

Abstract
Background
Improving our understanding of cancer and other complex diseases requires integrating diverse data sets and algorithms. Intertwining in vivo and in vitro data and in silico models are paramount to overcome intrinsic difficulties given by data complexity. Importantly, this approach also helps to uncover underlying molecular mechanisms. Over the years, research has introduced multiple biochemical and computational methods to study the disease, many of which require animal experiments. However, modeling systems and the comparison of cellular processes in both eukaryotes and prokaryotes help to understand specific aspects of uncontrolled cell growth, eventually leading to improved planning of future experiments. According to the principles for humane techniques milestones in alternative animal testing involve in vitro methods such as cell-based models and microfluidic chips, as well as clinical tests of microdosing and imaging. Up-to-date, the range of alternative methods has expanded towards computational approaches, based on the use of information from past in vitro and in vivo experiments. In fact, in silico techniques are often underrated but can be vital to understanding fundamental processes in cancer. They can rival accuracy of biological assays, and they can provide essential focus and direction to reduce experimental cost.
Main body
We give an overview on in vivo, in vitro and in silico methods used in cancer research. Common models as cell-lines, xenografts, or genetically modified rodents reflect relevant pathological processes to a different degree, but can not replicate the full spectrum of human disease. There is an increasing importance of computational biology, advancing from the task of assisting biological analysis with network biology approaches as the basis for understanding a cell’s functional organization up to model building for predictive systems.
Conclusion
Underlining and extending the in silico approach with respect to the 3Rs for replacement, reduction and refinement will lead cancer research towards efficient and effective precision medicine. Therefore, we suggest refined translational models and testing methods based on integrative analyses and the incorporation of computational biology within cancer research.
Keywords: Cancer research, Computational biology, Cancer bioinformatics, Integrative analysis, In silico modeling, In vitro methods, In vivo techniques, Ex vivo systems, Tumor growth, Alternative animal experimentation, 3Rs
Background
Cancer remains to be one of the top causes of disease-related death. World Health Organization (WHO) reported 8.8 million cancer-related deaths in 2015 [1]. Around one out of 250 people will develop cancer each year, and every fourth will die from it [2]. WHO estimates the number of new cases will rise by ∼ 70% over the next twenty years. Despite decades of research [3], mortality rates and recurrence remain high, and we have limited options for effective therapies or strategies regarding cancer prevention.
Tumor cells exhibit chaotic, heterogeneous and highly differentiated structures, which is determinative to the lack of effective anticancer drugs [4]. For that matter, predictive preclinical models that integrate in vivo, in vitro and in silico experiments, are rare but necessary for the process of understanding tumor complexity.
A biological system comprises a multiplicity of interconnected dynamic processes at different time and spatial range. The complexity often hinders the ability to detail relationships between cause and effect. Model-based approaches help to interprete complex and variable structures of a system and can account for biological mechanisms. Next to studying pathological processes or molecular mechanisms, they can be used for biomarker discovery, validation, basic approaches to therapy and preclinical testing. So far, preclinical research primarily involves in vivo models based on animal experimentation.
Intertwining biological experiments with computational analyses and modeling may help to reduce the number of experiments required, and improve the quality of information gained from them [5]. Instead of broad high-throughput screens, focused screens can lead to increased sensitivity, improved validation rates, and reduced requirements for in vitro and in vivo experiments. For Austria, the estimated number of laboratory animal kills per year was over 200 000 [6]. In Germany the number of animal experiments for research is estimated as 2.8 millions [7]. Worldwide, the quantity of killed animals for research, teaching, testing and experimentation exceeds 100 000 000 per year [6–14], as shown in Fig. 1.
Fig. 1.
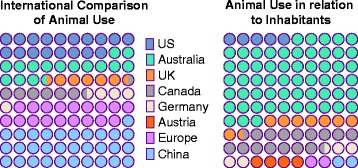
Worldwide use of animals for studies. International comparison in numbers of animals used for experimentation, such as toxicology testing for cosmetics, food, drugs, research, teaching and education [6–14]
Principles for humane techniques were classified as replacement, reduction and refinement, also known as the 3Rs [15]. While most countries follow recommendations of Research Ethics Boards [16], discussion of ethical issues regarding the use of animals in research continues [17]. So far, 3R principles have been integrated into legislation and guidelines how to execute experiments using animal models, still, rethinking of refined experimentation will ultimately lead to higher-quality science [18]. The 3R concept also implies economic, ethical and academic sense behind sharing experimental animal resources, making biomedical research data scientifically easily available [19]. The idea behind 3R has been implemented in several programs such as Tox21 and ToxCast also offering high throughput assay screening data on several cancer-causing compounds for bioactivity profiles and predictive models [20–22].
It is clear that no model is perfect, and is lacking some aspects of reality. Thus, one has to choose and use appropriate models to advance specific experiments. Cancer research relies on diverse data from clinical trials, in vivo screens and validation studies, and functional studies using diverse in vitro experimental methods, such as cell-based models, spheroid systems, and screening systems for cytotoxicity, mutagenicity and cancerogenesis [23, 24]. New technologies will advance in organ-on-a-chip technologies [25] but also include the in silico branch of systems biology with its goal to create the virtual physiological human [26]. The range of alternative methods has already expanded further towards in silico experimentation standing for “performed on a computer”. These computational approaches include storage, exchange and use of information from past in vitro and in vivo experiments, predictions and modeling techniques [27]. In this regard, the term non-testing methods has been introduced, which summarizes the approach in predictive toxicology using previously given information for risk assessment of chemicals [28]. Such methods generate non-testing data by the general approach of grouping, (quantitative) structure-activity relationships (QSAR) or comprehensive expert systems, which are respectively based on the similarity principle [29–31].
The regulation of the European Union for registration, evaluation, authorisation and restriction of chemicals (REACH) promotes adaptation of in vivo experimentation under the conditions that non-testing methods or in vitro methods provide valid, reliable, relevant information, adequate for the intended purpose, or in case that testing is technically impossible [30].
Generally, in vitro and in silico are useful resources for predicting several (bio)chemical and (patho)physiological characteristics of likewise potential drugs or toxic compounds, but have not been fit for full pharmacokinetic profiling yet [32]. In vitro as well as in silico models abound especially in the fields of toxicology and cosmetics, based on cell culture, tissues and simulations [33]. In terms of 3R, in vitro techniques allow to reduce, refine and replace animal experiments. Still, wet biomedical research requires numerous resources from a variety of biological sources. In silico methods can further be used to augment and refine in vivo and in vitro models. Validation of computational models will still require results from in vivo and in vitro experiments. Though, in the long run, integrative approaches incorporating computational biology will reduce laboratory work in the first place and effectively succeed in 3R.
Within the next sections, we summarize common methods and novel techniques regarding in vivo, in vitro and in silico cancer research, presented as overview in Fig. 2, and associated modeling examples listed in Table 1.
Fig. 2.
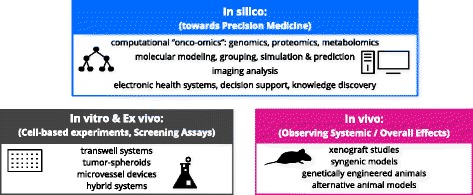
Preclinical techniques for cancer research. Examples for experiments on the computer (in silico), inside the living body (in vivo), outside the living body (ex vivo) as well as in the laboratory (in vitro)
Table 1.
