

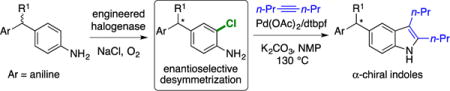
Abstract
Extensive effort has been devoted to engineering flavin-dependent halogenases (FDHs) with improved stability, expanded substrate scope, and altered regioselectivity. Here we show that variants of rebeccamycin halogenase (RebH) catalyze enantioselective desymmetrization of methylenedianilines via halogenation of these substrates distal to their pro-stereogenic center. Structure-guided engineering was used to increase the conversion and selectivity of these reactions, and the synthetic utility of the halogenated products was shown via conversion of to a chiral α-substituted indole. These results constitute the first reported examples of asymmetric catalysis by FDHs.
TOC image
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
Halogen substituents significantly impact the structure and function of organic compounds. Pharmaceuticals and agrochemicals, for example, frequently contain and owe their efficacy to halogenated aromatic moieties.1,2 Halogenated aromatics also play key roles as building blocks for chemical synthesis, particularly in metal-catalyzed cross-coupling reactions.3 The broad utility of halogenated compounds has driven extensive efforts to develop improved halogenation methods.4–6 Enzymatic halogenation has recently emerged as a promising tool for installing halogen substituents due to the high selectivity, mild conditions, and environmentally benign reagents associated with halogenase catalysis.7
Several flavin-dependent halogenases (FDHs) selectively functionalize C-H bonds on aromatic substrates, often at electronically disfavored positions.8 Moreover, the potential utility of these enzymes has been significantly improved via protein engineering.9 Our group, for example, has evolved variants of the tryptophan 7-halogenase RebH,10 an FDH from the rebeccamycin biosynthetic pathway, with improved thermostability and catalyst lifetime,11 expanded substrate scope,12 and altered regioselectivity13. Other groups have demonstrated that structure-guided mutagenesis can furnish improved variants of RebH and other FDHs.14–16
In addition to these protein engineering efforts, process optimization and metabolic engineering have been used to further expand the synthetic potential of FDHs. Multiple groups have shown that sequential or tandem halogenation/cross-coupling can be used to convert C-H bonds to C-C, C-N, and C-O bonds.17–21 Cross-linking RebH into aggregates has been shown to enable gram-scale halogenation,22 and incorporating FDHs into suitable host organisms has been used to halogenate natural products in vivo23,24.
To date, however, no examples of enantioselective halogenation catalyzed by FDHs have been reported. While aromatic halogenation does not lead to the formation of a stereogenic center, we envisioned that RebH variants developed in our laboratory could halogenate methylenedianilines to enable enantioselective desymmetrization of these compounds (Scheme 1A).25 This strategy was inspired by the work of Miller and co-workers, who demonstrated that 4,4′-methylenediphenols26 and diarylmethylamido bis(phenols)27 could be desymmetrized via peptide-catalyzed acetylation (Scheme 1B). Libraries of synthetic peptides were screened to identify a peptide that catalyzed this reaction with up to 95% enantiomeric excess (ee), despite the 6 Å separation between the site of functionalization and the pro-stereogenic center of the substrate (Scheme 1, R = t-Bu).28 Chiral methylenediarenes are commonly found in a wide range of pharmaceuticals, making methods for their asymmetric synthesis valuable.29 Moreover, the general challenge of asymmetric catalysis via functionalization distal to pro-stereogenic centers makes such desymmetrization processes fundamentally interesting.28
Scheme 1.
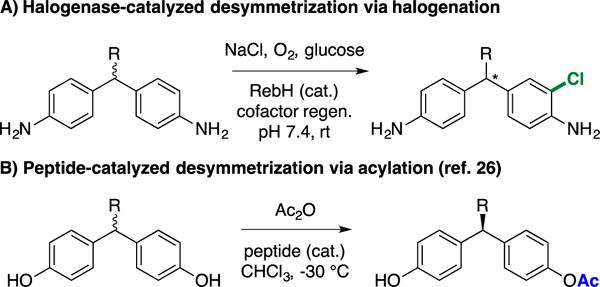
Catalytic methylenediarene desymmetrization
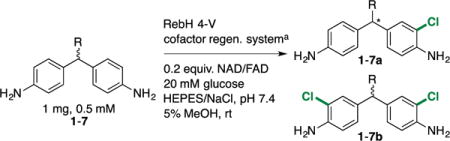
Because only t-Bu substituted methylenedianiline was desymmetrized high enantioselectivity using peptide catalysts (at cryogenic temperatures),28 halogenation of t-Bu substituted methylenedianiline (1) was prepared for our initial studies. RebH variant 4-V was first examined as a catalyst to halogenate this bulky substrate since it was evolved to functionalize large biologically active molecules.12 Indeed, 4-V catalyzed regioselective mono-halogenation of 1 ortho to its amine substituent, and provided essentially a single product enantiomer, 1a (99:1 e.r.) (Table 1, entry 1). A minor dihalogenated product (1b) was also formed via halogenation of the second aniline moiety.
