

Abstract
Cholesterol is a major lipid component of the mammalian plasma membrane. While much is known about its metabolism, its transport, and its role in atherosclerotic vascular disease, less is known about its role in neuronal pathophysiology. This study reveals an unexpected function of cholesterol in controlling pain transmission. We show that inflammation lowers cholesterol content in skin tissue and sensory DRG culture. Pharmacological depletion of cellular cholesterol entails sensitization of nociceptive neurons and promotes mechanical and thermal hyperalgesia through the activation of voltage‐gated Nav1.9 channels. Inflammatory mediators enhance the production of reactive oxygen species and induce partitioning of Nav1.9 channels from cholesterol‐rich lipid rafts to cholesterol‐poor non‐raft regions of the membrane. Low‐cholesterol environment enhances voltage‐dependent activation of Nav1.9 channels leading to enhanced neuronal excitability, whereas cholesterol replenishment reversed these effects. Consistently, we show that transcutaneous delivery of cholesterol alleviates hypersensitivity in animal models of acute and chronic inflammatory pain. In conclusion, our data establish that membrane cholesterol is a modulator of pain transmission and shed a new light on the relationship between cholesterol homeostasis, inflammation, and pain.
Keywords: analgesic, cholesterol, inflammation, Nav1.9 sodium channel, pain
Subject Categories: Immunology, Neuroscience
Introduction
Cholesterol fulfills crucial roles in cell function and viability. Most of the cellular cholesterol resides in the plasma membrane, where it constitutes 30–45% of lipid molecules. Cholesterol entails the formation of liquid‐ordered phases, raft‐like domains which are enriched in cholesterol and sphingomyelin. These raft domains coexist with liquid‐disordered membrane regions, poor in cholesterol (Silvius, 2003; Pike, 2006). Compelling evidence indicates that lipid rafts are involved in trafficking, in recycling, and in lateral segregation of many cell surface proteins forming dynamic signaling platforms (Simons & Toomre, 2000). In neurons, these domains are implicated in the regulation of many receptors and ion channels affecting their activation or inactivation parameters, open states, as well as their expression at the cell surface (Martens et al, 2000; Graziani et al, 2006; Dart, 2010; Levitan et al, 2010). Both indirect and direct interactions with cholesterol could account for these regulations. Indirect interactions rely on an increased energetic cost necessary for conformational changes of ion channels linked to the increased stiffening and thickness of the membrane, enforced by planar rings of cholesterol molecules. Direct interactions rely on the binding of cholesterol on specific domains at the surface or within a pocket of α‐helices of the proteins (Lee, 2004; Song et al, 2014).
Growing evidence suggests that pain sensation caused by tissue damage and inflammation is, in part, regulated by the pro‐ or anti‐nociceptive actions of lipid mediators, such as arachidonic acid or anandamide, on neural pathways (Piomelli & Sasso, 2014). Cholesterol/lipid rafts also have been shown to be involved in regulating nociceptive ion channels in DRGs (Gnanasekaran et al, 2011; Pristera et al, 2012; Jin et al, 2013; Sághy et al, 2015). Although these studies highlight interaction between cholesterol/lipid rafts and nociceptive signaling, they provide little information about whether change in cholesterol homeostasis contributes to inflammatory hyperalgesia.
Accordingly, we investigated whether change in plasma membrane cholesterol may contribute to the mechanisms of inflammatory nociceptor hypersensitization. We show that cellular cholesterol content is reduced in both mouse skin tissue and DRG cultures during inflammation. Depletion of membrane cholesterol by cholesterol transfer to methyl‐beta‐cyclodextrin (MβCD) or oxidation with cholesterol oxidase causes sensitization of nociceptive neurons and induces mechanical and thermal hyperalgesia in mice. The use of Nav1.8‐ and Nav1.9‐null mice provides evidence that cholesterol depletion‐induced pain depends on the activity of the voltage‐gated sodium channel Nav1.9 but not Nav1.8. Nav1.9 channels are almost exclusively expressed in nociceptive neurons, where they exert an important role in setting electrogenic properties of these neurons and in sustaining inflammatory pain (Priest et al, 2005; Amaya et al, 2006; Padilla et al, 2007; Maingret et al, 2008; Lolignier et al, 2011). We show that inflammation causes partitioning of Nav1.9 channels from lipid rafts to non‐raft membrane domains, concomitantly with a shift of the voltage dependence of Nav1.9 to more negative potentials, increasing neuronal excitability. We identify three peptide domains on the Nav1.9 channel subunit exhibiting cholesterol‐binding properties suggesting physical interaction. Finally, we demonstrate that cholesterol supply prevents upregulation of Nav1.9 activity by inflammatory mediators and alleviates pain hypersensitivity in persistent and chronic arthritic inflammatory pain models.
In conclusion, our data suggest that depletion of cellular cholesterol content is an important factor in determining sensitization of nociceptive neurons. Hence, far from the drawback effect of cholesterol emphasized in atherosclerosis (Maxfield & Tabas, 2005), these results pave the way for a novel approach for the treatment of pain based on topical delivery of cholesterol molecules.
Results
Inflammation decreases cholesterol level in tissues and sensory DRG neurons
To decipher the functions of cholesterol in inflammatory pain signaling, we first evaluated potential local changes of cholesterol level in λ‐carrageenan‐induced inflamed skin. Intraplantar injection of carrageenan triggered inflammatory response and mechanical hypersensitivity over 24 h (Fig 1A). The early inflammatory phase (0–4 h) is characterized by the release of inflammatory mediators including ATP, histamine, and bradykinin, while the late phase is linked with immune cell infiltration and prostaglandin production (Di Rosa et al, 1971; Posadas et al, 2004). We quantified the total cellular content of cholesterol from standardized mouse skin biopsies (1.8 mm3 made of stratum corneum, epidermis, and dermis; Fig 1B) 2 h after carrageenan injection, when mechanical allodynia was maximal. Skin biopsies taken from the inflamed ipsilateral plantar skin exhibited an 18 ± 4.5% reduction in cholesterol level compared with biopsies from the uninjected contralateral paws (Fig 1C). By contrast, animals injected with the saline solution did not exhibit a difference in the amount of skin cholesterol between ipsi‐ and contralateral paws (Fig 1C). Likewise, intraplantar injection of a cocktail of inflammatory mediators (made of histamine, bradykinin, ATP, prostaglandin E2, and norepinephrine; see Materials and Methods) caused pain hypersensitivity (Fig 1D) and a concomitant significant reduction in skin cholesterol level in the ipsilateral paw (Fig 1E).
Figure 1. Inflammation decreases cellular cholesterol in skin tissues and DRG neurons.
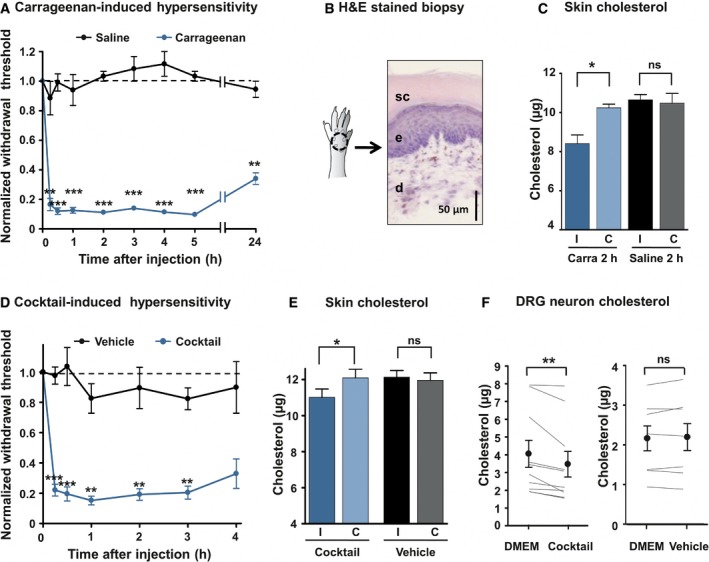
- Intraplantar injection of 3% λ‐carrageenan‐induced mechanical hypersensitivity (von Frey filaments, n = 7). Control animals were injected with saline solution (n = 6). Data are normalized to t = 0 score for each animal.
- Schematic drawing and hematoxylin–eosin staining of standardized skin biopsy specimen. sc: stratum corneum; e: epidermis; d: dermis.
- Skin biopsies from ipsilateral (I) and contralateral (C) paws of mice injected with the saline solution (Saline, n = 6) or with 3% λ‐carrageenan (Carra, n = 7) were analyzed. Cholesterol quantification was made 2 h after injection.
- Development of mechanical hypersensitivity induced by intraplantar injection of a 20× solution of a cocktail of pro‐inflammatory mediators (BK, PGE2, His, NE, ATP; n = 12). Control animals were injected with the vehicle (n = 7).
- Quantification of cholesterol level 2 h after injection of the inflammatory cocktail in the hind paw. The dosage was carried out as in (C); n = 8 for cocktail and n = 9 for vehicle.
- Dosage of cholesterol in DRG cultures. Paired neurons were treated for 15 min with DMEM or the 1× inflammatory cocktail (n = 10). In control experiment, paired neurons were treated with DMEM or the vehicle (n = 7).
Since pain is initiated by the stimulation of nociceptors innervating skin territories, we monitored cholesterol level of dorsal root ganglion (DRG) cultures enriched for neurons relative to glial cells (see Materials and Methods) and exposed to inflammatory mediators. Cholesterol level in DRG cultures treated for 15 min with the inflammatory cocktail was reduced by 16 ± 3.5% compared with cultures exposed to the vehicle (Fig 1F). Concomitant with the decrease in cholesterol level, we observed an overall increase in excitability of cultured DRG neurons exposed to the inflammatory cocktail. Indeed, 12‐min exposure of DRG neurons to the cocktail reduced the current threshold necessary to elicit an action potential (AP) by 32 ± 14% (Fig EV1A and B). The firing rate of DRG neurons in response to current injection was also increased twofold–threefold as described previously (Rush & Waxman, 2004; Maingret et al, 2008; Ostman et al, 2008) (Fig EV1D). Importantly, pre‐treatment with MβCD‐chol complex (20 mM), also named water‐soluble cholesterol (Zidovetzki & Levitan, 2007), for 10 min prevented any significant changes in current threshold for AP and excitability in DRG neurons exposed to the inflammatory cocktail (n = 7) compared with control neurons (Fig EV1A, C and E). Taken together, these data indicate that inflammation may produce a local decrease in cholesterol level in skin concomitantly with pain hypersensitivity and that a combination of inflammatory mediators is sufficient to decrease cholesterol in DRG cultures and promote neuronal hyperexcitability.
Figure EV1. Increased excitability induced by the inflammatory cocktail is prevented by soluble cholesterol.
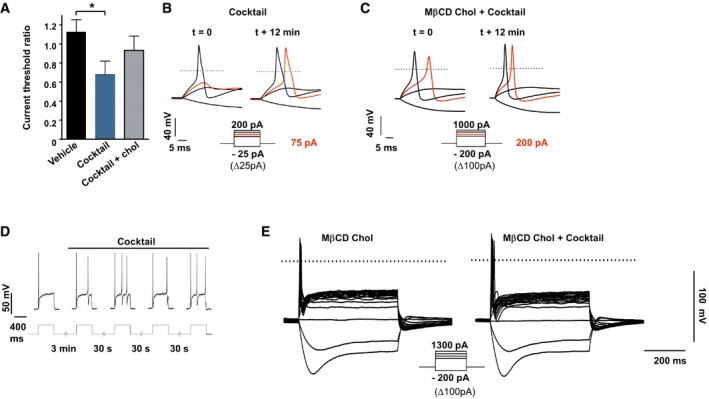
-
ARatio of current threshold for AP determined in DRG neurons treated with vehicle (n = 5) or with the inflammatory cocktail (1×) pre‐treated (n = 5) or not (n = 7) with MβCD‐chol (20 mM) for 10 min. For each cell, the current threshold was measured before (t = 0) and 12 min after bath application of the cocktail or its vehicle. Values are shown as mean ± standard error of the mean (SEM). Results were analyzed with a Mann–Whitney U‐test. *P < 0.05.
-
B, CRepresentative recordings of current threshold for AP before (t = 0) and 12 min after bath application of the inflammatory cocktail in DRG neurons pre‐treated (C) or not (B) with MβCD‐chol. For clarity's sake, not all traces were illustrated. The red traces represent the voltage response induced by the injected current necessary to induce an action potential at t = 12 min. The dashed lines indicate 0 mV.
-
DEnhanced firing of a DRG neuron (27 pF) after bath application of the inflammatory cocktail. Injected current: 100 pA.
-
EInhibition of inflammatory cocktail‐induced hyperexcitability in a DRG neuron pre‐treated with MβCD‐chol (20 mM).
