

Abstract
Background
The ginseng (Panax ginseng C.A. Meyer) is a perennial herbaceous plant that has been used in traditional oriental medicine for thousands of years. Ginsenosides, which have significant pharmacological effects on human health, are the foremost bioactive constituents in this plant. Having realized the importance of this plant to humans, an integrated omics resource becomes indispensable to facilitate genomic research, molecular breeding and pharmacological study of this herb.
Description
The first draft genome sequences of P. ginseng cultivar “Chunpoong” were reported recently. Here, using the draft genome, transcriptome, and functional annotation datasets of P. ginseng, we have constructed the Ginseng Genome Database http://ginsengdb.snu.ac.kr/, the first open-access platform to provide comprehensive genomic resources of P. ginseng. The current version of this database provides the most up-to-date draft genome sequence (of approximately 3000 Mbp of scaffold sequences) along with the structural and functional annotations for 59,352 genes and digital expression of genes based on transcriptome data from different tissues, growth stages and treatments. In addition, tools for visualization and the genomic data from various analyses are provided. All data in the database were manually curated and integrated within a user-friendly query page.
Conclusion
This database provides valuable resources for a range of research fields related to P. ginseng and other species belonging to the Apiales order as well as for plant research communities in general. Ginseng genome database can be accessed at http://ginsengdb.snu.ac.kr/.
Keywords: Panax ginseng, Genome database, Ginseng annotation, Ginseng genome browser
Background
Ginseng (Panax ginseng C.A. Meyer) is a perennial herb of the Panax genus in Araliaceae family and has widely been used as a traditional medicine in Eastern Asia and North America. The principle bioactive components in ginseng are ginsenosides (collectively a group of triterpene saponins), which are biosynthesized through the isoprenoid pathway [1]. Ginseng has various therapeutic effects on humans including for treatment of cancer, diabetes, cardiovascular and stress [2–6]. P. ginseng is known to be tetraploid (2n = 4× = 48), with an estimated genome size of approximately 3.6 Gbp [7, 8]. Its large, highly repetitive genome, which has experienced whole-genome duplication, has impeded the progress of whole-genome sequencing of P. ginseng [7]. In addition, the long generation time (4 years) and difficulty of maintenance in ginseng cultivation fields have limited the genetic study of P. ginseng. Nevertheless, with the advent of new sequencing technologies, expressed sequence tags (ESTs) and RNA-Seq data have been generated from various tissues and growth stages of P. ginseng [9–12], based on which a number of genes involved in ginsenoside biosynthesis pathway have been characterized [10, 11]. Recently, the complete chloroplast genome sequences of P. ginseng cultivars and related species were characterized [13, 14]. Furthermore, inter- and intra-species chloroplast genome diversity were also identified for authentication of ginseng cultivars and species [13–17].
At the outset of this project, a total of 17,773 ESTs from NCBI db-EST (as of January, 2017) and a database for adventitious root [9] were publicly available for ginseng. However, these data were insufficient to facilitate the functional and comparative genomics and molecular breeding of ginseng. There was no comprehensive database publicly available for ginseng despite its importance as a medicinal crop with high pharmacological value. Given the fact that ginseng shows numerous effects on human health, a genomic and transcriptomic database is vital for ginseng research communities and other close relatives in the Apiales order. It is also anticipated that an integrated database of genetic, genomic, and metabolomic resources of ginseng would serve as a valuable resource for translational genomics. Recently, we generated extensive genomic and transcriptomic data for P. ginseng cultivar “Chunpoong” [18].
In this study, we built a dynamic database that integrates a draft genome sequence, transcriptome profiles, and annotation datasets of ginseng. This Ginseng Genome Database is now publicly available (http://ginsengdb.snu.ac.kr/) for the use of scientific community around the globe for exploring the vast possibilities.
This user-friendly database will serve as a hub for mining gene sequences and their digital expression data of samples from various tissues, developmental stages, and treatments. Our database interface will facilitate the easy retrieval of gene families and associated functional annotations using InterPro, KEGG, BLAST and Gene Ontology (GO) databases. To expedite metabolomics in ginseng, we have made a separate section that categorizes the genes associated with various metabolic pathways including the ginsenoside biosynthesis pathway. In addition, we have included robust tools such as BLAST and genome browser (JBrowse) [19] for survey and visualization of ginseng genomic features. This database will be updated regularly with new genome sequences and information on annotation and will provide reference genomic information for research in P. ginseng as well as related species.