Overview of exemplary models for cancer research
| References | |
|---|---|
| In vivo | |
| Murine models | [37] |
| Genetically engineered mouse model | [36] |
| Zebra fish model | [42] |
| Drosophila model | [41] |
| Chick embryo model | [43, 44] |
| In vitro | |
| General 2D/3D in vitro models | [46, 53] |
| Transwell model | [48] |
| Spheroid system | [49] |
| Microfluidic system | [50] |
| Tissue-engineered microvessel model | [51] |
| In silico | |
| Sequence analysis | [63, 69, 74] |
| General pathway analysis and network inference | [132, 133, 135] |
| Pan-cancer | [62, 82, 134, 139] |
| Chemical perturbation mapping | [64, 66, 68] |
| Pharmacogenomic mapping | [99, 102, 117, 136] |
| Genome-phenotype mapping | [81] |
| Clinical data integration | [106] |
| Structure mapping | [102, 103] |
| Structure and activity | [100, 101] |
| Framework for key events and mode of action | [97, 98] |
| Image classification | [85, 87] |
| Growth prediction | [91–93] |
In vivo methods
Animals are the primary resource for research on the pathogenesis of cancer. Animal models are commonly used for studies on cancer biology and genetics as well as the preclinical investigation of cancer-therapy and the efficacy and safety of novel drugs [34]. Animal models represent the in vivo counterpart to cell-lines and suspension culture, while being superior in terms of physiological relevance offering imitation of parental tumors and a heterogeneous microenvironment as part of an interacting complex biochemical system.
In general, animal models primarily based on murine or rodent models can be subdivided into the following groups of (I) xenograft models, which refer to the heterotopic, subcutaneous intraperitoneal or orthotopic implantation into SCID (Severe Combined Immune Deficiency) or nude mice, (II) syngenic models involving the implantation of cells from the same strain into non-immunocompromised mice, and (III) genetically engineered models, which allow for RNA interference, multigenic mutation, inducible or reversible gene expression [35, 36].
Several engineered mouse models on cancer and related diseases have been developed so far [37]. In case of xenograft models, tumor-specific cells are transplanted into immunocompromised mice. Common tumor xenograft models lack the immune system response that can be crucial in tumor development and progression [38]. Xenograft models can be patient-derived, by transferation of a patient’s primary tumor cells after surgery into immunocompromised mice. The transplantation of immortalized tumor cell-lines represents a simplified preclinical model with limited clinical application possibilities [39]. For these reasons, there is a trend towards genetically engineered animal models, allowing for site-directed mutations on tumor-suppressor genes and proto-oncogenes as the basis for studies on oncogenesis [40].
Next to the gold standard of murine and rodent models, there are other animal model systems frequently used, such as the Drosophila melanogaster (fruit fly) or Danio rerio (zebra fish) [41, 42]. The fruit fly offers the advantage of low-cost handling and easy mutant generation while it holds a substantially high conservation of the human cancer-related signaling apparatus [41]. There are additional animal models, commonly referred to as alternatives, such as zebra fish models for angiogenesis studies and chick embryo CAM (chorioallantoic membrane) models, offering rapid tumor formation due to the highly vascularized CAM structure [40, 43, 44].
So far, preclinical model systems do not provide sufficient information on target validation, but aid in identifying and selecting novel targets, while new strategies offer a quantitative translation from preclinical studies to clinical applications [45].
In vitro methods
In vitro models offer possibilities for studying several cellular aspects as the tumor microenvironment using specific cell types, extracellular matrices, and soluble factors [46]. In vitro models are mainly based on either cell cultures of adherent monolayers or free-floating suspension cells [47]. They can be categorized into: (I) transwell-based models which include invasion and migration assays [48], (II) spheroid-based models involving non-adherent surfaces [49], hanging droplets and microfluidic devices [50], (III) tumor-microvessel models which come with predefined ECM (extracellular matrix) scaffolds and microvessel self-assemblies [51], and (IV) hybrid tumor models including embedded ex vivo tumor sections, 3D invasion through clusters embedded in gel, and 2D vacscular microfluidics [52].
Generally, such cell culture models focus on key aspects of metabolism, absorption, distribution, excretion of chemicals or other aspects of cell signaling pathways, such as aspects of metastasis under a controlled environment [53]. Scale-up systems attempt to emulate the physiological variability in order to extrapolate from in vitro to in vivo [54]. Advanced models as 3D culture systems more accurately represent the tumor environment [55]. Cell culture techniques include the formation of cell spheroids, which are frequently used in cancer research for approximating in vitro tumor growth as well as tumor invasion [56]. In particular, multicellular tumor spheroids have been applied for drug screening and studies on proliferation, migration, invasion, immune interactions, remodeling, angiogenesis and interactions between tumor cells and the microenvironment [46].
In vitro methods include studies on intercellular, intracellular or even intraorganellar processes, which determine the complexity of tumor growth to cancerogenesis and metastasis, based on several methods from the disciplines of biophysics, biochemistry and molecular biology [23].
Ex vivo systems offer additional possibilities to study molecular features. Such systems can be derived from animal and human organs or multiple donors. Thereby, ex vivo systems comprise the isolation of primary material from an organism, cultivation and storage in vitro and differentiation into different cell types [57]. In this regard, induced pluripotent stem cells, in particular cancer stem cell subpopulations, have been presented as in vitro alternative to xenograft experiments [58]. Moreover, ex vivo methods can be used to predict drug response in cancer patients [59]. These systems have been developed to improve basic in vitro cell cultures while overcoming shortcomings of preclinical animal models; thus, serving as more clinically relevant models [60].
In silico analysis
The term in silico was created in line with in vivo and in vitro, and refers to as performed on computer or via computer simulation [28]. In silico techniques can be summarized as the process of integrating computational approaches to biological analysis and simulation. So far, in silico cancer research involves several techniques including computational validation, classification, inference, prediction, as well as mathematical and computational modeling, summarized in Fig. 3. Computational biology and bioinformatics are mostly used to store and process large-scale experimental data, extract and provide information as well as develop integrative tools to support analysis tasks and to produce biological insights. Existing well-maintained databases provide, integrate and annotate ”information on various cancers [61], and are increasingly being used to generate predictive models, which in turn will inform and guide biomedical experiments. Table 2 lists several representative examples of such databases.
Fig. 3.
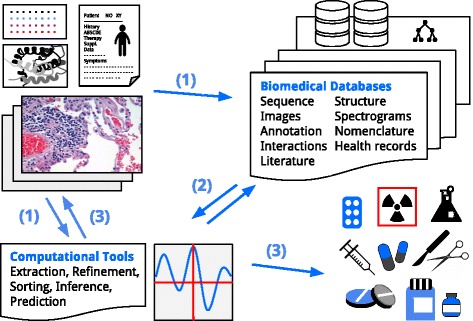
In silico pipeline. (1) Manual input into databases storing patient information, literature, images and experimental data, or direct data input into computational tools. (2) Refinement and retrieval over computational tools for classification, inference, validation and prediction. (3) Output for research strategies, model refinement, diagnosis, treatment and therapy. Note: Derivative elements have been identified as licensed under the Creative Commons, free to share and adapt
Table 2.
