCAGGC-3′.
Western blot analysis
Protein extracts were prepared as described (Bélanger et al., 2003). Total protein lysates (20 µg) were resolved on a denaturing 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and probed with rabbit monoclonal antibody against MEK1 (Clone E3442; Epitomics) and mouse monoclonal antibody against vinculin (clone hVIN-1; Sigma-Aldrich) used at 1/2000 and 1/5000, respectively (Nadeau et al., 2009). The most representative western blots are presented. The relative amount of MEK1 was obtained by densitometry analyses with the Fluor-S MAX MultiImager-captured images using ImageJ.
Mass spectrometry
For affinity-purification of MEK1 proteins, whole cell extracts were obtained by collecting cells from 7×150 mm Petri dishes having reached 90% confluence. Cells were scraped in ice-cold lysis buffer containing freshly added protease inhibitors (20 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA pH 8, 1% NP-40, 0.5% sodium deoxycholate, 10 mM ß-glycerophosphate, 10 mM sodium pyrophosphate, 50 mM NaF, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin) (1 ml per 150 mm Petri) and incubated for 30 min on ice (Beigbeder et al., 2016). Samples were centrifuged for 20 min at 20,000 g and supernatants (∼20 mg of protein) were pre-cleared by incubation with 75 µl protein G sepharose beads for 90 min at 4°C. After centrifugation, supernatants were incubated with 2 µg rabbit monoclonal MEK1 antibody with rotation for 1 h at 4°C followed by another incubation for 1 h at 4°C with 75 µl protein G sepharose beads. Affinity-purified MEK1 protein was washed three times in lysis buffer and twice in ‘light’ buffer (20 mM Tris pH 7.4, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml pepstatin) and processed for digestion as described (Beigbeder et al., 2016), except for the digestion that was performed with 5 µg endopeptidase GluC for 2 h at 37°C, followed by a 2-h incubation at room temperature with 5 μg trypsin. Digested peptides were concentrated by evaporation and resuspended in 2% acetonitrile, 0.05% TFA. PEPotecTM crude peptides for MEK1 WT (CNSPYIVGFYGAFYSDGE) and Y130C (CNSPYIVGFCGAFYSDGE) were synthesized with 13C(5),15N(1) isotopes on glutamic acid and carbamidomethyl cysteins (ThermoFisher Scientific). These isotope-labeled ‘heavy’ peptides were diluted 500× into peptide samples resulting from GluC/trypsin digestion. One third of each sample was analyzed by liquid chromatography-MS/MS on an Orbitrap Fusion mass spectrometer equipped with a nanoelectrospray ion source (ThermoFisher Scientific) and coupled to an UltiMate 3000 nanoRSLC chromatography system (Dionex). The system was operated in PRM mode. Briefly, peptides were trapped at 20 μl/min in 2% acetonitrile, 0.05% TFA on a 5 mm×300 μm C18 PepMap cartridge (Dionex). The pre-column was switch online upstream of a 50 cm, 75 μm Acclaim PepMap100 C18 column (Dionex), and peptides were eluted with a linear gradient of 5-40% solvent B (80% acetonitrile, 0.1% formic acid) over 30 min at 300 nl/min. MS2 spectra corresponding to the ‘heavy’ (synthetic) and ‘light’ (endogenous) targeted peptides were acquired during the whole gradient length using the XCalibur software version 3.0.63 (ThermoFisher Scientific). To achieve this, 1023.43(2+), 1026.43(2+), 1021.91(2+) and 1024.92(2+) m/z were successively isolated into the quadrupole analyzer in a window of 0.7 Da and fragmented by higher energy collision-induced dissociation (HCD) at 35% collision energy. The resulting fragments, in range of 120-2000 m/z, were detected by the orbitrap analyzer at a resolution of 30,000, with an automatic gain control target of 1e5 and a maximum injection time of 120 ms. Prior to each sample analysis, a blank run was performed using the same method (Fig. S2C,D).
