

Abstract
Adult mouse muscle satellite cells (MuSCs) are quiescent in uninjured muscles. Upon injury, MuSCs exit quiescence in vivo to become activated, re‐enter the cell cycle to proliferate, and differentiate to repair the damaged muscles. It remains unclear which extrinsic cues and intrinsic signaling pathways regulate quiescence exit during MuSC activation. Here, we demonstrated that inducible MuSC‐specific deletion of p110α, a catalytic subunit of phosphatidylinositol 3‐kinase (PI3K), rendered MuSCs unable to exit quiescence, resulting in severely impaired MuSC proliferation and muscle regeneration. Genetic reactivation of mTORC1, or knockdown of FoxOs, in p110α‐null MuSCs partially rescued the above defects, making them key effectors downstream of PI3K in regulating quiescence exit. c‐Jun was found to be a key transcriptional target of the PI3K/mTORC1 signaling axis essential for MuSC quiescence exit. Moreover, induction of a constitutively active PI3K in quiescent MuSCs resulted in spontaneous MuSC activation in uninjured muscles and subsequent depletion of the MuSC pool. Thus, PI3K‐p110α is both necessary and sufficient for MuSCs to exit quiescence in response to activating signals.
Keywords: FoxOs, mTORC1, muscle satellite cells, phosphatidylinositol 3‐kinase, quiescence exit
Subject Categories: Cell Cycle, Signal Transduction, Stem Cells
Introduction
In vertebrate skeletal muscles, adult muscle stem cells, also called muscle satellite cells (MuSCs), are quiescent and sandwiched between the sarcolemma of individual myofibers and the surrounding basal lamina (Brack & Rando, 2012; Yin et al, 2013; Sambasivan & Tajbakhsh, 2015). These adult MuSCs derive from the Pax3/Pax7‐positive progenitor cells in the dermomyotome during embryonic development and are characteristically marked by Pax7 expression in postnatal muscles (Buckingham & Relaix, 2015). MuSCs are responsible for injury‐induced muscle regeneration (Collins et al, 2005; Sacco et al, 2008; Lepper et al, 2011; Murphy et al, 2011; Sambasivan et al, 2011). Our current understanding of MuSCs during injury‐induced muscle regeneration is as follows: Upon muscle injury, the quiescent MuSCs (QSCs) first exit quiescence to become activated satellite cells (or ASCs), re‐enter the cell cycle to proliferate to increase in numbers (now called muscle precursor cells or myoblasts), and then differentiate and fuse to become multinucleated myotubes, eventually resulting in a full regeneration of the damaged muscles. During regeneration, a small number of the proliferative myoblasts can return back to quiescence to replenish the pool of QSCs (i.e., self‐renewal). Morphologically, the Pax7+ QSCs are small in size (~6 μm) with scarce cytoplasm and increased nuclear heterochromatin that is readily detectable by electron microscopy (Mauro, 1961; Fukada et al, 2007; Rodgers et al, 2014). In contrast, ASCs and proliferative myoblasts are bigger in size (~11 μm) with more cytoplasm and less heterochromatin and are molecularly characterized by the expression of MyoD (Yin et al, 2013), a member of the skeletal muscle‐specific myogenic regulatory factor (MRF) subfamily that also includes Myf5, MRF4, and myogenin (Tapscott, 2005).
Adult QSCs are tightly regulated. They remain quiescent in vivo with little turnover over a long period of time without being affected by routine daily muscular activities (Lepper et al, 2009; Chakkalakal et al, 2012; Pawlikowski et al, 2015). In response to muscle injury, it takes about an average of 24–36 h in vivo before most QSCs start to enter the first cell cycle as evidenced by the incorporation of a nucleoside analog (e.g., EdU or BrdU; Rocheteau et al, 2012; Rodgers et al, 2014). Similarly, it takes about 24–36 h before most of the freshly isolated MuSCs enter the first cell cycle in culture. A number of molecules and signaling pathways have been found to regulate MuSC quiescence. For example, Tie‐2, Sprouty1, miR‐489, Pten, eIF2α, and Suv4‐20 h1 were all shown to affect MuSC quiescence via various mechanisms (Abou‐Khalil et al, 2009, 2; Shea et al, 2010; Cheung et al, 2012; Boonsanay et al, 2016, 4; Zismanov et al, 2016, 2; Yue et al, 2017). Interference of any of these regulators compromises either quiescence maintenance in uninjured muscles or reestablishment of the QSC pool (i.e., self‐renewal) after muscle injury. In addition, the Notch pathway was found to be indispensable for maintaining MuSC quiescence. Inactivation of the Notch pathway results in spontaneous activation of MuSCs and subsequent depletion of the MuSC pool (Bjornson et al, 2012; Mourikis et al, 2012), while deliberate activation of the Notch signaling in MuSCs upregulates Pax7 expression and promotes the self‐renewal of MuSCs during injury‐induced muscle regeneration (Wen et al, 2012). However, it is unclear whether quiescence exit is actively regulated by a molecule or signaling pathway.
The phosphatidylinositol 3‐kinase (PI3K)/mechanistic target of rapamycin complex 1 (mTORC1) pathway is required for multiple cellular processes (Vanhaesebroeck et al, 2012; Thorpe et al, 2015; Saxton & Sabatini, 2017). This pathway is typically activated by various growth factors (e.g., epidermal growth factor) via distinct receptor tyrosine kinases (RTKs) on the plasma membrane. Mutations in several key components of the pathway that render the pathway constitutively active have been found in multiple human malignancies (Vanhaesebroeck et al, 2012; Thorpe et al, 2015). In myoblasts, the PI3K/mTOR pathway was found to promote myogenic differentiation in cell culture studies (Tamir & Bengal, 2000; Xu & Wu, 2000). In adult skeletal muscles, the pathway was also found to suppress muscle atrophy via mTORC1 and FoxOs (Sandri et al, 2004; Stitt et al, 2004; Bentzinger et al, 2008; Risson et al, 2009). Recently, mTORC1 was also found to promote a reversible “GAlert” state in adult MuSCs upon injury (Rodgers et al, 2014), while FoxO3a was found to regulate the self‐renewal of MuSCs after injury (Gopinath et al, 2014). The role of PI3K in regulating adult MuSCs has not been explored.
Here, we investigated the role of p110α of PI3K in adult MuSCs and muscle regeneration in vivo. By using different mouse models, we demonstrated that p110α of PI3K in adult MuSCs is both necessary and sufficient for injury‐induced quiescence exit and the cell cycle re‐entry. We further revealed that PI3K exerts its functions in MuSCs via two inter‐connected signaling axes: mTORC1‐c‐Jun and FoxOs.
Results
p110α of PI3K was indispensable for adult MuSC functions in vivo
Recent transcriptome analysis of adult MuSCs revealed that p110α mRNA is the most abundantly expressed catalytic subunit of PI3K in both QSCs and ASCs (Liu et al, 2013; Ryall et al, 2015). To examine the activation status of the PI3K pathway in adult MuSCs, we isolated single myofibers, fixed them either right away (for QSCs) or after 24 h in culture (for ASCs), and then performed immunostaining for the phosphorylated ribosomal S6 protein (p‐S6), a very sensitive marker for the activated PI3K/mTORC1 pathway (Park et al, 2008; Vanhaesebroeck et al, 2012; Rodgers et al, 2014; Saxton & Sabatini, 2017). We found that the obvious p‐S6 signal was only detected in ASCs (Fig EV1A). Consistently, p‐S6 was only detected in ASCs on muscle sections from acutely injured muscles in vivo (Fig EV1B). Our data indicated that the PI3K pathway was inactive in QSCs but became activated in ASCs. To reveal a functional role of PI3K in adult MuSCs in vivo, we first conditionally deleted p110α in Pax7‐expressing cells during development using a non‐inducible Pax7‐Cre line (as the germline knockout of p110α was embryonically lethal) (Bi et al, 1999; Keller et al, 2004; Zhao et al, 2006). In multiple litters we examined, no live homozygous mutant pups were found (Fig EV2A), indicating an indispensable role of PI3K in Pax7‐expressing cells during embryonic development. To bypass the embryonic lethality problem, we generated a MuSC‐specific and tamoxifen‐inducible p110α knockout (KO) mouse strain (i.e., p110α f/f:Pax7 CreER, also abbreviated as p110α‐iKO or iKO thereafter; Fig EV2B) (Murphy et al, 2011). The cardiotoxin (CTX)‐induced muscle injury‐regeneration model was employed to interrogate the in vivo function of MuSCs with or without p110α (Chargé & Rudnicki, 2004). At day 2 post‐injury, similar extent of injury as manifested by the appearance of numerous infiltrating immune cells (arrowheads) and degenerating myofibers (pink circles) was seen on the tibialis anterior (TA) muscle sections from both heterozygous control and p110α‐iKO mice (Fig 1A). By day 5 or day 7 post‐injury, multiple small regenerating myofibers (i.e., those with central nuclei) appeared on muscle sections from control mice. By contrast, very few such regenerating myofibers were found in muscles from the p110α‐iKO mice (Fig 1A). Consistently, by immunostaining for embryonic myosin heavy chain (eMHC), a protein that is only transiently expressed in early regenerating myofibers (d'Albis et al, 1988), we could detect numerous eMHC+ myofibers on muscle sections from the control mice. Yet, very few such myofibers were detected in muscles from the p110α‐iKO mice (Fig 1B and C). By day 14 post‐injury, while the injured TA muscles from the control mice were fully regenerated, those from the p110α‐iKO mice remained largely unrepaired. A few small regenerating myofibers (arrows) could be found in mutant muscles, which could derive from p110α + MuSCs that escaped Cre‐mediated recombination. Even at day 28 post‐injury, a large portion of TA muscles in p110α‐iKO mice remained unrepaired (Fig 1A), indicative of a severe block in muscle regeneration. Thus, p110α of PI3K was indispensable for MuSC functions in vivo.
