

Abstract
The nucleotide second messenger c‐di‐GMP nearly ubiquitously promotes bacterial biofilm formation, with enzymes that synthesize and degrade c‐di‐GMP being controlled by diverse N‐terminal sensor domains. Here, we describe a novel class of widely occurring c‐di‐GMP phosphodiesterases (PDE) that feature a periplasmic “CSS domain” with two highly conserved cysteines that is flanked by two transmembrane regions (TM1 and TM2) and followed by a cytoplasmic EAL domain with PDE activity. Using PdeC, one of the five CSS domain PDEs of Escherichia coli K‐12, we show that DsbA/DsbB‐promoted disulfide bond formation in the CSS domain reduces PDE activity. By contrast, the free thiol form is enzymatically highly active, with the TM2 region promoting dimerization. Moreover, this form is processed by periplasmic proteases DegP and DegQ, yielding a highly active TM2 + EAL fragment that is slowly removed by further proteolysis. Similar redox control and proteolysis was also observed for a second CSS domain PDE, PdeB. At the physiological level, CSS domain PDEs modulate production and supracellular architecture of extracellular matrix polymers in the deeper layers of mature E. coli biofilms.
Keywords: biofilm, DegP, DsbAB, EAL domain, second messenger
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
C‐di‐GMP is a nearly ubiquitous bacterial second messenger that in many species promotes the formation of biofilms which are multicellular aggregates of bacteria encased in a self‐produced protective matrix of biopolymers (Jenal & Malone, 2006; Römling et al, 2013; Jenal et al, 2017). Biofilms play an important role in chronic infections since bacteria within biofilms are more tolerant against antibiotics and better evade the immune system (Hall‐Stoodley et al, 2004). The cellular c‐di‐GMP level is the result of a dynamic balance between synthesis by diguanylate cyclases (DGC, with GGDEF domains) and degradation by specific phosphodiesterases (PDE, with EAL or HD‐GYP domains). There are cytoplasmic as well as membrane‐attached variants of these enzymes and many feature N‐terminal sensor domains—including PAS, GAF, REC as well as various membrane‐integrated domains—that control their enzymatic activities. In general, however, sensory input is one of the least understood aspects of these c‐di‐GMP‐related enzymes. Moreover, most bacteria possess multiple DGCs and PDEs with different sensor domains (Jenal & Malone, 2006; Hengge, 2009; Römling et al, 2013). Thus, the Gram‐negative bacterium Escherichia coli K‐12 has a complement of 29 GGDEF and/or EAL domain proteins that include 12 DGCs, 13 PDEs, and four “degenerate” proteins with alternative non‐enzymatic functions. In addition, two more DGCs and four more PDEs have been detected in various other commensal and pathogenic E. coli (Hengge et al, 2015; Povolotsky & Hengge, 2016).
A major target of c‐di‐GMP signaling in E. coli is the production of extracellular amyloid curli fibers and the exopolysaccharide cellulose (Zogaj et al, 2001; Barnhart & Chapman, 2006; Hufnagel et al, 2015). In macrocolony biofilms, these two components of the extracellular matrix assemble into a distinct supracellular architecture that confers stability, cohesion, and elasticity to the biofilm, that is, in essence tissue‐like properties that allow the biofilm to fold and buckle up into macroscopic morphological structures (Serra & Hengge, 2014). A key activator for matrix production is the transcription factor CsgD (Prigent‐Combaret et al, 2001; Brombacher et al, 2003). Via a complex cascade mechanism, the two PDE/DGC pairs PdeH/DgcE (formerly YhjH/YegE) and PdeR/DgcM (YciR/YdaM) can activate the transcription factor MlrA, which then drives the expression of CsgD (Pesavento et al, 2008; Lindenberg et al, 2013; Hengge, 2016). In this genetic switch mechanism, the functions of DgcE, DcgM, PdeH, and PdeR are now well defined. By contrast, much less is known about specific functions of the other DGCs and PDEs in E. coli and whether they somehow feed into this matrix control mechanism.
The present study was undertaken to characterize the mode of activation and physiological function of a group of five of the 13 PDEs in E. coli K‐12—PdeB (previously YlaB), PdeC (YjcC), PdeD (YoaD), PdeG (YcgG), and PdeN (Rtn)—that possess the same type of putative N‐terminal sensor domain. This protein region consists of two transmembrane segments (TM1 and TM2) flanking a periplasmic loop of about 200 amino acids that contains two highly conserved cysteine residues, one of which is part of a characteristic CSS motif (Appendix Fig S1). This suggested potential disulfide bond (DSB) formation in this periplasmic “CSS domain”, which might control the enzymatic activity of the cytoplasmic C‐terminal EAL domain. In general, the periplasmic space is an oxidizing environment with an intricate redox homeostasis that allows to use DSB/free thiol transitions as redox switches (Wouters et al, 2010). The DsbA/DsbB system helps to stabilize many periplasmic proteins in their native functional conformations by introducing DSBs, with electrons being delivered to the respiratory chain (Ito & Inaba, 2008). On the other hand, wrong DSBs that render periplasmic proteins non‐functional and that are generated in particular under conditions of oxidative stress can be resolved by the DsbD/DsbC/DsbG system, with electrons provided by the thioredoxin system (Cho & Collet, 2013). This homeostasis is complemented by the presence of periplasmic chaperones and proteases, which refold or remove misfolded proteins, respectively. In E. coli, major factors of this kind are the HtrA family proteases DegP, which can act as a chaperone or as a protease depending on the actual conditions, and its homolog DegQ (Clausen et al, 2011).
Given this context, are the conserved cysteine residues in the periplasmic CSS domain involved in controlling the enzymatic activity of these transmembrane PDEs? And if so, how is the redox state of the cysteine residues in the periplasm signaled to the EAL domain located at the inner side of the membrane? Does any of the Dsb systems have an impact on the redox state of the CSS domain and thereby contribute to control the activity of these PDEs? These questions were addressed here using one of the five CSS domain PDEs of E. coli K‐12, PdeC, as a representative of this widely occurring type of c‐di‐GMP PDE. The conserved cysteines and the DsbA/DsbB system are shown to play crucial roles in the control of PdeC activity. In addition, we demonstrate that the free thiol form of the CSS domain is a substrate for DegP/DegQ‐mediated proteolysis. Furthermore, the redox state and proteolytic processing in the periplasmic CSS domain control dimerization of the second transmembrane region that is associated with enzymatic activity of PdeC, which in its active state is shown to reduce biosynthesis of the extracellular matrix in the deeper zones of macrocolony biofilms of E. coli.
Results
Eliminating the conserved cysteine residues in the periplasmic loop domain activates PdeC
The morphology of macrocolonies provides an excellent indicator for the relative amount of curli fibers and cellulose in the matrix of this type of biofilm, which is exquisitely controlled by c‐di‐GMP (Lindenberg et al, 2013; Serra et al, 2013a,b). Thus, an E. coli strain (AR3110) that produces both components generates a spatially ordered pattern of ridges and wrinkles, a strain that makes curli fibers only (W3110) grows in a concentric ring pattern and a cellulose‐only producer (AR3110 csgBA) generates a multitude of tiny wrinkles (Appendix Fig S2A). A strain with elevated c‐di‐GMP levels that produces very high amounts of both curli fibers and cellulose (AR3110 pdeH) grows as flat, large, and rather stiff colonies that only occasionally buckle up into long and very high ridges, while regulatory mutations (rpoS, csgD) that eliminate curli and cellulose production result in unstructured colonies not stained with the matrix dye Congo red. Single or multiple mutations in any of the five genes encoding CSS domain PDEs did hardly affect macrocolony morphology, when introduced into a AR3110 background (Appendix Fig S2B), even though reporter fusions to these genes show low to moderate expression (Sommerfeldt et al, 2009). Only mutants carrying the pdeC deletion grew as slightly larger and flatter macrocolonies (Appendix Fig S2B), suggesting that at least PdeC can affect matrix production under these conditions.
In order to study the control of activity of a CSS domain PDE, we chose PdeC as a model system (Appendix Fig S3A). The pdeC gene was cloned onto a low copy number plasmid (pCAB18), with the tac promoter (ptac) driving its expression. Since ptac is relatively leaky, moderate expression is conveniently obtained in the absence of inducer (IPTG). In addition, ptac is a vegetative promoter (activated by RNAP containing the sigma factor RpoD or σ70), which—unlike the stationary phase σS, which naturally controls pdeC expression—drives expression also in the lower layer of macrocolonies where cells are in a post‐exponential growth mode (Serra & Hengge, 2014; Serra et al, 2015). Mutations were introduced into the pdeC coding sequence that eliminated either one or both of the two conserved cysteines (C75A; C106A or “ASS”, changing the CSS motif into ASS) or replaced the active site glutamate of the EAL motif by alanine (“AAL”), which eliminates PDE activity of EAL domain proteins (Schmidt et al, 2005; Tchigvintsev et al, 2010; Lindenberg et al, 2013).