Cholesterol depletion promotes neuronal hyperexcitability and pain behaviors
We next aimed to determine the causal relationship between inflammation‐induced cholesterol lowering and pain development. Inflammation‐induced decrease in membrane cholesterol could result from two non‐exclusive pathways: enhanced cholesterol efflux and increased oxidation of cholesterol into oxysterols. We pharmacologically tested both hypothesis with MβCD, an agent commonly used to remove cellular cholesterol (Ohtani et al, 1989; Ziblat et al, 2006; Zidovetzki & Levitan, 2007), and cholesterol oxidase, an enzyme that specifically oxidizes cholesterol into cholestenone, which shares with some oxysterols the ability to disrupt liquid‐ordered phase of the membrane bilayer (Lenne et al, 2006; Neuvonen et al, 2014). One hour after intraplantar injection of 40 mM MβCD and 3 h after 4 U/ml cholesterol oxidase injection, we measured 17 ± 4 and 15 ± 1.1% decrease in skin cholesterol level, respectively (Fig 2A). We showed that injection of MβCD produced a rapid development of mechanical and thermal hypersensitivity (Fig 2B and C). This effect was not due to the extraction of other lipids since intraplantar injection of 40 mM α‐cyclodextrin (αCD), which extracts phosphatidylcholine and sphingomyelin but not cholesterol (Ohtani et al, 1989; Monnaert et al, 2004), did not change mechanical or thermal sensitivities of animals (Fig 2B and C). Moreover, injection of 40 mM MβCD‐cholesterol (MβCD‐chol), a complex of MβCD saturated with cholesterol, did not modify mechanical withdrawal threshold of animals (Fig 2B). MβCD‐induced hypersensitivity was not due to an inflammatory response subsequent to cyclodextrin injection, since intraperitoneal injection 1 h prior to MβCD of the non‐steroidal anti‐inflammatory drug ibuprofen (NSAID, 75 mg/kg) failed to attenuate MβCD‐induced mechanical allodynia (Fig 2B), but was efficient in reducing carrageenan‐induced inflammatory pain (see Fig 8A). These results indicate that MβCD triggered pain signal by direct extraction of cholesterol and not by local inflammation. Consistently, depletion of membrane cholesterol by enzymatic oxidation caused slowly developing but severe mechanical hypersensitivity (Fig 2D). Collectively, these results indicate that extraction of membrane cholesterol as well as cholesterol oxidation causes pain hypersensitivity.
Figure 2. Pharmacological decrease in cholesterol enhances excitability of DRG neurons and causes hyperalgesia.
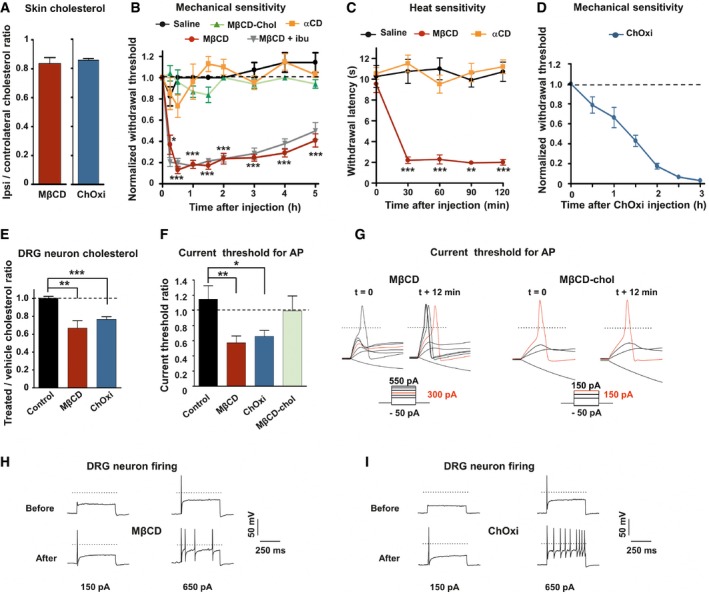
-
AQuantification of cholesterol content from skin paw biopsies of mice injected with MβCD (n = 6) or cholesterol oxidase (ChOxi; n = 8).
-
BIntraplantar injection of MβCD (n = 8), but not MβCD‐chol (n = 9), αCD (n = 7), or saline solution (n = 7), caused mechanical hypersensitivity. Effects of MβCD were not prevented by ibuprofen (75 mg/kg) injected intraperitoneally 1 h prior to intraplantar injection of MβCD (MβCD + ibu; n = 9).
-
CIntraplantar injection of MβCD (n = 8), but not αCD (n = 8) or saline solution (n = 8), caused heat hypersensitivity (Hargreaves radiant heat test).
-
DDevelopment of mechanical hypersensitivity induced by intraplantar injection of ChOxi (n = 6).
-
ECholesterol quantification in DRG cultures treated for 15 min with the saline (control, n = 7; MβCD, n = 9; or ChOxi, n = 7).
-
FCurrent threshold ratio for each DRG neuron was calculated by dividing the current threshold measured 12 min after drug treatment by that measured at t = 0 min (control, n = 7; MβCD, n = 7; ChOxi, n = 6; MβCD‐chol n = 5).
-
GRepresentative illustrations of current threshold in DRG neurons before (t = 0) and 12 min after bath application of MβCD and MβCD‐chol. Current steps were applied with 25‐pA increments. For clarity's sake, not all traces were illustrated. The red traces represent the voltage response induced by the injected current necessary to induce an action potential at t = 12 min. The dashed lines indicate 0 mV.
-
H, IRepresentative firing behavior of small DRG neurons before and after 12‐min treatment with MβCD (H) and ChOxi (I).
Figure 8. Topical supply of cholesterol alleviates subacute and chronic arthritis pain.
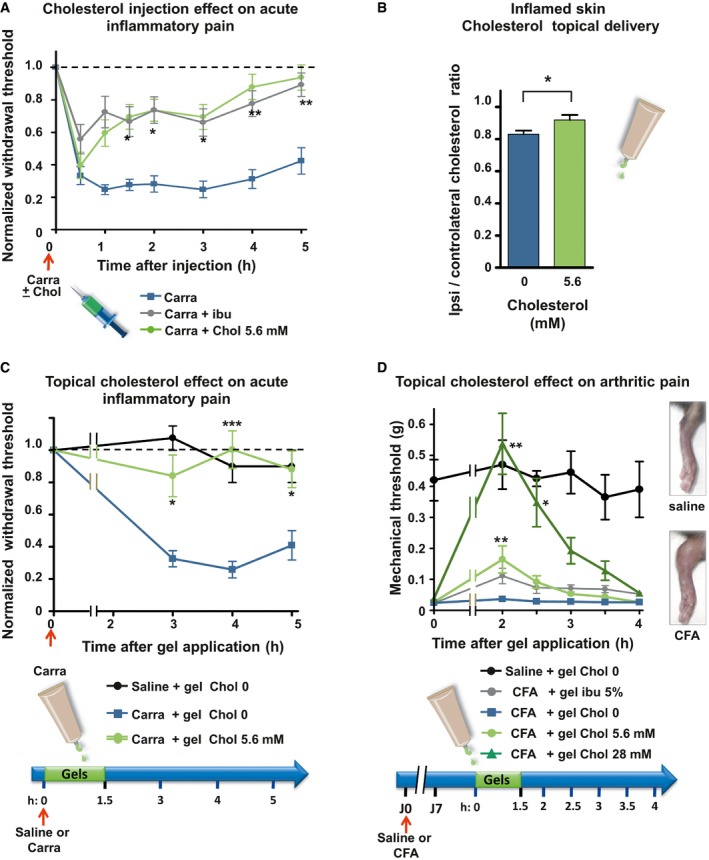
- Mechanical sensitivity of mice injected with 2% carrageenan (Carra; n = 13; blue line) or co‐injected with carrageenan and 5.6 mM of cholesterol (Carra + chol 5.6 mM; n = 12; green). Ibuprofen (75 mg/kg) was injected intraperitoneally 1 h prior to intraplantar injection of carrageenan into the hind paw (Carra + ibu; n = 15; gray line).
- Cholesterol content of ipsilateral and contralateral skin biopsies of mice intraplantarly injected with 3% carrageenan and treated for 1.5 h with either a topical application of an HEC gel containing 5.6 mM of cholesterol (n = 9) or a blank gel containing no cholesterol (0 mM; n = 8).
- Mechanical sensitivity of mice injected with 2% carrageenan and treated with topical application of HEC gels containing 0 mM (blue, n = 12) or 5.6 mM (green, n = 10) of cholesterol. Saline‐injected animals were treated with the blank gel (0 mM cholesterol, black, n = 4).
- Mechanical sensitivity of CFA‐induced arthritic mice. Hyperalgesic mice received topical HEC gels containing no cholesterol (blue, n = 6) or containing 5.6 mM of cholesterol (light green, n = 7) or 28 mM of cholesterol (dark green, n = 7) or containing 5% ibuprofen (gray, n = 6). Saline‐injected animals (black, n = 4), treated with the blank gel (0 mM cholesterol), were also included to ensure that anesthesia does not alter normal mechanical perception of mice. Right inset: 7 days after ipsilateral injections of CFA, mice exhibited severe swelling of the ankle compared with saline‐injected animals.
We next addressed the causal link between cholesterol depletion in DRG neurons and neuronal hyperexcitability. Incubation of DRG neurons with 20 mM MβCD for 15 min induced 33 ± 8% cellular cholesterol level decrease compared with twin culture treated with vehicle (Fig 2E). This short treatment did not change cell viability as the percentage of live DRG neurons using the trypan blue exclusion test was similar to that measured in control DRG cultures (89.6 ± 4.8% for MβCD, n = 2 cultures/4859 neurons; versus 86.8 ± 0.9% for control, n = 3 cultures/7857 neurons). Current‐clamp experiments in small‐diameter DRG neurons showed that acute application of MβCD decreased the current threshold for AP by 43 ± 9% (Fig 2F and G). In 2 out of 7 neurons tested, change in current threshold was also associated with a prominent increase in firing rate in response to depolarizing currents (Fig 2H). However, no significant change in the input resistance was seen during MβCD application (512 ± 83 MΩ before versus 600 ± 64 MΩ after treatment; P = 0.8). Importantly, exposure of DRG cultures to 20 mM MβCD‐chol had no effect on current threshold for AP (Fig 2F and G). Similarly, incubation with cholesterol oxidase (2 U/ml) reduced the amount of cellular cholesterol by 23.3 ± 3% in cultured DRG neurons (Fig 2E) and induced a reduction of 35 ± 7.7% in the current threshold for AP in DRG neurons (Fig 2F). Moreover, increase in firing rate was detected in 5 out of 14 neurons tested (Fig 2I). Collectively, these data demonstrate that depletion of cellular cholesterol, induced by cholesterol extraction or cholesterol oxidation, increases the excitability of DRG neurons. Although inflammatory mediators are known to sensitize nociceptors through multiple intracellular signaling pathways (Aley & Levine, 1999; Aley et al, 2000), our data raise the hypothesis that changes in membrane cholesterol level, possibly interfering with lipid raft domains and ion channel function (Dart, 2010; Levitan et al, 2014), contribute to the sensitization of nociceptors.
Cholesterol depletion‐induced pain depends on Nav1.9 but not on Nav1.8 channel function
Voltage‐gated sodium channels play a fundamental role in setting the excitability properties of neurons and therefore in the genesis and propagation of pain signals (Waxman, 2012). Among sodium channels expressed in adulthood, Nav1.8 and Nav1.9 channels are implicated in the sensitization of nociceptors underlying inflammatory pain. Nav1.8 has been shown to be associated with lipid raft domains and regulated by the level of cholesterol in DRG neurons (Pristera et al, 2012). Moreover, Nav1.9 is distributed in clusters along the peripheral fibers of trigeminal neurons (Padilla et al, 2007) and DRG sensory fibers of skin territories (Appendix Fig S1), which bears a resemblance to the distribution of Nav1.8 in supposed lipid rafts (Pristera et al, 2012). Thus, we tested a potential contribution of Nav1.8 and Nav1.9 channels to cholesterol depletion‐induced pain. We compared mechanical allodynia provoked by intraplantar injection of MβCD in both Nav1.8‐ and Nav1.9‐null mice and their respective wild‐type littermates. We found that MβCD‐induced mechanical allodynia was unchanged in Nav1.8 KO mice compared with wild‐type littermates (Fig 3A). In contrast, MβCD‐induced mechanical allodynia was half‐reduced in Nav1.9 KO mice compared with wild‐type littermates (Fig 3B). Residual MβCD‐induced allodynia in Nav1.9 KO mice was insensitive to intraperitoneal injection of ibuprofen (Fig 3C), indicating that, like in wild‐type animals, MβCD‐induced hypersensitivity in Nav1.9 KO animals was not due to Cox‐dependent local inflammation subsequent to cyclodextrin injection. We also confirmed that αCD did not alter withdrawal mechanical thresholds in Nav1.9 KO animals (Fig 3C). Similarly, MβCD‐chol injections did not change mechanical threshold of Nav1.9 KO mice (Fig 3C). Hyperalgesia induced by intraplantar injection of cholesterol oxidase was also substantially reduced in Nav1.9 KO mice compared with wild‐type littermates (Fig 3D). However, cholesterol oxidase‐induced hyperalgesia was similar in Nav1.8 KO mice compared with their wild‐type littermates (Appendix Fig S2).
Figure 3. Cholesterol depletion‐induced pain depends on Nav1.9 channels.