Construction and content
Whole-genome sequencing and assembly and gene models
The genome sequence data of P. ginseng were generated from an elite cultivar ‘Chunpoong’ using Illumina HiSeq platforms. A total of 746 Gb paired-end and 365 Gb mate-paired raw data were produced and assembled, yielding the draft genome sequence of about ~ 3.0 Gb in size. The repeat sequences were identified and masked using RepeatModeler [20] and RepeatMasker [21]. An automatic gene prediction was performed using evidence modeler (EVM) [22] with ab initio predictions (BRAKER 1 [23]), protein evidence, ESTs and RNA-Seq evidence [24]. After the removal of the transposon sequences, a total 59,352 putative protein coding genes were predicted. These genes were functionally annotated using InterPro [25], Blast2Go [26], KEGG [27] and BLASTP searches with known protein databases.
Transcriptome data
The transcriptome data were generated from various tissues and abiotic stress-treated samples of ginseng using Illumina HiSeq and PacBio platforms (http://ginsengdb.snu.ac.kr/transcriptome.php). Raw RNA-Seq reads of about 120 Gb were pre-processed in four steps to obtain high quality RNA reads. Initially, the bacterial contaminant reads were removed by read mapping against the available bacterial genomes using BWA [28]. After pre-processing, the duplicated reads were filtered out using FastUniq [29]. The third step is the removal of the ribosomal RNA (rRNA) reads using SortMeRNA [30]. Finally, the low-quality reads were removed using NGS QC Toolkit [31]. The high-quality RNA-Seq reads were used for de novo assembly by Trinity [32] and reference-guided assembly by HISAT & stringtie [33] and then for gene prediction on the draft genome sequence. In addition, high quality PacBio sequences were used to refine the predicted gene models.
Gene families and metabolic pathways
Genes were grouped based on protein domain (Pfam) and InterPro domain. Metabolic pathways were predicted with the KAAS server [27] using the reference information on gene annotation of Arabidopsis thaliana, Citrus sinensis, Glycine max, Vitis vinifera and Solanum lycopersicum. This information can be accessed at http://ginsengdb.snu.ac.kr/gene_family.php and http://ginsengdb.snu.ac.kr/metabolic_pathway.php.
Genome-scale metabolic network
Based on gene annotations, a compartmentalized genome-scale metabolic network was reconstructed providing the global overview of all metabolites, enzymes, reactions and pathways in ginseng. This network accounts for a total of 4946 genes, mapped to 2194 enzyme-catalyzed and protein-mediated transport reactions involving 2003 unique metabolites across six intracellular compartments. The global overview of ginseng genome-scale metabolic network can be accessed at http://ginsengdb.snu.ac.kr/network/index.html. This network can also be downloaded as a systems biology markup language (SBML) file.
Transcription factors
Transcription factors (TFs) are the key regulators for development and stimulus responses. TFs were identified based on the criteria of PlnTFDB [34] using iTAK (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi). A total of 4439 TF and transcription regulator genes were identified and classified into 94 TF families (http://ginsengdb.snu.ac.kr/tf_class.php).
Genes in the ginsenoside biosynthesis pathway
Ginsenosides are biosynthesized through the mevalonate (MVA) and 2-C-methyl-D-erythritol-4-phosphate (MEP) pathways [10, 35]. The number of genes that are involved in the biosynthesis of ginsenoside was identified based on KEGG as well as BLASTP annotations. UDP glycosyltransferase (UGT) genes, which are responsible for production of various types of ginsenosides in the final step of this pathway, were also identified based on InterPro ProSitePatterns (PS00375) and BLAST homology searches as well. The putative pathway and the related genes can be accessed at http://ginsengdb.snu.ac.kr/pathway.php.
Digital gene expression profiles
Digital gene expression profiles were determined using all of the RNA-Seq data. The FPKM values for all genes in each sample were calculated using RSEM [36]. Further, the expression data were normalized using Trimmed Mean of M values (TMM) to resolve the differences in the sequencing depth. The digital expression profiles can be accessed at http://ginsengdb.snu.ac.kr/gene_exp.php.
Utility and discussion
Database implementation
Ginseng Genome Database was established in the Linux (CentOS 6.6) operating system with an Apache HTTP server. PHP, HTML, JavaScript and Python scripts were used to build the user-friendly interface and design web pages. To visualize the genome, we included JBrowse version 1.11.6, which is JavaScript-based genome browser allowing visual analysis of the genome annotation [19]. We also included a BLAST server to perform homology searches with different data sets of ginseng. Moreover, we developed a Python-based tool to retrieve or download specific scaffolds and gene sequences. An overview of the ginseng genome database architecture is shown in Fig. 1.