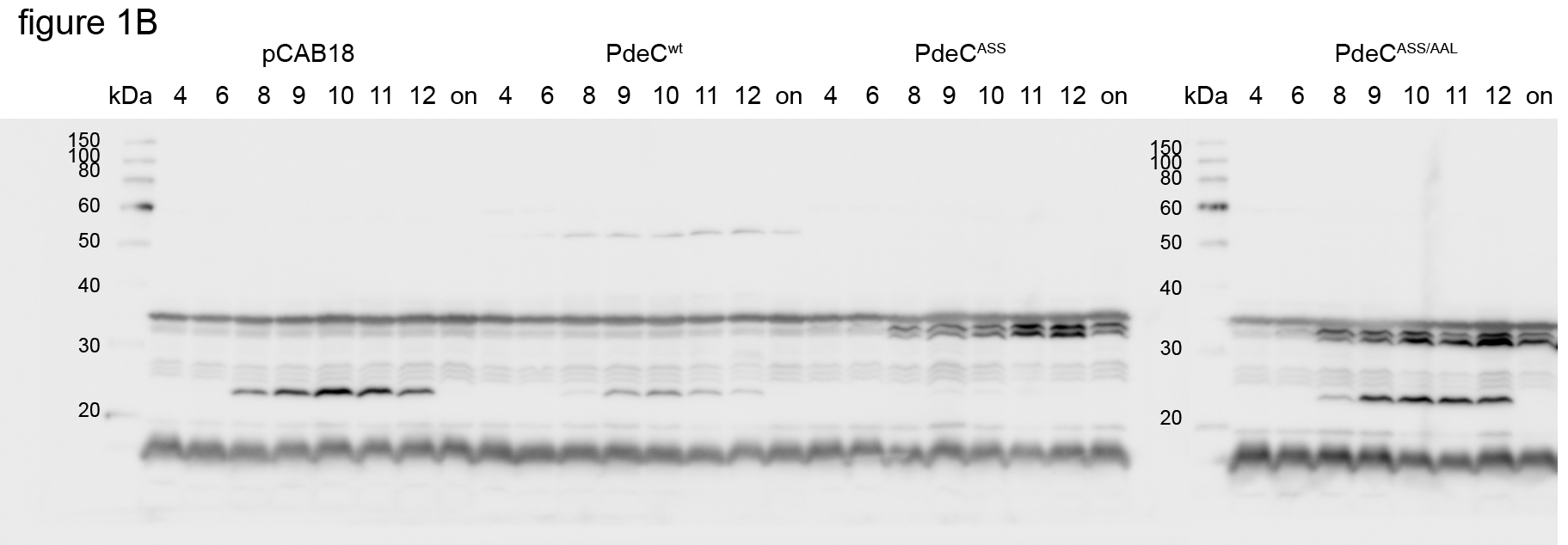
When introduced into strain AR3110, the wild‐type PdeC (PdeCwt) construct clearly reduced wrinkly macrocolony morphology, indicating less curli fiber and cellulose production (Fig 1A). The variants lacking one or both conserved cysteines had an even stronger effect, visible as a total lack of macrocolony structure and further reduced staining with the matrix dye Congo red. These effects were due to enhanced cellular c‐di‐GMP PDE activity, since they were completely suppressed by also introducing the EAL to AAL mutation. Effects were similar, no matter whether tested with strain AR3110 or derivatives lacking the chromosomal pdeC gene. These findings were confirmed by growing macrocolonies on YESCA medium (containing casamino acids instead of tryptone as in LB). Here, overall macrocolony morphology was less pronounced, with PdeCwt having hardly any effect, whereas the variants with either cysteine eliminated showed unstructured macrocolonies, reflecting enhanced PDE activity. These results were further corroborated by monitoring two c‐di‐GMP‐dependent functions: cellular levels of the matrix regulator CsgD (Fig 1B) and expression of the curli operon using a csgB::lacZ reporter fusion (Fig 1C) in liquid medium. Also here, PdeCwt reduced, while PdeCASS completely eliminated CsgD and csgB::lacZ expression. These effects equally depended on cellular PDE activity, since they were suppressed by the AAL mutation.
Figure 1. Macrocolony morphology and lower expression of CsgD and the curli operon indicates increased activity of PdeC variants unable to form disulfide bonds in the periplasmic loop of the CSS domain.
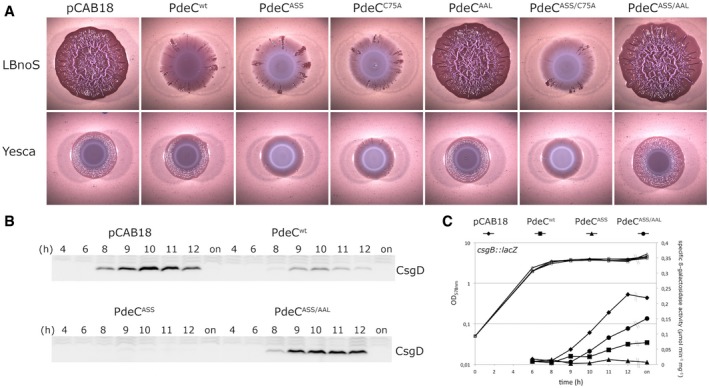
- Macrocolonies of Escherichia coli K‐12 strain AR3110 expressing the indicated PdeC versions under control of the tac promoter from the low copy number plasmid pCAB18 were grown on salt‐free LB (LBnoS) or YESCA agar plates supplemented with the matrix dye Congo red (CR) for 5 days at 28°C. For comparison with morphological patterns of macrocolonies of standard strains, see Appendix Fig S1.
- Derivatives of strain W3110 carrying a single copy csgB::lacZ reporter fusion and containing the indicated plasmids were grown in LB medium. Samples were taken during the growth of the cultures (numbers refer to hours of growth and respond to data sampling and measurement points shown in C; on, overnight). Cellular CsgD levels were determined by immunoblotting.
- From the same samples as in (B), specific β‐galactosidase activities expressed from csgB::lacZ were determined.
Source data are available online for this figure.
Thus, the presence of both Cys75 and Cys106 of the CSS domain results in a lower PDE activity of PdeC, whereas eliminating one or both of them promotes PdeC activity. This suggested a possible link to the potential of these cysteine residues to form a DSB. This observation also indicates transmembrane signaling from the periplasmic CSS domain to the cytoplasmic EAL domain. Moreover, c‐di‐GMP degradation by PdeC can clearly affect the major target of c‐di‐GMP regulation in E. coli, that is, biofilm matrix production.
Proteolytic processing of the periplasmic CSS domain of PdeC in response to the redox state of the conserved cysteine residues and a role for the DsbA/DsbB system
To control for expression, the different PdeC variants were generated with a His6 tag at their C‐terminal ends. To our surprise, detection of the proteins by immunoblotting also revealed changes in protein size: Whereas PdeCwt was visualized as a band of the expected size, PdeC variants lacking one or both cysteine residues in the periplasmic domain generated not only the full‐size band but also a prominent shorter fragment (Fig 2A), suggesting proteolytic degradation of the PdeC variants unable to form a periplasmic DSB. The size of this fragment (approximately 33 kD) indicated that proteolysis in the periplasmic loop generated a new N‐terminus close to the beginning of TM2 (confirmed by the slightly smaller size of a cloned TM2 + EAL fragment as shown in Fig 4A below). In addition, the PdeCASS‐derived fragment was associated with the membrane fraction of cell extracts (Appendix Fig S4). In overnight stationary phase cultures, in which de novo expression from the σ70‐driven ptac no longer occurs, the PdeC variants unable to form a DSB were completely degraded to this smaller fragment (Fig 2A). Moreover, PdeC variants with a single periplasmic cysteine only (PdeCASS or PdeCC75A)—in contrast to PdeCwt with its two cysteine residues or the periplasmic cysteine‐free variant PdeCASS/C75A—were observed to dimerize by intermolecular DSB formation via the single remaining periplasmic cysteine (Appendix Fig S5). In stationary phase cells, these dimers were resolved, with essentially all of the proteins accumulating as the small 33‐kD fragment.
Figure 2. Absence of a disulfide bond in the periplasmic loop of its CSS domain results in processing of PdeC to a 33‐kD C‐terminal fragment.
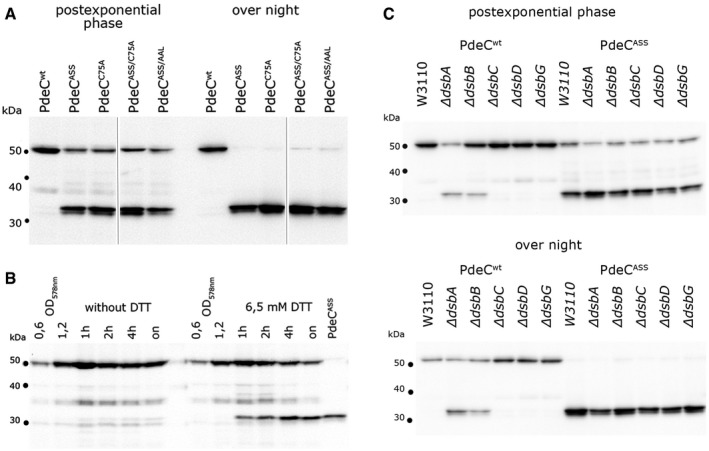
- Derivatives of strain W3110 containing the indicated plasmids were grown in LB medium, and samples were taken during post‐exponential growth and from overnight (“on”) cultures. PdeC variants (containing C‐terminal His6 tags) produced from the indicated plasmids were detected by immunoblotting.
- Strain W3110 containing the PdeCwt plasmid was grown to an OD578 of 1.2 and 6.5 mM DTT was added where indicated, with samples for immunoblotting taken at the times indicated. As a reference for the 33‐kD proteolytic fragment of PdeC, a sample obtained from the PdeCASS plasmid strain after overnight growth was included (last lane).
- Derivatives of strain W3110 carrying the indicated mutations in the different dsb genes and containing the plasmids expressing either PdeCwt or PdeCASS were grown in LB medium. Samples were taken during post‐exponential growth and from overnight cultures and PdeC variants expressed were visualized by immunoblotting.
Source data are available online for this figure.
Figure 4. The second transmembrane segment (TM2) is required for PDE activity of the EAL domain and promotes dimerization of PdeC.
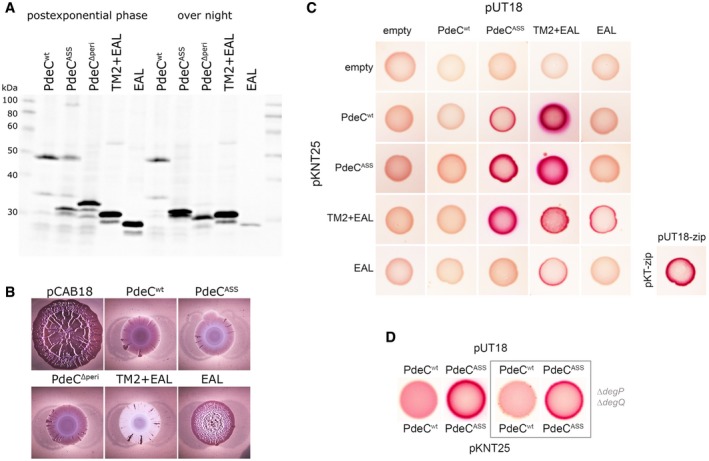
- Immunoblot analysis of strain W3110 expressing the indicated PdeC variants visualized via their His6 tags.
- Macrocolonies of strain AR3110 expressing the same PdeC variants as in (A) grown on salt‐free LB agar plates supplemented with Congo red (CR) for 5 days at 28°C.
- Protein–protein interactions of the indicated PdeC variants were tested using a bacterial two‐hybrid (2H) system that is based on the reconstitution of adenylate cyclase (Karimova et al, 1998) which allows the utilization of maltose as a C‐source in a cya strain of Escherichia coli (resulting in red color on MacConkey/maltose agar plates). As a positive control, the leucine zipper part (‘zip’) of the yeast GCN4 protein was used. For a similar two‐hybrid analysis with corresponding PdeC variants, in which TM2 was replaced by the second transmembrane region of LacY (TM2*), see Appendix Fig S9.