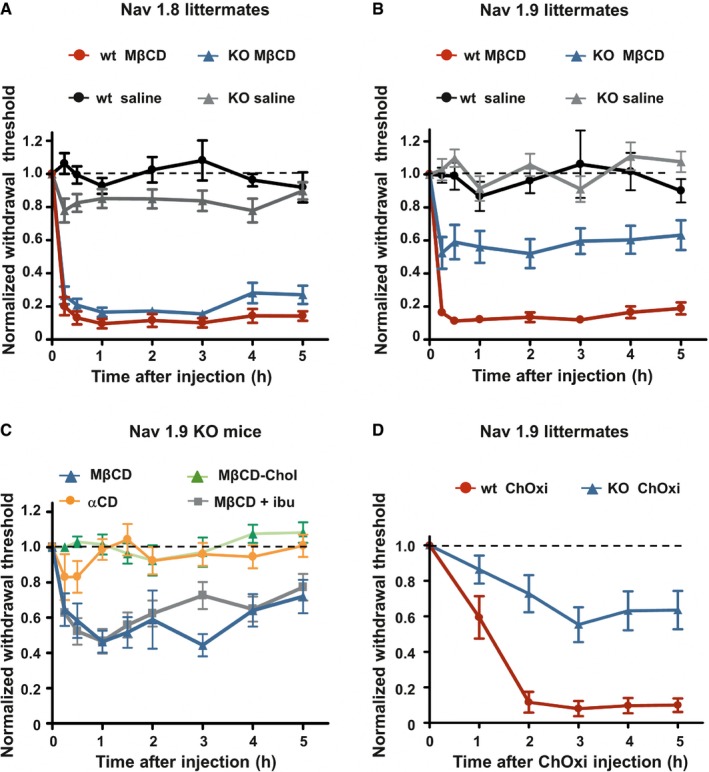
- Mechanical hypersensitivity caused by intraplantar injection of MβCD is similar in Nav1.8 KO mice (n = 8) compared with wt littermates (n = 10; P = 0.07). Animals injected with saline solution (n = 8 both KO and wt) did not exhibit modification of their mechanical threshold.
- Mechanical hypersensitivity caused by intraplantar injection of MβCD was clearly reduced in Nav1.9 KO mice (n = 10) compared with wt littermates (n = 9; P < 0.001). Saline‐injected animals (n = 9 both for KO and wt) did not exhibit modification of their mechanical threshold.
- Intraplantar injection of MβCD (n = 8), but not MβCD saturated with cholesterol (MβCD‐chol, n = 11), or αCD (n = 12) caused mechanical hypersensitivity in Nav1.9 KO animals. Effects of MβCD were not prevented by ibuprofen (75 mg/kg) injected intraperitoneally 1 h prior to intraplantar injection of MβCD (MβCD + ibu; n = 12).
- Mechanical hypersensitivity induced by intraplantar injection of cholesterol oxidase in Nav1.9 KO mice (ChOxi; n = 10) is significantly reduced compared with wt littermate (n = 5; P = 0.003).
These findings indicate that Nav1.9, but not Nav1.8, is involved in cholesterol depletion‐induced pain.
Pharmacological depletion of cholesterol alters the voltage dependence properties of Nav1.9 currents
We analyzed the consequences of low‐cholesterol conditions on electrophysiological properties of Nav1.9 currents. To isolate Nav1.9 currents, voltage‐clamp recordings were made using CsCl‐based pipette solution in DRG neurons from Nav1.8‐null mice, under conditions that minimize contamination by Ca2+ currents (Coste et al, 2007). We first tested the effects of acute application of MβCD and αCD on Nav1.9 currents recorded in small‐diameter DRG neurons. MβCD, but not αCD, induced a slow increase in Nav1.9 current (Appendix Fig S3). However, acute application of MβCD was not suitable for prolonged voltage‐clamp recordings, preventing complete biophysical characterization of Nav1.9 current modulation. Therefore, voltage‐dependent properties of Nav1.9 currents were examined isochronally 10 ± 1 min after patch rupture in DRG neurons pre‐treated with 20 mM MβCD or 2 U/ml of cholesterol oxidase. Nav1.9 currents had larger amplitude under these two conditions compared with those recorded in vehicle‐treated DRG neurons. For instance, current densities at −35 mV were −28.7 ± 3.2 pA/pF for control neurons versus −99.2 ± 13.9 pA/pF and −98.4 ± 21 pA/pF for neurons treated with MβCD and cholesterol oxidase, respectively (Fig 4A–C). Current–voltage relationship analysis showed a leftward shift of the voltage dependence of Nav1.9 currents without significant change in the maximum slope conductance (Fig 4B and C). The voltage dependence of activation was measured by plotting the normalized conductance against membrane potential and fitting the curve with the Boltzmann equation. The membrane potential at half‐maximal activation (V 0.5) was −33.65 ± 0.6 mV in MβCD‐treated neurons and −36.1 ± 0.9 mV in cholesterol oxidase‐treated neurons compared with −27.3 ± 0.2 mV in control neurons (Fig 4D). “Fast” inactivation V 0.5 values were also negatively shifted from −25.17 ± 1.14 mV in control DRG neurons to −34.8 ± 1.1 and −41.54 ± 0.38 mV in neurons pre‐treated with MβCD or ChOxi, respectively (Fig EV2). Together, these data suggest that depletion of cellular cholesterol induced by cholesterol extraction or cholesterol oxidation shifts the voltage dependence of Nav1.9 current to more negative potentials.
Figure 4. Cholesterol depletion modulates the voltage dependence of Nav1.9 activation.
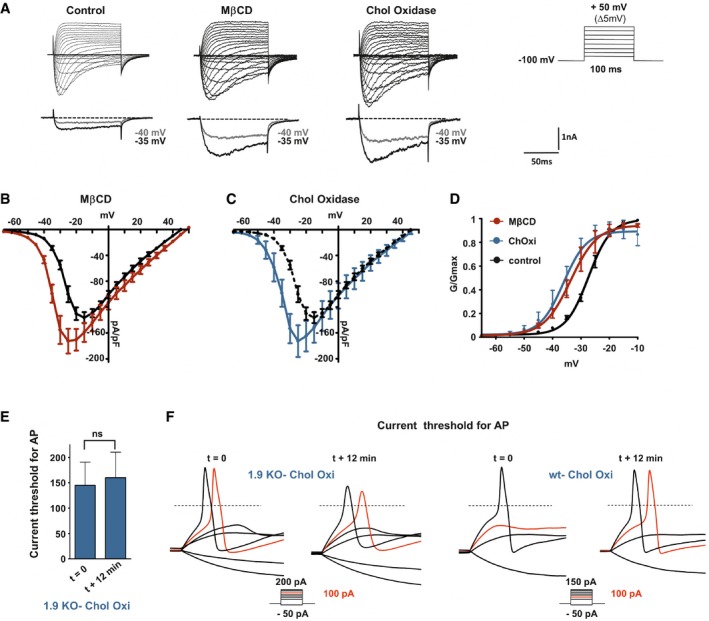
- Representative current traces evoked by 100‐ms depolarizing voltage steps ranged from −80 to +50 mV (Δ 5 mV, Vh = −100 mV). DRG neurons were pre‐incubated prior recording with DMEM alone (control) or DMEM supplemented with 20 mM MβCD or 2 U/ml cholesterol oxidase for 15 min at 37°C. Current traces were recorded 12 min after achieving whole‐cell configuration. For clarity's sake, current evoked at −40 and −35 mV is represented below each panel (gray and black traces, respectively).
- Current–voltage relationships of Nav1.9 current with (red curve, n = 10) or without (black curve, n = 5) pre‐incubation with MβCD.
- Current–voltage relationships of Nav1.9 current with (blue curve, n = 6) or without (black curve same as in B) cholesterol oxidase.
- Corresponding activation curves of Nav1.9 currents fitted with single Boltzmann equation. Values for V 0.5 of activation are −27.37 ± 0.2 mV, −33.65 ± 0.6 mV, and −36.18 ± 0.9 mV for control, MβCD, and cholesterol oxidase conditions, respectively.
- Current threshold of DRG neurons from Nav1.9 KO mice before treatment (t = 0 min; n = 5) and 12 min after drug treatment. Current thresholds were analyzed with a non‐parametric Wilcoxon signed‐rank test.
- Representative illustrations of current threshold in DRG neurons before (t = 0) and 12 min after bath application of cholesterol oxidase. Current steps were applied with 25‐pA increments. For clarity's sake, not all traces were illustrated. The red traces represent the voltage response induced by the injected current necessary to induce an action potential at t = 12 min. The dashed lines indicate 0 mV.
Figure EV2. Cholesterol depletion negatively shifts the “fast” inactivation voltage dependency of Nav1.9.
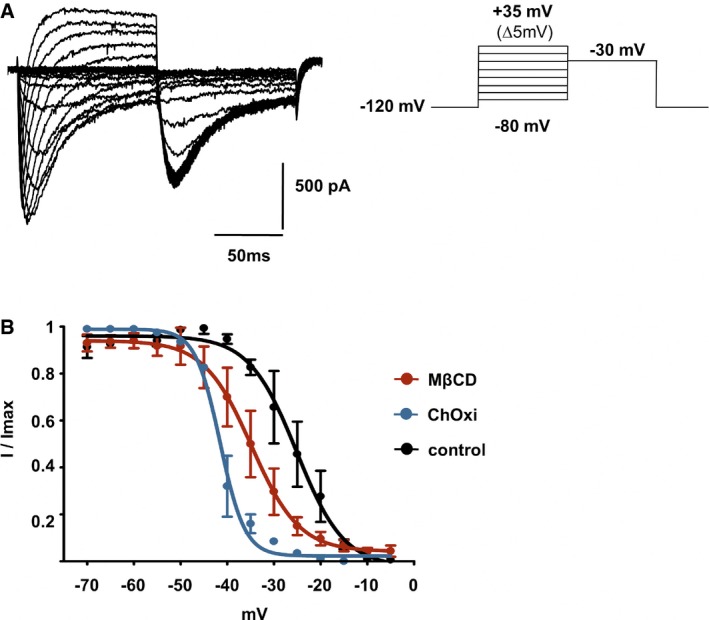
- Families of Nav1.9 current traces recorded after 10‐min treatment with 20 mM MβCD. The test pulse to −30 mV was preceded by a family of depolarizing pulses ranging from −80 to 35 mV for 100 ms, while the cell was held at −120 mV.
- Fast inactivation curves of Nav1.9 current in control DRG neurons (n = 3) and in neurons treated with 20 mM MβCD (n = 3) or 2 U/ml ChOxi (n = 2) for 10 min. Curves were fitted by using single Boltzmann equations, yielding V 0.5 values of −25.17 ± 1.14, −34.8 ± 1.1, and −41.54 ± 0.38 mV, respectively. Values are shown as mean ± standard error of the mean (SEM).
To evaluate the contribution of Nav1.9 channels on DRG neuron hyperexcitability observed upon cholesterol depletion, we measured current threshold necessary for AP generation in DRG neurons from Nav1.9 KO animals before and 12 min after acute application of cholesterol oxidase. Depletion in cholesterol had no effects on AP current threshold (Fig 4E and F) in Nav1.9 KO DRG neurons.
Taken together, our data demonstrate that decrease in cholesterol induces a shift of Nav1.9 voltage dependence to more negative potentials, increasing neuronal excitability. Therefore, Nav1.9 contributes to the neuronal hyperexcitability and mouse hyperalgesia induced by low‐cholesterol conditions.
Inflammatory mediators potentiate Nav1.9 through ROS‐dependent mechanisms
Oxidative stress and subsequent cholesterol oxidation may be a mechanism responsible for inflammation‐induced cholesterol depletion and Nav1.9 activation. We therefore tested whether the inflammatory cocktail could enhance ROS generation in DRG neuronal cultures. Treatment with the inflammatory cocktail increased intracellular reactive oxygen species (ROS) production in individual DRG neurons as detected using the ROS‐sensitive probe H2DCFDA (Fig EV3A and B). Increase in ROS production induced by the inflammatory cocktail was abolished by co‐incubation with the free radical spin‐trapping agent alpha‐phenyl t‐butyl nitrone (PBN) (Fig EV3B). In addition, the ROS scavenger N‐acetyl‐cysteine (NAC), a commonly used antioxidant, prevented the inflammatory cocktail to negatively shift Nav1.9 activation curve (Fig EV3C). The midpoint of activation curve was −35.02 ± 0.79 mV for cells exposed to the cocktail and −26.34 ± 1.5 mV for cells treated simultaneously with the cocktail and NAC. Consistently, intraplantar co‐injection of NAC with the inflammatory cocktail strongly reduced the mechanical hypersensitivity typically provoked by the inflammatory cocktail injected alone (Fig EV3D). Thus, these data indicate that inflammatory mediators modulate Nav1.9 via oxidative stress‐mediated signaling mechanisms.
Figure EV3. The inflammatory cocktail causes pain hypersensitivity through production of increased reactive oxygen species.