Figure EV1. The PI3K pathway was active in ASC but not QSC .

- Freshly isolated single myofibers from 2‐month‐old wild‐type mice were fixed either immediately or 24 h after culturing.
- TA muscles from uninjured or cardiotoxin (CTX)‐injured mice (2.5 days post‐injury) were dissected, cryosectioned, and fixed. Myofibers from (A) and TA muscle sections from (B) were then subjected to immunostaining for p‐S6 (green) together with Pax7 (A) and YFP (B) (red), respectively. The nuclei were counterstained with DAPI (blue). Arrowheads: YFP+ MuSCs. Scale bars: 25 μm.
Figure EV2. Generation of MuSC‐specific p110α knockout (KO) mice.

- Non‐inducible p110α KO mice. Top: the schematic showing the floxed exon at the p110α locus. Middle: the mating strategy to generate the non‐inducible p110α f/f:Pax7 Cre/+ mice. Bottom: the number of offspring of different genotypes obtained based on the mating strategy above.
- Tamoxifen‐inducible p110α KO mice. Top: the schematic showing the location of CreER in Pax7 CreER(GaKa) mice. Bottom: the mating strategy to generate the tamoxifen‐inducible p110α f/f:Pax7 CreER/CreER mice.
Figure 1. Inducible deletion of p110α in adult MuSCs severely impaired muscle regeneration.

- Top: the experimental scheme. Eight‐week‐old heterozygous (Ctrl) and iKO littermates (n = 3, in each group) were first treated with tamoxifen (Tmx) over 4 weeks followed by cardiotoxin (CTX) injection into tibialis anterior (TA) muscles. Mice were sacrificed 2, 5, 7, 14, and 28 days post‐injury, and TA muscles were subjected to cryosectioning and hematoxylin–eosin staining. Representative images of regenerating TA muscles are shown. The 2nd and 4th rows are enlarged images of the boxed areas shown above. Arrowheads: immune cells.
- Sections of TA muscles from D5 mice in (A) were immunostained for eMHC (red) and laminin (green). The nuclei were counterstained by DAPI (blue).
- Quantification of eMHC+ myofibers from (B) by examining multiple TA sections from three pairs of mice.
Deletion of p110α in adult MuSCs in uninjured muscles reduced Pax7 protein levels without obviously affecting the MuSC number
To understand what caused the regeneration failure in p110α‐iKO mice, we first examined the number of MuSCs in uninjured muscles from the mutant mice. By immunostaining for Pax7, a most commonly used marker for MuSCs, we found that there was a 90% decrease in the number of Pax7+ MuSCs on TA muscle sections from uninjured p110α‐iKO mice compared with that from the heterozygous controls (Fig 2A and B). Similarly, the number of Pax7+ MuSCs was also low on single myofibers freshly isolated from uninjured p110α‐iKO mice (Fig 2C). Moreover, in MuSCs freshly isolated by fluorescence‐activated cell sorting (FACS) based on Vcam1 expression (Liu et al, 2015), the percentage of Vcam1+/Sca1− MuSCs from p110α‐iKO mice was obviously reduced compared to that of the control mice (Fig 2D, top). Consistently, the majority of the MuSCs freshly isolated by FACS from the mutant mice was also negative for Pax7 staining (Fig 2D, bottom). It was unclear whether the results above reflected an actual decrease in the number of MuSCs or simply a reduction in the levels of Pax7 or Vcam1 proteins. To address this issue, we generated the mouse strains (i.e., heterozygous control: p110α f/+:Pax7 CreER/CreER:R26R YFP/YFP; and p110α‐iKO mice: p110α f/f:Pax7 CreER/CreER:R26R YFP/YFP) with a Pax7‐dependent reporter gene encoding yellow fluorescence protein (YFP) expressed from the Rosa26 locus so that YFP+ cells in muscles represented MuSCs irrespective of the expression status of Pax7 protein. When we isolated MuSCs by FACS based on YFP expression, unexpectedly, we found that there was not much reduction in the number of YFP+ MuSCs from uninjured muscles of p110α‐iKO mice (Fig 2E), indicating that it was the levels of Pax7 protein rather than the number of MuSCs that were obviously reduced in p110α‐iKO mice. To further prove the point, we isolated Vcam1+ MuSCs by FACS from p110α f/f:Pax7 CreER/CreER:R26R YFP/YFP mice and induced p110α deletion in culture by addition of 4‐hydroxytamoxifen (4‐OHT). We harvested the cells and analyzed the cell lysates by Western blot. We first confirmed that 4‐OHT induced efficient p110α deletion in culture (Fig 2F). Importantly, the protein levels of Pax7 were clearly reduced in p110α‐deleted cells (Fig 2F). Moreover, the protein levels of MyoD, but not Myf5, were also reduced in such cultured cells without p110α (Fig 2F). As inhibition of class I PI3K is known to induce autophagy (Mizushima et al, 2010), we next examined whether the reduction in Pax7 protein was due to enhanced autophagy. Firstly, by deleting p110α with 4‐OHT in cultured MuSCs, we showed that the reduced Pax7 protein levels in such cells could be partially restored by LY294002, a known inhibitor of autophagy (Fig EV3A; Mizushima et al, 2010). p62, a well‐established substrate of autophagy (Mizushima et al, 2010), was included as a control. Secondly, by knocking down Atg5 that encodes a key component of the autophagy machinery (Mizushima et al, 2010), we found that the reduced Pax7 protein levels were again partially restored (Fig EV3B). Thus, our data above demonstrated that inducible deletion of p110α in MuSCs of uninjured muscles did not obviously affect the maintenance of MuSCs but led to an obvious reduction in Pax7 protein levels via enhanced autophagy.
Figure 2. Ablation of p110α in adult MuSCs from uninjured muscles reduced Pax7 protein levels without obviously affecting the MuSC number.
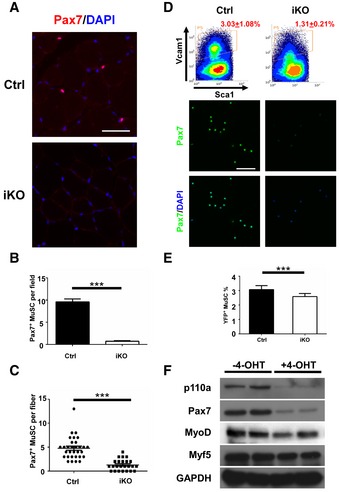
- Adult control and iKO mice (n = 3) were treated with Tmx as described in Fig 1. Seven days after the last injection of Tmx, mice were sacrificed and TA muscle sections were immunostained for Pax7 (red). The nuclei were counterstained by DAPI (blue).
- Quantification of Pax7+ MuSCs per field of TA muscle sections from (A) by examining multiple sections from three pairs of mice.
- Freshly isolated single myofibers (n > 20) from three pairs of Ctrl or iKO mice were immediately fixed and immunostained for Pax7.
- Top: representative FACS plots showing the percentage of Vcam1+ MuSCs (boxed areas) among the CD31−CD45−Sca1− cell populations from uninjured muscles of the Ctrl and iKO mice (n = 3 in each group). Bottom: Sorted YFP+ MuSCs were immediately fixed and immunostained for Pax7 (green). The nuclei were counterstained by DAPI (blue).
- Quantification of YFP+ MuSCs in whole hindlimb muscles by FACS from the Ctrl and iKO mice (n = 5).
- FACS‐isolated MuSCs from iKO mice (without Tmx) were cultured for 2 days followed by 2 days of 4‐OHT treatment. Whole‐cell lysate was subjected to Western blotting.
Figure EV3. Reduction in Pax7 protein in p110α‐null MuSCs was mediated by autophagy.