List of main databases and data resources in cancer research
| Name | Url | References |
|---|---|---|
| cBioPortal | http://www.cbioportal.org | [115, 127] |
| BioPreDyn-Bench | https://sites.google.com/site/biopredynbenchmarks/ | [104] |
| CGAP | https://cgap.nci.nih.gov/ | [62] |
| EPA Toxcast Screening Library | https://actor.epa.gov/dashboard/ | [20, 65] |
| EpiFactors | http://epifactors.autosome.ru/ | [76] |
| Human Protein Atlas | https://www.proteinatlas.org/ | [110, 111] |
| GDSC | http://www.cancerrxgene.org/ | [101, 103] |
| GDC/TCGA | https://portal.gdc.cancer.gov/ | [61, 138] |
| Gene Ontology | http://www.geneontology.org/ | [128] |
| KEGG | http://www.genome.jp/kegg/ | [130] |
| NCI-60 databases | https://discover.nci.nih.gov/cellminer/ | [115, 144] |
| Open TG-GATEs | http://toxico.nibiohn.go.jp/english/ | [66] |
| Reactome | https://reactome.org/ | [129] |
| pathDIP | http://ophid.utoronto.ca/pathDIP/ | [134] |
The Cancer genome project and Cancer Genome Atlas have generated an abundance of data on molecular alterations related to cancer [62]. The Cancer Genome Anatomy Project by the National Cancer Institute also provides information on healthy and cancer patient gene expression profiles and proteomic data with the objective to generate novel detection, diagnosis and treatment possibilities [63]. In this connection, analyzing molecular changes and collecting gene expression signatures of malignant cells is important for understanding cancer progression. As example, over a million profiles of genes, drugs and disease states have been collected as so-called cellular connectivity maps in order to discover new therapeutic targets for treating cancer [64]. Regarding the effect of small molecules on human health, computational toxicology has created in silico resources to organise, analyse, simulate, visualise, or predict toxicity as a measure of adverse effects of chemicals [31, 65]. Large-scale toxicogenomics data has been collected by multi-agency toxicity testing initiatives, for forecasting carcinogenicity or mutagenicity [20, 66–68]. Thereby, gene expression signatures and information on chemical pathway perturbation by carcinogenic and mutagenic compounds have been analyzed and incorporated into in silico models to predict the potential of hazard pathway activation including carcinogenicity to humans [20–22, 66].
The analysis of genomic and proteomic data largely focuses on comparison of annotated data sets, by applying diverse machine learning and statistical methods. Most genomic alterations comprise single nucleotide variants, short base insertions or deletions, gene copy number variants and sequence translocations [69]. Thereby, cancer genes are defined by genetic alterations, specifically selected from the cancer microenvironment, conferring an advantage on cancer cell growth. In this regard, the goal is set in characterizing driver genes. However, combination of such genes may provide prognostic signatures with clear clinical use. Integrating patterns of deregulated genome or proteome with information about biomolecular function and signaling cascades does in turn provide inside into underlying biological mechanism driving the disease.
Analysis of genomic and proteomic data relies on processing methods such as clustering algorithms [70]. Cluster analysis depicts the statistical process of group formation upon similarities, exemplary for exploratory data mining [71]. Understanding the heterogeneity of cancer diseases and the underlying individual variations requires translational personalized research such as statistical inference at the patient level [72]. Statistical inference represents the process of detailed reflections on data and deriving sample distributions, understanding large sample properties and concluding with scientific findings as knowledge discovery and decision making. This computational approach involving mathematical and biological modeling, allows to predict disease risk and progression [72].
Besides directly studying cancer genes and proteins, it is increasingly recognized that their regulators, not only involving so far known tumor suppressor genes and proto-oncogenes but also non-coding elements [73–75] and epigenetic factors in general can be highly altered in cancer [76, 77]. These include metabolic cofactors [78], chemical modifications such as DNA methylation [79], and microRNAs [80]. Another approach to studying cancer involves the view of dysregulated pathways instead of single genetic mutations [81]. The heterogeneous patient profiles are thereby analyzed for pathway similarities in order to define phenotypic subclasses related to genotypic causes to cancer. Next to elucidating novel genetic players in cancer diseases using genomic patient profiling, there are other studies focusing the underlying structural components of interacting protein residues in cancer [82]. This genomic-proteomic-structural approach is used to highlight functionally important genes in cancer. In this regard, studies on macromolecular structure and dynamics give insight into cellular processes as well as dysfunctions [83].
Image analysis and interpretation strongly benefit from diverse computational methods in general and within the field of cancer therapy and research. Computer algorithms are frequently used for classification purposes and assessment of images in order to increase throughput and generate objective results [84–86]. Image analysis via computerized tomography has been recently proposed for evaluating individualized tumor responses [87]. Pattern recognition describes a major example on extracting knowledge from imaging data. Recently, an algorithmic recognition approach of the underlying spatially resolved biochemical composition, within normal and diseased states, has been described for spectroscopic imaging [88]. Such an approach could serve as digital diagnostic resource for identifying cancer conditions, and complementing traditional diagnostic tests towards personalized medicine.
Computational biology provides resources and tools necessary for biologically-meaningful simulations, implementing powerful models of cancer using experimental data, supporting trend analysis, disease progression and strategic therapy assessment. Network models on cancer signaling have been build on the basis of time-course experiments measuring protein expression and activity in use of validating simulation prediction and testing drug target efficacy [89]. Simulations of metabolic events have been introduced with genome scale metabolic models for data interpretation, flux prediction, hypothesis testing, diagnostics, biomarker and drug target identification [90]. Mathematical and computational modeling have been further used to better understand cancer evolution [91–93].
Since the concept of 3R has its primary focus on replacing animal experimentation within the area of chemical assessment, several in silico methods have been or are being developed in the field of toxicology. So far, computational toxicology deals with the assessment of hazardous chemicals such as carcinogens rather than computational biomedicine and biological research concerning cancer. Still, underlying methods can be likewise integrated into both disciplines [94, 95]. Recently, toxicology has brought up the adverse outcome pathway (AOP) methodology, which is intended to collect, organise and evaluate relevant information on biological and toxicological effects of chemicals, more specifically, existing knowledge concerning biologically plausible and empirically supported links between molecular-level perturbation of a biological system and an adverse outcome at the level of biological organisation of regulatory concern [96, 97]. This framework is intended to focus humans as model organism on different biological levels rather than whole-animal models [95]. The International Program on Chemical Safety has also published a framework for analyzing the relevance of a cancer mode of action for humans, formerly assessed for carcinogenesis in animals [98]. The postulated mode of action comprises a description of critical and measurable key events leading to cancer. This framework has been integrated into the guidelines on risk assessment by the Environmental Protection Agency to provide a tool for harmonization and transparency of information on carcinogenic effect on humans, likewise intended to support risk assessors and also the research community. Noteworthy, next to frameworks, there are several common toxicological in silico techniques. Especially similarity methods play a fundamental role in computational toxicology with QSAR modeling as the most prominent example [28, 29]. QSARs mathematically relate structure-derived parameters, so-called molecular descriptors, to a measure of property or activity. Thereby, regression analysis and classification methods are used to generate a continuous or categorical result as qualitative or quantitative endpoint [29, 31]. Exemplary, models based on structure and activity data have been used to predict human toxicity endpoints for a number of carcinogens [22, 99–101]. Still, in order to predict drug efficacy and sensitivity, it is suggested to combine models on chemical features such as structure data with genomic features [102–104].
Combined, in silico methods can be used for both characterization and prediction. Thereby, simulations are frequently applied for the systematic analysis of cellular processes. Large-scale models on whole biological systems, including signal-transduction and metabolic pathways, face several challenges of accounted parameters at the cost of computing power [105]. Still, the complexity and heterogeneity of cancer as well as the corresponding vast amount of available data, asks for a systemic approach such as computational modeling and machine learning [106, 107]. Overall, in silico biological systems, especially integrated mathematical models, provide significant link and enrichment of in vitro and in vivo systems [108].
Computational cancer research towards precision medicine
Oncogenesis and tumor progression of each patient are characterized by multitude of genomic perturbation events, resulting in diverse perturbations of signaling cascades, and thus requiring thorough molecular characterization for designing effective targeted therapies [109]. Precision medicine customizes healthcare by optimizing treatment to the individual requirements of a patient, often based on the genetic profile or other molecular biomarkers. This demands state-of-the-art diagnostic and prognostic tools, comprehensive molecular characterization of the tumor, as well as detailed electronic patient health records [110].
Computational tools offer the possibility of identifying new entities in signaling cascades as biomarkers and promising targets for anticancer therapy. For example, the Human Protein Atlas provides data on the distribution and the expression of putative gene products in normal and cancer tissues based on immunohistochemical images annotated by pathologists. This database provides cancer protein signatures to be analysed for potential biomarkers [111, 112].