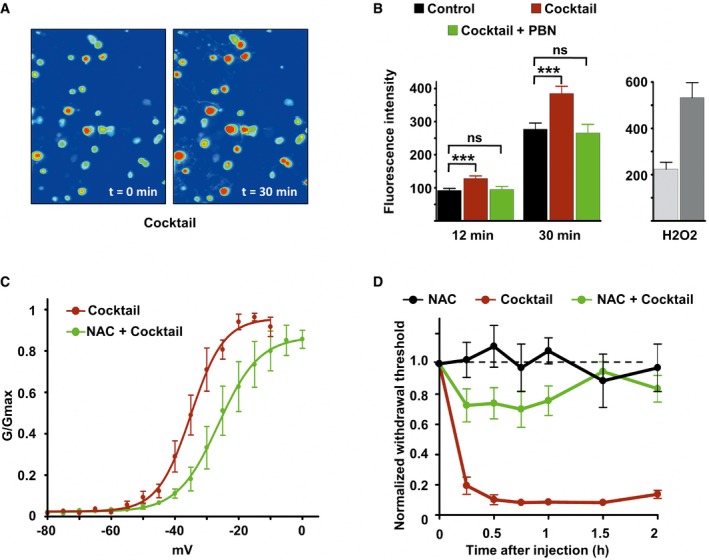
- Imaging of DRG neurons loaded with the ROS‐sensitive probe H2DCFDA before (t = 0) and 30 min after inflammatory cocktail application.
- Left panel: Fluorescence intensity of cultured DRG neurons loaded with ROS‐sensitive probe H2DCFDA in control neurons (black bars, n = 114), neurons treated with the inflammatory cocktail (red bars, n = 119), and neurons co‐treated with inflammatory cocktail and 4 mM of the ROS scavenger alpha‐phenyl‐N‐tert‐butyl nitrone (PBN, green bars, n = 95). Measure of fluorescence intensity was made 12 and 30 min after drug application. Right panel: positive control for ROS detection. DRG neurons (n = 15) were treated with 0.3% H2O2 and imaged at 12 (light gray) and 30 min (dark gray) after drug application. Fluorescence intensity (B) was analyzed with a non‐parametric Mann‐Whitney U‐test. ***P < 0.001.
- Activation curves of Nav1.9 current fitted with single Boltzmann equations giving V 0.5 values of −35.02 ± 0.79 mV (n = 3, black line) and −26.34 ± 1.5 mV (n = 8, green line) for cells treated with the cocktail or with cocktail + NAC, respectively. Current were recorded 10 min after adding drugs.
- Comparison of mechanical hypersensitivity induced by intraplantar injection of NAC (n = 5, 20 mM), cocktail (20×, n = 4), and NAC + cocktail (n = 9). Note that the effects of cocktail were strongly reduced by NAC when injected simultaneously.
Inflammatory mediators translocate Nav1.9 from lipid raft to non‐raft domains
We next analyzed the interaction between Nav1.9 channels and cholesterol. We first examined the possible segregation of Nav1.9 in lipid rafts which are microdomains of the membrane especially enriched in cholesterol. Lysates of acutely dissected DRGs were subjected to density gradient centrifugation assay. Based on resistance to solubilization by Triton X‐100 detergent at 4°C, and buoyancy at low‐density fractions (Brown & Rose, 1992), canonical lipid raft markers such as flotillin and caveolin were found to be enriched in the low‐density fractions 1–4 of the gradient. In contrast, the non‐raft protein, transferrin receptor (Tfr), was barely present in raft fractions. We showed that the vast majority of Nav1.9 channels co‐localized with lipid raft markers in detergent‐resistant membranes (DRM) (Fig 5A, left).
Figure 5. Inflammatory mediators modulate Nav1.9 partitioning in DRM raft‐like microdomains.
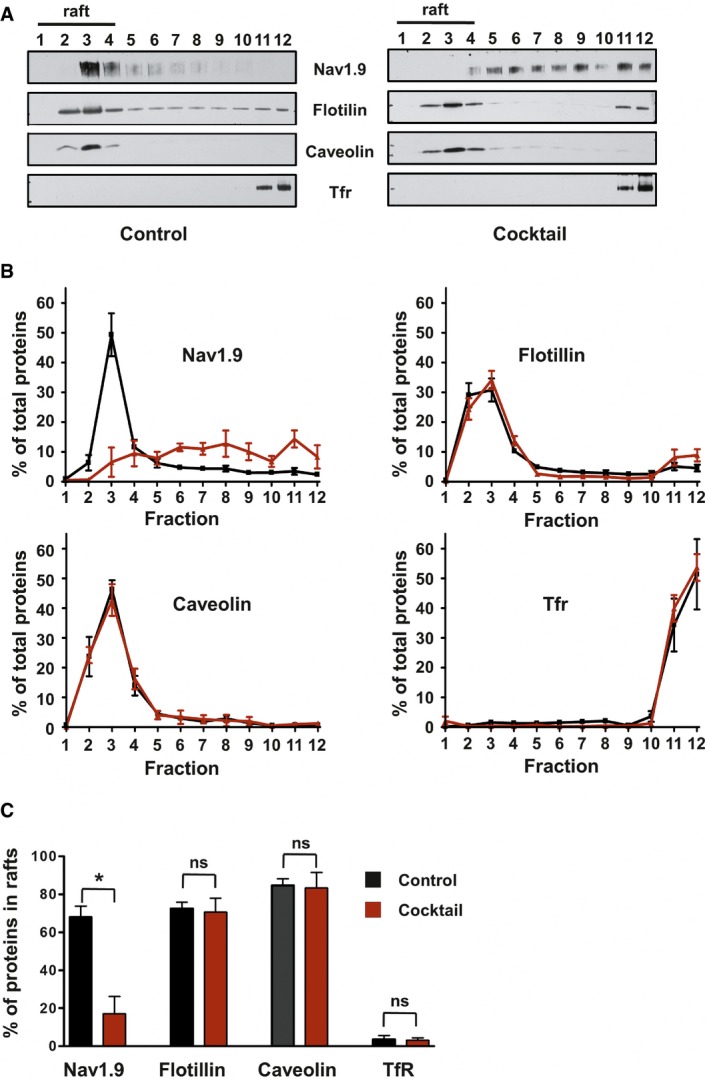
- Representative Western blot of DRM fractions of freshly isolated from DRGs incubated for 15 min at 37°C with DMEM (left panel) or DMEM + a cocktail of inflammatory mediators (right panel). Fraction 1 represents the top of the gradient and fraction 12 the bottom. Lipid rafts–DRM fractions (fractions 1 to 4) are revealed by the presence of flotillin and caveolin and the absence of Tfr proteins.
- Quantification of signals obtained for each separated fraction of DMEM‐treated control (black line; n = 3) or inflammatory cocktail‐treated (red line; n = 5) DRGs. Values are expressed as a percentage of the sum of total signals of all fractions.
- Quantification of protein in raft fractions (sum of fraction 1 to 4) of control or inflammatory cocktail‐treated DRGs (black and red bars, respectively; *P = 0.04, non‐parametric Mann–Whitney U‐test).
We then assessed the effect of inflammatory mediators on the preferential partitioning of Nav1.9 to raft microdomains. We analyzed extracts prepared from DRGs incubated for 15 min with the inflammatory cocktail. We found that Nav1.9 distribution in the presence of inflammatory mediators was markedly shifted toward higher density, non‐raft fractions (Fig 5A, right). Quantification of the immunoreactive signals revealed that 70 ± 7% of flotillin, 85 ± 3.6% of caveolin, and 3.6 ± 2% of Tfr were present in detergent‐resistant membrane (DRM) fractions and these proportions were almost unchanged upon cocktail treatment (Fig 5B and C). Analysis from the same DRG lysates showed that in control conditions, 68.2 ± 5.5% of the Nav1.9 signal was localized in DRM fractions 1–4 (Fig 5B and C). In contrast, after exposure of DRG neurons to cocktail, only 17.2 ± 9% of Nav1.9 signal remained localized in DRM–lipid raft fractions (Fig 5B and C). Since the partitioning of lipid raft markers (flotillin, caveolin) remained unchanged, inflammation appears to alter the distribution of Nav1.9, without disrupting the general organization of lipid rafts. Short treatment of DRG neurons with MβCD (40 mM), which resulted in partial destruction of lipid rafts, also caused a partial redistribution of Nav1.9 out of raft fractions (Appendix Fig S4).
These results indicate that upon stimulation by inflammatory mediators, Nav1.9 channels are relocalized from lipid rafts to non‐raft domains. This non‐raft cholesterol‐poor environment is predicted to be more permissive to conformational changes, which may explain the facilitated activation of Nav1.9 channels at more negative potentials.
Identification of cholesterol‐binding domains on Nav1.9 protein
We next looked for possible direct binding of cholesterol on Nav1.9 protein. Even though of a limited predictive value, several consensus cholesterol‐binding domains have emerged such as CRAC domains defined by the amino acid sequence (L/V) ‐ X1−5 ‐ (Y) ‐ X1−5 ‐ (K/R), CARC domains defined by (K/R) ‐ X1−5 ‐ (Y/F) ‐ X1−5 ‐ (L/V), and CCM domains defined by (R/K) ‐ X2–6 ‐ (I/V/L) ‐ X3 ‐ (W/Y) (Fantini & Barrantes, 2013; Song et al, 2014), where X could represent any amino acid (aa). Protein motif analysis indicated that Nav1.9 channel contains 29 consensus cholesterol‐binding domains (5 CRAC, 20 CARC, and 4 CCM) in transmembrane and juxtamembrane regions. Some of these sites were overlapping, limiting to 17 potential binding regions distributed in all four structural domains of the channel (Appendix Fig S5). We investigated the cholesterol‐binding capacity of some of these peptides by surface plasmon resonance (Sheng et al, 2012). Liposomes made of 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS), with or without cholesterol, were immobilized on a L1 lipophilic sensor chip, and 20 μM of peptide was injected in the microfluidic system. Three peptides exhibited a specific binding on cholesterol‐loaded liposomes (Fig 6B–D). Peptides 1 and 3 were located in the voltage‐sensor transmembrane segments S4 of domain I and domain III, respectively. Peptide 1 contains nine aa of a single CARC motif, and peptide 3 contains two CARC domains separated by three aa (Appendix Fig S5). Peptide 2, located in S1 segment of domain III, is 13 aa length overlapping a CRAC, a CARC, and a CCM domain (Fig 6A and Appendix Fig S5). We then analyzed the binding capacity of mutant peptides on cholesterol liposomes. For peptides 2 and 3, we mutated key residues of the CARC consensus motifs (Sheng et al, 2012; Nishio et al, 2014). For peptide 1, we introduced a single mutation which is the orthologous mutation of human disorder Arg225Cys (Zhang et al, 2013; Fig 6A). SPR experiments showed that the binding capacities of all three mutant peptides were significantly reduced on cholesterol‐loaded liposomes (Fig 6B–D). These results indicate that cholesterol directly binds to at least three domains of Nav1.9; however, whether these docking sites play a role in Nav1.9 modulation remains to be investigated.
Figure 6. Nav1.9 channels possess cholesterol‐binding domains.
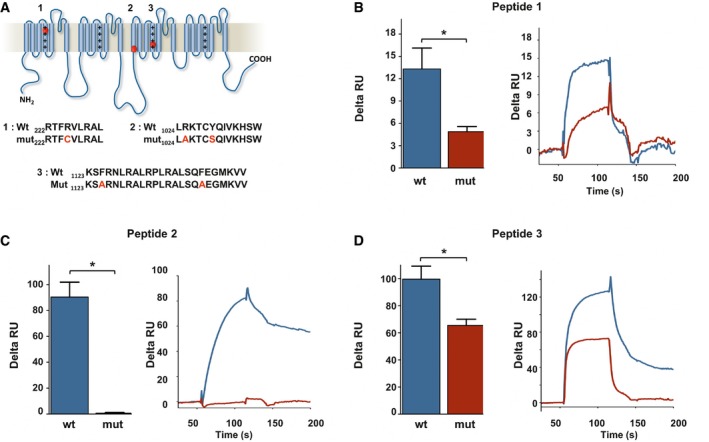
-
ALocalization of three potential cholesterol‐binding domains on Nav1.9 protein. Sequence of wt and mutant peptides tested by SPR is indicated below.
-
B–DCholesterol dependence of membrane binding of peptides 1–3. Peptides (20 μM) were injected on DOPC/DOPS/cholesterol liposomes captured on a L1 sensor chip. Results are presented as the difference of the resonance unit (RU) measured between cholesterol 60% and cholesterol 0% containing liposomes for each injection. Triplicate injections were performed for each independent experiment (n = 6 for wt peptides, blue; and n = 3 for mutant peptides, red). A representative sensorgram of each condition is illustrated on the right panels. Data are shown as mean ± standard error of the mean (SEM) and were analyzed with non‐parametric Mann–Whitney U‐test. *P < 0.05.