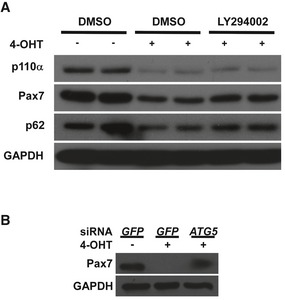
-
A, BFACS‐isolated MuSCs from iKO mice (without Tmx) were cultured for 2 days followed by 1 day of 4‐OHT treatment. Then, the cells were either treated with DMSO or LY294002 (10 μM) in (A) or transfected with siRNAs in (B) as indicated and grown in the presence of 4‐OHT for another day before harvest. Whole‐cell lysates were subjected to Western blotting.
p110α was indispensable for quiescence exit and the cell cycle re‐entry in adult MuSCs upon activation
To functionally characterize p110α‐null MuSCs, we first induced p110α deletion in adult MuSCs and then isolated MuSCs from the control and p110α‐iKO mice by FACS. The same number of freshly isolated MuSCs was then seeded in culture and grown for 2 days. 5‐ethynyl‐2′‐deoxyuridine (EdU) was added to the culture four hours before cell fixation and analysis. While nearly 60% of MuSC‐derived primary myoblasts from the control mice incorporated EdU, less than 20% of such cells from the mutant mice did so (Fig EV4A and B). To examine the proliferation potential of p110α‐null MuSCs in vivo, we injured the TA muscles of the control and p110α‐iKO mice (all carrying the R26R YFP reporter) with CTX and then examined YFP+ ASCs two and a half days after the injury. By immunostaining, multiple YFP+ cells were detected on TA muscle sections from the control mice, which was indicative of normal expansion of MuSCs upon muscle injury, but few YFP+ cells were found on TA muscle sections from p110α‐iKO mice (Fig 3A and B). As QSCs need to exit quiescence and re‐enter the cell cycle before they undergo proliferation, it was unclear whether the severe proliferation deficit seen in p110α‐null MuSCs was due to a defect in quiescence exit or the subsequent “cell cycling” step. To directly address this issue, we first isolated single myofibers from the control and p110α‐iKO mice and then suspended them in culture with EdU for 48 h. Under such conditions, nearly all MuSCs on myofibers from the control mice incorporated EdU, but very few mutant MuSCs did so (Fig 3C and D). Next, we directly examined the ability of p110α‐null MuSCs to incorporate EdU in vivo upon injury (Fig 3E, top). Consistent with the results in culture, while ~70% of the YFP+ MuSCs from the control mice already incorporated EdU two and a half days after the injury, less than 20% of the mutant MuSCs did so in vivo (Fig 3E and F).
Figure EV4. p110α‐null MuSCs were arrested in the G0 state.

- FACS‐isolated MuSCs from the tamoxifen (Tmx)‐treated Ctrl and iKO mice were cultured for 36 h followed by a 4‐h EdU pulse. Cells were fixed and stained for EdU (green) and DAPI (blue).
- Quantification of the EdU+ MuSCs from (A) by counting ˜600 MuSCs from triplicate wells.
- Histograms showing the mean FSC‐A values of MuSCs freshly isolated from the Ctrl and iKO mice.
- Freshly isolated single myofibers (n > 20) from the Ctrl and iKO mice (n = 3 in each group) were immediately fixed and stained for CD34 (red), YFP (green), and DAPI (blue). A representative image is shown.
- Quantification of the areas of MuSCs from (D) based on CD34 staining. More than 100 MuSCs were calculated for each group of mice.
- FACS‐isolated MuSCs from the Tmx‐treated Ctrl and iKO mice were cultured for 36 h followed by staining for MyoD (green) and DAPI (blue).
- Quantification of the MyoD+ ASCs from (F) by counting ˜600 ASCs in triplicate wells. Scale bars: 25 μm. In (B, E, G), the data are presented as mean ± s.d. ***P < 0.001.
Figure 3. p110α was indispensable for quiescence exit and the cell cycle re‐entry in adult MuSCs upon injury.
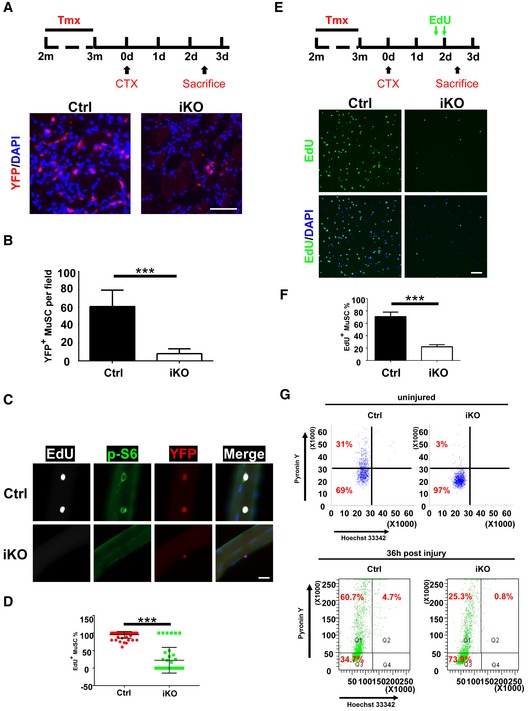
- Top: the experimental scheme. 2.5 days post‐injury, TA muscle sections were immunostained for YFP (red). The nuclei were counterstained by DAPI (blue). m: month; d: day.
- Quantification of the YFP+ MuSCs from (A) by counting ˜250 cells/mouse.
- Freshly isolated single fibers (n > 20) were cultured with EdU for 48 h. Cells were then fixed and subjected to staining for EdU (white), p‐S6 (green), and YFP (red). Representative images are shown.
- Quantification of the EdU+ MuSCs from (C) by counting > 20 fibers/mouse.
- Top: the experimental scheme. Lower hindlimb muscles were injured by CTX followed by intraperitoneal injection of two doses of EdU at 40 and 48 h post‐injury. MuSCs from lower hindlimb muscles were sorted by FACS, fixed immediately and stained for EdU (green). The nuclei were counterstained by DAPI (blue). The representative images are shown.
- Quantification of the EdU+ MuSCs from (E) by counting ˜500 MuSCs/mouse.
- FACS‐isolated MuSCs from uninjured or injured muscles (36 h post‐injury) of the control and iKO mice were stained with Hoechst 33342 and Pyronin Y and analyzed by FACS.
Although our data above clearly showed that p110α‐null MuSCs were defective in the cell cycle re‐entry, it was unclear whether they failed to exit quiescence or were blocked at the G1/S transition. QSCs have several hallmarks including the compact cell size with scarce cytoplasm and low RNA content, both of which increase upon activation of QSCs and could thus be used as indicators of quiescence exit (Fukada et al, 2007; Rodgers et al, 2014). On FACS plots, we found that the freshly isolated p110α‐null MuSCs were even smaller in size than the control MuSCs based on the mean forward scatter (FSC) values (Fig EV4C). This was most likely due to the fact that the control “MuSCs” was already partially activated during the isolation process (van den Brink et al, 2017; Machado et al, 2017; van Velthoven et al, 2017). Consistently, on freshly isolated myofibers, we found that the size of the mutant MuSCs was indeed smaller than that of the control MuSCs (Fig EV4D and E). By Pyronin Y staining for total RNA (Shapiro, 1981; Fukada et al, 2007; Rodgers et al, 2014), we found that more freshly isolated p110α‐null MuSCs were stuck in the G0 state with very low RNA content than the control MuSCs (Fig 3G, top). Furthermore, in freshly isolated ASCs 36 h after the CTX injury, while ~65.4% of the control ASCs already exit the G0, most (~73.9%) of the p110α‐null MuSCs remained in the G0 state (Fig 3G, bottom). In addition, the expression of MyoD protein also indicates quiescence exit as MyoD is only expressed in ASCs but not QSCs (Yin et al, 2013; Sambasivan & Tajbakhsh, 2015). When we put freshly isolated MuSCs in culture for 36 h, we found that nearly all control MuSCs were MyoD+ (Fig EV4F and G). In contrast, barely any p110α‐null MuSCs were MyoD+ at this time point. Thus, our data above indicated that loss of p110α in MuSCs prevented them from exiting quiescence and re‐entering the cell cycle upon activation.
mTORC1 is a key downstream mediator of p110α in regulating quiescence exit in adult MuSCs
PI3K can transmit signals via multiple downstream signaling molecules, with mTORC1 being the most prominent one (Saxton & Sabatini, 2017). To test whether mTORC1 functions downstream of PI3K to regulate quiescence exit in MuSCs, we decided to deliberately activate mTORC1 in p110α‐null MuSCs by further deleting Tsc1, a specific repressor of mTOCR1 (Saxton & Sabatini, 2017). To do so, we generated tamoxifen‐inducible MuSC‐specific p110α/Tsc1 double knockout mice with a YFP reporter expressed from the Rosa26 locus (i.e., Tsc1 f/f:p110α f/f:Pax7 CreER/CreER:R26R YFP/YFP; abbreviated as idKO thereafter). After tamoxifen treatment, we isolated MuSCs by FACS and cultured them for 2 days in the presence of EdU. By immunostaining, we showed that the p‐S6 signal, while barely detectable in p110α‐null MuSCs, was largely rescued in MuSCs from idKO mice to a level comparable to that in MuSCs from the control mice (Fig 4A and B), further confirming that mTORC1 was indeed re‐activated in MuSCs from idKO mice. Importantly, we found that the defective EdU incorporation in p110α‐null MuSCs was largely, but not fully, restored in MuSCs from idKO mice (Fig 4A and B). On FACS plots, we also noticed that the average size of the freshly sorted MuSCs from idKO mice was bigger than that from p110α‐iKO mice and similar to that from the control mice (Fig 4C, top). Moreover, we found that the reduced percentage of the Vcam1+ MuSCs in p110α‐iKO mice was largely rescued in idKO mice to a level close to that from the control mice (Fig 4C, bottom). Similarly, using 4‐OHT to induce p110α deletion in cultured MuSCs isolated from iKO and idKO mice (not treated with tamoxifen), we found that the reduced Pax7 expression in p110α‐null MuSCs was also largely restored when both p110α and Tsc1 were deleted (Fig 4D). By immunostaining for Pax7 using either freshly isolated myofibers or FACS‐isolated MuSCs, we further confirmed that the reduced expression of Pax7 in p110α‐null MuSCs was largely, but not fully, rescued in MuSCs from idKO mice (Fig 4E and F). As mTORC2 is also a known downstream mediator of PI3K (Saxton & Sabatini, 2017), we asked whether mTORC2 also plays a role in regulating quiescence exit in MuSCs. To test this, we used freshly sorted MuSCs from idKO mice and examined their EdU incorporation in culture with a control GFP‐siRNA or two specific siRNAs against Rictor, an indispensable component of the mTORC2 complex (Saxton & Sabatini, 2017). As shown in (Fig EV5), the restored EdU incorporation in MuSCs from idKO mice was not obviously affected by si‐Rictor, suggesting that mTORC2, unlike mTORC1, does not play a key role in regulating quiescence exit and the cell cycle re‐entry in adult MuSCs.