A different approach to the discovery of potential signaling targets is described by metabolomic profiling of biological systems which has been applied to find novel biomarkers for detection and prognosis of the disease [113–115].
Moreover, computational cancer biology and pharmacogenomics have been used for gene targeting by drug repositioning [116, 117]. Computational drug repositioning is another example for in silico cancer research, by identifying novel use for FDA-approved drugs, based on available genomic, phenotypic data with the help of bioinformatics and chemoinformatics [118–120]. Computer-aided drug discovery and development have improved the efficiency of pharmaceutical research and link virtual screening methods, homology and molecular modeling techniques [121, 122]. Pharmacological modeling of drug exposures helps to understand therapeutic exposure-response relationships [123]. Systems pharmacology integrates pharmacokinetic and pharmacodynamic drug relations into the field of systems biology regarding the multiscale physiology [124]. The discipline of pharmacometrics advances to personalized therapy by linking drug response modeling and health records [125]. Polypharmacological effects of multi-drug therapies render exclusive wet lab experimentation unfeasible and require modeling frameworks such as system-level networks [126]. Network pharmacology models involve phenotypic responses and side effects due to a multi-drug treatment, offering information on inhibition, resistance and on-/off-targeting. Moreover, the network approach allows to understand variations within a single cancer disease regarding heterogeneous patient profiles, and in the process, to classify cancer subtypes and to identify novel drug targets [81].
Tumorigenesis is induced by driver mutations and embeds passenger mutations that both can result in upstream or downstream dysregulated signaling pathways [127]. Computational methods have been used to distinguish driver and passenger mutations in cancer pathways by using public genomic databases available through collaborative projects such as the International Cancer Genome Consortium or The Cancer Genome Atlas (TCGA) [62] and others [128], together with functional network analysis using de novo pathway learning methods or databases on known pathways such as Gene Ontology [129], Reactome [130] or the Kyoto Encyclopedia of Genes and Genomes (KEGG) [131–134]. These primary pathway databases, based on manually curated physical and functional protein interaction data, are essential for annotation and enrichment analysis. To increase proteome coverage of such analyses, pathways can be integrated with comprehensive protein-protein interaction data and data mining approaches to predict novel, functional protein:pathway associations [135]. Importantly, this in silico approach not only expands information on already known parts of the proteome, it also annotates current “pathway orphans” such as proteins that currently do not have any known pathway association.
Comprehensive preclinical models on molecular features of cancer and diverse therapeutic responses have been built as pharmacogenomic resource for precision oncology [136, 137]. Future efforts will need to expand integrative approaches to combine information on multiple levels of molecular aberrations in DNA, RNA, proteins and epigenetic factors [62, 138], as well as cellular aspects of the microenvironment and tumor purity [139], in order to extend treatment efficacy and further refine precision medicine.
Conclusion
Informatics in aid to biomedical research, especially in the field of cancer research, faces the challenge of an overwhelming amount of available data, especially in future regards to personalized medicine [140]. Computational biology provides mathematical models and specialized algorithms to study and predict events in biological systems [141]. Certainly, biomedical researchers from diverse fields will require computational tools in order to better integrate, annotate, analyze, and extract knowledge from large networks of biological systems. This increasing need of understanding complex systems can be supported by “Executable Biology” [142], which embraces representative computational modeling of biological systems.
There is an evolution towards computational cancer research. In particular, in silico methods have been suggested for refining experimental programs of clinical and general biomedical studies involving laboratory work [143]. The principles of the 3Rs can be applied to cancer research for the reduction of animal research, saving resources as well as reducing costs spent on clinical and wet lab experiments. Computational modeling and simulations offer new possibilities for research. Cancer and biomedical science in general will benefit from the combination of in silico with in vitro and in vivo methods, resulting in higher specificity and speed, providing more accurate, more detailed and refined models faster. In silico cancer models have been proposed as refinement [143]. We further suggest the combination of in silico modeling and human computer interaction for knowledge discovery, gaining new insights, supporting prediction and decision making [144].
Here, we provided some thoughts as a motivator for fostering in silico modeling towards 3R, in consideration of refinement of testing methods, and gaining a better understanding of tumorigenesis as tumor promotion, progression and dynamics.
Acknowledgements
This work is based on research studies by students and members of the Holzinger group.
Funding
No specific funding was received for this study.
Availability of data and materials
The data-sets used and/or analysed during the current study are available from the corresponding author on reasonable request and data sources are referenced within figure legend and document text. Images were created by FJ and CJ under the terms of the Creative Commons license, stated in the Open Access subsection of Declarations. Figure 3 makes use of free to reuse pictures from pixabay and wikimedia licensed under Creative Commons and being free of known restrictions under copyright law, including all related and neighboring rights.
Abbreviations
- 3R
Refinement, reduction, replacement
- AOP
Adverse outcome pathway
- CAM
Chorioallantoic membrane
- ECM
Extracellular matrix
- FDA
Food and drug administration
- KEGG
Kyoto encyclopedia of genes and genomes
- pathDIP
Pathway data integration portal
- QSAR
Quantitative structure-activity relationship
- REACH
Registration, evaluation, authorisation and restriction of chemicals
- SCID
Severe combined immune deficiency
- TCGA
The cancer genome atlas
- WHO
World health organization
Authors’ contributions
CJ initiated and conceived the study, wrote the manuscript and contributed with biochemical expertise. FJ participated in the drafting of the manuscript and contributed with expertise in Informatics related to in silico modeling. IJ reviewed and refined the manuscript with expertise in integrative computational biology and cancer informatics. AH supervised the project and contributed with expertise in health informatics. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Claire Jean-Quartier, Email: c.jeanquartier@hci-kdd.org.
Fleur Jeanquartier, Email: fleur.jeanquartier@tugraz.at.
Igor Jurisica, Email: juris@ai.utoronto.ca.
Andreas Holzinger, Email: a.holzinger@hci-kdd.org.
References
- 1.World Health Organization. Cancer Fact sheet. Updated February 2017. http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed Mar 2017.
- 2.National Cancer Institute. Cancer Statistics at a Glance. https://www.cancer.gov/about-cancer/understanding/statistics. Last updated Dec 2015. Accessed Mar 2017.
- 3.Wagoner J. Occupational carcinogenesis: the two hundred years since percivall pott. Ann NY Acad Sci. 1976;271(1):1–4. [Google Scholar]
- 4.Stadler M, Walter S, Walzl A, Kramer N, Unger C, Scherzer M, Unterleuthner D, Hengstschläger M, Krupitza G, Dolznig H. Increased complexity in carcinomas: Analyzing and modeling the interaction of human cancer cells with their microenvironment. Semin Cancer Biol. 2015;35:107–24. doi: 10.1016/j.semcancer.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz AS, Yu J, Gardenour KR, Finley RLJ, Ideker T. Cost-effective strategies for completing the interactome. Nat Methods. 2009;6:55–61. doi: 10.1038/nmeth.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bundesministeriums für Wissenschaft, Forschung und Wirtschaft. Tierversuchsstatistik 2015. https://bmbwf.gv.at/das-ministerium/publikationen/forschung/statistiken/tierversuchsstatistiken. Accessed Nov 2017.
- 7.Senatskomission für tierexperimentelle Forschung der Deutschen Forschungsgemeinschaft . Tierversuche in der Forschung. Bonn: Deutsche Forschungsgemeinschaft; 2016. [Google Scholar]
- 8.Canadian Council on Animal Care . CCAC 2016 Animal Data Report. Ottawa: CCAC; 2016. [Google Scholar]
- 9.U.K. Government. Annual Statistics of Scientific Procedures on Living Animals Great Britain. London: Williams Lea Group; 2016. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/627284/ annual-statistics-scientificprocedures-living-animals-2016.pdf. Accessed Mar 2018.