Cholesterol delivery restores Nav1.9 channel properties
Altogether, our results suggest that decreased interactions with cholesterol facilitate Nav1.9 activation, inducing neuronal hyperexcitability and subsequent hyperalgesia. Therefore, we reasoned that delivery of cholesterol could reverse all these events. We first investigate the consequence of cholesterol delivery on DRG neurons treated with inflammatory cocktail. Properties of Nav1.9 currents were examined in voltage‐clamp experiments as previously described. Exposure of DRG neurons for 20 min to the inflammatory cocktail, which reduces neuronal cholesterol level (Fig 1F), produced a negative shift of the voltage dependence of Nav1.9 activation (Fig 7A–C). The potential for half‐maximal activation shifted from −27.3 ± 0.2 mV in control conditions (Fig 4D) to −34.6 ± 0.7 mV in the presence of inflammatory mediators, very much similar to what we observed after MβCD or cholesterol oxidase treatment (Figs 7D and 4D). We tested whether cholesterol enrichment could counteract the effect of the inflammatory soup. Indeed, pre‐incubation of 20 mM MβCD‐chol for 10 min fully prevented the effects of inflammatory mediators, with half‐activation potential close to normal value (V 0.5 = −26.7 ± 0.3 mV; Fig 7D). Of note, MβCD‐chol applied alone for 15 min had no significant effect on Nav1.9 activation potential (V 0.5 = −28.9 ± 0.7 and −27.3 ± 0.2 mV for cholesterol‐enriched and control neurons, respectively; Figs 7D and 4D). Importantly, MβCD‐chol also prevented the effects of GTPγS, a non‐hydrolyzable G protein‐activating analog of GTP, which bypasses inflammatory membrane receptors. V 0.5 values of Nav1.9 activation in the presence of internal GTPγS were −27.4 ± 0.7 and −49.2 ± 0.8 mV with and without MβCD‐chol treatment, respectively (Fig EV4). These results show that cholesterol supply to DRG neurons reduces the potentiation of Nav1.9 current triggered by inflammatory mediators and downstream G protein signaling pathways.
Figure 7. Cholesterol delivery prevents the modulation of Nav1.9 currents by inflammatory mediators.
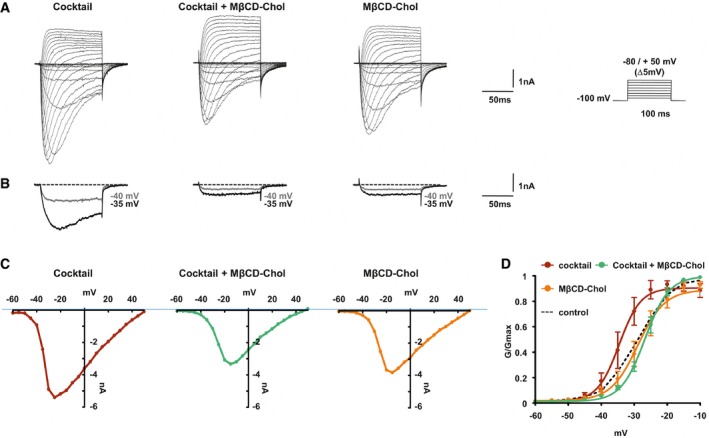
- Representative current traces evoked by 100‐ms depolarizing voltage steps ranged from −80 to +50 mV (Δ 5 mV, Vh = −100 mV) of DRG neurons treated with inflammatory cocktail, inflammatory cocktail plus MβCD‐chol, or MβCD‐chol alone. DRG neurons were pre‐incubated for 15 min at 37°C with DMEM alone or supplemented with 20 mM of MβCD‐chol; when used, the inflammatory cocktail was added to extracellular solution at room temperature (RT).
- Nav1.9 currents evoked at −40 and −35 mV (gray and black traces, respectively) extracted for clarity's sake from (A).
- Corresponding current–voltage relationships of Nav1.9 currents treated with an inflammatory cocktail, inflammatory cocktail plus MβCD‐chol, or MβCD‐chol alone.
- Mean activation curves of Nav1.9 currents recorded in the different conditions fitted with single Boltzmann equations. Values for V 0.5 of activation are −27.3 ± 0.2 mV in control conditions (n = 5, black dashed line, same as Fig 4B), −28.9 ± 1.6 mV for MβCD‐chol (n = 8, orange line), −34.5 ± 2 mV for inflammatory cocktail (n = 7, red line), and −26.7 ± 0.7 mV for MβCD‐chol + inflammatory cocktail (n = 11, green line). Values are shown as mean ± standard error of the mean (SEM).
Figure EV4. Activation of Nav1.9 by GTPγS, a non‐hydrolyzable G protein‐activating analog of GTP, is prevented by soluble cholesterol.
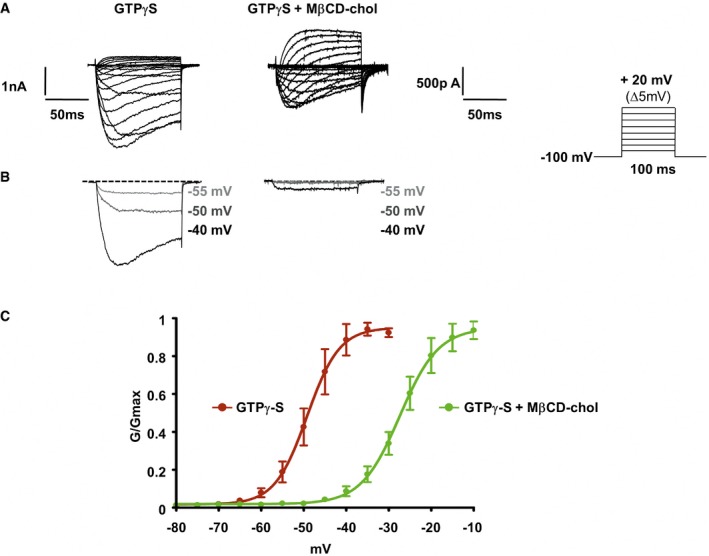
- Representative current traces evoked by 100‐ms depolarizing voltage steps from −80 to +20 mV (Δ5 mV, Vh = −100 mV). Nav1.9 currents were recorded 10 min after achieving whole‐cell recording configuration with patch pipette solution containing 400 μM of GTPγS. In the right panel, DRG neurons were pre‐incubated prior recording with MβCD‐chol (20 mM) for 10 min.
- Currents evoked at −55, −50, and −40 mV from (A) are illustrated.
- Corresponding activation curves of Nav1.9 current fitted with a single Boltzmann equation. Values for V 0.5 of activation are −27.4 ± 0.7 mV (n = 7, green line) and −49.2 ± 0.8 mV (n = 8, red line) with and without MβCD‐chol, respectively. Values are shown as mean ± standard error of the mean (SEM).
Topical application of cholesterol‐enriched gels eases inflammatory pain
Cholesterol was delivered by using MβCD‐chol complex, and further treatment concentrations are calculated based on cholesterol weight. We then tested the effect of cholesterol enrichment in vivo on inflammatory pain. In a first set of experiments, we co‐injected 5.6 mM MβCD‐chol complex with the inflammatory agent λ‐carrageenan. We observed that mechanical allodynia induced by λ‐carrageenan was significantly attenuated for at least 5 h (Fig 8A). MβCD‐chol turned out to be as potent as intraperitoneal injection of ibuprofen (75 mg/kg) at alleviating λ‐carrageenan‐induced pain behaviors (Fig 8A). The present results indicate that water‐soluble cholesterol possesses a local peripheral antalgic action in the carrageenan subacute inflammation model.
We further developed cholesterol transdermal gels with a capability for topical drug delivery. Our formulations were made of hydroxyethyl cellulose (HEC) polymer as a gelling agent, supplemented with two concentrations of soluble cholesterol. In vitro skin irritation tests on reconstituted human epidermis showed no toxicity of both formulas as tested by a MTT viability assay (Fig EV5A). The gel supplemented with the highest dose (28 mM) of cholesterol did not modify the overall structure of the treated skin (Fig EV5B). We then assessed the capability of cholesterol gels to restore normal levels of cholesterol of an inflamed skin. Thirty minutes after carrageenan injection, cholesterol‐free gel (chol 0) or gel containing 5.6 mM soluble cholesterol was applied to the site of inflammation for 1.5 h under anesthesia. Inflamed skin hind paw treated with the cholesterol‐free gel showed a typical reduction in cholesterol content (−17 ± 2.2% as compared to the contralateral paw; Fig 8B). On the contrary, when mice were treated with the 5.6 mM cholesterol gel, inflamed skin exhibited a minor reduction (−8 ± 3.1%) in cholesterol content, indicating efficient transcutaneous delivery of cholesterol to skin cells. We next evaluated the analgesic and anti‐inflammatory activities of cholesterol gels in the carrageenan‐induced pain model. A significant in vivo analgesic effect was produced by the gel containing 5.6 mM cholesterol for several hours, compared with cholesterol‐free gel (Fig 8C). However, no noticeable change in paw edema volume was observed, indicating that cholesterol formulation does not interfere with inflammatory process. Saline‐injected animals, treated with the blank gel, were also included to ensure that anesthesia did not modify normal mechanical sensitivity of mice (Fig 8C). We also checked that cholesterol gel does not induce anesthesia of naïve mice (Fig EV5C). We next aimed at evaluating the analgesic potential of cholesterol formulation in a model of rheumatoid arthritis (Chillingworth & Donaldson, 2003), using injection of complete Freund's adjuvant (CFA) into the left ankle. In this model of chronic inflammatory pain, the gels were applied 7 days after CFA injection, when mice exhibited severe swelling of the ankle and prominent mechanical allodynia (Fig 8D). We found that cholesterol formulations alleviated pain in a dose‐dependent manner. The gel containing 5.6 mM cholesterol had a similar antalgic action to that containing 5% ibuprofen, while application of the 28 mM cholesterol gel had a stronger effect over several hours and transiently restored withdrawal threshold to normal values (Fig 8D). Thus, our data show an antalgic effect of cholesterol supply on both subacute and chronic arthritic pain.
Figure EV5. Cholesterol gels do not impair cell viability, skin structure, and mechanical threshold.
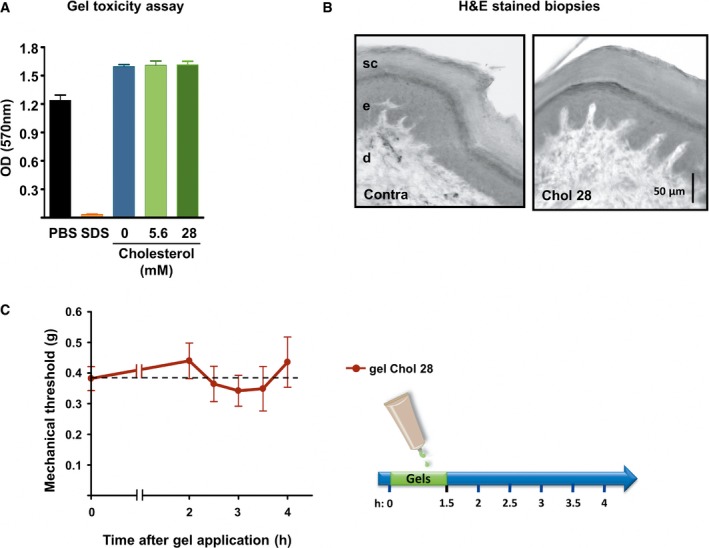
- Cell viability of reconstructed human epidermis measured with an MTT assay 48 h after exposure to PBS (non‐toxic control, n = 3), 5% SDS (toxic control, n = 3), HEC gel containing no cholesterol (0 mM, n = 3), and HEC gels containing soluble cholesterol at the concentration of 5.6 or 28 mM (n = 3 each).
- Hematoxylin–eosin staining of standardized skin biopsy specimen from paw treated for 1.5 h with an HEC gel containing 28 mM of cholesterol. sc: stratum corneum; e: epidermis; d: dermis.
- Mechanical withdrawal threshold of mice treated for 1.5 h with an HEC gel containing 28 mM of cholesterol (n = 9).
Discussion
The present study shows that cholesterol depletion occurs in inflamed skin tissues and that this decrease may trigger pain signaling. However, the global decrease (~18%) in cholesterol that was observed in inflamed skin biopsies suggests that cholesterol level may be lowered in keratinocytes, which is the predominant cell type (~90%) in the epidermis. We provide evidence to suggest that cholesterol depletion also occurs in DRG cultures (~16%) exposed to inflammatory mediators, and demonstrate that reducing membrane cholesterol level causes enhanced excitability of small‐diameter pain‐processing neurons. Although we cultured DRG cells under conditions that minimize the presence of fibroblasts and primary Schwann cells, such cultures contain heterogeneous populations of neurons and satellite cells, which makes difficult to ascertain the exact proportion of decrease in cholesterol to the neuronal population.