Figure 4. mTORC1 is a key downstream mediator of p110α in regulating quiescence exit in adult MuSCs.

- Age‐matched adult Ctrl, iKO, and idKO mice (n = 3 in each group) were first treated with Tmx as described in Fig 1. MuSCs were isolated by FACS and cultured with EdU for 36 h. Cells were fixed and subjected to staining for EdU (green) and p‐S6 (red). The nuclei were counterstained by DAPI (blue).
- Quantification of EdU+ or p‐S6+ MuSCs from (A) by counting ˜600 MuSCs/mouse.
- Representative histograms (top) showing the mean FSC‐A values and dot plots (bottom) showing the percentage of Vcam1+ MuSCs from the Ctrl, iKO, and idKO mice.
- FACS‐isolated MuSCs from the Ctrl, iKO, and idKO mice (without Tmx) were cultured for 2 days followed by 2 days of 4‐OHT treatment. Whole‐cell lysates were subjected to Western blotting.
- Freshly isolated single myofibers (n > 20) from the Ctrl, iKO, and idKO mice were fixed immediately and stained for Pax7. The mean number of the Pax7+ MuSCs/fiber was calculated.
- FACS‐isolated MuSCs were immediately fixed and stained for Pax7 (green) and DAPI (blue).
Figure EV5. mTORC2 was not required for the cell cycle re‐entry of MuSCs from idKO mice.
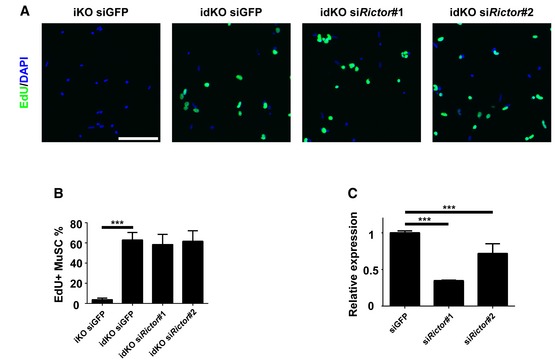
- FACS‐isolated MuSCs from iKO and idKO mice were transfected with siRNAs as indicated in the presence of EdU for 36 h followed by cell fixation and staining for EdU (green) and DAPI (blue).
- Quantification of the EdU+ MuSCs in (A) by counting ˜600 MuSCs in triplicate wells.
- Triplicate C2C12 cells were transfected with indicated siRNAs for 24 h. Total RNAs were extracted. The mRNA levels of Rictor were measured by RT–qPCR. Scale bar: 50 μm. In (B, C), the data are presented as mean ± s.d. ***P < 0.001.
p110α‐mTORC1 targeted c‐Jun to regulate quiescence exit and the cell cycle re‐entry
To uncover the mechanisms by which PI3K regulates quiescence exit in MuSCs, we aimed to identify PI3K target genes in MuSCs. To do so, we FACS‐isolated MuSCs from uninjured heterozygous control (i.e., p110α f/+:R26R YFP/YFP:Pax7 CreER/CreER) and p110α‐iKO (i.e., p110α f/f:R26R YFP/YFP:Pax7 CreER/CreER) mice, extracted the total RNA and subjected the samples to RNA‐seq analysis. The efficiency of tamoxifen‐induced p110α deletion was high (~95%) as judged by the absence of p110α exon 1‐derived sequences in mutant MuSCs based on our RNA‐seq data and the FACS analysis (Fig EV6A and B). Compared to the control, the mRNA levels of Pax7 and Myf5 were not obviously affected, but that of Myod was clearly downregulated, in p110α‐null MuSCs (Fig 5A). By gene set enrichment analysis (GSEA), we noticed that many of the downregulated genes in p110α‐null MuSCs belonged to the “AP1 transcription targets” (Fig 5B, top). Consistently, the mRNA levels of several AP‐1 family members including c‐Jun, Fos, and Fosb were all decreased in p110α‐null MuSCs (Fig 5B, bottom). The c‐Jun protein levels were also much lower in freshly isolated p110α‐null MuSCs (Fig 5C), but the Fos protein was undetectable in freshly isolated MuSCs. However, when we induced p110α deletion with 4‐OHT in cultured MuSCs, we could also detect reduced Fos protein levels in p110α‐null myoblasts (Fig EV7). To further examine whether c‐Jun or Fos is involved in regulating quiescence exit in MuSCs, freshly isolated p110α‐null MuSCs were separately infected with adenoviruses that expressed red fluorescence protein (RFP), c‐Jun, or Fos and then subjected to EdU incorporation assays in culture. We found that overexpression of c‐Jun but not Fos or RFP in p110α‐null MuSCs partially restored EdU incorporation (Fig 5D and E). Moreover, we found that the mRNA levels of c‐Jun, Fos, and Fosb, while repressed in p110α‐null MuSCs, were partially or fully de‐repressed in MuSCs from idKO mice (Fig 5F), suggesting that c‐Jun is also a transcriptional target of mTORC1. To further prove this point, we treated idKO mice with or without rapamycin, a potent inhibitor of mTORC1 (Saxton & Sabatini, 2017), by intraperitoneal injection, followed by fresh isolation of MuSCs by FACS, and detection of c‐Jun mRNA by RT–qPCR. Consistently, the elevated c‐Jun mRNA levels in MuSCs from idKO mice were re‐suppressed by rapamycin treatment (Fig 5G), confirming that c‐Jun is indeed a transcriptional target of mTORC1.
Figure EV6. Examination of the Cre‐mediated gene deletion efficiency.

- The genome browser view showing the expression of p110α exons in MuSCs from the Ctrl and iKO mice based on our RNA‐seq data. Note the absence of the transcripts from the 1st exon in MuSCs from four mutant iKO mice.
- The representative FACS plots showing the percentage of YFP+ MuSCs among the CD31−/CD45−/Sca1−/Vcam1+ cell population, which indirectly revealed the Cre‐mediated gene deletion efficiency at the p110α locus.
Figure 5. p110α‐mTORC1 targeted c‐Jun to regulate quiescence exit and the cell cycle re‐entry.

- Based on RNA‐seq, the mean FPKM values of Pax7, Myod, and Myf5 in MuSCs from the heterozygous Ctrl and iKO mice (n = 4) are shown.
- Top: the GSEA plot showing the enrichment of the “AP1 binding sites in the downregulated genes” in p110α‐null MuSCs. Bottom: the mean FPKM values of c‐Jun, Fos, and Fosb in MuSCs from the Ctrl and iKO mice (n = 4).
- FACS‐isolated MuSCs from Ctrl and iKO mice were immediately lysed, and whole‐cell lysates were subjected to Western blotting.
- FACS‐isolated MuSCs from the Ctrl and iKO mice (n = 3) were infected by the RFP‐, c‐Jun‐, and Fos‐expressing adenoviruses as indicated for 2 days in the presence of EdU. Cells were then fixed and stained for EdU (green). The nuclei were counterstained with DAPI (blue).
- Quantification of the EdU+ MuSCs from (D) by counting ˜600 MuSCs/mouse.
- Total RNAs were extracted from FACS‐isolated MuSCs from the Ctrl, iKO, and idKO mice (n = 3 in each group). The relative fold changes of c‐Jun, Fos, and Fosb were measured by RT–qPCR.
- Adult idKO mice were first treated with Tmx as described in Fig 1, followed by intraperitoneal injection of two doses of DMSO or rapamycin (4 nmol) at 12 and 1 h prior to sacrifice of mice (n = 3 in each group). The hindlimb muscles were digested by proteases and fixed in 2% paraformaldehyde before isolation of MuSCs by FACS. Total RNAs were extracted, and the relative fold change of c‐Jun was measured by RT–qPCR.
Figure EV7. Fos protein levels were reduced in cultured p110α‐null primary myoblasts.

FACS‐isolated MuSCs from iKO mice (without Tmx) were cultured for 2 days followed by 2 days of 4‐OHT treatment before harvest. Whole‐cell lysates were subjected to Western blotting.
