

Abstract
The protein kinase ATM is a master regulator of the DNA damage response but also responds directly to oxidative stress. Loss of ATM causes Ataxia telangiectasia, a neurodegenerative disorder with pleiotropic symptoms that include cerebellar dysfunction, cancer, diabetes, and premature aging. Here, we genetically separated DNA damage activation of ATM from oxidative activation using separation-of-function mutations. We found that deficiency in ATM activation by Mre11-Rad50-Nbs1 and DNA double-strand breaks resulted in loss of cell viability, checkpoint activation, and DNA end resection in response to DNA damage. In contrast, loss of oxidative activation of ATM had minimal effects on DNA damage-related outcomes but blocked ATM-mediated initiation of checkpoint responses after oxidative stress and resulted in deficiencies in mitochondrial function and autophagy. In addition, expression of ATM lacking oxidative activation generates widespread protein aggregation. These results indicate a direct relationship between the mechanism of ATM activation and its effects on cellular metabolism and DNA damage responses in human cells and implicates ATM in the control of protein homeostasis.
Introduction
Ataxia telangiectasia (A-T) is a disorder characterized by progressive cerebellar degeneration, predisposition to lymphoid malignancies, and diabetes that is caused by loss of the A-T mutated (ATM) kinase. Cells from A-T patients lack the ability to initiate DNA damage-induced checkpoints and are deficient in responses to DNA double strand breaks (DSBs) (1). ATM-deficient cells also exhibit abnormalities in responses to other forms of cellular stress, including oxidation (2, 3), hypoxia (4), hyperthermia (5), and hypotonic stress (6).
ATM was initially characterized solely as a regulator of the DNA damage response, a rapid initiation of checkpoints and DNA repair that requires the Mre11/Rad50/Nbs1 (MRN) complex to recruit and activate ATM at sites of double-strand breaks (1, 7–9). The importance of MRN in ATM activation is evident in the similar clinical phenotype of patients with A-T like disorder (ATLD) or Nijmegen breakage syndrome (NBS), caused by hypomorphic mutations in the Mre11, Rad50, or Nbs1 genes (10, 11). The MRN complex localizes to sites of DSBs, recruits ATM through interactions with Nbs1 and Mre11/Rad50, facilitates the conversion of inactive dimeric forms of ATM into active monomeric forms, and promotes the stable binding of ATM substrates for efficient phosphorylation (8). We have also demonstrated that ATM can be activated by oxidative stress independently of MRN or DNA damage (3). In this pathway, multiple disulfide bonds are formed within the ATM dimer that induce an active conformation. The disulfide formed at C2991 is particularly important, as mutation of this residue blocks the oxidation-mediated activation of ATM without affecting MRN/DNA-mediated activation.
ATM deficiency has been linked for many years with observations of high levels of reactive oxygen species (ROS) and inability to respond appropriately to oxidative conditions (2). For instance, A-T patients exhibit increased oxidative damage to lipids and DNA (12) and lower levels of antioxidants in their blood plasma (13). A-T fibroblasts show increased sensitivity to hydrogen peroxide and nitric oxide donors (14, 15), also suggesting a high basal level of oxidative stress. ATM-deficient mice exhibit a loss of hematopoietic stem cells that is attributable to high ROS (16), and the incidence of T-cell lymphomas in these mice is delayed and reduced by feeding with antioxidants (17–21). Our previous observations that ATM-deficient cells expressing the C2991L allele of ATM or the A-T patient allele R3047X exhibit high amounts of ROS and are resistant to peroxide-induced apoptosis (3) suggest that the activation of ATM by oxidation is causally linked to regulation of global redox homeostasis and to the A-T neurodegeneration phenotype.
After DNA damage, ATM phosphorylates many proteins including histone H2AX, Structural Maintenance of Chromosomes Protein 1 (SMC1), KRAB-associated Protein 1 (KAP1), Checkpoint kinase 2 (CHK2), and the transcription factor p53. However, H2AX and KAP1 are not phosphorylated in the presence of oxidative stress (3), presumably because ATM is not recruited to DNA sites where these substrates are located. ATM activation by cell cycle arrest during mitosis also does not result in the phosphorylation of SMC1 or p53, substrates which are known to be phosphorylated after ionizing radiation (22). These observations suggest that ATM may activate specific downstream effectors in addition to common substrates depending on which cellular stress is present.