How inflammatory reaction regulates membrane cholesterol content is currently unknown. However, two mechanisms, with possible synergistic action, may be at play: an imbalanced reverse cholesterol transport (RCT) and an induction of oxidative stress. RCT is initiated by the binding of cholesterol on ApoA protein and high‐density lipoprotein (HDL). In plasma membrane compartment, cholesterol is subsequently converted to cholesteryl ester and transported to the liver for elimination. In favor of this hypothesis, histamine and bradykinin, two inflammatory mediators, have been shown to enhance both transendothelial penetration of HDL into skin interstitial fluid and transfer of cholesterol into serum compartment (Kareinen et al, 2015). The second mechanism relies on enhanced production of ROS, nitric oxide (NO) synthesis, and the interplay of these molecules (Salvemini et al, 1996; Wang et al, 2004; Khattab, 2006; Gamper & Ooi, 2015). DRG sensory neurons and skin cells express a combination of enzymes implied in oxidative stress and activated by inflammatory factors such as NO synthase (NOS) isoforms (Levy et al, 2000; Cals‐Grierson & Ormerod, 2004; Boettger et al, 2007) and isoforms of the catalytic subunit of NADPH oxidase (Ibi et al, 2008; Valencia & Kochevar, 2008). An enhanced oxidative stress on membrane cholesterol would result in net decrease in cholesterol level together with the formation of various oxysterol bioactive molecules with potential destabilizing effect on raft compartments (Massey, 2006; Murphy & Johnson, 2008). Our data generated in vitro on DRG cultures support the oxidative stress hypothesis due to the absence of sources of ApoA1 and HDL and the increased ROS production observed upon exposure to the inflammatory cocktail. However, both mechanisms probably synergize in vivo.
We show that pain hypersensitivity mediated by cholesterol depletion was strongly reduced in Nav1.9 KO mice. Although not a proof for a direct regulation, this provides evidence that MβCD‐evoked pain hypersensitivity relies on Nav1.9 channel activity. Other channels/mechanisms are likely to operate as deletion of Nav1.9 did not fully abrogate the painful behavior of MβCD‐injected mice (Dart, 2010; Levitan et al, 2010; Pristera et al, 2012). Cholesterol efflux (MβCD) or cholesterol oxidation shifted the voltage dependence of activation and fast inactivation of Nav1.9 channels toward more negative potentials, hence promoting opening of the channels. Such modulation of Nav1.9 voltage dependence was also observed when DRG neurons were bathed with the inflammatory cocktail, mechanism of which depended on ROS production. Importantly, the effects of cholesterol efflux (MβCD), inflammatory mediators, and GTPγS were all strongly prevented by pre‐treatment with soluble cholesterol, arguing for a key role of cholesterol concentration in Nav1.9 modulation. Consistent with a functional relationship between cholesterol and Nav1.9, we found that Nav1.9 is enriched in lipid raft microdomains of DRG neuron membrane. This preferential partitioning was altered by the action of inflammatory mediators, which caused a redistribution of the main pool of Nav1.9 channels to non‐raft compartments. This suggests that Nav1.9 redeployment may represent an important regulatory mechanism favoring opening of Nav1.9 channels. Thus, the overall picture that emerges from our study is that cholesterol oxidation caused by inflammation/ROS production may lead to decreased levels of membrane cholesterol in the vicinity of Nav1.9 channels, promoting Nav1.9 channel relocation/opening, hyperexcitability, and pain signaling (Fig 9). However, we cannot rule out a direct oxidation of Nav1.9 channels by ROS, which may reduce channel's retention at rafts. Inflammation‐triggered cholesterol depletion pathway should be seen as one of the multiple pathways that concurrently lead to nociceptor sensitization during inflammation (Fig 9).
Figure 9. Overview of DRG neuron signaling pathways involved in inflammatory pain hypersensitivity.
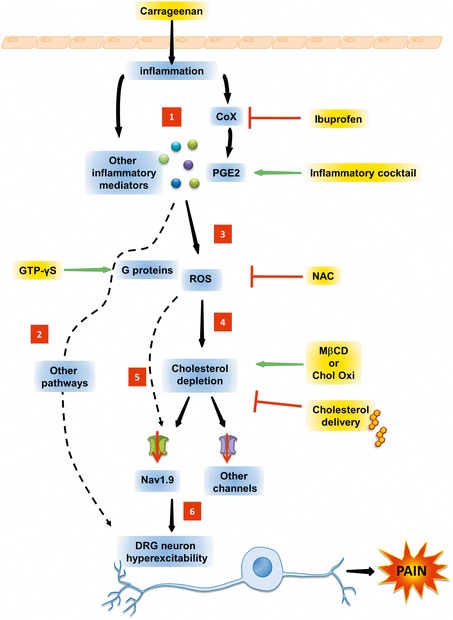
Injection of carrageenan stimulates the synthesis and/or secretion of inflammatory products (1) including PGE2, which in turn activate multiple membrane receptors/signaling pathways (2) and production of reactive oxygen species (ROS) (3). ROS may reduce the level of membrane cholesterol (4) and/or directly oxidate Nav1.9 channels, among others (5). This causes neuronal hypersensitization (6) that transforms nociceptor phenotype to pathophysiologic states of persistent nociceptor activation, lowered firing thresholds, and/or exaggerated pain responses.
Three different but not mutually exclusive hypotheses may be considered to explain the difference in the voltage‐dependent properties of Nav1.9 channels housed in raft and non‐raft domains (Dart, 2010; Levitan et al, 2010). First, the strong density of lipid head groups in rafts could lead to a modification of the transmembrane potential or a reshaping of the electric field profile through the voltage‐sensor domain of the channel. Second, regions of membrane enriched in cholesterol are known to be thicker and more rigid than the surrounding membrane (Andersen & Koeppe, 2007). Therefore, interactions between the lipids and the S4 basic residues may affect membrane mobility of the voltage‐sensor domain. Last, lipid rafts can provide a spatial and temporal meeting point for regulatory signaling molecules that might be transiently or permanently associated with Nav1.9 channels. Further experiments are needed to characterize precisely the cause(s) of Nav1.9 voltage‐dependent modulation.
Regarding the relationship between Nav1.9 and cholesterol, the present report suggests the possibility of a direct lipid–protein interaction. Analysis of mouse Nav1.9 sequence revealed the existence of several putative cholesterol‐binding domains (CRAC, CARC, or CCM) distributed along the four repeated structural domains of the channel, suggesting that Nav1.9 may interact with a belt of cholesterol molecules such as described for the nicotinic acetylcholine receptor (Baier et al, 2011) and the Kir2.1 channel (Rosenhouse‐Dantsker et al, 2011). Because of the loose stringency of these motifs, their predictive value has been questioned. However, proteins that unambiguously bind cholesterol like the benzodiazepine receptor, caveolin or ApoA1, contain these domains and site‐directed mutagenesis of these domains disrupts cholesterol‐binding capacity (Li & Papadopoulos, 1998; Epand et al, 2005; Dergunov, 2013). We provide evidence that at least three regions of the Nav1.9 channel do have cholesterol‐binding activity. Remarkably, two of these peptides, located in S4 domains of Nav1.9, encompass orthologous single‐point mutations discovered in human genetic disorders associated with neuropathic pain hypersensitivity syndrome and neuronal cell hyperexcitability (Zhang et al, 2013; Huang et al, 2014; Okuda et al, 2016). Given the difficulties inherent to Nav1.9 expression in heterologous systems (Vanoye et al, 2013; Lin et al, 2016), the generation of knock‐in mice harboring R225C and L1158P orthologous mutations will represent a genetic tool of choice for future studies to address the cholesterol dependency of these Nav1.9 point mutants.
Our study also raises the proof of concept that cholesterol formulation alleviates inflammatory pain. Transdermal drug delivery presents several advantages such as skin accessibility, exposure to the vascular and lymphatic networks, and non‐invasiveness of the delivery. It is particularly appropriate for drugs that may cause systemic side effects such as non‐steroidal anti‐inflammatory drugs. We developed cholesterol‐loaded gel formulation prototypes. We showed that permeation of the formulations across the skin prevents part of the decrease in cholesterol in inflamed skin. These data indicate that the formulations efficiently deliver cholesterol molecules to mouse skin in vivo. The formulations produced significant in vivo analgesic effects in the carrageenan and the CFA‐induced arthritis inflammation models, two inflammatory pain models in which Nav1.9 has an established role (Priest et al, 2005; Amaya et al, 2006; Lolignier et al, 2011). The low‐dose formulation appeared to be as potent as the NSAID ibuprofen for pain relief. High‐dose formulation was able to restore transiently mechanical threshold of arthritic mice. Improvement of long‐term controlled cholesterol delivery will be an interesting issue for pain therapy. Since gain of function of Nav1.9 currents has also been implicated in some neuropathic pain including diabetes (Hong & Wiley, 2006) and small‐fiber neuropathies (Zhang et al, 2013; Huang et al, 2014; Okuda et al, 2016), further characterization of cholesterol gel delivery in these pathological contexts is of potential interest.
In conclusion, our study brought to the light a novel role of cholesterol as an endogenous painkiller molecule. While depletion of cholesterol may be a critical initial event promoting pain, cholesterol supply has analgesic action in inflammatory pain models. The impact of cholesterol depletion on pain processing represents a mechanism to consider in the efforts aiming to understand nociceptor sensitization not only in inflammatory conditions, but also in some neuropathic contexts.
Materials and Methods
Animals
Animals were housed under controlled environmental conditions and kept under a 12‐/12‐h light/dark cycle, with food and water ad libitum. Mice used in this study were 6‐ to 7‐week‐old males from C57bl6, Nav1.8‐null, Nav1.9‐null genetic strains, and their respective wild‐type littermates (Akopian et al, 1999; Amaya et al, 2006). All procedures were in accordance with the directives of the French Ministry of Agriculture and Fisheries and the European Communities Council (86/609/EEC). Since pain might result from these experiments, the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain were followed. Behavioral tests and pain models used in this study were all approved by the ethics committee of Région PACA (France).
Inflammatory pain models and behavioral assays
Subacute paw inflammation was induced by intraplantar injection of 20 μl of saline solution supplemented with 2 or 3% λ‐carrageenan (Sigma‐Aldrich). Monoarthritis was induced under 2% isoflurane anesthesia by two subcutaneous injections of 15 μl of complete Freund adjuvant (CFA; prepared with 5 μg/μl heat‐killed Mycobacterium tuberculosis (Becton, Dickinson)), in each side of the tibio‐tarsal joint (Lolignier et al, 2011).
Mechanical sensitivity was assessed by von Frey filaments. Mice were let to settle down in a restricted area for 1 h before determining their basal mechanical threshold. Filaments of increasing force were applied five times each and pressed perpendicularly to the plantar surface of the hind paw until they bent. The first filament that evoked at least three withdrawal responses was assigned as the pain threshold in grams. Thermal sensitivity was assessed with a plantar test device (Hargreaves's method (Hargreaves et al, 1988)). Mice were let to settle down for 1.5 h in an open area, until cessation of exploratory behavior. Infrared (IR) source was set on 20%. Thermal threshold was expressed as the mean withdrawal latency of three measures at 5‐min intervals. A cutoff time of 20 s was applied to avoid injury. Basal threshold of animal was determined before drug application (t = 0).
DRG neuron cultures
Culture of DRG neurons has been previously described (Coste et al, 2007). Briefly, thoraco‐lumbar DRG ganglia were excised from intervertebral foramina, ventral and dorsal roots were then cut as close as possible, and connective tissues were removed to minimize Schwann cell and fibroblast contamination. Ganglia were incubated in enzyme solution containing 2 mg/ml of collagenase IA (Sigma‐Aldrich) for 45 min at 37°C before trituration in Hanks’ medium (Life Technologies). Culture medium was Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% heat‐inactivated fetal calf serum, 50 U/ml penicillin–streptomycin, 2 mM l‐glutamine, 25 mM glucose, 2 ng/ml glial‐derived neurotrophic factor (GDNF) (all from Life Technologies), and 25 ng/ml nerve growth factor (NGF, Millipore). Cells were seeded on 40‐mm culture dishes (Nunc) coated with 1 mg/ml laminin (Sigma‐Aldrich) and maintained in a humidified atmosphere (5% CO2, 37°C) for 16–24 h before experiments.
Drugs and chemicals
The 1× inflammatory cocktail solution contained 50 nM bradykinin (BK), 500 nM prostaglandin‐E2 (PGE2), 1 μM histamine (His), 500 nM norepinephrine (NE), and 2 μM ATP (all from Sigma‐Aldrich) (Maingret et al, 2008). The 1× control vehicle solution contained 0.005% ethanol, 0.0005% acetic acid, and 830 nM of HCl, used to dissolve PGE2, BK, and NE, respectively. MβCD‐chol also named water‐soluble cholesterol (Sigma‐Aldrich, or MP Bio) refers to a complex of MβCD saturated with cholesterol. The complex contains about 40 mg of cholesterol/g solid. Thus, the molar ratio of each molecule in the complex is about 7.1:1 for MβCD and cholesterol, respectively. Hydroxyethyl cellulose, MβCD, and αCD were from Sigma‐Aldrich. ChOxi is a flavoenzyme that catalyzes the oxidation and isomerization of cholesterol to cholest‐4‐en‐3‐one. The enzyme is extracellular and binds transiently to the membrane surface during catalysis (Vrielink, 2010). All drugs were prepared daily.
Intraplantar injection
For intraplantar injections, 20 μl of the following drugs diluted in saline solution was used: 40 mM MβCD; 40 mM αCD; 40 mM MβCD‐chol (based on MβCD concentration); 20× inflammatory cocktail; 4 U/ml cholesterol oxidase; and 20 mM N‐acetyl‐L‐cysteine (NAC; Sigma‐Aldrich). When cited, ibuprofen (75 mg/kg from a freshly prepared 7.5 mg/ml stock solution, Sigma‐Aldrich) was injected intraperitoneally, 1 h prior to λ‐carrageenan or MβCD intraplantar injection. Other mice of the same set of experiment were injected intraperitoneally with the same volume of saline solution 1 h prior to αCD, MβCD, MβCD‐chol, or λ‐carrageenan, intraplantar injection (Figs 2B, 3C and 8A).