FoxOs function as another class of key downstream effectors of PI3K to regulate quiescence exit in MuSCs
FoxOs are well‐established downstream effectors of PI3K and play essential roles in cell proliferation (Hay, 2011). Based on the RNA‐seq data generated by us and others, three FoxOs genes (i.e., FoxO1, FoxO3a, and FoxO4) are expressed in MuSCs. To test whether FoxOs function downstream of PI3K to regulate quiescence exit, we isolated p110α‐null MuSCs by FACS, individually knocked down three FoxOs, and then examined EdU incorporation in such cells. As shown in (Fig 6A and B), knockdown of any of the three FoxO genes in p110α‐null MuSCs partially restored their ability to incorporate EdU. As FoxOs are known to be nuclear in non‐cycling cells and cytoplasmic in proliferating cells (Hay, 2011), we next examined the subcellular localization of FoxO3a in MuSCs on isolated single myofibers suspended in culture for 36 h. By this time, most of the wild‐type MuSCs should be fully activated. Consistently, FoxO3a was mainly cytoplasmic or pan‐cellular in most of the MuSCs from the control mice (Fig 6C and D). In contrast, it was mainly nuclear in p110α‐null MuSCs. Interestingly, FoxO3a was cytoplasmic/pan‐cellular again in a large fraction of MuSCs from idKO mice (Fig 6C and D). As overexpression of c‐Jun partially restored EdU incorporation in p110α‐null MuSCs, it was unclear whether c‐Jun could also affect the subcellular localization of FoxOs. To test this, we first infected FACS‐isolated control or p110α‐null MuSCs with adenoviruses expressing RFP or c‐Jun, let cells grow in culture for 36 h, and then examined the subcellular localization of FoxO3a by immunostaining. While FoxO3a was mainly nuclear in p110α‐null MuSCs infected with the RFP‐expressing virus, it was cytoplasmic/pan‐cellular again in a large fraction of p110α‐null MuSCs infected with the c‐Jun‐expressing virus (Fig 6E and F). Our data above suggest that the mTORC1/c‐Jun axis cross‐talks with FoxOs downstream of PI3K.
Figure 6. FoxOs function as another key downstream effector of p110α in regulating quiescence exit in MuSCs.
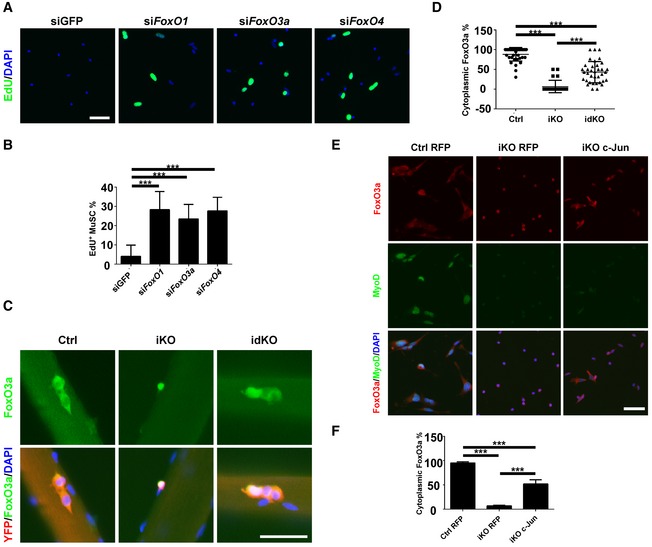
- FACS‐isolated MuSCs from iKO mice (n = 3) were transfected with siRNAs as indicated and cultured in the presence of EdU for 48 h. Cells were fixed and stained for EdU.
- Quantification of the EdU+ MuSCs from (A) by counting ˜600 MuSCs/mouse.
- Freshly isolated single myofibers (n > 20) from the Ctrl, iKO, and idKO mice were cultured for 36 h. Cells were fixed and stained for YFP (red), FoxO3a (green), and DAPI (blue).
- Quantification of the MuSCs with cytoplasmic FoxO3a from (C). ˜200 MuSCs on more than 20 myofibers/mouse were examined.
- FACS‐isolated MuSCs from the Ctrl and iKO mice (n = 3) were infected with the RFP‐ or c‐Jun‐expressing adenoviruses and cultured for 36 h before fixation followed by staining for FoxO3a (red), MyoD (green), and DAPI (blue).
- Quantification of the MuSCs with cytoplasmic FoxO3a from (E) by counting ˜600 MuSCs/mouse.
Induced expression of a constitutively active p110α in adult MuSCs led to their spontaneous exit from quiescence and gradual depletion
Our results so far had demonstrated that p110α was necessary and indispensable for MuSCs to exit quiescence upon activation. It was unclear whether a constitutively active p110α was sufficient for MuSCs to break quiescence in uninjured muscles in vivo. To test this, we generated another inducible MuSC‐specific R26R H1047R/YFP:Pax7 CreER/+ mouse line (abbreviated as ip110αca mice) that could be induced to express a constitutively active p110α mutant (i.e., p110αH1047R) in MuSCs from the Rosa26 locus (Samuels et al, 2004). A week after the last tamoxifen injection in adult mice, the control (i.e., R26R YFP/YFP:Pax7 CreER/+) and ip110αca mice were injected with EdU intraperitoneally for three consecutive days (Fig 7A, top). MuSCs were then isolated by FACS 3 days after the last EdU injection and subjected to staining analysis. While barely any MuSC from the control mice incorporated EdU, ~10% of MuSCs from ip110αca mice did so at this time (Fig 7A and B). Consistently, when we examined TA muscle sections, we could readily identify YFP+/EdU+ MuSCs from ip110αca mice, while such cells were barely detectable from the control mice (Fig EV8A). Notably, we could also detect EdU+/YFP− nuclei associated with myofibers from ip110αca mice (Fig EV8A, bottom panels). We suspected that such nuclei were from the mutant MuSCs that had differentiated and fused with the underlying myofibers, which could conceivably dilute the YFP signal and made it difficult to be visualized. As such EdU+/YFP− nuclei were closely associated with the peripherals of the laminin+ myofibers, it was unclear whether they were inside or outside of the sarcolemma of myofibers. To clarify this issue, we stained the sarcolemma for dystrophin that marks the inner side of the sarcolemma (Blake et al, 2002; Durbeej & Campbell, 2002). We found that such EdU+/YFP− nuclei were wrapped underneath the dystrophin+ sarcolemma (Fig EV8B and C), which supported our hypothesis that such EdU+ nuclei derived from MuSCs that were already differentiated and fused with the underlying myofibers. If we waited longer (e.g., 14 and 28 days) after the tamoxifen treatment, we noticed an obvious reduction in MuSC number on TA muscle sections from ip110αca mice compared to that from the control mice (Fig 7C and D). This was consistent with the findings in MuSC‐specific Pten‐null mice (Yue et al, 2017), which was expected as deletion of Pten led to constitutive activation of the PI3K pathway (Vanhaesebroeck et al, 2012). To further understand why the active PI3K promoted gradual depletion of MuSCs, we first examined the rate of the cell cycle re‐entry by mutant MuSCs. We isolated MuSCs from the control and ip110αca mice and left them in culture for 1 day followed by a 4‐h EdU pulse. While few control MuSCs incorporated EdU at this time, a large fraction of MuSCs from ip110αca mice already did so (Fig EV8D and E), suggesting an accelerated cell cycle re‐entry. We then examined the differentiation potentials of MuSCs from ip110αca mice. Total RNA was extracted from MuSCs that were freshly isolated by FACS from the control and ip110αca mice and subjected to RT–qPCR analysis. While the mRNA levels of Myod were comparable, those of myogenin, a key early pro‐differentiation gene (Tapscott, 2005), were obviously upregulated in p110αca+ MuSCs, suggesting precocious differentiation in such mutant MuSCs (Fig 7E). The pro‐differentiation effect of p110αca was further confirmed when we stained for the sarcomeric myosin heavy chain (MHC) in MuSCs that were cultured for 3 days (Fig EV8F and G): While a few control cells just started to express MHC, more bigger and brighter MHC+ myotubes (some were multinucleated) were already formed in p110αca+ cells.
Figure 7. Constitutive activation of p110α in MuSCs from uninjured muscles led to their spontaneous quiescence exit and gradual depletion.

- Top: the experimental scheme. Adult Ctrl and ip110αca mice (n = 3 in each group) were injected with Tmx and EdU as indicated and sacrificed 3 days after the last dose of EdU. FACS‐isolated MuSCs were fixed immediately and stained with EdU (green), Pax7 (red) and DAPI (blue). Arrowheads: EdU+ MuSCs.
- Quantification of the EdU+ MuSCs in (A) by counting ˜600 MuSCs/mouse.
- TA muscle sections from the Ctrl and ip110αca mice (n = 3) at different time points after the Tmx treatment were stained for YFP (red), laminin (green), and DAPI (blue). Representative images are shown. Arrows: YFP+ MuSCs.
- Quantification of the YFP+ MuSCs in (C) by counting ˜600 MuSCs/mouse.
- The relative mRNA levels of Myod and Myogenin in MuSCs from (A) were measured by RT–qPCR (n = 3).
- A schematic showing that PI3K regulates quiescence exit and the cell cycle re‐entry in MuSCs via both mTORC1‐c‐Jun and FoxOs.
Figure EV8. MuSCs from ip110αca mice proliferated and differentiated faster.

- TA muscle sections from the Ctrl and ip110αca mice as described in Fig 7A were stained for EdU (white), YFP (red), laminin (green), and DAPI (blue). Arrowheads: YFP+/EdU−, YFP−/EdU+, or YFP+/EdU+ cells.