Fig. 1.
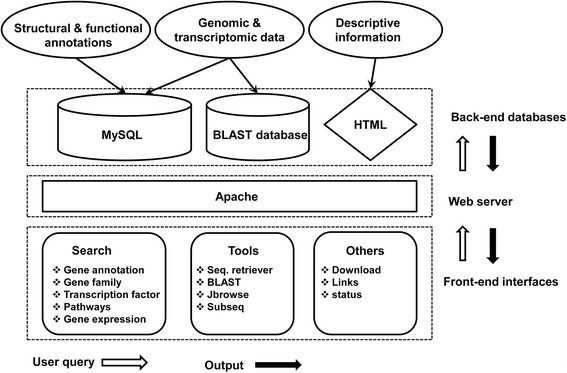
Overview of the architecture of the Ginseng Genome Database
Query search
Ginseng Genome Database provides two major panels, namely, a ‘Search’ panel and a ‘Tool’ panel, both of which comprise of all the information in an easy-to–use mode. Under the ‘Search’ panel, genes or gene families can be searched by gene ID, InterPro domain, Pfam domain, GO and KEGG orthology (KO) identifier (ID) and keywords (Fig. 2). Furthermore, users can browse gene families categorized using ‘InterPro’ and ‘Pfam’ domains. The ‘Gene family’ option provides a sub-menu to retrieve the group of genes related to user-defined functional domains or keywords. Users can download all coding sequences (CDSs) in a specific gene family or user-selected CDSs in FASTA format from the output page. The ‘Gene annotation’ section provides the detailed annotations including both structural and functional annotations of the user-queried genes (Fig. 3). In the output page, users can find the scaffold in which the specified genes, CDS and proteins were annotated and then can visualize those through JBrowse. Further, functional descriptions based on InterPro annotation including Pfam, Prositepatterns, and Superfamily, GO, KEGG and BLAST can also be browsed.
Fig. 2.
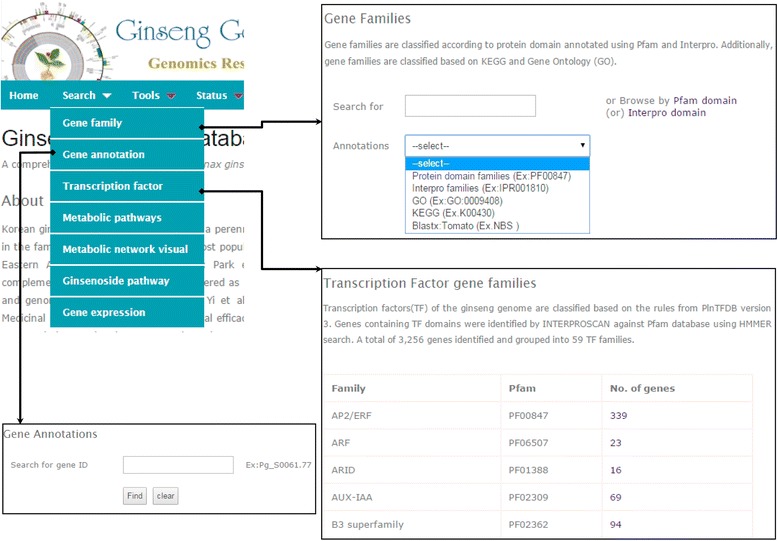
Query interface to retrieve information on gene annotations and transcription factors
Fig. 3.