Skin biopsies and cholesterol quantification
For skin cholesterol dosages, a 3.5‐mm punch biopsy specimen was taken from ipsi‐ and contralateral hind paws of each mouse and immediately processed for lipid extraction and fluorimetric assay according to the manufacturer's instructions (Sigma‐Aldrich). Mayer's eosin and hematoxylin (Sigma‐Aldrich) staining of biopsy specimen was performed on 10‐μm frozen tissue sections.
Cholesterol quantification of DRG neurons
Dissociated DRG neurons from a single mouse were equally plated into two culture dishes and let grown for 16 h. Both dishes were washed with DMEM in order to eliminate cellular debris. The control dish was incubated for 15 min at 37°C with 2 ml of DMEM + vehicle, while the twin dish was incubated in 2 ml DMEM supplemented with the drug of interest at the following concentrations: 1× cocktail; 20 mM MβCD; 20 mM MβCD‐chol (based on MβCD concentration); and 2 U/ml cholesterol oxidase. Cholesterol ratio refers to the amount of cholesterol in drug‐treated dishes over the amount of cholesterol in vehicle‐treated dishes. Cholesterol content was determined using enzymatic fluorimetric assay (Sigma‐Aldrich) according to the manufacturer's instructions.
ROS monitoring
ROS production was measured with the fluorescence indicator 5‐ (and 6‐)chloromethyl‐2′, 7′‐dichlorodihydrofluorescein diacetate acetyl ester (CM‐H2DCFDA, Molecular Probes, Inc) (Ma et al, 2009). DRG cultures were loaded with 1 μM of CM‐H2DCFDA in PBS for 30 min at 37°C. Cells were let to recover for 30 min in pre‐warmed PBS. Fluorescent imaging was performed on an Olympus cell R device and software with 480 ± 20/535 ± 20 nm excitation/emission filter. Images were acquired every 30 s for 60 min. The baseline correction for each cell was determined 5 min after the beginning of the recording just before drug application. Data are presented as the delta of mean gray value of fluorescence intensity in cells after background subtraction. 0.3% H2O2 was used as positive control of ROS production. N‐tert‐butyl‐α‐phenylnitrone (Sigma‐Aldrich) was used at 4 mM concentration as a ROS scavenger.
Detergent‐resistant lipid raft isolation
Freshly collected DRGs were incubated at 37°C with 1× inflammatory cocktail or 40 mM MβCD in DMEM solution. Control DRGs were incubated in DMEM solution. Ganglia were pelleted at 1,000 g for 1 min and resuspended in 140 μl of lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.5; 2 mM EDTA, 0.5% Triton X‐100 with protease inhibitor cocktail (Roche)) before homogenization with a Dounce homogenizer. All samples were incubated for 1 h at 4°C and then centrifugated at 11,000 g for 2 min. The supernatants were adjusted to a final Optiprep (Axis‐shield) concentration of 40%. The cell lysates were overlaid with 1.6 ml of 30% and 200 μl of 0% Optiprep step gradient and then subjected to 260,000 g (Beckman TLS‐55 rotor) for 4 h at 4°C. Fractions were collected from the top (fraction 1) to the bottom (fraction 12) of the gradient, in 150‐μl increments. 75 μl of 3× Laemmli buffer (supplemented with 0.1 M DTT) was added to each fraction. Samples were denatured for 5 min at 95°C. Equal volumes of each fraction were then electrophoresed on 4–12% gradient SDS–PAGE (Life Technologies). Proteins were transferred to nitrocellulose membranes (Whatman) and probed with rabbit anti‐Nav1.9 (Padilla et al, 2007), rabbit anti‐flotillin‐1 (Sigma‐Aldrich), rabbit anti‐caveolin‐1 (Santa Cruz), and mouse anti‐transferrin receptor (Life Technologies). Chemiluminescent detection was performed using Roche ECL reagents and Kodak BioMax MS film. DRGs treated for 15 min at 37°C with the inflammatory cocktail or DMEM were proceed as described above. Quantification of protein level was assessed using ImageJ software on unsaturated signals. Saturated black signal was attributed to 255 value and white to 0. For each picture, total signal of each fraction and three backgrounds were measured using a same rectangular selection. Averaged background (from the three background selections) was subtracted to signal of each fraction. For each experiment, signals of the 12 fractions were added and correspond to total protein quantity.
Patch‐clamp recording
Patch‐clamp recordings were achieved using an Axopatch 200B amplifier (Axon Instruments), filtered at 2 kHz, and digitally sampled at 20 kHz using PCLAMP 10 software. Currents were leak‐subtracted using a P/6 protocol. Voltage errors were minimized using 70–85% series resistance compensation. Patch pipettes had resistances of 2–3.5 MΩ. Extracellular solution (Kreb's) was (in mM) 131 NaCl, 10 glucose, 3 KCl, 1 MgCl2, 10 HEPES, and 2.5 CaCl2 (pH 7.35, 293 mOsm/l). For whole‐cell voltage‐clamp recordings of Na+ currents, intracellular pipette solution contained (mM) 130 CsCl, 10 HEPES, 8 NaCl, 5 EGTA, 2.4 CaCl2, 1 MgCl2, 4 MgATP, and 0.4 Na2GTP (pH 7.3, 298 mOsm/l). Extracellular solution was supplemented with 500 nM tetrodotoxin (TTX; from a stock solution 0.1 mM in water, Abcam), 1 mM amiloride hydrochloride (from a stock solution 1 M in 0.1% dimethylsulfoxide, Sigma‐Aldrich), and 50 μM La3+ (from a stock solution 1 M, Sigma‐Aldrich) in order to block TTX‐sensitive Na+ currents and Ca2+ currents (Coste et al, 2007). For current‐clamp recording, intracellular pipette solution contained (mM) 130 KCl, 10 HEPES, 8 NaCl, 5 EGTA, 2.4 CaCl2, 1 MgCl2, 4 MgATP, and 0.4 Na2GTP (pH 7.3, 298 mOsm/l). In Fig EV4, GTPγS (0.4 mM) was substituted for GTP. Extracellular media were exchanged using a gravity‐fed bath perfusion system at a flow rate of 2 ml/min, while bath solution was removed by continuous suction. All experiments were performed at room temperature.
Cell capacitance was estimated in the voltage‐clamp mode from the time constant of the decay phase of a current transient elicited by a 10‐mV hyperpolarizing step. DRG neurons used in this study had a cell membrane capacitance ranging from 15 to 37 pF. Current threshold was determined as the value of injected membrane current necessary to induce an action potential.
For voltage‐clamp experiments, DRG cultures were pre‐treated for 15 min at 37°C with either DMEM (control), 10–20 mM MβCD (in DMEM), 20 mM MβCD‐chol (based on MβCD concentration), or cholesterol oxidase (2 U/ml), excepted for Appendix Fig S3 where acute application of cyclodextrins was performed. The inflammatory cocktail ± 6 mM NAC or its vehicle (1×) was incubated in extracellular medium at room temperature. For current‐clamp experiments, acute application of drugs was performed in extracellular medium. In Fig EV1, 20 mM MβCD‐chol was first pre‐incubated for 10 min before recording and acute application of the inflammatory cocktail.
For data analysis, conductance–voltage curves were calculated for each cell according to the equation G = I/(V − E rev), where V is the test pulse potential and E rev is the reversal potential calculated according to the Nernst equation. The conductance–voltage curve was fitted using the Boltzmann function: G/G max= 1/(1+ exp[(V 0.5− V)/k]), where G/G max is the normalized conductance, V 0.5 is the potential of half‐maximum channel activation, and k is the steepness factor. PRISM 4.0 (GraphPad) software was used to perform linear and non‐linear fitting of data.
Cholesterol‐binding assays
Liposomes were prepared with different ratio of 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC), and cholesterol (chol) (all from Avanti Polar Lipids). Lipids were dissolved in chloroform (VWR chemicals). Lipid solutions were mixed to obtain 85% DOPC/15% DOPS, or 25% DOPC/15% DOPS/60% cholesterol (mol/mol), stabilized with 50 nM butylated hydroxytoluene (BHT; Sigma‐Aldrich), and then evaporated under N2. 850 nmoles of dried lipids was used in each condition. Lipid films were hydrated with phosphate‐buffered saline (100 mM phosphate buffer, 2.7 mM potassium chloride, and 137 mM sodium chloride, pH 7.4) to yield a lipid concentration of 1.7 mM and heated to 60°C. The dispersed lipids (0.5 ml) were sonicated on ice for 40 s. Liposomes were then performed by 19 passes through 100‐nm polycarbonate filters using a Lipofast basic apparatus (Avestin, Ottawa) and stored at 4°C.
SPR experiments were performed at 25°C on a Biacore 3000 system (GE Healthcare, Uppsala, Sweden) using an L1 sensor chip (GE Healthcare) and 0.22‐μm‐filtered and degassed PBS (Sigma‐Aldrich) as running buffer. Surface was conditioning with two 30‐s injections (5 μl/min) of 40 mM n‐octyl‐β‐glucopyranoside (Euromedex). Cholesterol containing liposomes were injected (5 μl/min) into the experimental flow cells and liposomes without cholesterol into the control flow cell to yield the same resonance units (around 4,500 RU) of bound liposomes. Peptides were diluted in running buffer and injected at 50 μl/min for 60 s. Binding signals measured on control flow cell were subtracted from binding signals measured on experimental flow cells to give specific binding signal on cholesterol. Chips were regenerated with 10 mM NaOH between each injection.
Transdermal delivery of cholesterol and toxicity tests
For transdermal delivery, 2% hydroxyethyl cellulose (Sigma‐Aldrich) dissolved in pure water was supplemented with water‐soluble cholesterol (5.6 or 28 mM based on cholesterol concentration; Sigma‐Aldrich, or MP Bio) and let polymerize for 1 h to 1 h 30 min at room temperature under 360° vertical rotation at 30 rpm. Small amount of gel, typically 1 ml, was prepared daily just prior utilization. 100 μl of gel was applied on mouse hind paw for carrageenan experiment or on the hind paw and the ankle for the CFA experiment, under 2% isoflurane anesthesia for 1.5 h. Skin paw was then carefully cleaned with saline solution, and recovery from anesthesia was allowed for 30 min before assessment of mechanical thresholds.
Cutaneous irritation tests followed the OECD TG439 guideline for the testing of chemicals. Briefly, reconstructed human epidermis (SkinEthic) was exposed to either a 5% SDS solution (toxic control), PBS (non‐toxic control), or HEC gels with 0, 5.6, or 28 mM of soluble cholesterol, for 42 min. Analysis of cell viability was performed 42 h later using a MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) viability assay.
Immunohistochemistry and imaging
Immunofluorescence staining was performed as previously described (Padilla et al, 2007). Briefly, 14‐μm‐thick skin tissues were incubated with primary antibodies (anti‐Nav1.9 L23, 1/400 dilution; anti‐peripherin, 1/400 dilution (Chemicon)). Goat anti‐rabbit IgG Alexa Fluor488 (1/200, Molecular Probes) and goat anti‐mouse IgG Alexa Fluor546 (1/400, Molecular Probes) were used as secondary antibodies. Confocal image acquisition was performed on a Leica TCS SP2 laser‐scanning microscope (Leica Microsystems, Mannheim, Germany) using the 488‐nm band of an Ar laser and the 543‐nm band of an He–Ne laser for excitation of Alexa 488 (spectral detection 500–560 nm) and Alexa 546 (spectral detection 560–660 nm), respectively. High‐magnification images were acquired using a 40× oil immersion objective (numerical aperture 1.25). Pinhole was adjusted to the first Airy disk resulting in a calculated lateral and axial resolution of approximately 156–175 nm and 471 nm, respectively. Pixel size was adjusted to 61 nm by adjusting the electronical zoom. For double‐staining, light emitted from the two fluorophores was detected sequentially. Three‐dimensional stacks of confocal high‐magnification images were obtained using a 120‐nm step. Images were reconstructed by projection of Z‐series optical sections. To enhance resolution, the images were deconvolved based on a theoretical point‐spread function (PSF) using AutoQuant X (Media Cybernetics, Rockville, USA). Image editing was performed using Adobe Photoshop (Adobe Systems, San Jose, CA).
Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad). For the experiments of skin cholesterol quantification, since the contralateral paw served as control for the paw in which the drug was applied, no randomization was used. When different substances were tested in vivo, groups of animals from the same cage were randomly injected with drugs or vehicle, so variation in mean baseline sensory thresholds was minimized.
Data from behavioral experiments were analyzed using two‐way ANOVA test. Other experiments used non‐parametric, two‐tailed P‐values with 95% confidence limits. Depending on the design of the experiment, paired (Wilcoxon signed‐rank test) or unpaired analysis (Mann–Whitney U‐test) was used as stated in the figure legends. All values are shown as mean ± standard error of the mean (SEM). P‐values were illustrated with * symbol with the following representation: *** for P < 0.001, ** for P < 0.01, and * for P < 0.05. P < 0.05 was considered statistically significant.