The 10 most intense b or y fragment ions derivating from each parent mass were extracted from the raw files with Skyline software v3.6 (PMID 20147306) to allow the reconstruction of elution peaks. The superposition of fragments traces and the co-elution of ‘heavy’ and ‘light’ peptides were both utilized to confirm the detection of targeted peptides. Peptide quantification was performed using the area under the elution curve, as calculated via Skyline. For each peptide, the areas of the three most intense ions of the targeted endogenous peptide were summed and normalized relative to the area of the ‘heavy’ synthetic peptide (Table S2).
MEF isolation and pERK induction
MEFs were obtained from E13.5 embryos, cultured and immortalized as described (Giroux et al., 1999). Cells were starved in medium containing 0.1% FBS overnight before treatment with 20% FBS, EGF or FGF2 at 2 ng/ml for 0, 3, 5, 10, 30 and 60 min. Protein extracts were prepared as described (Bélanger et al., 2003). Total protein lysates (20 µg) were resolved on a denaturing 10% SDS-PAGE and probed with rabbit monoclonal antibody against phospho-ERK1/2 (1/5000; Cell Signaling Technology), and with mouse monoclonal antibody against vinculin (1/5000; Sigma-Aldrich) as a loading control. The relative amount of phospho-ERK1/2 was obtained by densitometry analyses with the Fluor-S MAX MultiImager-captured images using ImageJ software. Experiments were performed in triplicate.
Pulmonary stenosis histological analysis
Paraffin-embedded E13.5 embryos were sectioned at 4 μm. Morphology was analyzed following Hematoxylin and Eosin (H&E) staining. Pulmonary artery lumen surface area was measured every 8 µm along the left and right pulmonary arteries (∼20 measures per specimen).
Skeletal analysis
Whole-mount skeletons were stained with Alcian Blue for cartilage and Alizarin Red for bone. Mice were first skinned, eviscerated and fixed overnight at 4°C in 95% EtOH. The next day, specimens were put into Alcian Blue 0.015%; 20% acetic acid prepared in 95% EtOH for 7 days at 37°C. Skeletons were then rinsed for 1 h in 95% EtOH, clarified overnight in 2% KOH and stained overnight in 0.003% Alizarin Red prepared in 1% KOH. Specimens were transferred in 1% KOH:glycerol (1:1) for 7 days, and kept at room temperature in glycerol:EtOH (1:1) until analysis.
Brain immunostaining
Mice were anesthetized, transcardially perfused with 4% paraformaldehyde and postfixed. Brain tissue was extracted and vibratome sectioned for immunolabeling. Brain sections were incubated for 2 days with primary antibody: rabbit anti-GFAP (1:1000; Abcam ab7260), rabbit anti-Olig2 (1:1000; Millipore ab9610), mouse anti-NeuN (1:1000; Millipore MAB377) in 1× PBS 0.2% Triton with 5% normal donkey serum. Tissue was then washed 3× for 20 min in 1× PBS 0.2% Triton and incubated for 2 days in Alexa Fluor dye-conjugated secondary antibodies and DAPI. Confocal images of immunolabeled brain slices were acquired on a Zeiss LSM 800 confocal microscope. Three anatomically matched regions of primary sensory cortex and hippocampal CA1 from three independent sections were imaged per animal. Cortical and hippocampal regions of 1-2 mm2 were manually defined and assessed for the relative density of GFAP+ and Olig2+ cells. At least three mutants and three littermate controls were analyzed.
Statistical analyses
Samples were statistically compared using the Student's t-test and analysis of variance on linear models (ANOVA), when appropriate. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
We thank Drs L. Jeannotte and J. Mansfield for critical comments, A. McMahon for Sox2Cre mice, and Florence Roux-Dalvai for technical assistance with mass spectrometry. Mass spectrometry was performed at the CHU de Québec – Université Laval Proteomics Platform.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.A., B.D.Y., K.A.R., N.B., J.N., J.C.; Methodology: R.A., B.D.Y., J.C.; Validation: R.A.; Formal analysis: R.A., J.C.; Investigation: R.A., N.H., K.L.-T., M.H., K.J., S.R.K., J.C.; Resources: L.C.; Writing - original draft: R.A., M.H., K.J., N.B., J.N., J.C.; Writing - review & editing: R.A., M.H., K.J., K.A.R., N.B., J.N., J.C.; Supervision: J.C.; Funding acquisition: B.D.Y., K.A.R., N.B., J.N., J.C.