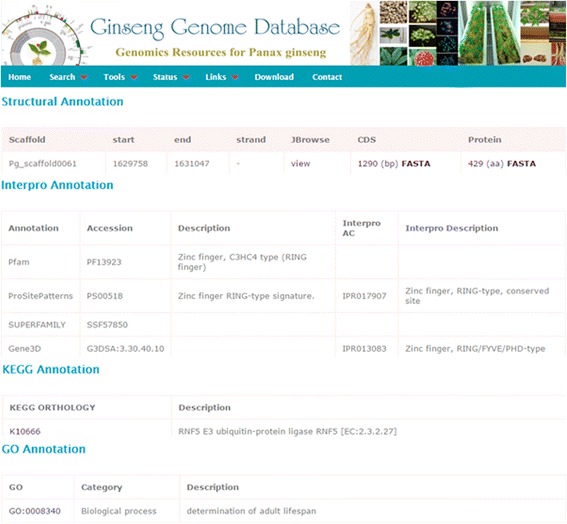
A detailed snapshot of the structural and functional annotations of a queried gene
A list of annotated TF families is included in the ‘Transcription factors’ section (Fig. 2). Users can explore the TF genes related to specific TF families and download the corresponding CDSs. Under the ‘Metabolic pathways’ section, the users can simply enter a pathway name or click browse pathways to retrieve the genes involved in a particular pathway. Our database also provides links, so that users can check the ‘enzyme commission (EC) number’ for the corresponding genes and the complete pathway from the KEGG database. The known pathway of biosynthesis of ginsenosides and genes corresponding to each enzyme are listed under the ‘Ginsenoside pathway’ section. In the ‘Gene expression’ section, users can input a specific gene identifier and can choose to compare expressions between ginseng plant tissues or between abiotic stresses. This will return the expression data in bar-chart form using different colors.
Sequence retriever
We incorporated a ‘sequence retrieving tool’ using Python script. This can be utilized by entering single or batch gene (CDSs and peptides) or scaffold IDs in the input query box. We customized the output options with ‘view’ and ‘download’. Users can view single or multiple sequences in FASTA format on the web page by choosing the ‘view’ option. In case of many sequences, the user may select the ‘download’ option to download the sequences in FASTA format.
BLAST
This database also offers a homology search tool, ‘BLAST’, which was embedded in the database using the ncbi-wwwblast package (v2.2.26) to provide a graphic interface for the users. A BLAST-able databases of the whole draft genome sequence, coding sequences (CDSs), and protein sequences were made for BLAST searches. Additionally, the transcriptome data from various tissues and abiotic stress treatments of ginseng generated for the whole-genome study, the RNA-Seq assembly that were previously published and the ESTs were provided for BLAST searches. The users can perform BLAST searches by directly pasting the query sequences in the ‘query text box’, by choosing the appropriate search program (BLASTN, BLASTP, BLASTX, TBLASTN or TBLASTX), where BLASTP and TBLASTN are queried only against amino acid sequences. Options to filter low complexity and to set the E-value are available under the ‘Other options’ section. The result format can also be customized using the options under ‘Result options’.
JBrowse
Under the ‘Tools’ panel, ‘JBrowse’ was included to visualize the genomic features of ginseng. All of the assembled scaffolds and the predicted genes were used in constructing the genome browser. The main page of ‘JBrowse’ contains several tracks under different sub-sections. Users can choose the ‘scaffold’ (only 30 scaffolds can be seen in the drop-down menu) or type the name of the scaffold with or without a location in the search box. Users can visualize various genomic features such as ‘gene models’, ‘ab initio gene models’ generated for the gene annotation pipeline, ‘assembled transcriptome structure’ and ‘repeats’. In addition, the alignments of RNA-Seq reads to the genome sequence generated directly from Binary Alignment/MAP (BAM) and PacBio contig alignment using GMAP [37] were also incorporated to perk up the structural annotation of the gene. Furthermore, protein sequences of non-coding genes including microRNA (miRNA) and long non-coding RNAs (lncRNA) can be seen along with their gene features. Apart from Panax ginseng, we have incorporated the genome-guided transcriptome assembly of other Panax species, namely, P. notoginseng and P. quinquefolius which would aid in comparing the gene structure or find missing genes any other Panax species.
Downloads
All the assembled genomic and transcriptomic sequences are available at http://ginsengdb.snu.ac.kr/data.php. Our database provides HTTP links to download the draft genome sequences (v1) and putative CDSs and protein sequences (v1.1) in FASTA format. The gene and repeat structure annotations are available in Generic File Format (GFF3). The list of data files including de novo and reference-guided transcriptome assembly generated for whole genome study as well as the previously published transcriptome sequences generated from our research are also accessible in FASTA format. Besides, the filtered RNA-Seq data used for genome analysis and the genome-scale metabolic network of ginseng can also be downloaded as a SBML file.
Conclusions
Ginseng Genome Database, the original, all-inclusive database for ginseng, is built on the most recent information of its draft genome sequence and accurate annotations. It serves as an open-access interface to retrieve genomic information from genome to gene level and to visualize all diverse components of the genome. The Ginseng Genome Database will form a valuable resource enhancing various research fields like functional/comparative genomics, metabolomics, molecular breeding, and evolutionary analysis of ginseng.