To delineate which activities of ATM are specific to its oxidation or DNA damage-related functions, it is essential to have separation-of-function alleles that affect each pathway specifically. The C2991L allele of ATM serves this purpose for the oxidative pathway as described above, but no mutants have been described that specifically alter the DNA damage-dependent pathway. Here we identify such an allele and characterize the responses of ATM that rely on MRN-dependent activation of ATM. These results show that there are in fact two distinct and independent pathways to activate ATM: one that operates through MRN that is induced by double-strand breaks and regulates checkpoint functions and DNA repair, and one that is activated by oxidative stress and governs ROS abundance and protein homeostasis. Global phosphoproteomic analysis of human cells expressing these separation-of-function alleles indicates widespread changes with loss of ATM activation by oxidative stress, largely due to deficiency in phosphorylation events normally catalyzed by the protein kinase CK2. Here, we found that CK2 aggregated in the absence of functional ATM and that global increases in protein aggregation were associated with the inability to activate ATM via oxidative stress. These studies provide novel insights into the mechanisms of ATM activation and the DNA-damage independent functions of ATM in human cells and indicate the pleiotropic nature of the signaling deficiencies in ATM-deficient cells that stem from the loss of oxidation-induced activation.
Results
The 2RA ATM mutant is deficient in activation by MRN and DNA
The mTOR protein kinase, which is a member of the phosphatidylinositol-3 kinase (PI3K) related kinase (PIKK) family and is structurally related to ATM, is regulated by phosphatidic acid (PA) (23). Although ATM responds to different stimuli, we hypothesized that mutations that disrupt PA stimulation of mTOR may serve as a guide for the location of mutations in ATM that might disrupt its regulation. We therefore looked for residues in ATM located a similar distance from the conserved kinase active site as the Arg2109 residue in mTOR that affects PA-mediated signaling (23). In ATM, there are two arginine residues (R2579 and R2580), which are analogous in their position to Arg2109 on mTOR and located at the end of the FAT domain in ATM (Fig. 1A). We mutated these sites to alanine (R2579A/R2580A, “2RA”) in the human ATM gene and expressed and purified the recombinant protein as previously described (24) (Fig. S1). The mutant protein was tested in comparison to the wild-type protein in an in vitro assay using the N-terminus of p53 fused to GST as a substrate and monitoring phosphorylation of Ser15 by quantitative Western blot. This comparison showed that the 2RA mutations result in complete loss of ATM activation by MRN and DNA (Fig. 1B, C). We also tested for activation by oxidation using hydrogen peroxide, which activates ATM through a DNA-independent mechanism (3). The 2RA ATM mutant still exhibits H2O2-mediated stimulation similar to the wild-type protein (Fig. 1D).
Fig. 1. Disruption of MRN- and DNA-dependent ATM activation by R2579A/R2580A mutations of ATM.
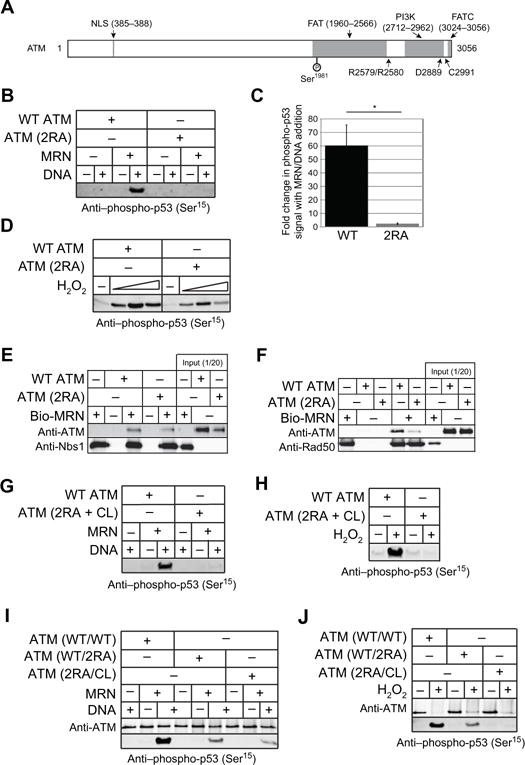