Table 1.
4-V-catalyzed halogenation of alkyl-substituted 4,4′-methylenedianilines.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | R | Time (h) | Yield (%)c | e.r.c | Cl:Cl2c |
| 1b | t-Bu (1) | 16 | 80 | 99:1 | 9.3:1 |
| 2 | Me (2) | 16 | 17 | 84:16 | 0.5:1 |
| 3 | n-Pr (3) | 16 | 67 | 81:19 | 5.7:1 |
| 4 | n-Pr (3) | 2 | 76 | 78:22 | 18.3:1 |
| 5 | n-Pr (3) | 6 | 72 | 77:23 | 6.8:1 |
| 6 | n-Pr (3) | 48 | 54 | 82:18 | 2.8:1 |
The cofactor regeneration system consisted of 0.5 mol% MBP-RebF and 50 U/ml glucose dehydrogenase. NAD = nicotinamide adenine dinucleotide, FAD = flavin adenine dinucleotide, MBP = maltose binding protein.
10 mg reaction.
Yield and e.r. of mono-chlorinated product determined by HPLC relative to internal standard.
The activity of 4-V on methyl (2) and n-propyl (3) substituted methylenedianilines was next examined (Table 1, entries 2 and 3). Unfortunately, however, lower yield and selectivity was observed for both substrates relative to 1 (entry 1). We therefore considered possible relationships between the observed yields and selectivities that could help guide the development of improved enzymes for the desired desymmetrization.30 For example, the enantioselectivity of 4-V-catalyzed halogenation could result from either enantiotopic group selection or from a secondary kinetic resolution of the monochlorinated product.28 In the latter case, selective halogenation of the minor enantiomer of the monochlorinated product would enrich the major enantiomer at the expense of yield. Non-selective secondary halogenation would simply reduce the yield of mono-halogenated product.
To distinguish these possibilities, halogenation of 3 was monitored over time (Table 2, entries 3-6). Despite a substantial increase in the quantity of dichlorinated product, e.r. increased from only 78:22 to 82:18, indicating that a secondary kinetic resolution cannot be the major source of enantioinduction. This process could be further characterized by measuring krel values for halogenation of racemic 1-7a,28 but this was not pursued due to its small contribution to the observed enantioselectivity. Instead, the enantioselectivity of FDH-catalyzed desymmetrization results primarily from enantiotopic group selection, and the lower yields for 2 and 3 appear to result from poorly-selective dihalogenation of both monohalogenated product enantiomers.26
Table 2.
Conversion and selectivity data for desymmetrization reactions catalyzed by RebH variantsa
| e.r., % yield (fold improvement in conversion over RebH)b |
||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Enzymec
|
4-V | 6-TL | 3-SS | DA | AA | YD |
| Substrate | ||||||
| t-Bu (1) | 99:1, 80 (12.1) | - | 90:10, 54 (8.1) | 99:1, 76 (11.4) | 95:5, 80 (12.2) | 93:7, 83 (12.5) |
| Me (2) | 84:16, 16 (1.9) | 85:15, 34 (4.1) | 95:5, 21 (2.6) | 85:15, 17 (2.0) | 82:18, 18 (2.2) | - |
| n-Pr (3) | 81:19, 25 (2.8) | 76:24, 45 (5.2) | - | 71:29, 41 (4.8) | 72:28, 61 (7.0) | - |
| CH2CO2Et (4) | 69:31, 12 (3.3) | 78:22, 22 (6.1) | 65:35, 11 (3.2) | - | 51:49, 17 (4.6) | 68:32, 16 (4.3) |
| Benzyl (5) | 88:12, 75 (9.8) | 53:47, 65 (8.5) | 83:17, 48 (6.3) | 88:12, 98 (12.9) | 95:5, 91 (11.8) | 94:6, 89 (11.7) |
| m-OMe-Bn (6) | 76:24, 21 (7.0) | 66:34, 32 (10.9) | - | 75:25, 30 (10.1) | - | 87:13, 30 (10.2) |
| p-CF 3-Bn (7) | 93:7, 24 (10.8) | - | - | 95:5, 82 (37.4) | 92:8, 61 (27.8) | 86:14, 50 (22.6) |