- To assess for dimerization in the absence of proteolytic processing of PdeCASS, an otherwise isogenic degP degQ cya recipient strain was used for the two‐hybrid analysis.
In order to test whether this proteolysis also occurs with PdeCwt under strongly reducing conditions, DTT was added to the growing culture, which indeed resulted in the appearance of the fragment (Fig 2B). Furthermore, this fragment derived from PdeCwt was also detected in dsbA and dsbB mutant backgrounds (Fig 2C), indicating that DSB formation between Cys75 and Cys106 in the periplasmic loop of PdeC is promoted by the DsbA/DsbB system. In addition, we tested whether knocking out the periplasmic DSB‐reducing DsbD/DsbC/DsbG system had any effect on PdeC (Fig 2C). This was not the case in growing cells, but in stationary phase cells (overnight), cellular levels of PdeCwt were reproducibly lower in a wild‐type background than in dsbD, dsbC, and dsbG mutants (although the smaller fragment did not accumulate), while mutations in dsbD, dsbC, and dsbG did not make a difference with the DSB‐free PdeCASS. This suggests that the DsbD/DsbC/DsbG system may contribute to slow destabilization of PdeCwt in stationary phase cells by occasionally attacking the DSB in the CSS domain of PdeCwt.
Taking together the results concerning function (Fig 1) and redox‐dependent proteolytic processing (Fig 2) of PdeCwt and the different PdeC variants unable to form DSBs, we conclude that (i) PdeC is a c‐di‐GMP‐degrading PDE that can down‐regulate matrix formation in macrocolony biofilms; (ii) PdeC is highly active when its periplasmic CSS domain is in the free thiol state, that is, in the absence of a DSB linking Cys75 and Cys106; (iii) DSB formation is promoted by the DsbA/DsbB system; and (iv) the absence of the DSB promotes proteolytic cleavage to a shorter membrane‐associated polypeptide which consists of TM2 and the EAL domain.
Role of the periplasmic proteases DegP and DegQ in PdeC processing and the relationship between proteolysis and enzymatic activity of the free thiol form of PdeC
Proteolytic cleavage in the absence of the DSB in the CSS domain raised novel interesting questions. What is the periplasmic protease(s) involved? Is the absence of the DSB sufficient or is also proteolytic cleavage required to stimulate PDE activity of PdeC? In order to separate these two processes genetically, either a non‐cleavable PdeC mutant or a knockout of the relevant protease(s) would be required. The cloned TM2 + EAL fragment (which begins with Ser231 after a starting methionine; see Appendix Fig S3 and Fig 4A) was about 2 kD smaller than the proteolytic fragment, which placed the N‐terminus of the proteolytic fragment around amino acid 215 of PdeCASS. A series of single amino acid exchanges were introduced into this region of PdeCASS, but none altered its processing pattern (Appendix Fig S6). This suggested that the protease involved does not recognize a specific amino acid motif, but rather attacks a polypeptide region that is unfolded (due to the absence of the stabilizing DSB) and/or is a processive protease that stops to move along the polypeptide chain at a certain minimal distance from the membrane‐inserted TM2 that is determined by the molecular structure and function of the protease. Proteases that operate in this manner are DegP, a major periplasmic chaperone/protease (Krojer et al, 2002; Meltzer et al, 2009; Clausen et al, 2011), as well as its homolog DegQ. We therefore tested whether knockout mutations in degP and degQ affected the appearance of PdeCASS in immunoblots (Fig 3A). While the single mutations showed partial effects (less processed fragment and more intermolecular dimers of the full‐size protein), no processed fragment was observed in the degP degQ double mutant; that is, these two proteases are responsible for processing of PdeCASS in a redundant manner. PdeCwt was not affected in the degP degQ double mutant. In a dsbA knockout background, however, production of the 33‐kD fragment from PdeCwt also depended on DegP and DegQ (Appendix Fig S7).
Figure 3. The periplasmic proteases DegP and DegQ are responsible for proteolytic processing of PdeC, but processing is not a prerequisite for PDE activity of PdeC.
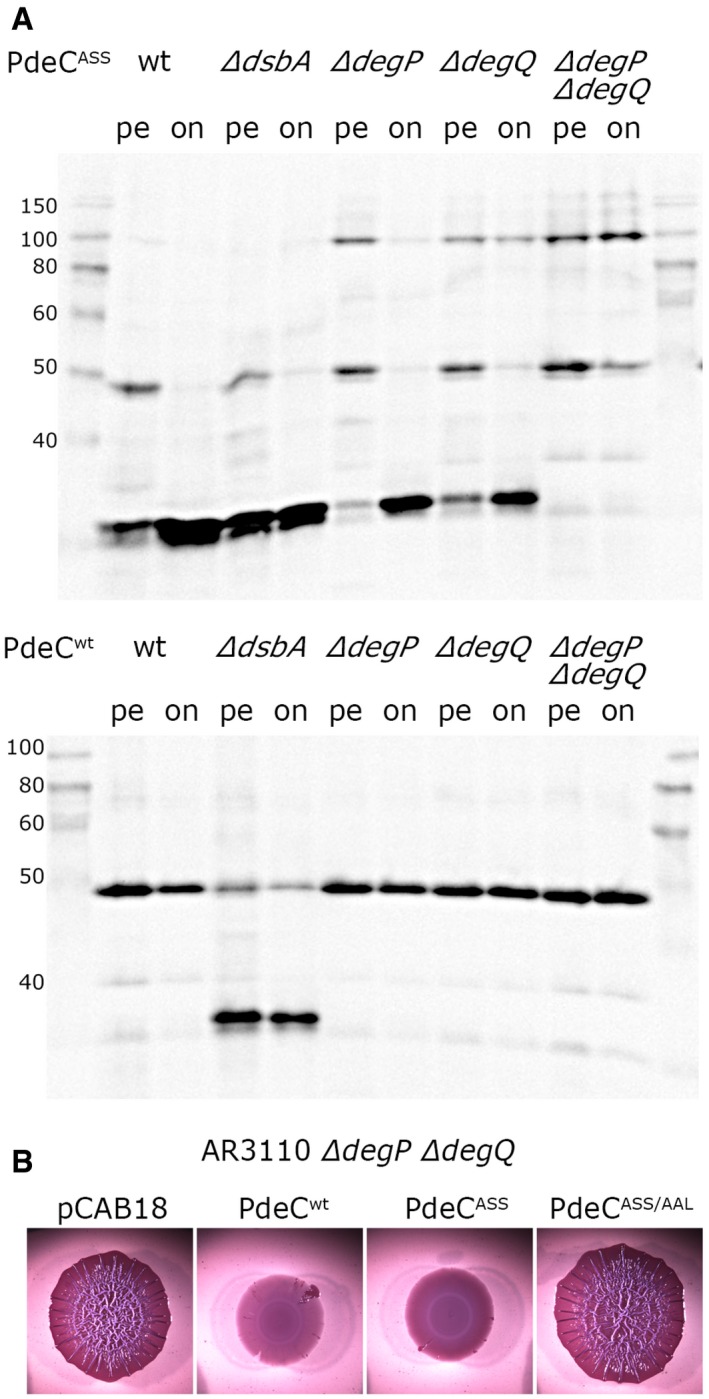
- Derivatives of strain W3110 carrying single or double mutations in degP and degQ and containing the plasmids expressing either PdeCwt or PdeCASS were grown in LB. Samples were taken during post‐exponential growth (pe) or overnight (on), and PdeC variants expressed were visualized by immunoblotting. The dsbA mutant was included to show the position of the 33‐kD processed fragment of PdeC.
- Macrocolony morphology of the degP degQ derivative of AR3110 carrying the indicated plasmids indicates high PDE activity of PdeCASS; that is, processing to the 33‐kD fragment is not required for high PDE activity of PdeCASS.
Source data are available online for this figure.
The absence of cleavage of PdeCASS in the degP degQ mutant allowed to address the question whether proteolytic processing is required or whether the absence of the DSB in the CSS domain is sufficient for PDE activity of the EAL domain of PdeC. The non‐wrinkled macrocolony morphotype of the degP degQ mutant expressing PdeCASS (Fig 3B) clearly indicated that also the full‐size form PdeCASS is active as a PDE. We conclude that while proteolytic processing of PdeC occurs in the absence of the intramolecular DSB in the CSS domain, it is not required to activate PdeC as a PDE.
The second transmembrane segment TM2 is required for the activity of the EAL domain of PdeC and acts as a dimerization domain
In order to further dissect the roles of the periplasmic CSS domain and the transmembrane regions in the activation process, additional PdeC derivatives with precise deletions were constructed (with C‐terminal His6 tags as in the full‐size versions): (i) PdeCΔperi, which lacks the periplasmic CSS domain (Thr44 to Arg234) and thus has a TM1 + TM2 + EAL architecture with only a very short periplasmic loop between TM1 and TM2; (ii) TM2 + EAL (beginning with Ser231 after an initial methionine); and (iii) the EAL domain alone [beginning with Arg263 after an initial Met‐Ser sequence, with serine introduced for protein stability according to the N‐end rule (Mogk et al, 2007)]. On immunoblots, these partially deleted PdeC constructs appeared as protein fragments of the expected sizes (Fig 4A). Thus, PdeCΔperi is longer and TM2 + EAL is slightly shorter than the proteolytic fragment generated from PdeCASS, which confirms that the latter contains TM2. The observed size of PdeCΔperi also confirmed that TM1 is an integral part of PdeC rather than a cleaved signal sequence (in addition, TM1 does not feature a cleavage site for leader peptidase (Appendix Fig S3) nor did PdeC generate a larger protein when expressed in a secA ts mutant at non‐permissive temperature; Appendix Fig S8). The EAL domain alone was soluble, whereas PdeCwt, the PdeCASS‐derived proteolytic fragment, PdeCΔperi with its two TM regions and the TM2 + EAL polypeptide were all found in the membrane fraction (Appendix Fig S4). Especially for TM2 + EAL, this is noteworthy, because it means that TM2 alone (in the absence of TM1) can insert into the membrane. Interestingly, overnight stationary phase cells, which no longer synthesize these proteins (since ptac is inactive), revealed additional proteolytic degradation of these proteins (Fig 4A): PdeCΔperi was further shortened indicating that TM1 and part of TM2 get slowly removed, TM2 + EAL in general produced also a minor band corresponding in size almost to the free EAL domain suggesting that TM2 is slowly cleaved off at the inner side of the membrane and the free EAL domain almost completely disappeared in stationary phase cells.