(A) Schematic diagram of ATM structure; mutations analyzed in this study are indicated. Features and domains within ATM consist of the nuclear localization signal (NLS), the FRAP/ATM/TRRAP (FAT), the Kinase domain (PI3-K), and the FAT c-terminal (FATC) domain. (B) Kinase assays with 1.35 nM dimeric wild-type or 2RA ATM, 9.6 nM MRN, 6.25 nM GST-p53 substrate, and ~140 nM linear DNA, probed with antibody to phosphoserine 15 of p53. (C) Quantitation of 3 independent experiments comparing wild-type ATM to 2RA ATM, showing the fold change in phospho-signal with MRN/DNA compared to the reactions without MRN/DNA. Error bars indicate standard deviation. (D) Kinase assays with 2.7 nM dimeric wild-type or 2RA ATM and 12.5 nM GST-p53 substrate in the presence of H2O2 (0.27, 0.81, and 2.4 mM). (E and F) Binding assays with wild-type or 2RA ATM and biotinylated MRN or MR as indicated. Biotinylated MRN or MR (20nM) were incubated with 50 nM ATM and isolated with streptavidin-coated magnetic beads. Bound proteins were visualized by western blotting with anti-ATM and anti-Nbs1 or anti-Rad50 antibodies. (G and H) Kinase assays as in (A) and (B) except with triple mutant ATM (R2579A/R2580A/C2991L, “2RA+CL”). (I and J) Kinase assays as in (A) and (B) except using heterodimeric ATM (wild-type/2RA and 2RA/CL) as indicated.
Because ATM can interact with MRN through both Nbs1 and Mre11/Rad50 (MR) (25), we performed binding assays with ATM and biotinylated MRN or MR to investigate the mechanism of ATM inactivation in the 2RA mutant. The 2RA ATM mutant bound to MRN similarly to wild-type ATM but was deficient in binding to MR (Fig. 1E, F). These results suggest that interaction of ATM and MR in addition to interaction with Nbs1 is necessary to activate ATM kinase activity and that the 2RA mutations disrupt this interaction.
We have previously shown that mutation of a conserved cysteine residue in the FATC domain of ATM generates a separation-of-function mutant (C2991L) that cannot be activated by oxidative stress but shows normal activity in the presence of MRN and DNA (3). Here we generated a combination mutant (“2RA+CL”) containing both the 2RA and C2991L (CL) mutations. As expected, the 2RA+CL ATM double mutant was completely deficient in both MRN/DNA-dependent and H2O2-dependent ATM activation, as monitored by p53 phosphorylation in vitro (Fig. 1G, H). To determine whether the 2RA mutations are dominant, we then co-expressed the 2RA mutant with either wild-type ATM or the CL mutant, using Flag and HA tags on the proteins to purify heterodimeric ATM complexes of WT/2RA, 2RA/CL, and WT/WT purified in the same manner for comparison. The heterodimer combinations of wild-type and 2RA (WT/2RA) or 2RA and CL (2RA/CL) each showed partial activity in the presence of MRN and DNA in vitro compared to the combination of two wild-type ATM proteins (WT/WT), indicating that the 2RA mutations do not block the function of another ATM monomer in trans (Fig. 1I). This is expected considering that the ATM is initially in dimeric form but becomes monomeric in response to DNA damage (6). In contrast, the 2RA/CL ATM heterodimer showed a complete loss of activity in the presence of H2O2 (Fig. 1J), consistent with our previous finding that the C2991 residue must be intact in both subunits of the ATM dimer for activation by oxidation (3). In contrast, the 2RA/WT heterodimeric mutant did exhibit oxidative activation although with lower efficiency than the wild-type complex, similar to our result with the 2RA homodimer complex (Fig. 1D). These results together confirm that two distinct mechanisms exist for the activation of ATM and can be genetically separated.