These results suggested that improved binding of substrate (rather than one enantiomer of a mono-halogenated product) could be used to improve desymmetrization yield and enatioselectivity. Tryptophan binds to RebH via a number of interactions between its amino acid moiety and active site residues, including Tyr455 and Glu461 (Fig. 1a, Fig. S1a).31 Docking simulations suggested that substrate 2 could bind to 4-V in poses consistent with the observed halogenation regioselectivity13 by projecting its methyl substituent into the amino acid binding pocket (Fig. 1b). Notably, poses consistent with both product enantiomers were identified in 4-V, and both were predicted to involve only hydrophobic interactions between the spectator aniline substituent and the enzyme (Fig. S1b). We speculated that engineering this pocket to bind the spectator aniline substituent of 2 instead of the methyl substituent might lead to better discrimination between enantiomeric poses. Several variants of 4-V were therefore modeled by mutating Tyr455 and Glu461 to Ala or Asp with the goal of accommodating the bulk of the spectator aniline substituent and allowing hydrogen bonding or ion pairing with the aniline amine group. Docking simulations conducted on one of these variants, 4-V Y455D/E461A (thus abbreviated DA), suggested that the desired binding could occur, but enantiomeric poses were still identified, despite the fact that one of the two poses (cyan) was now predicted to hydrogen bond to the designed Asp455 residue (Fig. 1c, Fig. S1c). Interestingly, however, substrate 7, which has a bulkier p-CF3-benzyl substituent, was predicted to bind in only one pose consistent with the halogenase mechanism (Fig. 1d). In this pose, the spectator aniline is predicted to hydrogen bond to Gly112, while the benzyl substituent was predicted to occupy the engineered amino acid binding pocket. In this case, the additional space in the engineered pocket, rather than hydrogen bonding interactions, appears critical for altered binding.
Figure 1.
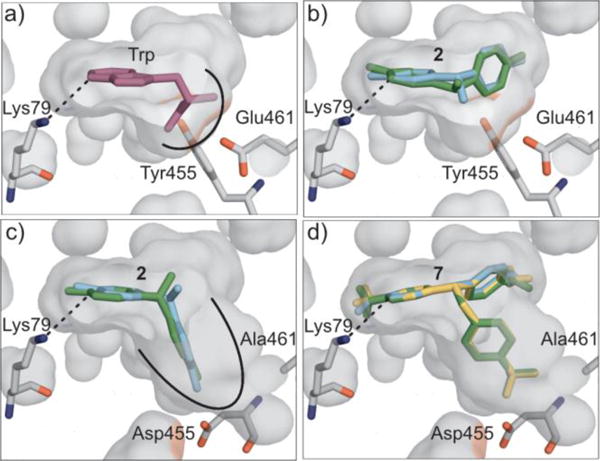
Active site models of 4-V (a/b) and DA (c/d) built from the crystal structure of wild-type RebH containing bound Trp (PDB entry 2OA131). a) 4-V model showing Trp substrate from RebH structure in magenta. b/c) enantiomeric poses of 2 identified via docking simulations in models of 4-V (b) and DA (c). d) three lowest energy poses for 7 overlaid in DA. Site of reaction relative to Lys79 is indicated with a dashed line (a-d) and the amino acid binding pocket is denoted with an arc (a/c).
Encouraged by the predicted binding differences between different amino acid binding pocket variants, we evaluated the activity of four such variants (DA, AA, YD, and ND named using the above convention) along with three other RebH variants previously engineered in our laboratory (4-V, 6-TL, and 3-SS)12,13 toward a small panel of methylenedianilines (Table 2). In general, significantly better conversion of all substrates investigated was observed using engineered variants relative to wt RebH. Variants previously evolved in our laboratory showed the highest enantioselectivity for desymmetrization of methylenedianiline substrates 1-4 (Table 2, entries 1-4), which have relatively small methylene substituents. These variants were obtained via directed evolution efforts aimed at improving RebH activity on large substrates (4-V and 3-SS)12 or altering RebH selectivity (6-TL)13. Variant ND, not shown in Table 2 because it showed otherwise unremarkable selectivity, provided a slight preference (47:53, 18% yield) for the opposite product enantiomer using substrate 4. This variant could therefore serve as a viable starting point for evolving halogenases with the opposite sense of enantioinduction on 4 and other substrates. The amino acid binding pocket variants DA, AA, and YD provided significantly higher activity and enantioselectivity on benzyl substituted substrates 5-7 (Table 2, entries 5-7). These substrates were halogenated with 10-37-fold higher conversion relative to wt RebH, clearly showing the impact of amino acid binding pocket mutations on desymmetrization activity and selectivity.