Phosphodiesterase activity of the shortened PdeC variants was tested using macrocolony morphology (Fig 4B). PdeCΔperi was clearly an active PDE; that is, the periplasmic CSS domain is not required for PDE activity, and close spatial vicinity of the two transmembrane regions does not inhibit PDE activity. The cloned TM2 + EAL fragment was highly active, with macrocolonies on CR plates being not only unstructured but of essentially white color, that is, not even a basal level of Congo red‐binding matrix was produced. However, in pronounced contrast, the isolated EAL domain alone showed very low PDE activity not sufficient to prevent the formation of wrinkled macrocolonies (Fig 4B). This means that the presence of TM2 and thus also membrane attachment of the EAL domain of PdeC are crucial for PDE activity.
We then addressed the question whether TM2 is just a topogenic sequence, which ensures proper localization of the EAL domain at the cytoplasmic side of the membrane, or whether TM2 also contributes in a more specific way to controlling PdeC activity. Structure‐function analyses have indicated that PDEs are active as dimers (Barends et al, 2009; Minasov et al, 2009; Rao et al, 2009; Robert‐Paganin et al, 2012; Sundriyal et al, 2014). If PdeC activation depends on dimer formation, TM2 could act as a specific dimerization domain that may help the EAL domain to form a stable active dimer. We therefore tested the different PdeC variants for dimer formation in a bacterial two‐hybrid (2H) system that is amenable to membrane proteins (Karimova et al, 1998). Interaction in this 2H system leads to the reconstitution of adenylate cyclase (AC) from two AC fragments that in our experiments were fused to the cytoplasmic C‐termini of the potentially interacting proteins.
This approach demonstrated that the soluble EAL domain alone as well as the less active PdeCwt did not dimerize (Fig 4C). By contrast, the highly active PdeCASS, which generates the 33‐kD proteolytic fragment, as well as the cloned TM2 + EAL fragment showed strong dimerization. Moreover, the TM2 + EAL fragment interacted equally strongly with PdeCASS, thereby also confirming that TM2 in the TM2 + EAL construct is properly inserted and oriented within the membrane even though it is not synthesized with TM1 as an initial topogenic sequence as present in the naturally synthesized PdeCASS polypeptide. Notably, PdeCASS with its 33‐kD fragment as well as the cloned TM2 + EAL fragment seemed to “force” PdeCwt into interaction (although detectable only in one vector combination). Thus, the interaction of a free, that is, structurally less confined TM2 region in these fragments with TM2 in full‐size PdeCwt seems strong enough to overcome the structural cohesion and constraints generated by DSB formation in the CSS domain of PdeCwt.
These data indicate that TM2 promotes dimerization which is likely to stimulate PDE activity of the EAL domain which on its own does not dimerize. In order to test whether dimerization via TM2 is sequence‐specific, TM2 was substituted in all 2H constructs by a heterologous transmembrane segment of similar hydrophobicity and transmembrane orientation. For this purpose, the second transmembrane region of the lactose permease LacY (termed TM2*; Appendix Fig S3) was chosen. In pronounced contrast to the findings with the original PdeC variants, the TM2* constructs did not show interactions (Appendix Fig S9). Only the TM2* + EAL fragment was found to self‐interact under conditions of strong overproduction which may reflect oligomerization via the hydrophobic TM2* in micelles. We conclude that the interactions of the various PdeC constructs shown in Fig 4C are specific, i.e. depend on the amino acid sequence of TM2.
Finally, we asked whether TM2‐promoted dimerization also occurs in the non‐processed, yet active form of PdeCASS in the degP degQ double mutant background. Also in this configuration, dimerization was observed (Fig 4D); that is, dimerization is linked to the activation of PdeC no matter whether the active form is a full‐size free thiol form of PdeC or a TM2 + EAL fragment (generated either by DegP/DegQ‐mediated processing or artificially by direct cloning). In conclusion, the TM2 region in PdeC promotes dimerization, with DSB formation in the periplasmic CSS domain interfering with this dimerization. However, when the CSS domain is in the free thiol conformation, dimerization can occur, which is associated with increased PDE activity of the EAL domain.
Redox control and proteolysis of PdeC and a second CSS domain PDE, PdeB, under conditions of physiological expression
For the experiments reported above, PdeC and its derivatives were moderately overproduced from a σ70‐driven promoter. Therefore, we wanted to confirm the results with PdeC physiologically expressed from its chromosomal gene. In addition, we chose PdeB (Appendix Fig S1), which like PdeC is under σS control and is the most highly expressed of all five CSS domain PDEs of E. coli K‐12 (Sarenko et al, 2017), to test whether a second CSS domain PDE shows similar redox control of activity and proteolysis. When cells expressing chromosomally encoded C‐terminally Flag‐tagged PdeC or PdeB were exposed to reducing conditions by adding DTT during growth, not only PdeC but also PdeB generated shorter proteolytical fragments as previously seen with overproduced PdeC (Fig 5A, left panels). The same fragments were also observed in the dsbA mutant background, and their appearance depended on DegP and DegQ (Fig 5A, right panels). Thus, DsbA‐dependent DSB formation and DegP/DegQ‐mediated proteolytic processing of the free thiol form are more general features of CSS domain PDEs expressed at physiological levels.
Figure 5. Chromosomally encoded PdeC shows redox control and DegP/DegQ‐mediated proteolysis and a second CSS domain PDE, PdeB, is controlled similarly.
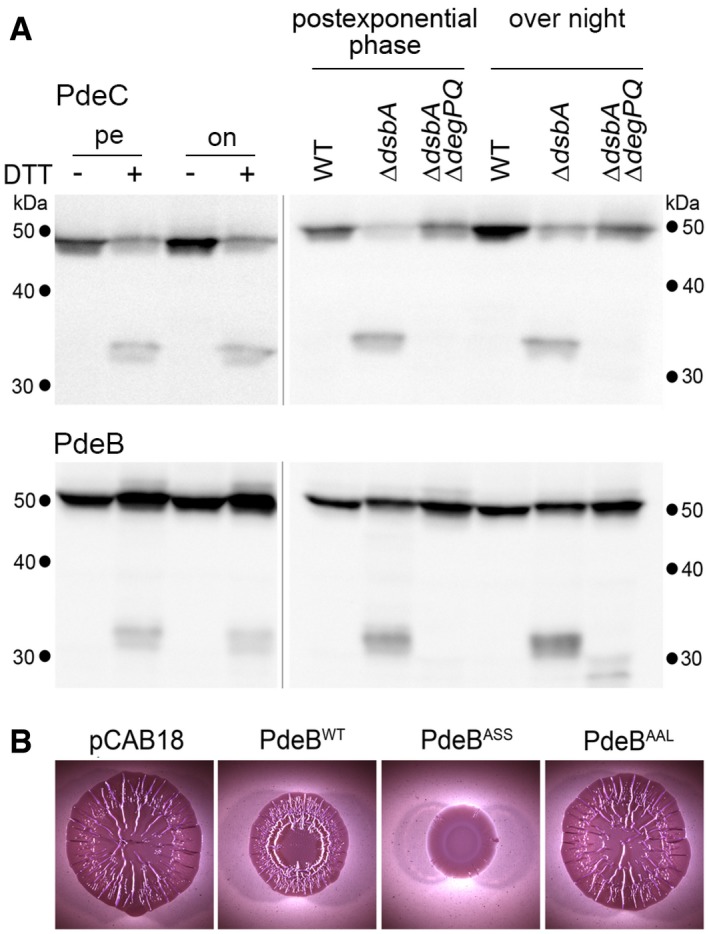
- Derivatives of strain W3110 in which the chromosomal pdeC and pdeB genes carry 3′‐inserts encoding Flag tags were grown in LB medium. DTT treatment of the cultures (left panels) was as described in the legend to Fig 2. Strains shown in the right panels also carried the indicated mutations. Samples were taken during post‐exponential phase (at an OD578 of 3–3.5) and from overnight cultures and subject to immunoblot analysis with Flag tag antibodies.
- Macrocolonies of Escherichia coli K‐12 strain AR3110 expressing the indicated PdeB variants under control of the tac promoter from the low copy number plasmid pCAB18 were grown on salt‐free LB (LBnoS) with the matrix dye Congo red (CR) for 5 days at 28°C.
Source data are available online for this figure.
In order to see whether this redox control of PdeB has similar functional consequences as for PdeC, we generated mutant variants PdeBASS and PdeBAAL expressed from the low copy number plasmid pCAB18 in analogy to the PdeC variants described above. As apparent from macrocolony morphology, the non‐DSB variant PdeBASS clearly interfered more strongly with biofilm matrix production (Fig 5B). Thus, PdeBASS has higher PDE activity than PdeBwt; that is, the potential to form a DSB in the CSS domain is associated with reduced activity as described above for PdeC.
Redox controlled in vitro PDE activity of purified PdeC reconstituted into nanodiscs
With all genetic data shown above indicating that PdeC with a free thiol CSS domain shows higher activity than when in the oxidized DSB form, we tested whether this redox regulation can also be detected with the purified protein in vitro. As a transmembrane protein with a cytoplasmic activity controlled by redox events on the periplasmic side of the protein, we purified detergent‐solubilized PdeC (with a C‐terminal His6 tag) and reconstituted it in the quasi‐native membrane environment of nanodiscs. This system consists of self‐assembled discoidal phospholipid bilayers stabilized by a thread‐like membrane scaffold protein (MSP; derived from human apolipoprotein A‐I) which wraps around the phospholipid bilayer that can incorporate a transmembrane protein (Bayburt et al, 2002; Borch & Hamann, 2009; Ritchie et al, 2009). Soluble reconstituted PdeC‐containing nanodiscs, prepared in the absence of any reducing agent, were purified by affinity chromatography (via the His6 tag on PdeC). PDE activity was then determined in the presence or absence of DTT using radiolabeled c‐di‐GMP and thin‐layer chromatography to detect c‐di‐GMP and the reaction product 5′‐pGpG (Fig 6A). PDE activity was indeed only detectable in the presence of DTT (Fig 6A). However, besides the two cysteine residues in the periplasmic CSS domain, PdeCwt contains three cysteines in its cytoplasmic EAL domain, which—in contrast to in vivo conditions in the reducing cytoplasm—may form functionally inhibitory DSBs during purification under aerobic conditions. To circumvent this problem, we engineered the following proteins: (i) a PdeC variant (PdeCEALΔC), in which all three cytoplasmic cysteines (C285, C299, and C489) were replaced by serine residues, and (ii) a chimeric protein (PdeC/N) that contains the TM1‐CSS‐TM2 part of PdeC followed by the naturally cysteine‐free cytoplasmic EAL domain of PdeN, which is yet another one of the five E. coli K‐12 CSS domain PDEs (Appendix Fig S1). Although less active than PdeCwt, PdeCEAL∆C and PdeC/N had PDE activity in vivo as indicated by macrocolony morphology (Fig 6C). When purified and reconstituted into nanodiscs, PdeCEALΔC and PdeC/N showed basal PDE activity in the absence of DTT, which confirmed that illegitimate DSB formation of the cytoplasmic cysteines inhibited in vitro activity of PdeCwt (Fig 6A). Most importantly, PDE activity of PdeCEAL∆C and PdeC/N was clearly stimulated by adding DTT (Fig 6A, with quantification shown in Fig 6B), indicating that reduction in the CSS domain of PdeC to the free thiol form stimulated the enzymatic activity of the EAL domain.