Author contributions
MA, FP, and PD performed electrophysiological recordings. MA, CP, and FP performed DRG cholesterol quantification and behavioral analysis. GF performed SPR experiments. All authors discussed the results. FP and PD conceived the project and wrote the manuscript.
Conflict of interest
F.P., M.A., and P.D have filed a patent for inhibitors of Na(v)1.9 channel activity and uses thereof for treating pain (no. PCT/EP2014/053390).
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Agence Nationale de la Recherche (ANR‐08‐MNPS‐025‐02, ANR‐09‐MNPS‐037‐01 to PD), the Fondation pour la Recherche Médicale (Équipes FRM 2013 DEQ20130326482 to PD), and the SATT Sud Est (to FP). We thank Dr B. Coste and M. Crest for critical reading. We thank A. Fernandez from the animal facility for technical assistance.
The EMBO Journal (2018) 37: e97349
See also: https://doi.org/10.15252/embj.201899231 (April 2018)
References
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN (1999) The tetrodotoxin‐resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2: 541–548 [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1999) Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19: 2181–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly‐Rosen D, Levine JD (2000) Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 20: 4680–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ (2006) The voltage‐gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci 26: 12852–12860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE II (2007) Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 36: 107–130 [DOI] [PubMed] [Google Scholar]
- Baier CJ, Fantini J, Barrantes FJ (2011) Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep 1: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettger MK, Uceyler N, Zelenka M, Schmitt A, Reif A, Chen Y, Sommer C (2007) Differences in inflammatory pain in nNOS‐, iNOS‐ and eNOS‐deficient mice. Eur J Pain 11: 810–818 [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK (1992) Sorting of GPI‐anchored proteins to glycolipid‐enriched membrane subdomains during transport to the apical cell surface. Cell 68: 533–544 [DOI] [PubMed] [Google Scholar]
- Cals‐Grierson MM, Ormerod AD (2004) Nitric oxide function in the skin. Nitric Oxide 10: 179–193 [DOI] [PubMed] [Google Scholar]
- Chillingworth NL, Donaldson LF (2003) Characterisation of a Freund's complete adjuvant‐induced model of chronic arthritis in mice. J Neurosci Methods 128: 45–52 [DOI] [PubMed] [Google Scholar]
- Coste B, Crest M, Delmas P (2007) Pharmacological dissection and distribution of NaN/Nav1.9, T‐type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol 129: 57–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart C (2010) Lipid microdomains and the regulation of ion channel function. J Physiol 588: 3169–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergunov AD (2013) Mutation mapping of apolipoprotein A‐I structure assisted with the putative cholesterol recognition regions. Biochim Biophys Acta 1834: 2030–2035 [DOI] [PubMed] [Google Scholar]
- Di Rosa M, Giroud JP, Willoughby DA (1971) Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J Pathol 104: 15–29 [DOI] [PubMed] [Google Scholar]
- Epand RM, Sayer BG, Epand RF (2005) Caveolin scaffolding region and cholesterol‐rich domains in membranes. J Mol Biol 345: 339–350 [DOI] [PubMed] [Google Scholar]
- Fantini J, Barrantes FJ (2013) How cholesterol interacts with membrane proteins: an exploration of cholesterol‐binding sites including CRAC, CARC, and tilted domains. Front Physiol 4: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Ooi L (2015) Redox and nitric oxide‐mediated regulation of sensory neuron ion channel function. Antioxid Redox Signal 22: 486–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanasekaran A, Sundukova M, van den Maagdenberg AM, Fabbretti E, Nistri A (2011) Lipid rafts control P2X3 receptor distribution and function in trigeminal sensory neurons of a transgenic migraine mouse model. Mol Pain 29: 7–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziani A, Rosker C, Kohlwein SD, Zhu MX, Romanin C, Sattler W, Groschner K, Poteser M (2006) Cellular cholesterol controls TRPC3 function: evidence from a novel dominant‐negative knockdown strategy. Biochem J 396: 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88 [DOI] [PubMed] [Google Scholar]
- Hong S, Wiley JW (2006) Altered expression and function of sodium channels in large DRG neurons and myelinated A‐fibers in early diabetic neuropathy in the rat. Biochem Biophys Res Commun 339: 652–660 [DOI] [PubMed] [Google Scholar]
- Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib‐Hajj SD, Merkies IS, Waxman SG (2014) Gain‐of‐function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137: 1627–1642 [DOI] [PubMed] [Google Scholar]
- Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe‐Nishimura C (2008) Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci 28: 9486–9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, Zhang H, Gamper N (2013) Activation of the Cl‐ channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal 6: ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kareinen I, Cedo L, Silvennoinen R, Laurila PP, Jauhiainen M, Julve J, Blanco‐Vaca F, Escola‐Gil JC, Kovanen PT, Lee‐Rueckert M (2015) Enhanced vascular permeability facilitates entry of plasma HDL and promotes macrophage‐reverse cholesterol transport from skin in mice. J Lipid Res 56: 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab MM (2006) TEMPOL, a membrane‐permeable radical scavenger, attenuates peroxynitrite‐ and superoxide anion‐enhanced carrageenan‐induced paw edema and hyperalgesia: a key role for superoxide anion. Eur J Pharmacol 548: 167–173 [DOI] [PubMed] [Google Scholar]
- Lee AG (2004) How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta 1666: 62–87 [DOI] [PubMed] [Google Scholar]
- Lenne PF, Wawrezinieck L, Conchonaud F, Wurtz O, Boned A, Guo XJ, Rigneault H, He HT, Marguet D (2006) Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J 25: 3245–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan I, Fang Y, Rosenhouse‐Dantsker A, Romanenko V (2010) Cholesterol and ion channels. Subcell Biochem 51: 509–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan I, Singh DK, Rosenhouse‐Dantsker A (2014) Cholesterol binding to ion channels. Front Physiol 5: 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Tal M, Hoke A, Zochodne DW (2000) Transient action of the endothelial constitutive nitric oxide synthase (ecNOS) mediates the development of thermal hypersensitivity following peripheral nerve injury. Eur J Neurosci 12: 2323–2332 [DOI] [PubMed] [Google Scholar]
- Li H, Papadopoulos V (1998) Peripheral‐type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139: 4991–4997 [DOI] [PubMed] [Google Scholar]
- Lin Z, Santos S, Padilla K, Printzenhoff D, Castle NA (2016) Biophysical and pharmacological characterization of Nav1.9 voltage dependent sodium channels stably expressed in HEK‐293 cells. PLoS ONE 11: e0161450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolignier S, Amsalem M, Maingret F, Padilla F, Gabriac M, Chapuy E, Eschalier A, Delmas P, Busserolles J (2011) Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE 6: e23083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Zhang L, Westlund KN (2009) Reactive oxygen species mediate TNFR1 increase after TRPV1 activation in mouse DRG neurons. Mol Pain 5: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Coste B, Padilla F, Clerc N, Crest M, Korogod SM, Delmas P (2008) Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol 131: 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JR, Navarro‐Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM (2000) Differential targeting of Shaker‐like potassium channels to lipid rafts. J Biol Chem 275: 7443–7446 [DOI] [PubMed] [Google Scholar]
- Massey JB (2006) Membrane and protein interactions of oxysterols. Curr Opin Lipidol 17: 296–301 [DOI] [PubMed] [Google Scholar]
- Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438: 612–621 [DOI] [PubMed] [Google Scholar]
- Monnaert V, Tilloy S, Bricout H, Fenart L, Cecchelli R, Monflier E (2004) Behavior of alpha‐, beta‐, and gamma‐cyclodextrins and their derivatives on an in vitro model of blood‐brain barrier. J Pharmacol Exp Ther 310: 745–751 [DOI] [PubMed] [Google Scholar]
- Murphy RC, Johnson KM (2008) Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J Biol Chem 283: 15521–15525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuvonen M, Manna M, Mokkila S, Javanainen M, Rog T, Liu Z, Bittman R, Vattulainen I, Ikonen E (2014) Enzymatic oxidation of cholesterol: properties and functional effects of cholestenone in cell membranes. PLoS ONE 9: e103743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabarti P (2014) CH‐pi hydrogen bonds in biological macromolecules. Phys Chem Chem Phys 16: 12648–12683 [DOI] [PubMed] [Google Scholar]
- Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J (1989) Differential effects of alpha‐, beta‐ and gamma‐cyclodextrins on human erythrocytes. Eur J Biochem 186: 17–22 [DOI] [PubMed] [Google Scholar]
- Okuda H, Noguchi A, Kobayashi H, Kondo D, Harada KH, Youssefian S, Shioi H, Kabata R, Domon Y, Kubota K, Kitano Y, Takayama Y, Hitomi T, Ohno K, Saito Y, Asano T, Tominaga M, Takahashi T, Koizumi A (2016) Infantile pain episodes associated with novel Nav1.9 mutations in familial episodic pain syndrome in Japanese families. PLoS ONE 11: e0154827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostman JA, Nassar MA, Wood JN, Baker MD (2008) GTP up‐regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J Physiol 586: 1077–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla F, Couble ML, Coste B, Maingret F, Clerc N, Crest M, Ritter AM, Magloire H, Delmas P (2007) Expression and localization of the Nav1.9 sodium channel in enteric neurons and in trigeminal sensory endings: implication for intestinal reflex function and orofacial pain. Mol Cell Neurosci 35: 138–152 [DOI] [PubMed] [Google Scholar]
- Pike LJ (2006) Rafts defined: a report on the keystone symposium on lipid rafts and cell function. J Lipid Res 47: 1597–1598 [DOI] [PubMed] [Google Scholar]
- Piomelli D, Sasso O (2014) Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci 17: 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posadas I, Bucci M, Roviezzo F, Rossi A, Parente L, Sautebin L, Cirino G (2004) Carrageenan‐induced mouse paw oedema is biphasic, age‐weight dependent and displays differential nitric oxide cyclooxygenase‐2 expression. Br J Pharmacol 142: 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, Liberator P, Iyer LM, Kash SF, Kohler MG, Kaczorowski GJ, MacIntyre DE, Martin WJ (2005) Contribution of the tetrodotoxin‐resistant voltage‐gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci USA 102: 9382–9387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pristera A, Baker MD, Okuse K (2012) Association between tetrodotoxin resistant channels and lipid rafts regulates sensory neuron excitability. PLoS ONE 7: e40079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse‐Dantsker A, Logothetis DE, Levitan I (2011) Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J 100: 381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Waxman SG (2004) PGE2 increases the tetrodotoxin‐resistant Nav1.9 sodium current in mouse DRG neurons via G‐proteins. Brain Res 1023: 264–271 [DOI] [PubMed] [Google Scholar]
- Sághy É, Szőke É, Payrits M, Helyes Z, Börzsei R, Erostyák J, Jánosi TZ, Sétáló G Jr, Szolcsányi J (2015) Evidence for the role of lipid rafts and sphingomyelin in Ca2+‐gating of transient receptor potential channels in trigeminal sensory neurons and peripheral nerve terminals. Pharmacol Res 100: 101–116 [DOI] [PubMed] [Google Scholar]
- Salvemini D, Wang ZQ, Wyatt PS, Bourdon DM, Marino MH, Manning PT, Currie MG (1996) Nitric oxide: a key mediator in the early and late phase of carrageenan‐induced rat paw inflammation. Br J Pharmacol 118: 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng R, Chen Y, Yung Gee H, Stec E, Melowic HR, Blatner NR, Tun MP, Kim Y, Kallberg M, Fujiwara TK, Hye Hong J, Pyo Kim K, Lu H, Kusumi A, Goo Lee M, Cho W (2012) Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain‐containing scaffold proteins. Nat Commun 3: 1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvius JR (2003) Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim Biophys Acta 1610: 174–183 [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31–39 [DOI] [PubMed] [Google Scholar]
- Song Y, Kenworthy AK, Sanders CR (2014) Cholesterol as a co‐solvent and a ligand for membrane proteins. Protein Sci 23: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A, Kochevar IE (2008) Nox1‐based NADPH oxidase is the major source of UVA‐induced reactive oxygen species in human keratinocytes. J Invest Dermatol 128: 214–222 [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Kunic JD, Ehring GR, George AL Jr (2013) Mechanism of sodium channel NaV1.9 potentiation by G‐protein signaling. J Gen Physiol 141: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrielink A (2010) Cholesterol oxidase: structure and function. Subcell Biochem 51: 137–158 [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D (2004) A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther 309: 869–878 [DOI] [PubMed] [Google Scholar]
- Waxman SG (2012) Sodium channels, the electrogenisome and the electrogenistat: lessons and questions from the clinic. J Physiol 590: 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Wen J, Yang W, Wang C, Gao L, Zheng LH, Wang T, Ran K, Li Y, Li X, Xu M, Luo J, Feng S, Ma X, Ma H, Chai Z, Zhou Z, Yao J, Zhang X, Liu JY (2013) Gain‐of‐function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 93: 957–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziblat R, Lirtsman V, Davidov D, Aroeti B (2006) Infrared surface plasmon resonance: a novel tool for real time sensing of variations in living cells. Biophys J 90: 2592–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 1768: 1311–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File