- TA muscle sections as described above were stained for EdU (green) and dystrophin (red).
- Quantification of the EdU+ cells underneath the dystrophin+ sarcolemma in (B). Three pairs of mice were examined including five TA muscle sections per mouse and three microscopic fields per section.
- FACS‐isolated MuSCs from the Ctrl or ip110αca mice were cultured for 24 h followed by a 4‐h EdU pulse.
- Quantification of the EdU+ ASCs in (D) by counting ˜600 ASCs in triplicate wells.
- FACS‐isolated MuSCs from the Ctrl or ip110αca mice were cultured for 3 days followed by cell fixation and staining for MHC (red), p‐S6 (green), and DAPI (blue).
- Quantification of the MHC+ cells in (F) by counting ˜600 nuclei in triplicate wells. Scale bars: 25 μm. In (C, E, G), the data are presented as mean ± s.d. **P < 0.01; ***P < 0.001.
Discussion
p110α of PI3K specifically regulates quiescence exit in adult MuSCs upon muscle injury
Upon muscle injury, MuSCs exit quiescence and re‐enter the cell cycle to proliferate. However, it was unclear whether quiescence exit is actively regulated by any intracellular signaling pathways. Here, we demonstrated that p110α of PI3K and the PI3K‐mediated signaling pathway exert such functions in MuSCs. Unlike MuSC‐specific deletion of p110α using Pax7‐Cre, which resulted in embryonic lethality (Fig EV2A), that of p110β of PI3K (with a Myf5‐Cre line) had no obvious deleterious effects on mouse development and survival, as well as muscle mass and strength in vivo (Matheny et al, 2015). This is likely due to the fact that p110α is the predominant form of PI3K expressed in QSCs, while p110β is barely detectable in QSCs based on the RNA‐seq data from us and others (Liu et al, 2013; Ryall et al, 2015). Although PI3K is known to regulate cycling cells (Vanhaesebroeck et al, 2012; Thorpe et al, 2015), unexpectedly, we find that a main point of action for PI3K in adult MuSCs is at a critical step before MuSCs re‐enter the cell cycle. Thus, our work here has uncovered a previously unrecognized checkpoint in QSCs: Loss of PI3K arrests MuSCs in quiescence, while its activation induces quiescence exit and the cell cycle re‐entry. Our findings here do not exclude additional roles of PI3K in subsequent steps after MuSCs exit quiescence, including MuSC proliferation, differentiation, and self‐renewal. To address these issues, different Cre driver strains are needed in order to specifically delete p110α at specific stages of MuSC progression.
Loss of p110α of PI3K in adult MuSCs has profound impact on the expression of key myogenic and growth‐promoting transcription factors. We showed that Pax7 protein, instead of its mRNA, levels were greatly reduced in p110α‐null MuSCs (Figs 2F and 5A and C). We found that enhanced autophagy induced by loss of p110α was mainly responsible for Pax7 degradation (Fig EV3). In addition, both mRNA and protein levels of Myod, but not Myf5, were also obviously downregulated in p110α‐null MuSCs (Figs 2F and 5A). Moreover, we showed that c‐Jun, Fos, and Fosb, all members of the AP1 family, were previously unrecognized transcription targets of the PI3K‐mTORC1 axis in MuSCs. Recent work from several groups showed that the mRNA levels of these AP1 family genes were very low in QSCs (i.e., those from pre‐fixed muscles), but they were quickly induced during MuSC isolation without prior fixation, suggesting that these AP1 family genes, along with other immediate–early genes (e.g., Egr1‐3, ATF3, etc.), promote quiescence exit in MuSCs (van den Brink et al, 2017; Machado et al, 2017; van Velthoven et al, 2017). We hypothesized that failure of these immediate–early genes to be quickly induced in our p110α‐null MuSCs underlies their defects in quiescence exit. However, it is technically challenging to experimentally prove that some of these gene products indeed exert key functions in quiescence exit as most freshly isolated MuSCs are already partially activated during the isolation process. Moreover, pre‐fixed MuSCs are not suitable for subsequent experimental manipulation (e.g., gene overexpression or knockdown). The failure of p110α‐null MuSCs to exit quiescence provides an excellent platform for us to test the functions of some AP1 family members in promoting quiescence exit. We showed that overexpression of c‐Jun, but not Fos, in p110α‐null MuSCs largely rescued their defects in quiescence exit and the cell cycle re‐entry (Fig 5D and E). Consistently, in fibroblasts lacking c‐Jun or Fos, only loss of c‐Jun but not Fos severely compromised cell growth (Shaulian & Karin, 2001). Moreover, in serum‐starved cells, c‐Jun is also known to be essential for the serum‐induced cell cycle re‐entry (Shaulian & Karin, 2001). Thus, our findings here establish c‐Jun as a key downstream mediator of the PI3K‐mTORC1 axis that drives quiescence exit and the cell cycle re‐entry during early activation of QSCs.
In addition to c‐Jun, we show that FoxOs are also involved in regulating quiescence exit and the cell cycle re‐entry in MuSCs (Fig 6). Although FoxOs are generally thought to be directly regulated by the PI3K‐Akt axis without the direct involvement of mTORC1 (Hay, 2011; Saxton & Sabatini, 2017), we found that there is obvious cross‐talk between the mTORC1‐c‐Jun signaling axis and FoxOs: In MuSCs from p110α/Tsc1 idKO mice, FoxO3a was cytoplasmic/pan‐cellular in a large fraction of the cells (Fig 6C and D). Similarly, overexpression of c‐Jun in p110α‐null MuSCs also promoted the cytoplasmic translocation of FoxO3a (Fig 6E and F). In both cases above, the cytoplasmic translocation of FoxOs correlated with the ability of the mutant MuSCs to exit quiescence and to re‐enter the cell cycle. Although the activated FoxOs are known to inhibit mTORC1 via various mechanisms (Hay, 2011), it remains unclear how the activated mTORC1 or c‐Jun triggers cytoplasmic translocation (i.e., inactivation) of FoxOs.
An evolutionally conserved role for the PI3K/mTORC1/FoxOs pathway in regulating quiescence exit in diverse somatic stem cells
During Drosophila development, neural stem cells (NSC or neuroblasts) in the central brain are quiescent between the embryonic and larval phases of growth. In response to IGF1‐like peptides from the neighboring glial cells, the PI3K/Akt/Tor pathway in neuroblasts is activated, which drives neuroblasts out of quiescence to enter the larval phase of growth (Chell & Brand, 2010; Sousa‐Nunes et al, 2011). Similarly, in mouse forebrain NSC, mTORC1 was found to be essential for the expansion of the transit‐amplifying progenitor cells from the quiescent stem cell pool (Paliouras et al, 2012). Consistently, FoxOs were also found to be essential for the maintenance of the quiescent NSC pool in mice (Paik et al, 2009; Renault et al, 2009). Loss of FoxOs promotes spontaneous quiescence exit of NSC and gradual depletion of the adult NSC pool. Furthermore, in mouse quiescent naïve T cells, antigen stimulation promotes quiescence exit and the cell cycle re‐entry, which results in subsequent clonal expansion. This process is also shown to be absolutely dependent on mTORC1, as deletion of Tsc1 promotes spontaneous quiescence exit and reduction of peripheral T cells, while deletion of Raptor (i.e., genetic inactivation of mTORC1) arrests naïve T cells in the quiescent state and prevents them from quiescence exit (Yang et al, 2011, 2013). Thus, in different somatic stem cells that can transit between quiescence and proliferation, the PI3K/mTORC1/FoxOs pathway has a conserved role in critically regulating quiescence exit. Yet, there are unique features in MuSCs. Firstly, MuSC‐specific interference of mTORC1 activity alone (e.g., by deletion of Tsc1 or Raptor) mainly affected the establishment of the GAlert state without severely impairing MuSC functions during muscle regeneration (Rodgers et al, 2014). Secondly, unlike what occurred in mouse NSC, deletion of FoxO3a alone in MuSCs was insufficient to cause spontaneous quiescence exit (Gopinath et al, 2014). Instead, loss of FoxO3a only affected MuSC self‐renewal upon injury. Our findings here suggest that the potent effect of PI3K on quiescence exit and the cell cycle re‐entry in MuSCs most likely lies in its ability to simultaneously activate two inter‐connected signaling axes: one consisting of the mTORC1‐c‐Jun axis and the other FoxOs (Fig 7F). Interference of either axis alone only has minor impacts on MuSCs, but that of both axes severely compromises quiescence exit in MuSCs.
In summary, we demonstrate here that p110α of PI3K is both necessary and sufficient to promote quiescence exit and the cell cycle re‐entry in MuSCs upon activation. It does so via key downstream effectors including the mTORC1‐c‐Jun axis and FoxOs. Our findings also provide a clue to a long‐standing mystery regarding the extrinsic signals that activate QSCs upon muscle injury: those RTK‐engaging ligands (e.g., insulin‐like growth factor, epidermal growth factor) that are known to activate the PI3K‐mTORC1‐c‐Jun/FoxOs pathway are promising candidates worthy of further investigation in the future.