Figure 6. Redox control of activity of purified PdeC reconstituted in nanodiscs.
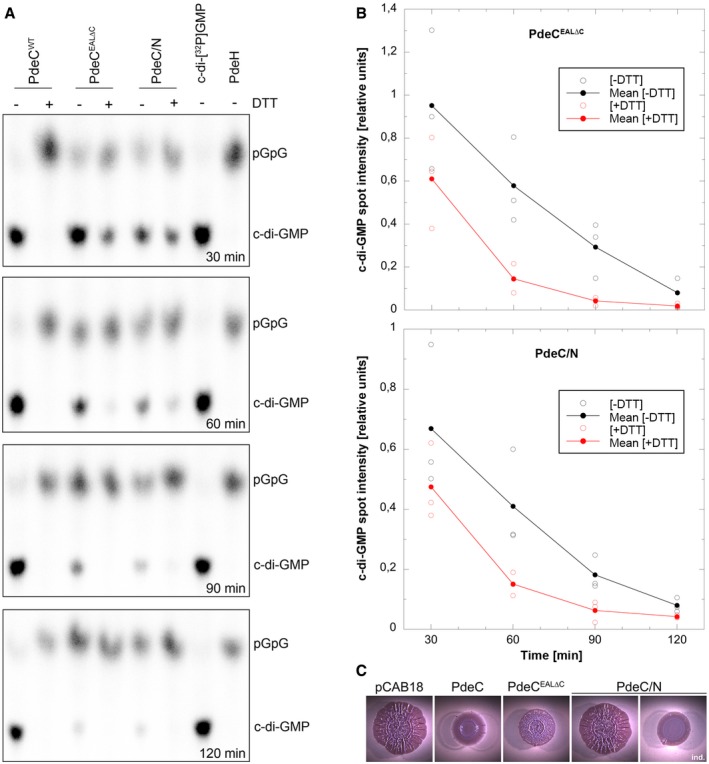
- The detergent‐solubilized and purified PdeC variants as indicated were reconstituted into nanodiscs and assayed for PDE activity in the absence or presence of 6.5 mM DTT. Samples were taken at the times indicated (from top to bottom) and the remaining radiolabeled substrate c‐di‐GMP and the enzymatic product 5′‐pGpG were detected by thin‐layer chromatography and autoradiography. The purified soluble and highly active Escherichia coli PDE PdeH was used to generate radiolabeled 5′‐pGpG as a reference. The experiment was performed three times independently, with a representative example shown here.
- Quantification of radiolabeled c‐di‐GMP spots as visualized in (A) was performed for the three independent experiments (open symbols for single data points) and averaged (closed symbols) to show enzymatic turnover of c‐di‐GMP by purified and nanodisc‐reconstituted PdeCEAL∆C and PdeC/N in the absence (black) and presence of DTT (red symbols).
- Macrocolonies of strain AR3110 expressing the indicated PdeC variants from the low copy number plasmid pCAB18 were grown on salt‐free LB with the matrix dye Congo red (CR) for 5 days at 28°C. PdeC/N activity could be detected only upon induction with 0.1 mM IPTG (ind.).
CSS domain PDEs affect matrix production and architecture in the deeper zones of macrocolony biofilms
Macrocolony morphology was only weakly affected by knocking out pdeC or even all five genes encoding CSS domain PDEs (Appendix Fig S2). Also cellular levels of the matrix synthesis activating regulator CsgD were unchanged in a ΔpdeC mutant. However, the introduction of a chromosomal pdeC ASS allele reduced CsgD levels during growth in liquid culture (Fig 7A) and reduced wrinkle formation in the central areas of macrocolony biofilms (Fig 7B). In order to detect biofilm‐associated effects of these chromosomal mutations in greater detail, macrocolonies grown in the presence of the fluorescent matrix dye thioflavin S were fixed, vertically cryosectioned and analyzed by fluorescence microscopy. While none of these mutations changed the disordered matrix in the upper layer of the outer, relatively young macrocolony areas (Appendix Fig S10), the more structured matrix architecture in the central older area was clearly affected (Fig 7C; corresponding phase‐contrast and fluorescence/phase‐contrast overlay images shown in Appendix Fig S11). As described previously (Serra & Hengge, 2014), the parental strain AR3110 shows a dense arrangement of a curli–cellulose composite matrix all around the small starving cells in the upper layer, which in deeper layers is followed first by “vertical pillars” of matrix interspaced with non‐matrix‐producing regions, then by a loose horizontal network of matrix‐covered cells and finally by a matrix‐free cell layer at the bottom of the macrocolony (Fig 7C). With the strain lacking all five CSS domain PDEs and, to a minor extent, also with the ∆pdeC mutant, the patches of densely matrix‐surrounded cells extended deeper into the macrocolonies (into the “vertical pillar” zone; especially visible after 3 days of growth) and more matrix was produced in the deeper zone. By contrast, the pdeC ASS mutant clearly produced less matrix as particularly visible in the deeper zone of the matrix‐producing layer (Fig 7C and Appendix Fig S11). This indicated that CSS domain PDEs are not inactive under standard conditions but contribute to shaping biofilm matrix architecture by selectively reducing matrix production in the deeper layers of mature macrocolony biofilms. The observation that chromosomally encoded PdeCASS did so in a more pronounced manner (Fig 7C) again confirmed that the free thiol form of PdeC has increased PDE activity.
Figure 7. CSS domain PDEs affect matrix production and architecture in deeper zones of mature macrocolony biofilms.
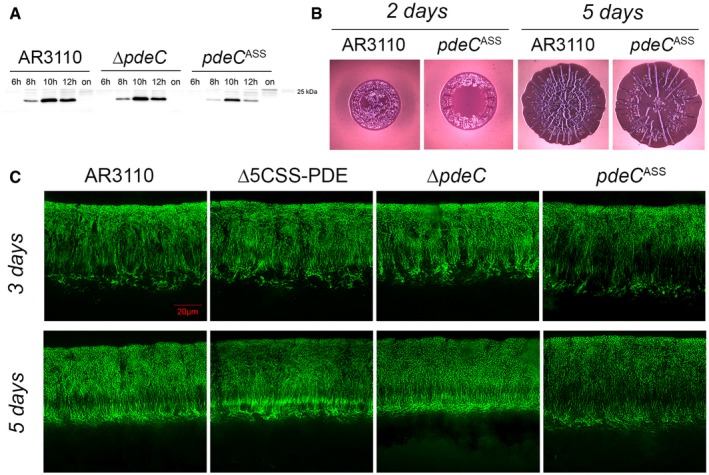
- Strain AR3110 and its derivatives carrying the indicated chromosomal mutations were grown in LB medium. Samples were taken during the growth of the cultures (numbers refer to hours of growth; on, overnight). Cellular CsgD levels were determined by immunoblotting.
- Macrocolonies of strain AR3110 and its chromosomal pdeC ASS derivative were grown on salt‐free LB (LBnoS) with the matrix dye Congo red (CR) at 28°C for 2 and 5 days.
- Fluorescence micrographs of vertical cryosections through the central flat regions of macrocolony biofilms of strain AR3110 and the indicated mutant derivatives were obtained after growth on salt‐free LB for 3 and 5 days at 28°C. Agar plates contained the fluorescent matrix dye thioflavin S which allows to visualize details of the extracellular matrix architecture present in the upper 45–50 μm of the macrocolonies (the entire height of Escherichia coli macrocolonies is approximately 65 μm; cells in the lower layer do not produce matrix but a mesh of entangled flagella; Serra et al, 2013a). Here, only fluorescent images are shown, the corresponding phase‐contrast and merged fluorescent/phase‐contrast images are presented in Appendix Fig S11.
Source data are available online for this figure.
Discussion
The transmembrane CSS domain phosphodiesterase PdeC controls c‐di‐GMP by a novel redox control mechanism
In this study, we demonstrate that the c‐di‐GMP phosphodiesterase PdeC—as a representative of the five CSS domain PDEs of E. coli K‐12—is controlled by a disulfide‐based redox switch in its N‐terminal periplasmic CSS domain. Summarizing our results, we come to the following model for the function of PdeC (Fig 8): Flanked by two transmembrane regions (TM1 and TM2), the CSS domain loops into the periplasm, where its two highly conserved cysteine residues can form an intramolecular DSB—a process that is promoted by the DsbA/DsbB system and is associated with reduced PdeC activity. On the other hand, the free thiol form of the CSS domain promotes enzymatic activity of the cytoplasmic EAL domain. In addition, this free thiol form also gets attacked by the major periplasmic HtrA‐type proteases DegP and DegQ, which generate a smaller transmembrane PdeC fragment consisting essentially of TM2 and the C‐terminal cytoplasmic EAL domain. Also this fragment shows high enzymatic activity, with activity of both the full‐size form and the fragment being associated with dimerization promoted in a sequence‐specific manner by the TM2 transmembrane segment. The proteolytic PdeC fragment shows additional slow shortening—by a still unidentified protease(s)—between TM2 and the EAL domain. The soluble EAL domain alone is inactive, does not dimerize, and slowly disappears by complete proteolysis. A similar role of DSB formation in the CSS domain and DsbA, DegP, and DegQ in the control of PdeB (Fig 5) indicates that this mechanism may in general control the enzymatic activity of this widespread, but hitherto uncharacterized type of bacterial transmembrane c‐di‐GMP phosphodiesterases.