ATM is activated by two independent pathways in cells
To confirm the in vitro results with the 2RA mutant, we established an inducible expression system to test the effects of ATM mutations in human adherent cells. We used a human osteosarcoma cell line (U2OS) containing an FRT recombination site integrated into the genome adjacent to a CMV promoter blocked by two copies of the Tet operator (Invitrogen Flp-In™ T-Rex™ system). Vector only, or the wild-type, 2RA, or CL alleles of ATM were transfected into the cells with a construct expressing Flp recombinase and stable clones were generated. In each cell line we depleted endogenous ATM with lentivirus containing shRNA specific for ATM before inducing ATM expression with doxycycline. The cells were then treated with DNA damaging agents including ionizing radiation (IR) and camptothecin (CPT) and oxidizing agents, including hydrogen peroxide (H2O2) and sodium arsenite, to examine the effects of the ATM mutations on substrate phosphorylation in cells. We used arsenite here because work from other groups has indicated that arsenite stimulates checkpoint activation responses that are dependent on ATM (26) and that ATM is required for survival of human cells to arsenite even though arsenite does not detectably generate DNA double-strand breaks (Fig. S2)(27). Arsenite is thought to induce ROS through activation of NADPH oxidase (Nox) isoforms as well as through the mitochondria (28, 29) and can produce both superoxide as well as hydrogen peroxide in the cytoplasm. Arsenite is stable in serum-containing media over long periods unlike exogenously added hydrogen peroxide (30, 31) and has been reported to induce checkpoint responses mediated by caffeine-sensitive protein kinases (26). As expected, ATM depletion resulted in a complete defect in the phosphorylation of known ATM substrates including KAP1 and CHK2 after each treatment we used (Fig. 2).
Fig. 2. Separation-of-function mutations in ATM dictate responses to DNA damage and oxidative stress.
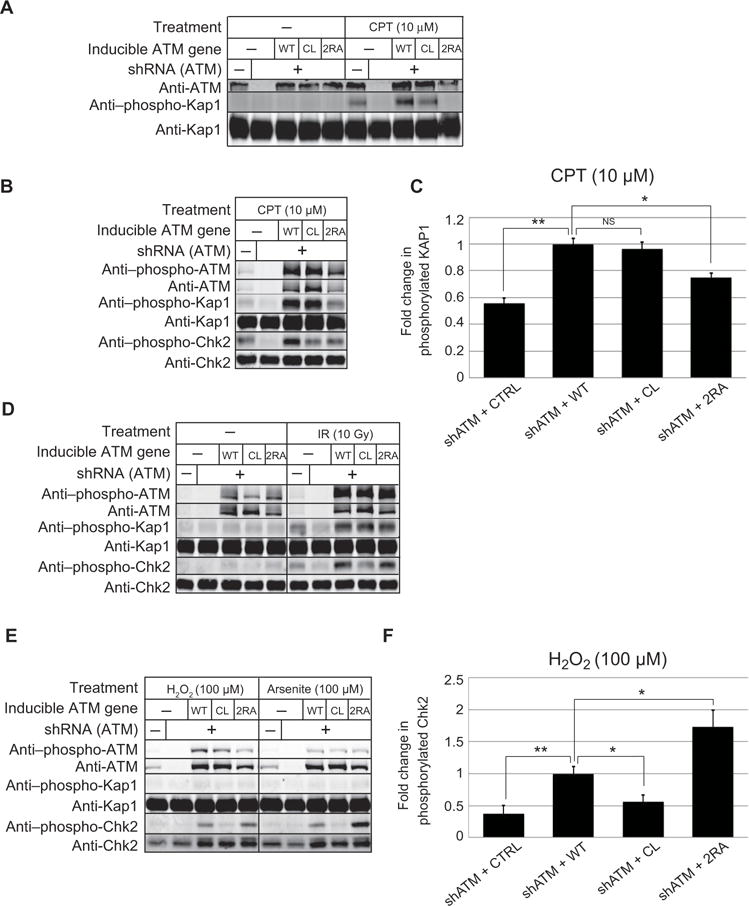
Human U2OS osteosarcoma cells were depleted for endogenous ATM using shRNA and induced to express various ATM alleles as indicated. (A) Cells were induced for ATM expression with doxycycline (10 ng/ml) and exposed to CPT (10 μM) for 1hr. ATM activity was examined using antibodies directed against phospho-KAP1 Ser824 and ATM levels were assessed with anti-ATM antibody. (B) U2OS cells were treated as in (A) except expression was induced with doxycycline (1 μg/ml). ATM activity was assessed with antibodies directed against ATM, phospho-ATM Ser1981, KAP1, phospho-KAP1 Ser824, CHK2, and phospho-CHK2 Thr68 as indicated. (C) U2OS cells expressing shRNA against ATM (shATM) and either vector (CTRL) or various ATM alleles as in (B) were treated with 10 μM CPT for 1hr and the levels of phosphorylated KAP1 were determined, in comparison to total KAP1 protein, and normalized with the phosphorylated signal from wild-type ATM expressing cells. (D) U2OS cells were depleted of endogenous ATM and induced for recombinant ATM expression as in (B) but were exposed to 10 Gy IR followed by 1hr recovery. Phosphorylation was assessed as in (B). (E) U2OS cells were treated as in (B) but exposed to 100 μM H2O2 or arsenite for 1hr in the presence of 1 μg/ml of doxycycline. Phosphorylation was assessed as in (B). (F) U2OS cells expressing various ATM alleles as in (B) were treated with 100 μM H2O2 for 1hr in serum-free media and the amout of phosphorylated CHK2 was quantitated in comparison to total CHK2 protein and normalized with the phosphorylated signal from wild-type ATM expressing cells. Data are means ± S.D. from 3 independent experiments. and indicate comparisons in which *p<0.05 and **p<0.005, by XX test.