Materials and Methods
Animals
p110α flox/flox (Stock No: 017704), Tsc1 flox/flox (Stock No: 005680), and R26R‐p110α H1047R (Stock No: 016977), Pax7 CreERT(GaKa)/+ (Stock No: 017763), and R26R‐EYFP (Stock No: 006148) mice were all from the Jackson Laboratory (Bar Harbor, ME, USA). In order to delete p110α and Tsc1 or induce EYFP and p110α H1047R (at the Rosa26 locus) in adult mice, 75 μg of tamoxifen (Tmx) dissolved in corn oil per gram of body weight was intraperitoneally injected into mice for five consecutive days followed by 7–8 doses of Tmx every 2–3 days or indicated otherwise. All the mice were maintained and handled according to the protocols approved by the Animal Ethics Committee at HKUST.
Injury‐induced muscle regeneration
Adult mice were anaesthetized with Avertin (0.5 mg/g of body weight) by intraperitoneal injection. 30 μl of 10 μM cardiotoxin (CTX) was injected into tibialis anterior (TA) muscles to induce acute injury. TA muscles were dissected at various time points after the injury followed by histology and staining (Zhu et al, 2016).
Isolation of adult muscle stem cells (MuSCs) by FACS
Hindlimb muscles dissected from mice were minced, digested by collagenase II (800 U/ml) in Ham's F10 with 10% horse serum (washing medium) at 37°C for 90 min. Digested muscles were triturated and washed in the washing medium once and then subjected to further digestion with collagenase II (80 U/ml) and dispase (1 U/ml) for 30 min. The suspensions were passed through a 20‐Gauge needle attached to a syringe for 15 times and filtered through a 40‐μm cell strainer (BD Biosciences). Single‐cell suspension was then incubated with the following antibodies: Alexa 647 anti‐CD31 (BioLegend), Alexa 647 anti‐CD45 (BioLegend), Pacific Blue anti‐Sca1 (BioLegend), biotin anti‐Vcam1 (BD Biosciences) and PE/Cy7 streptavidin (BioLegend). MuSCs (CD31−/CD45−/Sca1−/Vcam1+) were sorted by a BD FACSAria II cell sorter (BD Biosciences). MuSCs genetically labeled with EYFP were sorted based on their autofluorescence.
Immunostaining
Cultured cells, single myofibers or TA muscle sections were fixed in ice‐cold methanol or 4% PFA for 5 min, washed with 0.1% PBST, permeabilized by 0.5% PBST for 20 min, and then blocked with 4% IgG‐free BSA in PBST for 1 h. For Pax7 and eMHC staining, antigen retrieval was required by boiling samples in 0.01 M citric acid at 90°C for 5 min before blocking. Samples were incubated with primary antibodies at 4°C overnight and washed for three times followed by incubation with a secondary antibody for 1 h. The nuclei were counterstained with 100 ng/ml 4′,6‐diamidino‐2‐phenylindole (DAPI). The EdU staining was performed with a Click‐iT™ imaging kit (Thermo Fisher Scientific). The primary antibodies and the dilution factors used were listed below: anti‐GFP (Abcam, 1:400), anti‐Pax7 (Hybridoma Bank, 1:100), anti‐eMHC (Hybridoma Bank, 1:200), anti‐p‐S6 (S235/236) (Cell Signaling, 4858S, 1:300), anti‐FoxO3a (Cell Signaling, #2497S, 1:300), anti‐MyoD (Santa Cruz, sc760, 1:100), anti‐CD34 (eBioscience, 14‐0341‐82, 1:400), anti‐laminin (Sigma, L9393, 1:500), and anti‐dystrophin (Abcam, ab15277, 1:500).
RNA extraction and RT–qPCR
Total RNA was extracted from cells with the TRIzol reagent (Invitrogen) following the manufacturer's instructions. Total RNA was reverse‐transcribed into cDNAs with an oligo‐dT primer by the ImProm‐II Reverse Transcription System (Promega). Selected target genes were amplified by qPCR. The relative gene expression was calculated by the 2−ΔΔCt method. The qPCR primers (5′–3′) were listed below:
Myod: (F) 5′‐CGCTCCAACTGCTCTGATG‐3′; (R) 5′‐TAGTAGGCGGTGTCGTAGCC‐3′
Myf5: (F) 5′‐ACAGCAGCTTTGACAGCATC‐3′; (R) 5′‐AAGCAATCCAAGCTGGACAC‐3′
c‐Jun: (F) 5′‐CCTTCTACGACGATGCCCTC‐3′; (R) 5′‐GGTTCAAGGTCATGCTCTGTTT‐3′
Fos: (F) 5′‐CGGGTTTCAACGCCGACTA‐3′; (R) 5′‐TTGGCACTAGAGACGGACAGA‐3′
Fosb: (F) 5′‐TTTTCCCGGAGACTACGACTC‐3′; (R) 5′‐GTGATTGCGGTGACCGTTG‐3′
Rictor: (F) 5′‐ACAGTTGGAAAAGTGGCACAA‐3′; (R) 5′‐GCGACGAACGTAGTTATCACCA‐3′
Cell culture
Primary myoblasts were cultured in Ham's F10 medium with 20% fetal bovine serum supplemented with 100 ng/ml basic fibroblast growth factor (bFGF) and 10 μM p38 inhibitor (SB202190).
Western blot
We followed the same procedures as described in Zhu et al (2016). The primary antibodies and the dilution factors used were listed below: anti‐GAPDH (Ambion, AM4300, 1:20,000), anti‐Pax7 (Hybridoma Bank, 1:1,000), anti‐Myf5 (Santa Cruz, sc302, 1:1,000), anti‐p110α (Cell Signaling, #4249S, 1:2,000), anti‐c‐Jun (Cell Signaling, #9165S, 1:2,000), and anti‐Fos (Santa Cruz, sc52, 1:1,000).
siRNA transfection
siRNAs were transiently transfected into primary myoblasts at the concentration of 30 nM by Lipofectamine RNAiMax reagent (Thermo Fisher Scientific) following manufacturers’ recommendations. siRNAs used were listed below:
Gfp: 5′‐GCTGACCCTGAAGTTCATC‐3′
FoxO1: 5′‐GGGAGAATGTTCGCTTTCT‐3′
FoxO3a: 5′‐GTGCCCTACTTCAAGGATA‐3′
FoxO4: 5′‐CACGCTCAGTGAAGATCTA‐3′
Rictor#1: 5′‐GCCCTCCATTGCAACAATA‐3′
Rictor#2: 5′‐GGAGGAGTTTGAACAGTTA‐3′
Adenoviral infection
For adenovirus production, 293A cells were transfected at 100% confluence by Lipofectamine with 1 μg of pAd/BLOCK‐iT™‐DEST (Thermo Fisher Scientific) vectors carrying RFP, mouse c‐Jun or Fos cDNAs. Ten days after transfection, the viral supernatants were harvested. Cells were infected with the adenovirus for 6 h. After the removal of the virus‐containing media and washing, the cells were grown in the growth medium (F10 with 10% HS).
G0‐G1 separation
Cells were incubated with 10 μg/ml of Hoechst 33342 at 37°C for 45 min. Pyronin Y was then directly added to the cells to a final concentration of 0.5 μg/ml for 15 min at 37°C followed by FACS analysis.
RNA‐seq and data analysis
Four pairs of adult littermates of the heterozygous control and p110α iKO mice were treated with Tmx as described in Fig 1. Seven days after the last Tmx injection, MuSCs were freshly isolated by FACS based on YFP expression, and the total RNAs from each mouse were purified separately with the NucleoSpin® RNA XS kit from Macherey–Nagel (Düren, Germany). The sequencing libraries were prepared following the same procedures as described in Zhu et al (2016). Over 50 million paired‐end sequencing reads were obtained for each sample. Raw sequencing reads were aligned to the mouse genome assembly (mm10) using the STAR aligner. The aligned fragments were assembled into transcript using the Tuxedo suit (Cufflinks and Cuffmerge) followed by statistical tests on differential expression (Cuffdiff2), in which the significance cutoff was set at 0.05 (FDR‐adjusted p_value). To visualize the sequencing data in the genome browser (IGV), Bedtools were used to convert the aligned bam files into bigwig files. GSEA was performed with the dataset generated from the Cuffdiff output and the gene set obtained from the Molecular Signature Database (MSigDB).
Data availability
Raw data of our RNA‐seq experiment were deposited to GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109472, accession number: GSE109472).
Statistical analysis
Error bars in all figures represented standard deviation (s.d.). The unpaired Student's t‐test was employed to evaluate the statistical significance with P < 0.05 considered statistically significant. No statistical methods were used to pre‐determine the sample size.
Author contributions
GW and ZW designed all the experiments, analyzed data, and wrote the manuscript. GW performed all the experiments with the help from HZ, CS, LH, and YY. THC and KL provided mice and helped analyze the data. All authors discussed the data and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by research grants from the Hong Kong Research Grant Council (GRF16102615, C6015‐14G, AoE/M‐09/12, and AoE/M‐604/16), the Center for Systems Biology & Human Health, the Center for Stem Cell Research, and the State Key Laboratory of Molecular Neuroscience at HKUST.