Funding
This work was supported by the Canadian Institutes of Health Research (MOP-97801 to J.C.; MOP-130335 to N.B.) and the National Institutes of Health (R01-NS097537 to J.N.; R01-AR062165 to K.A.R.).
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.031278.supplemental
References
- Anastasaki C. and Gutmann D. H. (2014). Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 23, 6712-6721. 10.1093/hmg/ddu389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadi C., Cheung L.-K., Giblett S., Patel B., Jin H., Mercer K., Kamata T., Lee P., Williams A., McMahon M. et al. (2012). The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 26, 1945-1958. 10.1101/gad.193458.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoidi R., Maltais A. and Charron J. (2016). Functional redundancy of the kinases MEK1 and MEK2: rescue of the Mek1 mutant phenotype by Mek2 knock-in reveals a protein threshold effect. Sci. Signal. 9, ra9 10.1126/scisignal.aad5658 [DOI] [PubMed] [Google Scholar]
- Aoki Y., Niihori T., Inoue S.-I. and Matsubara Y. (2016). Recent advances in RASopathies. J. Hum. Genet. 61, 33-39. 10.1038/jhg.2015.114 [DOI] [PubMed] [Google Scholar]
- Araki T., Mohi M. G., Ismat F. A., Bronson R. T., Williams I. R., Kutok J. L., Yang W., Pao L. I., Gilliland D. G., Epstein J. A. et al. (2004). Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10, 849-857. 10.1038/nm1084 [DOI] [PubMed] [Google Scholar]
- Beigbeder A., Velot L., James D. A. and Bisson N. (2016). Sample preparation for mass spectrometry analysis of protein-protein interactions in cancer cell lines and tissues. Methods Mol. Biol. 1458, 339-347. 10.1007/978-1-4939-3801-8_23 [DOI] [PubMed] [Google Scholar]
- Bélanger L.-F., Roy S., Tremblay M., Brott B., Steff A.-M., Mourad W., Hugo P., Erikson R. and Charron J. (2003). Mek2 is dispensable for mouse growth and development. Mol. Cell. Biol. 23, 4778-4787. 10.1128/MCB.23.14.4778-4787.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M. R., Rizvi T. A., Karyala S., McKinnon R. D. and Ratner N. (2003). Aberrant growth and differentiation of oligodendrocyte progenitors in neurofibromatosis type 1 mutants. J. Neurosci. 23, 7207-7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonauth V., Roy S., Gravel M., Guillemette S. and Charron J. (2006). Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis. Development 133, 3429-3440. 10.1242/dev.02526 [DOI] [PubMed] [Google Scholar]
- Boucherat O., Chakir J. and Jeannotte L. (2012). The loss of Hoxa5 function promotes Notch-dependent goblet cell metaplasia in lung airways. Biol. Open 1, 677-691. 10.1242/bio.20121701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. A., Diggs-Andrews K. A., Gianino S. M. and Gutmann D. H. (2012). Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell. Neurosci. 49, 13-22. 10.1016/j.mcn.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalanotti F., Solit D. B., Pulitzer M. P., Berger M. F., Scott S. N., Iyriboz T., Lacouture M. E., Panageas K. S., Wolchok J. D., Carvajal R. D. et al. (2013). Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin. Cancer Res. 19, 2257-2264. 10.1158/1078-0432.CCR-12-3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-C., Wakimoto H., Conner D., Araki T., Yuan T., Roberts A., Seidman C. E., Bronson R., Neel B. G., Seidman J. G. et al. (2010). Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J. Clin. Invest. 120, 4353-4365. 10.1172/JCI43910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-H., Gianino S. M. and Gutmann D. H. (2015). Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev. 29, 1677-1682. 10.1101/gad.261677.