- 10.Animal and Plant Health Inspection Service. Annual Report Animal Usage by Fiscal Year: United States Department of Agriculture; 2016. Updated August 2016, Accessed March 2018. https://www.aphis.usda.gov/animal_welfare/downloads/reports/Annual-Report-Animal-Usage-by-FY2016.pdf.
- 11.Leary SA. The Exclusion of Mice, Rats, and Birds: The American Anti-Vivisection Society; 2017, pp. 12–13. http://aavs.org/assets/uploads/2017/08/2017-1_av-magazine_exclusion-mice-rats-birds.pdf?x82509.
- 12.MacLaughlin K. China finally setting guidelines for treating lab animals. 2016. https://doi.org/10.1126/science.aaf9812. http://www.sciencemag.org/news/2016/03/china-finally-setting-guidelines-treating-lab-animals. Accessed Feb 2017.
- 13.Humane Research Australia. Australian Statistics of Animal Use in Research and Teaching. 2015. http://www.humaneresearch.org.au/statistics/statistics_2015. Accessed Mar 2018.
- 14.Bundesministerium für Ernährung und Landwirtschaft. Verwendung von Versuchstieren im Jahr 2016. 2016. https://www.bmel.de/DE/Tier/Tierschutz/_texte/TierschutzTierforschung.html?docId=10323474. Accessed Mar 2018.
- 15.Russell W, Burch R. The Principles of Humane Experimental Technique. Reprinted 1992 ed. UK: Wheathampstead: Universities Federation for Animal Welfare; 1959. [Google Scholar]
- 16.Latham SR. U.s. law and animal experimentation: A critical primer. Hast Cent Rep. 2012;42(6):35–9. doi: 10.1002/hast.107. [DOI] [PubMed] [Google Scholar]
- 17.Tannenbaum J, Rowan AN. Rethinking the morality of animal research. Hast Cent Rep. 1985;15:32–43. [PubMed] [Google Scholar]
- 18.Sneddon LU, Halsey LG, Bury NR. Considering aspects of the 3rs principles within experimental animal biology. J Exp Biol. 2017;220(17):3007–16. doi: 10.1242/jeb.147058. [DOI] [PubMed] [Google Scholar]
- 19.Morrissey B, Blyth K, Carter P, Chelala C, Jones L, Holen I, Speirs V. The sharing experimental animal resources, coordinating holdings (search) framework: Encouraging reduction, replacement, and refinement in animal research. PLoS Biol. 2017;15(1):2000719. doi: 10.1371/journal.pbio.2000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benfenati E, Benign R, DeMarini DM, Helma C, Kirkland D, Martin TM, Mazzatorta P, Ouèdraogo-Arras G, Richard AM, Schilter B, Schoonen WGEJ, Snyder RD, Yang C. Predictive models for carcinogenicity and mutagenicity: Frameworks, state-of-the-art, and perspectives. J Environ Sci Health C. 2009;27:57–90. doi: 10.1080/10590500902885593. [DOI] [PubMed] [Google Scholar]
- 21.Kleinstreuer NC, Yang J, Berg EL, Knudsen T, Richard AM, Martin M, Reif DM, Judson R, Polokoff M, Dix DJ, et al. Phenotypic screening of the toxcast chemical library to classify toxic and therapeutic mechanisms. Nat Biotechnol. 2014;32(6):583–91. doi: 10.1038/nbt.2914. [DOI] [PubMed] [Google Scholar]
- 22.Huang R, Xia M, Sakamuru S, Zhao J, Shahane SA, Attene-Ramos M, Zhao T, Austin CP, Simeonov A. Modelling the tox21 10 k chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun. 2016;7:10425. doi: 10.1038/ncomms10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tritthart HA. In vitro test systems in cancer research. ALTEX. 1996;13(3):118–24. [PubMed] [Google Scholar]
- 24.Teicher BA. In vivo/ex vivo and in situ assays used in cancer research: a brief review. Toxicol Pathol. 2009;37(1):114–22. doi: 10.1177/0192623308329473. [DOI] [PubMed] [Google Scholar]
- 25.Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. 2015;14:248–60. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viceconti M HP. The virtual physiological human: Ten years after. Annu Rev Biomed Eng. 2016;18:103–23. doi: 10.1146/annurev-bioeng-110915-114742. [DOI] [PubMed] [Google Scholar]
- 27.Zurlo J, Rudacille D, Goldberg AM. The three R’s: The way forward. Environ Health Perspect. 1996;104(8):878. doi: 10.1289/ehp.96104878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raunio H. In silico toxicology - non-testing methods. Front Pharmacol. 2011;2(33):33. doi: 10.3389/fphar.2011.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.European Chemicals Agency. Guidance on Information Requirements and Chemical Safety Assessment. 2008. http://echa.europa.eu/.
- 30.European Chemicals Agency. Guidance on Information Requirements and Chemical Safety Assessment. 2011. http://echa.europa.eu/.
- 31.Raies AB, Bajic VB. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci. 2016;6(2):147–72. doi: 10.1002/wcms.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Workman P, Aboagye E, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DAH, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA, ad hoc committee of the National Cancer Research Institute Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–77. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein S, Maggioni S, Bucher J, Mueller D, Niklas J, Shevchenko V, Mauch K, Heinzle E, Noor F. In silico modeling for the prediction of dose and pathway-related adverse effects in humans from in vitro repeated-dose studies. Toxicol Sci. 2015;149:55–66. doi: 10.1093/toxsci/kfv218. [DOI] [PubMed] [Google Scholar]
- 34.Yee NS, Ignatenko N, Finnberg N, Lee N, Stairs D. Animal models of cancer biology. Cancer Growth Metastasis. 2015;8(Suppl 1):115–8. doi: 10.4137/CGM.S37907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.House CD, Hernandez L, Annunziata CM. Recent technological advances in using mouse models to study ovarian cancer. Front Oncol. 2014;4(26):26. doi: 10.3389/fonc.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med. 2017;9(2):137–53. doi: 10.15252/emmm.201606857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gengenbacher N, Singhal M, Augustin HG. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat Rev Cancer. 2017;17:751–65. doi: 10.1038/nrc.2017.92. [DOI] [PubMed] [Google Scholar]
- 38.Vitale G, Gaudenzi G, Circelli L, Manzoni M, Bassi A, Fioritti N, Faggiano A, Colao A, NIKE group Animal models of medullary thyroid cancer: state of the art and view to the future. Endocr Relat Cancer. 2017;24(1):1–12. doi: 10.1530/ERC-16-0399. [DOI] [PubMed] [Google Scholar]
- 39.Rubio-Viqueira B, Hidalgo M. Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clin Pharmacol Ther. 2009;85:217–21. doi: 10.1038/clpt.2008.200. [DOI] [PubMed] [Google Scholar]
- 40.Kariyil BJ. In vivo models in cancer research. Int J Curr Res. 2015;7:24399–404. [Google Scholar]
- 41.Yadav AK, Srikrishna S, Gupta SC. Cancer drug development using drosophila as an in vivo tool: From bedside to bench and back. Trends Pharmacol Sci. 2016;37:789–806. doi: 10.1016/j.tips.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 42.Astone M, Dankert EN, Alam SkK, Hoeppner LH. Fishing for cures: The allure of using zebrafish to develop precision oncology therapies. npj Precis Oncol. 2017;1(39):39. doi: 10.1038/s41698-017-0043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deryugina EIA, Quigley JP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. 2008;130(6):1119–30. doi: 10.1007/s00418-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kain KH, Miller JW, Jones-Paris CR, Thomason RT, Lewis JD, Bader DM, Barnett JV, Zijlstra A. The chick embryo as an expanding experimental model for cancer and cardiovascular research. Dev Dyn. 2014;243(2):216–28. doi: 10.1002/dvdy.24093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gould SE, Junttila MR, de Sauvage FJ. Translational value of mouse models in oncology drug development. Nat Med. 2015;21:431–9. doi: 10.1038/nm.3853. [DOI] [PubMed] [Google Scholar]
- 46.Katt ME, Placone AL, Wong AD, Xu ZS, Searson PC. In vitro tumor models: Advantages, disadvantages, variables, and selecting the right platform. Front Bioeng Biotechnol. 2016;4:12. doi: 10.3389/fbioe.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cavanaugh H, Haier J. Basic Tissue and Cell Culture in Cancer Research. NY: Wiley; 2005. [Google Scholar]
- 48.Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. journal of visualized experiments. JoVE. 2014;88:e51046. doi: 10.3791/51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raghavan S, Mehta P, Horst EN, Ward MR, Rowley KR, Mehta G. Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity. Oncotarget. 2016;7(13):16948–61. doi: 10.18632/oncotarget.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee H, Park W, Ryu H, Jeon NL. A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics. 2014;8(5):054102. doi: 10.1063/1.4894595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong AD, Searson PC. Mitosis-mediated intravasation in a tissue-engineered tumor-microvessel platform. Cancer Res. 2017;77(22):6453–61. doi: 10.1158/0008-5472.CAN-16-3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jijakli K, Khraiwesh B, Fu W, Luo L, Alzahmi A, Koussa J, Chaiboonchoe A, Kirmizialtin S, Yen L, Salehi-Ashtiani K. The in vitro selection world. Methods. 2016;106:3–13. doi: 10.1016/j.ymeth.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Pouliot N, Pearson HB, Burrows A. Investigating metastasis using in vitro platforms. Landes Biosci. 2013. http://www.ncbi.nlm.nih.gov/books/NBK100379/.