Figure 8. Model of the redox state of the periplasmic CSS domain controlling PDE activity of PdeC in Escherichia coli .
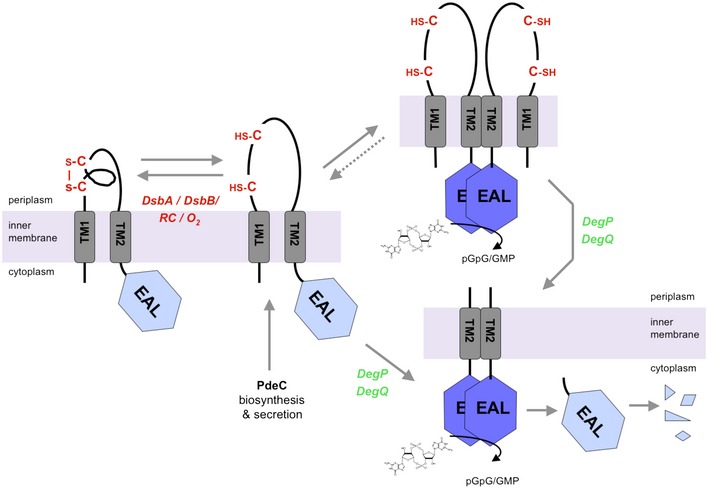
Newly synthesized and membrane‐inserted PdeC has several options: (i) DSB formation (promoted by DsbA/DsbB, with electrons being delivered to oxygen via the respiratory chain (RC)), (ii) dimerization, and/or (iii) DegP/DegQ‐mediated processing to the shorter still membrane‐inserted TM2 + EAL form that also dimerizes. Dimeric forms of full‐size or processed PdeC exhibit increased PDE activity (symbolized by dark blue EAL domains), which reduces c‐di‐GMP and thereby inhibits production of biofilm matrix components. Standard aerobic growth conditions favor DSB formation rather than dimerization of the full‐size protein and thus keep PdeC in a less active or inactive form. Proteases involved in further processing of the DegP/DegQ‐generated TM2 + EAL fragment of PdeC have yet to be identified. The data (shown in Fig 5) for a second CSS domain PDE of E. coli K‐12, PdeB, indicate that activity of PdeB is controlled in a similar way. For further details, see Discussion.
In general, this novel redox control mechanism of CSS domain PDEs should allow for multiple signal input via its accessory components. In principle, a newly synthesized and membrane‐inserted PdeC molecule in the still loosely folded free thiol form can follow several paths (Fig 8): (i) Facilitated by the DsbA/DsbB system (which requires an active respiratory chain and oxygen as a terminal electron acceptor), PdeC can form the periplasmic DSB, which results in a tightly folded CSS domain and low or no enzymatic activity of the EAL domain; (ii) the free thiol form can dimerize to an active PDE; or (iii) the free thiol form can be processed to an equally dimerizing and active TM2 + EAL fragment, whose activity can be eliminated by further degradation only. As discussed in detail below, the “molecular fate” of newly synthesized PdeC should therefore depend on several parameters: (i) the activity of the DSB‐modulating system, that is, DsbA/DsbB and possibly DsbD/DsbC/DsbG, and (ii) the cellular levels and availability of the proteases DegP and DegQ.
Role of the DsbA/DsbB system in redox control of PdeC activity
In wild‐type E. coli growing under standard aerobic laboratory conditions, the DsbA/DsbB system promotes intramolecular DSB formation between Cys75 and Cys106 in PdeC. This DSB formation is likely to promote tighter folding of the periplasmic CSS domain, since it protects against attack by DegP and DegQ which are proteases that recognize unfolded regions in periplasmic proteins (Clausen et al, 2011; Figs 2, 3, and 5). DSB formation in the CSS domain also interferes with dimerization via TM2 (Fig 4) and reduces enzymatic activity of the cytoplasmic EAL domain of PdeC. The latter was manifested in vivo as reduced CsgD and extracellular matrix production by mutants unable to form this DSB in PdeC (Figs 1 and 7) as well as in vitro as enhanced PDE activity of purified PdeC reconstituted in nanodiscs upon DTT treatment (Fig 6). Importantly, this DTT‐mediated stimulation of PDE activity was also found in two PdeC variants (PdeCEALΔC and PdeC/N) that featured only the two CSS domain cysteines and where constructed to avoid loss of PDE activity due to illegitimate in vitro DSB formation involving the three cytoplasmic cysteine residues of PdeC (representing generally non‐conserved amino acids in EAL domains). Taken together, these data characterize the periplasmic CSS domain as a novel sensory input domain that controls the activity of c‐di‐GMP‐specific PDEs by a DSB/thiol switch mechanism.
While a DSB/thiol switch is a novelty for a bacterial second messenger‐controlling enzyme, it is partially reminiscent of the regulation of the transmembrane transcription factors ToxR and TcpP in Vibrio cholerae. However, these factors, which are structurally unrelated to CSS‐PDEs, have an opposite transmembrane orientation, with DNA‐binding HTH domains at their N‐termini and C‐terminal DSB‐forming domains in the periplasm (Miller et al, 1987). In addition, ToxR and TcpP, which are involved in virulence and porin regulation of V. cholerae, are activated by DSB formation in their C‐terminal periplasmic domains (Fengler et al, 2012; Fan et al, 2014). For TcpP, it has been shown that covalent dimerization via an intermolecular DSB results in activation (Yang et al, 2013). For PdeC, however, intermolecular DSB formation can only be observed when one of the two cysteine residues in the CSS domain is missing (as in PdeCASS or PdeCC75A), indicating that for PdeC intramolecular DSB is physiologically relevant. Despite the structural differences and opposite functional consequences of DSB formation in their periplasmic domains, PdeC, ToxR, and TcpP all show susceptibility to proteolysis in their periplasmic domains. Yet, this degradation involves different sets of proteases (Almagro‐Moreno et al, 2015; Teoh et al, 2015). Notably, ToxR and TcpP can escape degradation with the help of the accessory periplasmic proteins ToxS and TcpH, respectively (Almagro‐Moreno et al, 2015; Morgan et al, 2015). Taken together, PdeC, ToxS, and TcpH demonstrate the striking versatility of transmembrane signaling that combines DSB/free thiol switching with proteolytic processing.
Proteolysis of the free thiol form of PdeC—reversible versus irreversible activation and final inactivation
PdeCASS is highly active and dimerizes also in a degP degQ mutant background, indicating that DegP/DegQ‐mediated cleavage is not essential to activate PdeC (Fig 3). Nevertheless, cleavage readily occurs both with the reduced form of PdeCwt (in the presence of DTT) as well as with PdeCASS (Figs 2 and 5). In addition, also a cloned TM2 + EAL fragment that mimics the processed PdeC fragment has high PDE activity (Fig 4). This means that activation of the C‐terminal EAL domain occurs in two interlinked modes (Fig 8): (i) transition into the free thiol form of full‐size PdeC, which may eventually be inactivated again by DSB formation in the CSS domain, and (ii) proteolytic processing to a C‐terminal fragment that is still membrane‐inserted via TM2 (Appendix Fig S4).
While proteolysis by DegP/DegQ may just be a by‐product of partial unfolding of the CSS domain that comes along with reducing the DSB, its occasional occurrence is also a way of achieving slow turnover of PdeC and other CSS domain PDEs. Notably, four of the five CSS domain PDEs of E. coli K‐12 are under σS control; that is, they are mainly expressed in slowly growing and/or stationary phase cells (Sommerfeldt et al, 2009; Sarenko et al, 2017), in which proteins are no longer subject to dilution by cell division. However, proteolysis should be a relatively slow process to allow newly synthesized and membrane‐integrated PdeC to become oxidized by the DsbA/DsbB system. On the other hand, once PdeC becomes proteolytically processed, it is also irreversibly activated which calls for further proteolysis so that cells eventually can get rid of the active protein. Our data indeed suggest terminal proteolytic removal of processed active PdeC, since all PdeC fragments generated in this study showed further shortening in overnight cells (well detectable since de novo protein synthesis from the tac promoter no longer occurs). The stand‐alone EAL domain even almost completely disappeared with no shorter fragments accumulating. Moreover, also the cellular amount of full‐size PdeCwt protein was reduced in overnight cells—provided that the DSB‐reducing DsbD/DsbC/DsbG system was intact, suggesting that occasional removal of the DSB initiates slow degradation also for PdeCwt. However, with this processing being occasional only, this does not lead to the accumulation of visible proteolytic intermediates as with PdeCASS, which has no means to escape efficient attack by DegP/DegQ.
In conclusion, activity control of PdeC is tightly interlinked with a complex proteolytic pathway, which involves DegP/DegQ on the periplasmic side as well as additional proteases that cleave on the inner side of the membrane and in the cytoplasm, which will have to be identified in future studies (Fig 8). Overall, this sequential proteolysis is conceptually related to the degradative inactivation of RseA, the anti‐sigma factor for σE (RpoE) in the cell envelope stress response—even though transmembrane orientation of RseA (C‐terminus in the periplasm) and the actual mechanism including signal input and the periplasmic protease involved (DegS) are all different (Alba & Gross, 2004; Kroos & Akiyama, 2013). It seems that sequential transmembrane proteolysis is such a powerful control mechanism that bacteria “invented” it repeatedly for very different types of transmembrane proteins (anti‐sigma factors, TFs, PDEs) by combining different modules available (signal input mechanisms, proteases).
While it is obvious that sequential proteolysis can depend on a specific initial signal input that affects the structure or accessibility of the protein substrate, another possibility for regulation relies on the levels and availability of the different proteases involved. DegP expression is under control of the σE‐mediated as well as the Cpx‐dependent cell envelope stress responses (Connolly et al, 1997). Moreover, general proteases (like chaperones) can be sequestered when unfolded and aggregated proteins, which are also substrates, suddenly accumulate. Thus, it will be interesting to study whether imposing cell envelope stresses or heat shock has any impact on the activity and degradation patterns of PdeC. While our temperature shift experiment (with a secA ts strain) was initially aimed at addressing the question whether TM1 is a cleavable signal sequence (which is not the case; Appendix Fig S7), it also showed that shift of wild‐type cells to 42°C indeed seemed to increase processing of PdeC to the 33‐kD fragment (Appendix Fig S7).