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R. L., Sill M. W., Thaker P. H., Bender D. P., Street D., McGuire W. P., Johnston C. M. and Rotmensch J. (2015). A phase II evaluation of selumetinib (AZD6244, ARRY-142886), a selective MEK-1/2 inhibitor in the treatment of recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 138, 30-35. 10.1016/j.ygyno.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentici M. L., Sarkozy A., Pantaleoni F., Carta C., Lepri F., Ferese R., Cordeddu V., Martinelli S., Briuglia S., Digilio M. C. et al. (2009). Spectrum of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and genotype-phenotype correlations. Eur. J. Hum. Genet. 17, 733-740. 10.1038/ejhg.2008.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. (2016). Dabrafenib plus Trametinib: a review in advanced melanoma with a BRAF (V600) mutation. Target Oncol. 11, 417-428. 10.1007/s11523-016-0443-8 [DOI] [PubMed] [Google Scholar]
- Ehrman L. A., Nardini D., Ehrman S., Rizvi T. A., Gulick J., Krenz M., Dasgupta B., Robbins J., Ratner N., Nakafuku M. et al. (2014). The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse telencephalon. J. Neurosci. 34, 3767-3778. 10.1523/JNEUROSCI.3515-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty K. T., Infante J. R., Daud A., Gonzalez R., Kefford R. F., Sosman J., Hamid O., Schuchter L., Cebon J., Ibrahim N. et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694-1703. 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S., Duriez E., Crone C., Kellmann M., Moehring T. and Domon B. (2012). Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709-1723. 10.1074/mcp.O112.019802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier A. S., Furstoss O., Araki T., Chan R., Neel B. G., Kaplan D. R. and Miller F. D. (2007). Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron 54, 245-262. 10.1016/j.neuron.2007.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux S., Tremblay M., Bernard D., Cadrin-Girard J.-F., Aubry S., Larouche L., Rousseau S., Huot J., Landry J., Jeannotte L. et al. (1999). Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 9, 369-376. 10.1016/S0960-9822(99)80164-X [DOI] [PubMed] [Google Scholar]
- Gutmann D. H., Loehr A., Zhang Y., Kim J., Henkemeyer M. and Cashen A. (1999). Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene 18, 4450-4459. 10.1038/sj.onc.1202829 [DOI] [PubMed] [Google Scholar]
- Haura E. B., Ricart A. D., Larson T. G., Stella P. J., Bazhenova L., Miller V. A., Cohen R. B., Eisenberg P. D., Selaru P., Wilner K. D. et al. (2010). A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 16, 2450-2457. 10.1158/1078-0432.CCR-09-1920 [DOI] [PubMed] [Google Scholar]
- Hayashi S., Lewis P., Pevny L. and McMahon A. P. (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr. Patterns 2, 93-97. 10.1016/S0925-4773(02)00292-7 [DOI] [PubMed] [Google Scholar]
- Hegedus B., Dasgupta B., Shin J. E., Emnett R. J., Hart-Mahon E. K., Elghazi L., Bernal-Mizrachi E. and Gutmann D. H. (2007). Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 1, 443-457. 10.1016/j.stem.2007.07.008 [DOI] [PubMed] [Google Scholar]
- Hernández-Porras I., Fabbiano S., Schuhmacher A. J., Aicher A., Cañamero M., Cámara J. A., Cussó L., Desco M., Heeschen C., Mulero F. et al. (2014). K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. USA 111, 16395-16400. 10.1073/pnas.1418126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Porras I., Jiménez-Catalán B., Schuhmacher A. J. and Guerra C. (2015). The impact of the genetic background in the Noonan syndrome phenotype induced by K-Ras(V14I). Rare Dis. 3, e1045169 10.1080/21675511.2015.1045169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschleb J., Ananiev G. and Schwartz D. C. (2007). Pulsed-field gel electrophoresis. Nat. Protoc. 2, 677-684. 10.1038/nprot.2007.94 [DOI] [PubMed] [Google Scholar]