- 54.European Food Safety Authority Modern methodologies and tools for human hazard assessment of chemicals. EFSA J. 2014;12(3638):87. [Google Scholar]
- 55.Lovitt CJ, Shelper TB, Avery VM. Advanced cell culture techniques for cancer drug discovery. Biology. 2014;3:345–67. doi: 10.3390/biology3020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahamer H, Devaney TTJ, Tritthart HA. Fractal dimension for a cancer invasion model. Fractals Compl Geom Patt Scaling Nat Soc. 2001;9(01):61–76. [Google Scholar]
- 57.Turin I, Schiavo R, Maestri M, Luinetti O, Bello BD, Paulli M, Dionigi P, Roccio M, Spinillo A, Ferulli F, Tanzi M, Maccario R, Montagna D, Pedrazzoli P. In vitro efficient expansion of tumor cells deriving from different types of human tumor samples. Med Sci. 2014;2(2):70–81. [Google Scholar]
- 58.Mackenzie I. New in vitro assays for studying the biology of cancer stem cells. In: NC3Rs research portfolio. London: National Centre for the Replacement Refinement & Reduction of Animals in Research: 2011. p. 37–40. https: //www.nc3rs.org.uk/sites/default/files/documents/Corporate_publications/Research_Reviews/Research\%20Review\%.
- 59.Vaira V, Fedele G, Pyne S, Fasoli E, Zadra G, Bailey D, Snyder E, Faversani A, Coggi G, Flavin R, Bosari S, Loda M. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. PNAS. 2010;107(18):8352–8356. doi: 10.1073/pnas.0907676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aref A, Barbie D. Ex Vivo Engineering of the Tumor Microenvironment. Berlin: Springer; 2017. [Google Scholar]
- 61.Jeanquartier F, Jean-Quartier C, Schreck T, Cemernek D, Holzinger A. Integrating open data on cancer in support to tumor growth analysis. Inf Tech Bio Med Inf Lect Notes Comput Sci Lect Notes Comput Sci LNCS. 2016;9832(Information Technology in Bio- and Medic):49–66. [Google Scholar]
- 62.Weinstein JN, Collisson EA, Mills GB, Shaw KM, Ozenberger BA, Ellrott K, Cancer Genome Atlas Research Network The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riggins G J SRL. Genome and genetic resources from the cancer genome anatomy project. Hum Mol Genet. 2001;7:663–7. doi: 10.1093/hmg/10.7.663. [DOI] [PubMed] [Google Scholar]
- 64.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, Lahr DL, Hirschman JE, Liu Z, Donahue M, Julian B, Khan M, Wadden D, Smith IC, Lam D, Liberzon A, Toder C, Bagul M, Orzechowski M, Enache OM, Piccioni F, Johnson SA, Lyons NJ, Berger AH, Shamji AF, Brooks AN, Vrcic A, Flynn C, Rosains J, Takeda DY, Hu R, Davison D, Lamb J, Ardlie K, Hogstrom L, Greenside P, Gray NS, Clemons PA, Silver S, Wu X, Zhao W-N, Read-Button W, Wu X, Haggarty SJ, Ronco LV, Boehm JS, Schreiber SL, Doench JG, Bittker JA, Root DE, Wong B, Golub TR. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.The Humane Society of the United States and Procter & Gamble. AltTox: Toxicity testing overview. Last updated: February 19, 2016. http://alttox.org/mapp/toxicity-testing-overview/. Accessed Mar 2017.
- 66.Judson R, Houck K, Martin M, Knudsen T, Thomas RS, Sipes N, Shah I, Wambaugh J, Crofton K. In vitro and modelling approaches to risk assessment from the us environmental protection agency toxcast programme. Basic Clin Pharmacol Toxicol. 2014;115(1):69–76. doi: 10.1111/bcpt.12239. [DOI] [PubMed] [Google Scholar]
- 67.Igarashi Y, Nakatsu N, Yamashita T, Ono A, Ohno Y, Urushidani T, Yamada H. Open tg-gates: a large-scale toxicogenomics database. Nucleic Acids Res. 2015;43(Database issue):921–7. doi: 10.1093/nar/gku955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mulas F, Li A, Sherr DH, Monti S. Network-based analysis of transcriptional profiles from chemical perturbations experiments. BMC Bioinformatics. 2017;18(Suppl 5):130. doi: 10.1186/s12859-017-1536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hofree M, Carter H, Kreisberg JF, Bandyopadhyay S, Mischel PS, Friend S, Ideker T. Challenges in identifying cancer genes by analysis of exome sequencing data. Nat Commun. 2016;7:12096. doi: 10.1038/ncomms12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Handl J, Knowles J, Kell DB. Computational cluster validation in post-genomic data analysis. Bioinformatics. 2005;21(15):3201–12. doi: 10.1093/bioinformatics/bti517. [DOI] [PubMed] [Google Scholar]
- 71.Kaufman L, Rousseeuw PJ. Finding Groups in Data: an Introduction to Cluster Analysis, vol 344. NY: Wiley; 2009. [Google Scholar]
- 72.Geman D, Ochs M, Price ND, Tomasetti C, Younes L. An argument for mechanism-based statistical inference in cancer. Hum Genet. 2015;134(5):479–95. doi: 10.1007/s00439-014-1501-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16(8):483–93. doi: 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- 74.Piraino SW, Furney SJ. Beyond the exome: the role of non-coding somatic mutations in cancer. Ann Oncol. 2016;27(2):240–8. doi: 10.1093/annonc/mdv561. [DOI] [PubMed] [Google Scholar]
- 75.Cuykendall TN, Rubin MA, Khurana E. Non-coding genetic variation in cancer. Curr Opin Syst Biol. 2017;1(Supplement C):9–15. doi: 10.1016/j.coisb.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morrow JJ, Scacheri PC. Epigenetics, enhancers, and cancer. In: Berger NA, editor. Energy Balance and Cancer. Switzerland: Springer; 2016. [Google Scholar]
- 77.Medvedeva YA, Lennartsson A, Ehsani R, Kulakovskiy IV, Vorontsov IE, Panahandeh P, Khimulya G, Kasukawa T, FANTOM Consortium. DrablÃs F. Epifactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015;2015:bav067. doi: 10.1093/database/bav067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett. 2015;356(2):209–314. doi: 10.1016/j.canlet.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, Lin CY, Royo R, Sanchez-Mut JV, Martinez R, Gut M, Torrents D, Orozco M, Gut I, Young RA, Esteller M. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17:11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ryan BM, Robles AI, Harris CC. Genetic variation in microrna networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim Y-A, Cho D-Y, Przytycka TM. Understanding genotype-phenotype effects in cancer via network approaches. PLoS Comput Biol. 2016;12(3):e1004747. doi: 10.1371/journal.pcbi.1004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Porta-Pardo E, Garcia-Alonso L, Hrabe T, Dopazo J, Godzik A. A pan-cancer catalogue of cancer driver protein interaction interfaces. PLOS. 2015;11:e1004518. doi: 10.1371/journal.pcbi.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maximova T, Mofatt R, Ma B, Nussinov R, Shehu A. Principles and overview of sampling methods for modeling macromolecular structure and dynamics. PLOS Comput Biol. 2016;12:e1004619. doi: 10.1371/journal.pcbi.1004619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smolle J, Hofmann-Wellenhof R, Kofler R, Cerroni L, Haas J, Kerl H. Computer simulations of histologic patterns in melanoma using a cellular automaton provide correlations with prognosis. J Investig Dermatol. 1995;105:797–801. doi: 10.1111/1523-1747.ep12326559. [DOI] [PubMed] [Google Scholar]
- 85.Patil SA, Kuchanur MB. Lung cancer classification using image processing. Int J Eng Innov Technol. 2012;2(30):37–42. [Google Scholar]