Acknowledgments
Funding
This study was supported by a grant from the Next-Generation BioGreen21 Program (PJ01311901, PJ01334605), Rural Development Administration, Republic of Korea and Synthetic Biology Initiative of the National University of Singapore (DPRT/943/09/14).
Availability of data and materials
Datasets in ginseng genome database are freely accessible for research purposes for non-profit and academic organizations at http://ginsengdb.snu.ac.kr. Further, all sequence data were deposited to the National Agricultural Biotechnology Information Center (NG-0858-000001~NG-0858-009845) (http://nabic.rda.go.kr). The database is optimized for Internet Explorer, Mozilla Firefox, Google Chrome and Safari.
Abbreviations
- bp
Base pair
- CDS
Coding sequence
- EC
Enzyme commission
- EST
Expressed sequence tag
- EVM
Evidence modeler
- FPKM
Fragments per kilobase of exon per million fragments
- GFF
General feature format
- kb
Kilobase pair
- lncRNA
Long noncoding RNA
- NGS
Next generation sequencing
- TF
Transcription factor
- TMM
Trimmed mean of M values
Authors’ contributions
JM and TJY developed the methodology and conducted the study. JM, BSC, SCL, NK, WJ and ML participated in the database and bioinformatics tool development. JM, DYL, JYP, SVM and TJY drafted the manuscript, which was revised by all authors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Murukarthick Jayakodi, Email: murukarthick@snu.ac.kr.
Beom-Soon Choi, Email: bschoi@phyzen.com.
Sang-Choon Lee, Email: sclee0923@hanmail.net.
Nam-Hoon Kim, Email: namhoonkim@snu.ac.kr.
Jee Young Park, Email: jypark74@snu.ac.kr.
Woojong Jang, Email: woojong424@gmail.com.
Meiyappan Lakshmanan, Email: Meiyappan_Lakshmanan@bti.a-star.edu.sg.
Shobhana V. G. Mohan, Email: vgshobhana@gmail.com
Dong-Yup Lee, Email: dongyuplee@skku.edu.
Tae-Jin Yang, Phone: +82-2-880-4547, Email: tjyang@snu.ac.kr.
References
- 1.Wang J, Gao WY, Zhang J, Zuo BM, Zhang LM, Huang LQ. Advances in study of ginsenoside biosynthesis pathway in Panax ginseng C. A. Meyer. Acta Physiol Plant. 2012;34(2):397–403. doi: 10.1007/s11738-011-0844-3. [DOI] [Google Scholar]
- 2.Saito H, Yoshida Y, Takagi K. Effect of Panax Ginseng root on exhaustive exercise in mice. Jpn J Pharmacol. 1974;24(1):119–127. doi: 10.1254/jjp.24.119. [DOI] [PubMed] [Google Scholar]
- 3.Peng D, Wang H, Qu C, Xie L, Wicks SM, Xie J. Ginsenoside Re: its chemistry, metabolism and pharmacokinetics. Chin Med. 2012;7:2. doi: 10.1186/1749-8546-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol. 1999;58(11):1685–1693. doi: 10.1016/S0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- 5.Shang W, Yang Y, Zhou L, Jiang B, Jin H, Chen M. Ginsenoside Rb1 stimulates glucose uptake through insulin-like signaling pathway in 3T3-L1 adipocytes. J Endocrinol. 2008;198(3):561–569. doi: 10.1677/JOE-08-0104. [DOI] [PubMed] [Google Scholar]
- 6.Radad K, Gille G, Liu L, Rausch WD. Use of ginseng in medicine with emphasis on neurodegenerative disorders. J Pharmacol Sci. 2006;100(3):175–186. doi: 10.1254/jphs.CRJ05010X. [DOI] [PubMed] [Google Scholar]
- 7.Choi HI, Waminal NE, Park HM, Kim NH, Choi BS, Park M, Choi D, Lim YP, Kwon SJ, Park BS, et al. Major repeat components covering one-third of the ginseng (Panax ginseng C.A. Meyer) genome and evidence for allotetraploidy. Plant J. 2014;77(6):906–916. doi: 10.1111/tpj.12441. [DOI] [PubMed] [Google Scholar]
- 8.Waminal NE, Park HM, Ryu KB, Kim JH, Yang TJ, Kim HH. Karyotype analysis of Panax ginseng C.A.Meyer, 1843 (Araliaceae) based on rDNA loci and DAPI band distribution. Comp Cytogenet. 2012;15:425–441. doi: 10.3897/CompCytogen.v6i4.3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayakodi M, Lee SC, Park HS, Jang W, Lee YS, Choi BS, Nah GJ, Kim DS, Natesan S, Sun C, et al. Transcriptome profiling and comparative analysis of Panax ginseng adventitious roots. J Ginseng Res. 2014;38(4):278–288. doi: 10.1016/j.jgr.