- Inoue S.-I., Moriya M., Watanabe Y., Miyagawa-Tomita S., Niihori T., Oba D., Ono M., Kure S., Ogura T., Matsubara Y. et al. (2014). New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum. Mol. Genet. 23, 6553-6566. 10.1093/hmg/ddu376 [DOI] [PubMed] [Google Scholar]
- Ishii A., Furusho M. and Bansal R. (2013). Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J. Neurosci. 33, 175-186. 10.1523/JNEUROSCI.4403-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal G. A., Goyal Y., Burdine R. D., Rauen K. A. and Shvartsman S. Y. (2015). RASopathies: unraveling mechanisms with animal models. Dis. Model. Mech. 8, 769-782. 10.1242/dmm.020339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y. H., Patnaik B. B., Kang S. W., Chae S.-H., Oh S., Kim D. H., Noh M. Y., Seo G. W., Jeong H. C., Noh J. Y. et al. (2013). Analysis of the genome of a Korean isolate of the Pieris rapae granulovirus enabled by its separation from total host genomic DNA by pulse-field electrophoresis. PLoS ONE 8, e84183 10.1371/journal.pone.0084183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. N., Krantz B. A. and Wilkinson K. D. (1998). Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry 37, 3358-3368. 10.1021/bi972274d [DOI] [PubMed] [Google Scholar]
- Lee Y.-S., Ehninger D., Zhou M., Oh J.-Y., Kang M., Kwak C., Ryu H.-H., Butz D., Araki T., Cai Y. et al. (2014). Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci. 17, 1736-1743. 10.1038/nn.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Newbern J. M., Wu Y., Morgan-Smith M., Zhong J., Charron J. and Snider W. D. (2012). MEK is a key regulator of gliogenesis in the developing brain. Neuron 75, 1035-1050. 10.1016/j.neuron.2012.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Fallon L., Lashuel H. A., Liu Z. and Lansbury P. T. Jr (2002). The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111, 209-218. 10.1016/S0092-8674(02)01012-7 [DOI] [PubMed] [Google Scholar]
- Lugowska I., Kosela-Paterczyk H., Kozak K. and Rutkowski P. (2015). Trametinib: a MEK inhibitor for management of metastatic melanoma. Onco. Targets Ther. 8, 2251-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lush M. E., Li Y., Kwon C.-H., Chen J. and Parada L. F. (2008). Neurofibromin is required for barrel formation in the mouse somatosensory cortex. J. Neurosci. 28, 1580-1587. 10.1523/JNEUROSCI.5236-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J. and Hol E. M. (2011). GFAP in health and disease. Prog. Neurobiol. 93, 421-443. 10.1016/j.pneurobio.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Mizuguchi R., Sugimori M., Takebayashi H., Kosako H., Nagao M., Yoshida S., Nabeshima Y.-I., Shimamura K. and Nakafuku M. (2001). Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan-neuronal and subtype-specific properties of motoneurons. Neuron 31, 757-771. 10.1016/S0896-6273(01)00413-5 [DOI] [PubMed] [Google Scholar]
- Moriya M., Inoue S. I., Miyagawa-Tomita S., Nakashima Y., Oba D., Niihori T., Hashi M., Ohnishi H., Kure S., Matsubara Y. et al. (2015). Adult mice expressing a Braf Q241R mutation on an ICR/CD-1 background exhibit a cardio-facio-cutaneous syndrome phenotype. Hum. Mol. Genet. 10.1093/hmg/ddv435 [DOI] [PubMed] [Google Scholar]
- Nadeau V. and Charron J. (2014). Essential role of the ERK/MAPK pathway in blood-placental barrier formation. Development 141, 2825-2837. 10.1242/dev.107409 [DOI] [PubMed] [Google Scholar]
- Nadeau V., Guillemette S., Bélanger L.-F., Jacob O., Roy S. and Charron J. (2009). Map2k1 and Map2k2 genes contribute to the normal development of syncytiotrophoblasts during placentation. Development 136, 1363-1374. 10.1242/dev.031872 [DOI] [PubMed] [Google Scholar]