The EMBO Journal (2018) 37: e98239
See also: https://doi.org/10.15252/embj.201899297 (April 2018)
References
- Abou‐Khalil R, Le Grand F, Pallafacchina G, Valable S, Authier F‐J, Rudnicki MA, Gherardi RK, Germain S, Chretien F, Sotiropoulos A, Lafuste P, Montarras D, Chazaud B (2009) Autocrine and paracrine angiopoietin 1/Tie‐2 signaling promotes muscle satellite cell self‐renewal. Cell Stem Cell 5: 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Albis A, Couteaux R, Janmot C, Roulet A, Mira JC (1988) Regeneration after cardiotoxin injury of innervated and denervated slow and fast muscles of mammals. Myosin isoform analysis. Eur J Biochem 174: 103–110 [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Romanino K, Cloëtta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Rüegg MA (2008) Skeletal muscle‐specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8: 411–424 [DOI] [PubMed] [Google Scholar]
- Bi L, Okabe I, Bernard DJ, Wynshaw‐Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3‐kinase. J Biol Chem 274: 10963–10968 [DOI] [PubMed] [Google Scholar]
- Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA (2012) Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30: 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE (2002) Function and genetics of dystrophin and dystrophin‐related proteins in muscle. Physiol Rev 82: 291–329 [DOI] [PubMed] [Google Scholar]
- Boonsanay V, Zhang T, Georgieva A, Kostin S, Qi H, Yuan X, Zhou Y, Braun T (2016) Regulation of skeletal muscle stem cell quiescence by Suv4‐20 h1‐dependent facultative heterochromatin formation. Cell Stem Cell 18: 229–242 [DOI] [PubMed] [Google Scholar]
- Brack AS, Rando TA (2012) Tissue‐specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 10: 504–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson‐Maduro J, Baron CS, Robin C, van Oudenaarden A (2017) Single‐cell sequencing reveals dissociation‐induced gene expression in tissue subpopulations. Nat Methods 14: 935 [DOI] [PubMed] [Google Scholar]
- Buckingham M, Relaix F (2015) PAX3 and PAX7 as upstream regulators of myogenesis. Semin Cell Dev Biol 44: 115–125 [DOI] [PubMed] [Google Scholar]
- Chakkalakal JV, Jones KM, Basson MA, Brack AS (2012) The aged niche disrupts muscle stem cell quiescence. Nature 490: 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargé SBP, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84: 209–238 [DOI] [PubMed] [Google Scholar]
- Chell JM, Brand AH (2010) Nutrition‐responsive glia control exit of neural stem cells from quiescence. Cell 143: 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA (2012) Maintenance of muscle stem‐cell quiescence by microRNA‐489. Nature 482: 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE (2005) Stem cell function, self‐renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122: 289–301 [DOI] [PubMed] [Google Scholar]
- Durbeej M, Campbell KP (2002) Muscular dystrophies involving the dystrophin‐glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev 12: 349–361 [DOI] [PubMed] [Google Scholar]
- Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe‐Suzuki Y, Takeda S (2007) Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 25: 2448–2459 [DOI] [PubMed] [Google Scholar]
- Gopinath SD, Webb AE, Brunet A, Rando TA (2014) FOXO3 promotes quiescence in adult muscle stem cells during the process of self‐renewal. Stem Cell Rep 2: 414–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N (2011) Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta 1813: 1965–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Hansen MS, Coffin CM, Capecchi MR (2004) Pax3: Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev 18: 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Conway SJ, Fan C‐M (2009) Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Partridge TA, Fan C‐M (2011) An absolute requirement for Pax7‐positive satellite cells in acute injury‐induced skeletal muscle regeneration. Development 138: 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, Hurgo BMC, Leavitt T, Shih J, Brunet A, Rando TA (2013) Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep 4: 189–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, Rando TA (2015) Isolation of skeletal muscle stem cells by fluorescence‐activated cell sorting. Nat Protoc 10: 1612–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado L, Esteves de Lima J, Fabre O, Proux C, Legendre R, Szegedi A, Varet H, Ingerslev LR, Barrès R, Relaix F, Mourikis P (2017) In situ fixation redefines quiescence and early activation of skeletal muscle stem cells. Cell Rep 21: 1982–1993 [DOI] [PubMed] [Google Scholar]
- Matheny RW, Riddle‐Kottke MA, Leandry LA, Lynch CM, Abdalla MN, Geddis AV, Piper DR, Zhao JJ (2015) Role of phosphoinositide 3‐OH kinase p110β in skeletal myogenesis. Mol Cell Biol 35: 1182–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9: 493–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140: 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourikis P, Sambasivan R, Castel D, Rocheteau P, Bizzarro V, Tajbakhsh S (2012) A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30: 243–252 [DOI] [PubMed] [Google Scholar]
- Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G (2011) Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138: 3625–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, Chae S‐S, Zheng H, Ying H, Mahoney J, Hiller D, Jiang S, Protopopov A, Wong WH, Chin L, Ligon KL, DePinho RA (2009) FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5: 540–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliouras GN, Hamilton LK, Aumont A, Joppé SE, Barnabé‐Heider F, Fernandes KJL (2012) Mammalian target of rapamycin signaling is a key regulator of the transit‐amplifying progenitor pool in the adult and aging forebrain. J Neurosci 32: 15012–15026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322: 963–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski B, Pulliam C, Betta ND, Kardon G, Olwin BB (2015) Pervasive satellite cell contribution to uninjured adult muscle fibers. Skelet Muscle 5: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan AA, Salih DAM, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, Palmer TD, Butte AJ, Brunet A (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5: 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, Richard‐Bulteau H, Vignaud A, Baas D, Defour A, Freyssenet D, Tanti J‐F, Le‐Marchand‐Brustel Y, Ferrier B, Conjard‐Duplany A, Romanino K, Bauché S, Hantaï D, Mueller M, Kozma SC et al (2009) Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 187: 859–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai C‐R, Goodell MA, Rando TA (2014) mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 510: 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheteau P, Gayraud‐Morel B, Siegl‐Cachedenier I, Blasco MA, Tajbakhsh S (2012) A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 148: 112–125 [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell'Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez‐Cruz G, Fulco M, Sartorelli V (2015) The NAD(+)‐dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16: 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM (2008) Self‐renewal and expansion of single transplanted muscle stem cells. Nature 456: 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud‐Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A (2011) Pax7‐expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138: 3647–3656 [DOI] [PubMed] [Google Scholar]
- Sambasivan R, Tajbakhsh S (2015) Adult skeletal muscle stem cells. Results Probl Cell Differ 56: 191–213 [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304: 554 [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 117: 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro HM (1981) Flow cytometric estimation of DNA and RNA content in intact cells stained with Hoechst 33342 and pyronin Y. Cytometry 2: 143–150 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2001) AP‐1 in cell proliferation and survival. Oncogene 20: 2390–2400 [DOI] [PubMed] [Google Scholar]
- Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA, Brack AS (2010) Sprouty1 regulates reversible quiescence of a self‐renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 6: 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa‐Nunes R, Yee LL, Gould AP (2011) Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila . Nature 471: 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403 [DOI] [PubMed] [Google Scholar]
- Tamir Y, Bengal E (2000) Phosphoinositide 3‐kinase induces the transcriptional activity of MEF2 proteins during muscle differentiation. J Biol Chem 275: 34424–34432 [DOI] [PubMed] [Google Scholar]
- Tapscott SJ (2005) The circuitry of a master switch: myod and the regulation of skeletal muscle gene transcription. Development 132: 2685–2695 [DOI] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15: 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, Hawkins P (2012) PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13: 195–203 [DOI] [PubMed] [Google Scholar]
- van Velthoven CTJ, de Morree A, Egner IM, Brett JO, Rando TA (2017) Transcriptional profiling of quiescent muscle stem cells in vivo . Cell Rep 21: 1994–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Bi P, Liu W, Asakura A, Keller C, Kuang S (2012) Constitutive notch activation upregulates Pax7 and promotes the self‐renewal of skeletal muscle satellite cells. Mol Cell Biol 32: 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wu Z (2000) The insulin‐like growth factor‐phosphatidylinositol 3‐kinase‐Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma‐derived RD cells. J Biol Chem 275: 36750–36757 [DOI] [PubMed] [Google Scholar]
- Yang K, Neale G, Green DR, He W, Chi H (2011) The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol 12: 888–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Shrestha S, Zeng H, Karmaus PWF, Neale G, Vogel P, Guertin DA, Lamb RF, Chi H (2013) T cell exit from quiescence and differentiation into Th2 cells depend on Raptor‐mTORC1‐mediated metabolic reprogramming. Immunity 39: 1043–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, Rudnicki MA (2013) Satellite cells and the muscle stem cell niche. Physiol Rev 93: 23–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, Gavin TP, Kuang S (2017) Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun 8: 14328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM (2006) The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci USA 103: 16296–16300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Xiao F, Wang G, Wei X, Jiang L, Chen Y, Zhu L, Wang H, Diao Y, Wang H, Ip NY, Cheung TH, Wu Z (2016) STAT3 regulates self‐renewal of adult muscle satellite cells during injury‐induced muscle regeneration. Cell Rep 16: 2102–2115 [DOI] [PubMed] [Google Scholar]
- Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas AE, Crist C (2016) Phosphorylation of eIF2α is a translational control mechanism regulating muscle stem cell quiescence and self‐renewal. Cell Stem Cell 18: 79–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
Raw data of our RNA‐seq experiment were deposited to GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109472, accession number: GSE109472).