- 86.Vargas A, Angeli M, Pastrello C, McQuaid R, Li H, Jurisicova A, Jurisica I. Robust quantitative scratch assay. Bioinformatics. 2016;2(9):1439–40. doi: 10.1093/bioinformatics/btv746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu W, Wang J, Zhang HH. Computerized PET/CT image analysis in the evaluation of tumour response to therapy. Br J Radiol. 2015;88(1048):20140625–20140625. doi: 10.1259/bjr.20140625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tiwari S, Bhargava R. Extracting knowledge from chemical imaging data using computational algorithms for digital cancer diagnosis. Yale J Biol Med. 2015;88:131–43. [PMC free article] [PubMed] [Google Scholar]
- 89.Christopher R, Dhiman A, Fox J, Gendelman R, Haberitcher T, Kagle D, Spizz G, Khalil I, Hill C. Data-driven computer simulation of human cancer cell. Ann N Y Acad Sci. 2004;1020:132–53. doi: 10.1196/annals.1310.014. [DOI] [PubMed] [Google Scholar]
- 90.Nilsson A, Nielsen J. Genome scale metabolic modeling of cancer. Metab Eng. 2016. https://doi.org/10.1016/j.ymben.2016.10.022. [DOI] [PubMed]
- 91.Beerenwinkel N, Greenman CD, Lagergren J. Computational cancer biology: An evolutionary perspective. PLoS Comput Biol. 2016;12(2):1004717. doi: 10.1371/journal.pcbi.1004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benzekry S, Lamont C, Beheshti A, Tracz A, Ebos JM, Hlatky L, Hahnfeldt P. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol. 2014;10(8):1003800. doi: 10.1371/journal.pcbi.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rejniak KA, Anderson AR. Hybrid models of tumor growth. Wiley Interdiscip Rev Syst Biol Med. 2011;3(1):115–25. doi: 10.1002/wsbm.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kohonen P, Ceder R, Smit I, Hongisto V, Myatt G, Hardy B, Spjuth O, Grafström R. Cancer biology, toxicology and alternative methods development go hand-in-hand. Basic Clin Pharmacol Toxicol. 2014;115:50–8. doi: 10.1111/bcpt.12257. [DOI] [PubMed] [Google Scholar]
- 95.Langley GR, Adcock IM, Busquet F, Crofton KM, Csernok E, Giese C, Heinonen T, Herrmann K, Hofmann-Apitius M, Landesmann B, et al. Towards a 21st-century roadmap for biomedical research and drug discovery: consensus report and recommendations. Drug Discov Today. 2017;22(2):327–39. doi: 10.1016/j.drudis.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 96.Organisation de Coopération et de Développement Économiques. Series on testing and assessment: Guidance document on developing and assessing adverse outcome pathways. Technical Report 184, Environment, OECD, Health and Safety Publications. 2017.
- 97.Perkins EJ, Antczak P, Burgoon L, Falciani F, Garcia-Reyero N, Gutsell S, Hodges G, Kienzler A, Knapen D, McBride M, Willett C. Adverse outcome pathways for regulatory applications: Examination of four case studies with different degrees of completeness and scientific confidence. Toxicol Sci. 2015;148:14–25. doi: 10.1093/toxsci/kfv181. [DOI] [PubMed] [Google Scholar]
- 98.Boobis AR, Cohen SM, Dellarco V, McGregor D, Vickers MEBMC, Willcocks D, Farland W. Ipcs framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol. 2006;36:781–92. doi: 10.1080/10408440600977677. [DOI] [PubMed] [Google Scholar]
- 99.Kotlyar M, Fortney K, Jurisica I. Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods. 2012;57(4):499–507. doi: 10.1016/j.ymeth.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 100.Fjodorova N, Vračko M, Novič M, Roncaglioni A, Benfenati E. New public QSAR model for carcinogenicity. Chem Cent J. 2010;4(supplement 1):S3. doi: 10.1186/1752-153X-4-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Malik A, Singh H, Andrabi M, Husain SA, Ahmad S. Databases and qsar for cancer research. Cancer Informat. 2006;2:99–111. [PMC free article] [PubMed] [Google Scholar]
- 102.Menden M, Iorio F, Garnett M, McDermott U, Benes C, Ballester PJ, Saez-Rodriguez J. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE. 2013;8(4):61318. doi: 10.1371/journal.pone.0061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ghersi D, Singh M. Interaction-based discovery of functionally important genes in cancers. Nucl Acids Res. 2014;42(3):e18. doi: 10.1093/nar/gkt1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber D, Stratton M, Benes C, McDermott U, Garnett M. Genomics of drug sensitivity in cancer (gdsc): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):955–61. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Villaverde AF, Henriques D, Smallbone K, Bongard S, Schmid J, Cicin-Sain D, Crombach A, Saez-Rodriguez J, Mauch K, Balsa-Canto E, Mendes P, Jaeger J, Banga JR. Biopredyn-bench: a suite of benchmark problems for dynamic modelling in systems biology. BMC Syst Biol. 2015;9:8. doi: 10.1186/s12918-015-0144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hochheiser H, Castine M, Harris D, Savova G, Jacobson RS. An information model for computable cancer phenotypes. BMC Med Inform Decis Mak. 2016;16(1):121. doi: 10.1186/s12911-016-0358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jeanquartier F, Jean-Quartier C, Kotlyar M, Tokar T, Hauschild A-C, Jurisica I, Holzinger A. Machine Learning for Health Informatics. Cham: Springer; 2016. Machine learning for in silico modeling of tumor growth. [Google Scholar]
- 108.Gonçalves E, Bucher J, Ryll A, Niklas J, Mauch K, Klamt S, Rocha M, Saez-Rodriguez J. Bridging the layers: Towards integration of signal transduction, regulation and metabolism into mathematical models. Mol BioSyst. 2013;9:1576–83. doi: 10.1039/c3mb25489e. [DOI] [PubMed] [Google Scholar]
- 109.Carels N, Spinassé LB, Tilli TM, Tuszynski JA. Toward precision medicine of breast cancer. Theor Biol Med Model. 2016;13:7. doi: 10.1186/s12976-016-0035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mirnezami R, Nicholson J, Darzi A. Preparing for precision medicine. N Engl J Med. 2012;366:489–91. doi: 10.1056/NEJMp1114866. [DOI] [PubMed] [Google Scholar]
- 111.Uhlén M, Björling E, Agaton C, Szigyarto C, Amini B, Andersen E, Andersson A, Angelidou P, Asplund A, Asplund C, Berglund L, Bergström K, Brumer H, Cerjan D, Ekström M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Björklund M, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, I O, E O, J O, L P, A P, Rimini R, Rockberg J, Runeson M, Sivertsson A, Sköllermo A, J S, M S, Sterky F, Strömberg S, Sundberg M, Tegel H, Tourle S, Wahlund E, Waldén A, Wan J, Wernérus H, Westberg J, Wester K, U W, Xu L, Hober S, Pont’´en F. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4(12):1920–32. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 112.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist P-H, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Sci (New York, NY). 017;357(6352). https://doi.org/10.1126/science.aan2507. [DOI] [PubMed]
- 113.Locasale JW, Melman T, Song S, Yang X, Swanson KD, Cantley LC, Wong ET, Asara JM. Metabolomics of human cerebrospinal fluid identifies signatures of malignant glioma. Mol Cell Proteomics. 2012;11(6):M111.014688. doi: 10.1074/mcp.M111.014688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McDunn JE, Li Z, Adam K-P, Neri BP, Wolfert RL, Milburn MV, Lotan Y, Wheeler TM. Metabolomic signatures of aggressive prostate cancer. Prostate. 2013;3(73):1547–60. doi: 10.1002/pros.22704. [DOI] [PubMed] [Google Scholar]