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayakodi M, Lee SC, Lee YS, Park HS, Kim NH, Jang W, Lee HO, Joh HJ, Yang TJ. Comprehensive analysis of Panax ginseng root transcriptomes. BMC Plant Biol. 2015;15(1):138. doi: 10.1186/s12870-015-0527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y, Park HS, Lee DK, Jayakodi M, Kim NH, Koo HJ, Lee SC, Kim YJ, Kwon SW, Yang TJ. Integrated transcriptomic and metabolomic analysis of five Panax ginseng cultivars reveals the dynamics of ginsenoside biosynthesis. Front Plant Sci. 2017;8:1048. doi: 10.3389/fpls.2017.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Zhu Y, Guo X, Sun C, Luo H, Song J, Li Y, Wang L, Qian J, Chen S. Transcriptome analysis reveals ginsenosides biosynthetic genes, microRNAs and simple sequence repeats in Panax ginseng C. A. Meyer. BMC Genomics. 2013;14:245. doi: 10.1186/1471-2164-14-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim K, Lee SC, Lee J, Lee HO, Joh HJ, Kim NH, Park HS, Yang TJ. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS One. 2015;10:e0117159. [DOI] [PMC free article] [PubMed]
- 14.Kim K, Lee SC, Lee J, Yu Y, Yang K, Choi BS, Koh HJ, Waminal NE, Choi HI, Kim NH. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci Rep. 2015;5:15655. doi: 10.1038/srep15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Nguyen VB, Dong JZ, Wang Y, Park JY, Lee SC, Yang TJ. Evolution of the Araliaceae family inferred from complete chloroplast genomes and 45S nrDNAs of 10 Panax-related species. Sci Rep. 2017;7:4917. doi: 10.1038/s41598-017-05218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen VB, Park HS, Lee SC, Lee J, Park JY, Yang TJ. Authentication markers for five major Panax species developed via comparative analysis of complete chloroplast genome sequences. J Agric Food Chem. 2017;65(30):6298–6306. doi: 10.1021/acs.jafc.7b00925. [DOI] [PubMed] [Google Scholar]
- 17.Sarwat M, Yamdagni MM. DNA barcoding, microarrays and next generation sequencing: recent tools for genetic diversity estimation and authentication of medicinal plants. Crit Rev Biotechnol. 2016;36(2):191–203. doi: 10.3109/07388551.2014.947563. [DOI] [PubMed] [Google Scholar]
- 18.Kim NH, Jayakodi M, Lee SC, Choi BS, Jang W, Lee J, Kim HH, Waminal NE, Lakshmanan M, Binh NV, et al. Genome and evolution of the shade-requiring medicinal herb Panax ginseng. Plant Biotechnol J. 2018; 10.1111/pbi.12926. [DOI] [PMC free article] [PubMed]
- 19.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19(9):1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smit A, Hubley R. Repeat masker website. 2010. RepeatModeler Open-1.0. [Google Scholar]
- 21.Smit AF, Hubley R, Green P. RepeatMasker Open-3.0. 1996. [Google Scholar]
- 22.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2015;32:767–769. doi: 10.1093/bioinformatics/btv661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith Jr RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 27.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, Chen S. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 2012;7:e52249. doi: 10.1371/journal.pone.0052249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28(24):3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 31.Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin J, Zhang H, Kong L, Gao G, Luo J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2013;42:D1182–D1187. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao S, Wang L, Liu L, Liang Y, Sun Y, Wu J. Both the mevalonate and the non-mevalonate 5pathways are involved in ginsenoside biosynthesis. Plant Cell Rep. 2014;33(3):393–400. doi: 10.1007/s00299-013-1538-7. [DOI] [PubMed] [Google Scholar]
- 36.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Datasets in ginseng genome database are freely accessible for research purposes for non-profit and academic organizations at http://ginsengdb.snu.ac.kr. Further, all sequence data were deposited to the National Agricultural Biotechnology Information Center (NG-0858-000001~NG-0858-009845) (http://nabic.rda.go.kr). The database is optimized for Internet Explorer, Mozilla Firefox, Google Chrome and Safari.