A physiological role for PdeC and potentially additional CSS domain PDEs in biofilm formation
Finally, our study addressed the question of the physiological function of PdeC and potentially additional CSS domain PDEs in E. coli biofilms, where synthesis of the extracellular matrix is a major target of c‐di‐GMP signaling. What could be natural conditions that decrease DSB formation in PdeC and thereby activate PdeC and possibly other CSS domain PDEs? Our observation (Fig 7C) that mutants lacking PdeC or all five CSS domain PDEs show a denser matrix architecture specifically in the deeper layer of mature macrocolony biofilms, whereas a chromosomal pdeC ASS allele shows the opposite phenotype, indicates that a condition that can activate PdeC and similar CSS domain PDEs is low oxygen supply. Within macrocolonies a steep vertical oxygen gradient is generated as a result of diffusion (from the air interface above) and cellular consumption (by respiratory energy metabolism). Thus, within similar macrocolonies of Pseudomonas aeruginosa, the oxygen content was found to be reduced to 10% of atmospheric oxygen at a depth of 45–50 μm from the surface, with essentially anoxic conditions below 60 μm (Dietrich et al, 2013). Notably, the height of macrocolonies of E. coli AR3110 is approximately 60–70 μm (Serra et al, 2013a; see also Appendix Fig S11), suggesting microaerobic conditions in the deeper layers where CSS domain PDEs are active. Low oxygen content in these deeper zones (in the absence of alternative electron acceptors) is likely to become limiting for the activity of the DsbA/DsbB system which has to deliver its electrons to the respiratory chain (Ito & Inaba, 2008). As a consequence, DSB formation of CSS domain PDEs would slow down with the other options, namely dimerization‐associated activation and eventually processing by DegP/DegQ (Fig 8), becoming more efficient.
Interestingly, the DsbA/DsbB pathway also affects the activity of a diguanylate cyclase, DgcN (formerly YfiN), which also has a transmembrane configuration and is inactivated by YfiR, a periplasmic protein whose direct inhibitory interaction with DgcN requires DsbA/DsbB‐dependent DSB formation in YfiR (Hufnagel et al, 2014). Since in general c‐di‐GMP‐controlled functions are the result of a dynamic activity balance between DGCs and PDEs (Jenal et al, 2017), it could be an interesting hypothesis for future studies that also DgcN may be activated by reduced DsbA/DsbB activity and act as an antagonistic DGC that negotiates the actual matrix density in the microaerobic deeper zones of macrocolony biofilms with CSS domain PDEs. Such a role is consistent with both DgcN and CSS domain PDEs being present in slowly growing cells (as in a dense biofilm), since DgcN is expressed from a σ70‐driven promoter and is present throughout all growth phases, whereas four of the five CSS domain PDEs are under σS control and accumulate in cells that grow slowly or not at all (Sarenko et al, 2017). On the other hand, in rapidly growing cells DgcN would be predominant and may be part of a reducing stress response that allows cells to generate a protective matrix independently of the σS/CsgD pathway (Hufnagel et al, 2014).
Outlook—potential adaptive variations in function and regulation of multiple CSS‐PDEs in single species
Escherichia coli K‐12 has five CSS domain PDEs (PdeB, PdeC, PdeD, PdeG, and PdeN) and certain EHEC and EPEC strains even possess another one (PdeT/VmpA) (Branchu et al, 2012; Hengge et al, 2015; Povolotsky & Hengge, 2016). For PdeB, we have shown here a control of activity by DsbA‐mediated DSB formation and DegP/DegQ‐mediated proteolytic processing similar to PdeC (Fig 5). But also the other proteins feature the characteristic two cysteine residues at similar positions in their CSS domains (Appendix Fig S1) and thus probably can form a periplasmic DSB. In addition, all five CSS domain PDEs of E. coli K‐12 are expressed under standard laboratory conditions (Sarenko et al, 2017). Why does E. coli have so many CSS domain PDEs, with four of them (PdeB, PdeC, PdeD, and PdeN) belonging to the most highly conserved PDEs present in > 90% of a set of 61 strains that include all phylogroups and pathotypes (Povolotsky & Hengge, 2016)?
It is conceivable that DSB formation of the many CSS domain PDEs differs slightly depending on specific redox potentials and specific recognition by the oxidizing DsbA/DsbB system and/or the reducing DsbD/DsbC/DsbG system. In addition, in some case the oxidized DSB form may also be enzymatically active, while reduction to the free thiol form may trigger proteolysis and irreversible inactivation. Alternatively, if processing leads to a highly active form, as shown here for PdeC, the efficiency of proteolytic processing as well as the lifetime of the processed form could be different for other CSS domain PDEs and thereby affect their activities. While details of these potential variations will have to be investigated in future studies, CSS domain PDEs and their mechanisms of control seem a highly versatile system that may rapidly diversify and evolve to adapt to different conditions and niches. In addition, some CSS domain PDEs could also have specialized functions, possibly by specifically interacting with other DGCs, PDEs, and/or distinct effector/target systems in “local signaling modules” (Sarenko et al, 2017). Future comparative analyses of mechanisms of activity control and protein interaction partners will shed further light on this intriguing novel class of c‐di‐GMP signaling enzymes.
Materials and Methods
Bacterial strains and growth conditions
Escherichia coli strains used in this study are derivatives of the K‐12 strain W3110. Construction of strains, mutations, and plasmids are described in detail in the Appendix, with all oligonucleotide primers used listed in Appendix Table S1. Macrocolony biofilms were grown on salt‐free LB or YESCA agar plates containing Congo red (for the detailed procedure, see Appendix Supplementary Methods). Liquid cultures were grown in LB medium and started with an optical density at 578 nm (OD578) of 0.05 (Serra et al, 2013a,b). Macrocolonies to be compared were grown on the same agar plate, with photographs (see below) being taken separately to achieve higher image resolution. Since cellulose and curli fiber expression occurs only below 30°C in E. coli K‐12 derivatives, cultures were grown at 28°C unless otherwise indicated. The term “post‐exponential growth” in liquid LB culture refers to an OD578 of 2.8–3.5 (reached after 8–10 h growth under our standard conditions). Overnight cultures have an OD578 of 4.8–5.2.
Standard molecular biological and biochemical procedures
Cloning procedures (including a description of the low copy number ptac expression vector and sequences of oligonucleotide primers), mutagenesis techniques, immunoblotting of proteins, cell extract fractionation and overexpression and purification of proteins are described in the Appendix Supplementary Methods.
In vivo protein–protein interaction studies
Protein–protein interactions were assayed using an adenylate cyclase‐based bacterial two‐hybrid system (Karimova et al, 1998). Oligonucleotide primers for cloning onto pUT18 and pKNT25 plasmids to generate fusion proteins are listed in the Appendix. Plasmids were co‐transformed into a ∆cya derivative of strain W3110 and incubated on MacConkey agar plates supplemented with maltose (1%), ampicillin (100 μg/ml), and kanamycin (50 μg/ml) for 48 h at 28°C. Red colonies indicate utilization of maltose which depends on cAMP production due to direct interaction of the proteins fused to the otherwise separate adenylate cyclase domains. All colonies to be compared were grown together on a single agar plate (140 mm diameter), but separate close‐up photographs were taken.
Reconstitution of purified PdeC variants into lipid bilayer nanodiscs
C‐terminally His6‐tagged versions of PdeCwt, PdeCEAL∆C, and PdeC/N were solubilized using n‐dodecyl β‐D‐maltoside (DDM) as a detergent and purified by a standard protocol for membrane proteins (Hebbeln et al, 2007) with minor modifications as described in the Appendix Supplementary Methods. The solubilized proteins were reconstituted into nanodiscs (Bayburt et al, 2002; Borch & Hamann, 2009) using chloroform‐solubilized E. coli total lipid extract (Avanti Polar Lipids) and the purified membrane scaffold protein MSP1E3D1. Nanodiscs containing the desired PdeC variants were purified on Ni‐NTA agarose (Macherey‐Nagel) using the His6 tag on the inserted proteins, concentrated with Amicon Ultra‐4 columns (Merck Millipore) and used within 7 days. The detailed procedure is described in the Appendix Supplementary Methods.
Stereomicroscopy
Escherichia coli macrocolony biofilms were visualized at 10× magnification with a Stemi 2000‐C stereomicroscope (Zeiss, Oberkochen, Germany). Digital images were captured with an AxioCamICC3 digital camera coupled to the stereomicroscope, operated via the AxioVision 4.8 software (Zeiss).
Cryosectioning of macrocolony biofilms and fluorescence microscopy
The procedure and materials used for cryomicrotomy and fluorescence microscopy of macrocolony biofilms grown in the presence of thioflavin S (TS), which binds to both amyloid curli fibers and cellulose, have been described in detail (Serra & Hengge, 2017). The exact protocol for obtaining 5‐μm‐thick sections perpendicular to the plane of macrocolonies can be found in the Appendix Supplementary Methods. Samples were visualized at 630× magnification. All digital images were captured using an AxioCam 506 mono digital camera coupled to the Axio Observer.Z1 microscope and Zen 2 software (Zeiss). All cryosectioning and fluorescence microscopy experiments were done in two biological replicates.
Statistics
Unless otherwise stated, experiments reported in this study were done in at least three biological replicates. In photographic documentations, representative results are shown.
Author contributions
Concept of the study: RH; design of experiments: all authors; experiments were performed by SH, ML, OS, TKLN, and TJ; interpretation of data: all authors; the paper was written by RH; all authors read and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 1
{kind=link}
Source Data for Figure 2
{kind=link}
Source Data for Figure 3
{kind=link}
Source Data for Figure 4
{kind=link}
Source Data for Figure 5
Source Data for Figure 7
{kind=link}
Acknowledgements
We thank Heidi Landmesser and Friedrich Finkenwirth for valuable advice on nanodisc experiments and for providing membrane scaffold protein. Financial support was provided by the European Research Council under the European Union's Seventh Framework Programme (ERC‐AdG 249780 to RH) and the Deutsche Forschungsgemeinschaft (He 1556/17‐1 and He 1556/21‐1 to RH).