In addition to demonstrating the utility of FDHs for enantioselective catalysis, desymmetrization of methylenedianilines via halogenation provides a handle for subsequent Pd-catalyzed transformations.21 We envisioned that a Larock-type cyclization32 would be particularly useful for generating enantioenriched 5-substituted indoles, which have been explored as monoamine uptake inhibitors and glucocorticoid receptor modulators.33,34 This sequence would leave an aniline substituent on the resulting product (Ar in Scheme 2) that could be further functionalized via diazotization. In practice, heating enantioenriched 1a with 4-octyne in the presence of a Pd catalyst35 led to formation of the desired indole with no loss of enantiopurity (Scheme 2). While diazotization of 1b was not pursued, diazotization/reduction of halogenated 4 (4a, Fig. S2) allowed for comparison of the absolute stereochemistry of the resulting 3,3-diarylpropanoate to the same compound previously synthesized via asymmetric conjugate reduction.36 Based on this comparison, the major enantiomer of 4a was assigned the S-configuration, which is consistent with the expected isomer based on docking of substrate 7.
Scheme 2.
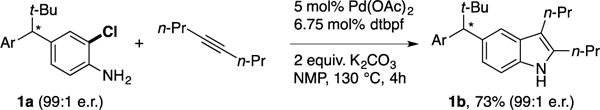
Indole synthesis via halogenation/annulation.
In summary, we found that RebH variant 4-V, an enzyme engineered in our laboratory to halogenate large, biologically active indoles and carbazoles,12 also catalyzes remote desymmetrization of a methylenedianinelines with high enantioselectivity. To our knowledge, this is the first report of an FDH catalyzing an enantioselective halogenation reaction. A small panel of 4-V variants was generated based on docking simulations of methylenedianiline binding in the enzyme active site. These variants, along with others previously engineered in our laboratory, provided improved activity and selectivity on a number of methylenedianiline substrates, illustrating how targeted mutagenesis can be used to readily improve halogenase enantioselectivity in analogy to previous efforts focused on substrate scope and regioselectivity.14,15 One of the resulting enantioenriched, halogenated methylenedianiline products was converted to 5-substituted indole with no loss in enantioselectivity at the α-stereogenic center. While only a few substrates were evaluated for this work, we anticipate, given the broad substrate scope of FDHs,37 that future research efforts will further expand the potential applications of these halogenases in the preparation of a range of useful, bioactive compounds. For instance, similar reactions of 3,3′- or 2,2′-methyelenedianilines would give rise to alternate indole regioisomers, and the amine substituent on the unfunctionalized aniline could be used as a handle for additional substitution. Both of these variations are the subject of ongoing investigations in our group. Moreover, other enantioselective halogenation reactions could also be catalyzed by these powerful enzymes. Investigations toward this end are currently ongoing in our laboratory.
Supplementary Material
Acknowledgments
This work was supported by the NIH (1R01GM115665). J.T.P. was supported by an NIH Chemistry and Biology Interface training grant (T32 GM008720).
Footnotes
Supporting Information. Procedures and full characterization of new compounds are provided (PDF).