- 115.Farshidfar F, Weljie AM, Kopciuk KA, Hilsden R, McGregor SE, Buie WD, MacLean A, Vogel HJ, Bathe OF. A validated metabolomic signature for colorectal cancer: exploration of the clinical value of metabolomics. Br J Cancer. 2016;115:848–57. doi: 10.1038/bjc.2016.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Belizário JE, Sangiuliano BA, Perez-Sosa M, Neyra JM, Moreira DF. Using pharmacogenomic databases for discovering patient-target genes and small molecule candidates to cancer therapy. Front Pharmacol. 2016;7:312. doi: 10.3389/fphar.2016.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fortney K, Griesman J, Kotlyar M, Pastrello C, Angeli M, Sound-Tsao M, Jurisica I. Prioritizing therapeutics for lung cancer: An integrative meta-analysis of cancer gene signatures and chemogenomic data. PLoS Comput Biol. 2015;11(3):e1004068. doi: 10.1371/journal.pcbi.1004068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shim JS LJ. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10(7):654–63. doi: 10.7150/ijbs.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiao M, Liu G, Xue Y, Ding C. Computational drug repositioning for cancer therapeutics. Curr Top Med Chem. 2015;15(8):767–75. doi: 10.2174/1568026615666150302105831. [DOI] [PubMed] [Google Scholar]
- 120.Li J, Zheng S, Chen B, Butte A, Swamidass S, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform. 2016;17(1):2–12. doi: 10.1093/bib/bbv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang S. Computer-aided drug discovery and development. Methods Mol Biol. 2011;716:23–38. doi: 10.1007/978-1-61779-012-6_2. [DOI] [PubMed] [Google Scholar]
- 122.Guedes RA, Serra P, Salvador JA, Guedes RC. Computational approaches for the discovery of human proteasome inhibitors: An overview. Molecules. 2016;21(7):927. doi: 10.3390/molecules21070927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamazaki S, Spilker M, Vicini P. Translational modeling and simulation approaches for molecularly targeted small molecule anticancer agents from bench to bedside. Expert Opin Drug Metab Toxicol. 2016;12(3):253–6. doi: 10.1517/17425255.2016.1141895. [DOI] [PubMed] [Google Scholar]
- 124.Li X, Oduola W, Qian L, Dougherty E. Integrating multiscale modeling with drug effects for cancer treatment. Cancer Informat. 2016;14(Suppl 5):21–31. doi: 10.4137/CIN.S30797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buil-Bruna N, López-Picazo J, Martín-Algarra S, Trocóniz I. Bringing model-based prediction to oncology clinical practice: A review of pharmacometrics principles and applications. Oncologist. 2016;21(2):220–32. doi: 10.1634/theoncologist.2015-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tang J, Aittokallio T. Network pharmacology strategies toward multi-target anticancer therapies: from computational models to experimental design principles. Curr Pharm Des. 2014;20(1):23–36. doi: 10.2174/13816128113199990470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nussinov R, Jang H, Tsai C-J. The structural basis for cancer treatment decisions. Oncotarget. 2014;5(17):7285–302. doi: 10.18632/oncotarget.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cbio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disc. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.The Gene Ontology Consortium Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):331–8. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Res. 2017. https://doi.org/10.1093/nar/gkx1132.
- 131.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. Kegg: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):353–61. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Merid SK, Goranskaya D, Alexeyenko A. Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinformatics. 2014;15:308. doi: 10.1186/1471-2105-15-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dimitrakopoulos CM, Beerenwinkel N. Computational approaches for the identification of cancer genes and pathways. Wiley Interdiscip Rev Syst Biol Med. 2016;9(1):e1364. doi: 10.1002/wsbm.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, Lawrence MS, Gonzalez-Perez A, Tamborero D, Cheng Y, Ryslik GA, Lopez-Bigas N, Getz G, Ding L, Raphael BJ. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47(2):106–14. doi: 10.1038/ng.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rahmati S, Abovsky M, Pastrello C, Jurisica I. pathDIP: an annotated resource for known and predicted human gene-pathway associations and pathway enrichment analysis. Nucleic Acids Res. 2017;45(D1):D419–D426. doi: 10.1093/nar/gkw1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iorio F, Knijnenburg T, Vis D, Bignell G, Menden M, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan R, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray N, Haber D, Stratton M, Benes C, Wessels L, Saez-Rodriguez J, McDermott U, MJ G. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–54. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schubert M, Klinger B, Klünemann M, Garnett M, Blüthgen N, Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. bioRxiv. 2016;:065672. http://biorxiv.org/content/early/2016/08/28/065672.full.pdf. [DOI] [PMC free article] [PubMed]
- 138.Holzinger I, Jurisica A. Knowledge Discovery and Data Mining in Biomedical Informatics: The Future Is in Integrative, Interactive Machine Learning Solutions Vol. 8401. Heidelberg: Springer; 2014. [Google Scholar]
- 139.Aran D, Sirota M, Butte A. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;4(6):8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nass SJ, Wizemann T, et al. Informatics Needs and Challenges in Cancer Research: Workshop Summary. Washington (DC): National Academies Press (US); 2012. [PubMed] [Google Scholar]
- 141.Kitano H. Computational systems biology. Nature. 2002;420(6912):206–10. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 142.Fisher J, Henzinger TA. Executable cell biology. Nat Biotechnol. 2007;25(11):1239–49. doi: 10.1038/nbt1356. [DOI] [PubMed] [Google Scholar]
- 143.Trisilowati & Mallet DG. In silico experimental modeling of cancer treatment. ISRN Oncol. 2012;2012:828701. doi: 10.5402/2012/828701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jeanquartier F, Jean-Quartier C, Cemernek D, Holzinger A. In silico modeling for tumor growth visualization. BMC Syst Biol. 2016;10:59. doi: 10.1186/s12918-016-0318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y. Cellminer: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the nci-60 cell line set. Cancer Res. 2012;72(14):3499–511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data-sets used and/or analysed during the current study are available from the corresponding author on reasonable request and data sources are referenced within figure legend and document text. Images were created by FJ and CJ under the terms of the Creative Commons license, stated in the Open Access subsection of Declarations. Figure 3 makes use of free to reuse pictures from pixabay and wikimedia licensed under Creative Commons and being free of known restrictions under copyright law, including all related and neighboring rights.