The EMBO Journal (2018) 37: e97825
References
- Alba BM, Gross CA (2004) Regulation of the Escherichia coli sigmaE‐dependent envelope stress response. Mol Microbiol 52: 613–619 [DOI] [PubMed] [Google Scholar]
- Almagro‐Moreno S, Root MZ, Taylor RK (2015) Role of ToxS in the proteolytic cascade of virulence regulator ToxR in Vibrio cholerae . Mol Microbiol 98: 963–976 [DOI] [PubMed] [Google Scholar]
- Barends TR, Hartmann E, Griese JJ, Beitlich T, Kirienko NV, Ryjenkov DA, Reinstein J, Shoeman RL, Gomelsky M, Schlichting I (2009) Structure and mechanism of a bacterial light‐regulated cyclic nucleotide phosphodiesterase. Nature 18: 1015–1018 [DOI] [PubMed] [Google Scholar]
- Barnhart MM, Chapman MR (2006) Curli biogenesis and function. Annu Rev Microbiol 60: 131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Grinkova YV, Sligar SG (2002) Self‐assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett 2: 853–856 [Google Scholar]
- Borch J, Hamann T (2009) The nanodisc: a novel tool for membrane protein studies. Biol Chem 390: 805–814 [DOI] [PubMed] [Google Scholar]
- Branchu P, Hindré T, Fang X, Thomas R, Gomelsky M, Claret L, Harel J, Gobert AP, Martin C (2012) The c‐di‐GMP phosphodiesterase VmpA absent in Escherichia coli K12 strains affects motility and biofilm formation in the enterohemorrhagic O157:H7 serotype. Vet Immunol Immunopathol 152: 132–140 [DOI] [PubMed] [Google Scholar]
- Brombacher E, Dorel C, Zehnder AJB, Landini P (2003) The curli biosynthesis regulator CsgD co‐ordinates the expression of both positive and negative determinants for biofilm formation in Escherichia coli . Microbiology 149: 2847–2857 [DOI] [PubMed] [Google Scholar]
- Cho S‐H, Collet J‐F (2013) Many roles of the bacterial envelope reducing pathways. Antioxid Redox Signal 18: 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T, Kaiser M, Huber R, Ehrmann M (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12: 152–162 [DOI] [PubMed] [Google Scholar]
- Connolly L, De las Penas A, Alba BM, Gross CA (1997) The response to extracytoplasmic stress n Escherichia coli is controlled by partially overlapping pathways. Genes Dev 11: 2012–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich LE, Okegbe C, Price‐Whelan A, Sakhtah H, Hunter RC, Newman DK (2013) Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol 195: 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Liu Z, Jabeen N, Birdwill LD, Zhu J, Kan B (2014) Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen‐limiting conditions. Infect Immun 82: 1676–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengler VHI, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J (2012) Disulfide bond formation and ToxR activity in Vibrio cholerae . PLoS One 7: e47756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall‐Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2: 95–108 [DOI] [PubMed] [Google Scholar]
- Hebbeln P, Rodionov DA, Alfandega A, Eitinger T (2007) Biotin uptake in prokaryotes by solute transporters with an optional ATP‐binding cassette‐containing module. Proc Natl Acad Sci USA 104: 2909–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R (2009) Principles of cyclic‐di‐GMP signaling. Nat Rev Microbiol 7: 263–273 [DOI] [PubMed] [Google Scholar]
- Hengge R, Galperin MY, Ghigo J‐M, Gomelsky L, Green J, Hughes KT, Jenal U, Landini P (2015) Systematic nomenclature for GGDEF and EAL domain‐containing c‐di‐GMP turnover proteins of Escherichia coli . J Bacteriol 198: 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R (2016) Trigger phosphodiesterases as a novel class of c‐di‐GMP effector proteins. Philos Trans R Soc Lond B Biol Sci 371: 20150498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel DA, DePas WH, Chapman MR (2014) The disulfide bonding system suppresses CsgD‐independent cellulose production in Escherichia coli . J Bacteriol 196: 3690–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel DA, DePas WH, Chapman MR (2015) The biology of the Escherichia coli extracellular matrix. In Microbial Biofilms, Ghannoum M, Parsek M, Whiteley M, Mukherjee PK. (eds), 2nd edn. Washington, DC: ASM Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Inaba K (2008) The disulfide bond formation (Dsb) system. Curr Opin Struct Biol 18: 450–458 [DOI] [PubMed] [Google Scholar]
- Jenal U, Malone J (2006) Mechanisms of cyclic‐di‐GMP signaling in bacteria. Annu Rev Genet 40: 385–407 [DOI] [PubMed] [Google Scholar]
- Jenal U, Reinders A, Lori C (2017) Cyclic‐di‐GMP: second messenger extraordinaire. Nat Rev Microbiol 15: 271–284 [DOI] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D (1998) A bacterial two‐hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA 95: 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krojer T, Garrido‐Franco M, Huber R, Ehrmann M, Clausen T (2002) Crystal structure of DegP (HtrA) reveals a new protease‐chaperone machine. Nature 416: 455–459 [DOI] [PubMed] [Google Scholar]
- Kroos L, Akiyama Y (2013) Biochemical and structural insights into intramembrane metalloprotease mechanisms. Biochim Biophys Acta 1828: 2873–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenberg S, Klauck G, Pesavento C, Klauck E, Hengge R (2013) The EAL domain phosphodiesterase YciR acts as a trigger enzyme in a c‐di‐GMP signaling cascade in E. coli biofilm control. EMBO J 32: 2001–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer M, Hasenbein S, Mamant N, Merdanovic M, Poepsel S, Hauske P, Kaiser M, Huber R, Krojer T, Clausen T, Ehrmann M (2009) Structure, function and regulation of the conserved serine proteases DegP and DegS of Escherichia coli . Res Microbiol 160: 660–666 [DOI] [PubMed] [Google Scholar]
- Miller VL, Taylor RK, Mekalanos JJ (1987) Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48: 271–279 [DOI] [PubMed] [Google Scholar]
- Minasov G, Padavattan S, Shuvalova L, Brunzelle JS, Miller DJ, Baslé A, Massa C, Collart FR, Schirmer T, Anderson WF (2009) Crystal structures of YkuI and its complex with second messenger cyclic di‐GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. J Biol Chem 284: 13174–13184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Schmidt R, Bukau B (2007) The N‐end rule pathway for regulated proteolysis: prokaryotic and eukaryotic strategies. Trends Cell Biol 17: 165–172 [DOI] [PubMed] [Google Scholar]
- Morgan SJ, French EL, Thomson JJ, Seaborn CP, Shively CA, Krukonis ES (2015) Formation of an intramolecular periplasmic disulfide bond in TcpP protects TcpP and TcpH from degradation in Vibrio cholerae . J Bacteriol 198: 498–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento C, Becker G, Sommerfeldt N, Possling A, Tschowri N, Mehlis A, Hengge R (2008) Inverse regulatory coordination of motility and curli‐mediated adhesion in Escherichia coli . Genes Dev 22: 2434–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povolotsky TL, Hengge R (2016) Genome‐based comparison of c‐di‐GMP signaling in commensal and pathogenic Escherichia coli . J Bacteriol 198: 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent‐Combaret C, Brombacher E, Vidal O, Ambert A, Lejeune P, Landini P, Dorel C (2001) Complex regulatory network controls initial adhesion and biofilm formation in Escherichia coli via regulation of the csgD gene. J Bacteriol 183: 7213–7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Qi Y, Chong HS, Kotaka M, Li B, Li J, Lescar J, Tang K, Liang ZX (2009) The functional role of a conserved loop in EAL domain‐based cyclic di‐GMP‐specific phosphodiesterase. J Bacteriol 191: 4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG (2009) Chapter 11 – Reconstitution of membrane proteins in phospholipid nanodiscs. Methods Enzymol 464: 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert‐Paganin J, Nonin‐Lecomte S, Réty S (2012) Crystal structure of an EAL domain in complex with reaction product 5′‐pGpG. PLoS One 7: e52424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U, Galperin MY, Gomelsky M (2013) Cyclic‐di‐GMP: the first 25 years of a universal bacterial second messenger. Microb Mol Biol Rev 77: 1–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, Kaever V, Hengge R (2017) More than enzymes that make and break c‐di‐GMP ‐ the protein interaction network of GGDEF/EAL domain proteins of Escherichia coli . MBio 8: e01639–01617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AJ, Ryjenkov DA, Gomelsky M (2005) The ubiquitous protein domain EAL is a cyclic diguanylate‐specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187: 4774–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, Richter AM, Hengge R (2013a) Cellulose as an architectural element in spatially structured Escherichia coli biofilms. J Bacteriol 195: 5540–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, Richter AM, Klauck G, Mika F, Hengge R (2013b) Microanatomy at cellular resolution and spatial order of physiological differentiation in a bacterial biofilm. MBio 4: e00103–e00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, Hengge R (2014) Stress responses go three‐dimensional – the spatial order of physiological differentiation in bacterial macrocolony biofilms. Environ Microbiol 16: 1455–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, Klauck G, Hengge R (2015) Vertical stratification of matrix production is essential for physical integrity and architecture of macrocolony biofilms of Escherichia coli . Environ Microbiol 17: 5073–5088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra DO, Hengge R (2017) Experimental detection and visualization of the extracellular matrix in macrocolony biofilms In c‐di‐GMP signaling: methods & protocols – methods in molecular biology, Sauer K. (ed), pp 133–145. New York, NY: Humana Press, Springer Nature; [DOI] [PubMed] [Google Scholar]
- Sommerfeldt N, Possling A, Becker G, Pesavento C, Tschowri N, Hengge R (2009) Gene expression patterns and differential input into curli fimbriae regulation of all GGDEF/EAL domain proteins in Escherichia coli . Microbiology 155: 1318–1331 [DOI] [PubMed] [Google Scholar]
- Sundriyal A, Massa C, Samoray D, Zehender F, Sharpe T, Jenal U, Schirmer T (2014) Inherent regulation of EAL domain‐catalyzed hydrolysis of second messenger cyclic di‐GMP. J Biol Chem 289: 6978–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchigvintsev A, Xu X, Singer A, Chang C, Brown G, Proudfoot M, Cui H, Flick R, Anderson WF, Joachimiak A, Galperin MY, Savchenko A, Yakunin AF (2010) Structural insight into the mechanism of c‐di‐GMP hydrolysis by EAL domain phosphodiesterases. J Mol Biol 402: 524–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh WP, Matson JS, DiRita VJ (2015) Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail‐specific protease (Tsp). Mol Microbiol 97: 822–831 [DOI] [PubMed] [Google Scholar]
- Wouters MA, Fan SW, Haworth NL (2010) Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid Redox Signal 12: 53–91 [DOI] [PubMed] [Google Scholar]
- Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J (2013) Bile salt‐induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci USA 110: 2348–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogaj X, Nimtz M, Rohde M, Bokranz W, Römling U (2001) The multicellular morphotypes of Salmonella typhimurium and Escherichia coli produce cellulose as the second component of the extracellular matrix. Mol Microbiol 39: 1452–1463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7