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Jeschke P. Pest Manag Sci. 2010;66:10–27. doi: 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]
- 2.Hernandes M, Cavalcanti S, Moreira D, de Azevedo Junior W, Leite A. Current drug targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- 3.Diederich F, Stang P. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; 2008. [Google Scholar]
- 4.Podgoršek A, Zupan M, Iskra J. Angew Chem Int Ed. 2009;48:8424–8450. doi: 10.1002/anie.200901223. [DOI] [PubMed] [Google Scholar]
- 5.Vigalok A, Kaspi AW. In: Topics in Organometallic Chemistry. Vigalok A, editor. Vol. 31. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 19–38. [Google Scholar]
- 6.Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weichold V, Milbredt D, van Pée K-H. Angew Chem Int Ed. 2016;55:6374–6389. doi: 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- 8.van Pée K-H, Patallo E. Appl Microbiol Biot. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 9.Payne JT, Andorfer MC, Lewis JC. Engineering Flavin-Dependent Halogenases. 1st. Vol. 575. Elsevier Inc; 2016. pp. 93–126. (Methods in Enzymology). [DOI] [PubMed] [Google Scholar]
- 10.Yeh E, Garneau S, Walsh CT. Proc Natl Acad Sci USA. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poor CB, Andorfer MC, Lewis JC. ChemBioChem. 2014;15:1286–1289. doi: 10.1002/cbic.201300780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Payne JT, Poor CB, Lewis JC. Angew Chem Int Ed. 2015;54:4226–4230. doi: 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andorfer MC, Park H-J, Vergara-Coll J, Lewis JC. Chem Sci. 2016;7:3720–3729. doi: 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shepherd SA, Karthikeyan C, Latham J, Struck A-W, Thompson ML, Menon BRK, Styles MQ, Levy C, Leys D, Micklefield J. Chem Sci. 2015;6:3454–3460. doi: 10.1039/c5sc00913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glenn WS, Nims E, O’Connor SE. J Am Chem Soc. 2011;133:19346–19349. doi: 10.1021/ja2089348. [DOI] [PubMed] [Google Scholar]
- 16.Lang A, Polnick S, Nicke T, William P, Patallo EP, Naismith JH, van Pée K-H. Angew Chem Int Ed. 2011;50:2951–2953. doi: 10.1002/anie.201007896. [DOI] [PubMed] [Google Scholar]
- 17.Roy AD, Grüschow S, Cairns N, Goss RJM. J Am Chem Soc. 2010;132:12243–12245. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]
- 18.Runguphan W, O’Connor SE. Org Lett. 2013;15:2850–2853. doi: 10.1021/ol401179k. [DOI] [PubMed] [Google Scholar]
- 19.Latham J, Henry J-M, Sharif HH, Menon BRK, Shepherd SA, Greaney MF, Micklefield J. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frese M, Schnepel C, Minges H, Voß H, Feiner R, Sewald N. ChemCatChem. 2016;8:1799–1803. [Google Scholar]
- 21.Durak LJ, Payne JT, Lewis JC. ACS Catal. 2016;6:1451–1454. doi: 10.1021/acscatal.5b02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frese M, Sewald N. Angew Chem Int Ed. 2014;54:298–301. doi: 10.1002/anie.201408561. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez C, Zhu L, Brana A, Salas A, Rohr J, Mendez C, Salas JA. Proc Natl Acad Sci USA. 2005;102:461–466. doi: 10.1073/pnas.0407809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Runguphan, W.; Qu, X.; O’Connor, S. E. 2010, 468, 461–464.
- 25.García-Urdiales E, Alfonso I, Gotor V. Chem Rev. 2005;105:313–354. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]
- 26.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2006;128:16454–16455. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- 27.Hurtley AE, Stone EA, Metrano AJ, Miller SJ. J Org Chem. 2017;82:11326–11336. doi: 10.1021/acs.joc.7b02339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2008;130:16358–16365. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mondal S, Panda G. RSC Advances. 2014;4:28317–28358. [Google Scholar]
- 30.Schreiber SL, Schreiber TS. J Am Chem Soc. 1987;109:1525–1529. [Google Scholar]
- 31.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN. Proteins. 2008;70:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 32.Zeni G, Larock RC. Chem Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]
- 33.Lucas MC, Carter DS, Cai H-Y, Lee EK, Schoenfeld RC, Steiner S, Villa M, Weikert RJ, Iyer PS. Bioorg Med Chem Lett. 2009;19:4630–4633. doi: 10.1016/j.bmcl.2009.06.076. [DOI] [PubMed] [Google Scholar]
- 34.Xiao H-Y, Wu D-R, Sheppeck JE, II, Habte SF, Cunningham MD, Somerville JE, Barrish JC, Nadler SG, Dhar TGM. Bioorg Med Chem Lett. 2013;23:5571–5574. doi: 10.1016/j.bmcl.2013.08.049. [DOI] [PubMed] [Google Scholar]
- 35.Shen M, Li G, Lu BZ, Hossain A, Roschangar F, Farina V, Senanayake CH. Org Lett. 2004;6:4129–4132. doi: 10.1021/ol048114t. [DOI] [PubMed] [Google Scholar]
- 36.Itoh K, Tsuruta A, Ito J-I, Yamamoto Y, Nishiyama H. J Org Chem. 2012;77:10914–10919. doi: 10.1021/jo302357b. [DOI] [PubMed] [Google Scholar]
- 37.Andorfer MC, Grob JE, Hajdin CE, Chael JR, Siuti P, Lilly J, Tan KL, Lewis JC. ACS Catal. 2017;7:1897–1904. doi: 10.1021/acscatal.6b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.