- Nava C., Hanna N., Michot C., Pereira S., Pouvreau N., Niihori T., Aoki Y., Matsubara Y., Arveiler B., Lacombe D. et al. (2007). Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype phenotype relationships and overlap with Costello syndrome. J. Med. Genet. 44, 763-771. 10.1136/jmg.2007.050450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niihori T., Aoki Y., Narumi Y., Neri G., Cavé H., Verloes A., Okamoto N., Hennekam R. C. M., Gillessen-Kaesbach G., Wieczorek D. et al. (2006). Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38, 294-296. 10.1038/ng1749 [DOI] [PubMed] [Google Scholar]
- Nordlund M. L., Rizvi T. A., Brannan C. I. and Ratner N. (1995). Neurofibromin expression and astrogliosis in neurofibromatosis (type 1) brains. J. Neuropathol. Exp. Neurol. 54, 588-600. 10.1097/00005072-199507000-00013 [DOI] [PubMed] [Google Scholar]
- Novitch B. G., Chen A. I. and Jessell T. M. (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773-789. 10.1016/S0896-6273(01)00407-X [DOI] [PubMed] [Google Scholar]
- Nowaczyk M. J. M., Thompson B. A., Zeesman S., Moog U., Sanchez-Lara P. A., Magoulas P. L., Falk R. E., Hoover-Fong J. E., Batista D. A. S., Amudhavalli S. M. et al. (2014). Deletion of MAP2K2/MEK2: a novel mechanism for a RASopathy? Clin. Genet. 85, 138-146. 10.1111/cge.12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa Y., Wang Y.-L., Osaka H., Aoki S. and Wada K. (2001). Cloning, expression, and mapping of a mouse gene, Uchl4, highly homologous to human and mouse Uchl3. Biochem. Biophys. Res. Commun. 283, 627-633. 10.1006/bbrc.2001.4841 [DOI] [PubMed] [Google Scholar]
- Paquin A., Hordo C., Kaplan D. R. and Miller F. D. (2009). Costello syndrome H-Ras alleles regulate cortical development. Dev. Biol. 330, 440-451. 10.1016/j.ydbio.2009.04.010 [DOI] [PubMed] [Google Scholar]
- Pekny M., Wilhelmsson U. and Pekna M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30-38. 10.1016/j.neulet.2013.12.071 [DOI] [PubMed] [Google Scholar]
- Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S. and Coon J. J. (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475-1488. 10.1074/mcp.O112.020131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J., Puzerey P. A., Karlo J. C., Galan R. F. and Landreth G. E. (2012). Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J. Neurosci. 32, 8663-8677. 10.1523/JNEUROSCI.1107-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen K. A. (2013). The RASopathies. Annu. Rev. Genomics Hum. Genet. 14, 355-369. 10.1146/annurev-genom-091212-153523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen K. A., Huson S. M., Burkitt-Wright E., Evans D. G., Farschtschi S., Ferner R. E., Gutmann D. H., Hanemann C. O., Kerr B., Legius E. et al. (2015). Recent developments in neurofibromatoses and RASopathies: management, diagnosis and current and future therapeutic avenues. Am. J. Med. Genet. A 167, 1-10. 10.1002/ajmg.a.36793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J., Adjei A. A., Lorusso P. M., Waterhouse D., Hecht J. R., Natale R. B., Hamid O., Varterasian M., Asbury P., Kaldjian E. P. et al. (2004). Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456-4462. 10.1200/JCO.2004.01.185 [DOI] [PubMed] [Google Scholar]
- Rizvi T. A., Akunuru S., de Courten-Myers G., Switzer R. C. III, Nordlund M. L. and Ratner N. (1999). Region-specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res. 816, 111-123. 10.1016/S0006-8993(98)01133-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A., Allanson J., Jadico S. K., Kavamura M. I., Noonan J., Opitz J. M., Young T. and Neri G. (2006). The cardiofaciocutaneous syndrome. J. Med. Genet. 43, 833-842. 10.1136/jmg.2006.042796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P. and Rauen K. A. (2008). Biochemical characterization of novel germline BRAF and MEK mutations in cardio-facio-cutaneous syndrome. Methods Enzymol. 438, 277-289. 10.1016/S0076-6879(07)38019-1 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Tetsu O., Tidyman W. E., Estep A. L., Conger B. A., Cruz M. S., McCormick F. and Rauen K. A. (2006). Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311, 1287-1290. 10.1126/science.1124642 [DOI] [PubMed] [Google Scholar]
- Seth S., Biswas T., Biswas B., Roy A. and Datta A. K. (2016). Cardiofaciocutaneous syndrome: case report of a rare disorder. J. Clin. Diagn. Res. 10, SD01-SD02. 10.1111/crj.12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul Y. D. and Seger R. (2007). The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta 1773, 1213-1226. 10.1016/j.bbamcr.2006.10.005 [DOI] [PubMed] [Google Scholar]
- Signorelli J. and Shah Gandhi A. (2016). Cobimetinib: a novel MEK inhibitor for metastatic melanoma. Ann. Pharmacother. 51, 146-153. 10.1177/1060028016672037 [DOI] [PubMed] [Google Scholar]
- Sofroniew M. V. and Vinters H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7-35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer J. L. (1996). Repeated seizures increase GFAP and vimentin in the hippocampus. Brain Res. 717, 147-153. 10.1016/0006-8993(96)00059-5 [DOI] [PubMed] [Google Scholar]
- Sun Y., Liu W.-Z., Liu T., Feng X., Yang N. and Zhou H.-F. (2015). Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 35, 600-604. 10.3109/10799893.2015.1030412 [DOI] [PubMed] [Google Scholar]
- Tidyman W. E. and Rauen K. A. (2016a). Expansion of the RASopathies. Curr. Genet. Med. Rep. 4, 57-64. 10.1007/s40142-016-0100-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman W. E. and Rauen K. A. (2016b). Pathogenetics of the RASopathies. Hum. Mol. Genet. 25, R123-R132. 10.1093/hmg/ddw191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic J., Sauzeau V., Soto-Montenegro M. L., Reig S., Desco M., Wright E. M. B., Canamero M., Mulero F., Ortega S., Bustelo X. R. et al. (2011). Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc. Natl. Acad. Sci. USA 108, 5015-5020. 10.1073/pnas.1016933108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. A., Kaplan M. P., Brensinger C. M., Guo W. and Abel T. (2005). Ubiquitin C-terminal hydrolase L3 (Uchl3) is involved in working memory. Hippocampus 15, 610-621. 10.1002/hipo.20082 [DOI] [PubMed] [Google Scholar]
- Wu X., Simpson J., Hong J. H., Kim K.-H., Thavarajah N. K., Backx P. H., Neel B. G. and Araki T. (2011). MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 121, 1009-1025. 10.1172/JCI44929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L., Larsen R. S., Bjorklund G. R., Li X., Wu Y., Philpot B. D., Snider W. D. and Newbern J. M. (2016). Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. eLife 5, e11123 10.7554/eLife.11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon G., Rosenberg J., Blaser S. and Rauen K. A. (2007). Neurological complications of cardio-facio-cutaneous syndrome. Dev. Med. Child Neurol. 49, 894-899. 10.1111/j.1469-8749.2007.00894.x [DOI] [PubMed] [Google Scholar]
- Zhou Q., Choi G. and Anderson D. J. (2001). The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 31, 791-807. 10.1016/S0896-6273(01)00414-7 [DOI] [PubMed] [Google Scholar]
- Zhu Y., Harada T., Liu L., Lush M. E., Guignard F., Harada C., Burns D. K., Bajenaru M. L., Gutmann D. H. and Parada L. F. (2005). Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development 132, 5577-5588. 10.1242/dev.02162 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.