Induction of ATM expression with low levels of doxycycline (10 ng/ml) resulted in approximately equivalent levels of recombinant ATM compared with the endogenous ATM protein in the U2OS cell line (Fig. 2A). Under these conditions, treatment of cells with CPT resulted in phosphorylation of KAP1 on ser824, which is ATM-dependent (32). Comparison of the mutant alleles revealed that the wild-type and CL expressing cell lines efficiently phosphorylate KAP1, whereas the 2RA expressing line is completely deficient, similar to our results with purified ATM in vitro.
We also analyzed the responses of the cell lines with higher amounts of doxycycline induction (1 μg/ml) which yields higher abundance of recombinant ATM and found a similar deficit in KAP1 phosphorylation in the 2RA cell line (Fig. 2B). In three biological replicates, the fold change in KAP1 phosphorylation was reproducibly diminished in cells expressing the 2RA allele in comparison to cells expressing the wild-type allele (Fig. 2C, S3). In contrast, the cells expressing the CL mutant showed wild-type levels of KAP1 phosphorylation.
Unlike CPT treatment, exposure of the cell lines to ionizing radiation (10 Gy) resulted in equivalent phosphorylation of KAP1 and Chk2 by the 2RA ATM mutant in comparison to wild-type, although cells expressing the CL allele failed to fully restore phosphorylation of Chk2 (Fig. 2D). Since ionizing radiation induces formation of reactive oxygen species (ROS) as well as DNA double-strand breaks (DSBs), we considered the possibility that both modes of activation are operating when this form of DNA damage is induced. To test this idea directly, we compared the responses of the U2OS cell lines to two different forms of ROS induction. We have previously shown that treatment of cells with low levels of hydrogen peroxide induces ATM activation via the oxidation pathway independent of DNA damage (3). Here we compared peroxide to an alternative method of inducing ROS with sodium arsenite (26) and found that both treatments induce ATM-dependent phosphorylation of CHK2 on thr68 but not KAP1 phosphorylation (Fig. 2E, F). In this case only the cells expressing the CL allele fail to phosphorylate CHK2, consistent with our previous results. It is important to note that the peroxide and arsenite treatments used here do not induce double-strand breaks, as measured by γ-H2AX phosphorylation (Fig. S2). Even though the 2RA ATM appears to be less efficient than the wild-type protein in responding to peroxide in vitro, the mutant exhibits wild-type or even higher levels of activity in response to the oxidative agents in cells.
MRN-dependent ATM activation is critical for cell survival after DNA damage but not oxidative stress
To understand the effects of the 2RA and CL ATM mutations on the responses of human cells after DNA damage and oxidative stress, we performed clonogenic cell survival assays after ionizing radiation, CPT, or arsenite exposure. Cultures of cells with shRNA depletion of ATM showed significantly lower rates of survival after treatment with each of these agents compared to control shRNA treated cells, and this sensitivity was partially recovered by expression of wild-type ATM (Fig. 3). After treatment of the U2OS cells expressing the ATM mutant alleles with the DNA damaging agents IR or CPT, cells expressing CL ATM showed similar or higher survival rates compared to cells expressing wild-type ATM (Fig. 3A, B). The cells expressing 2RA ATM showed significantly lower survival after exposure to these DNA damaging agents compared to the wild-type ATM and the CL ATM expressing cells, although the survival was generally intermediate between ATM depleted cells and cells expressing wild-type ATM, suggesting that 2RA ATM has some residual activity or that there is some contribution of the oxidative pathway under these conditions (Fig. 3A, B). For comparison, we also tested cells expressing the kinase-deficient D2889A (DA) mutant allele of ATM (33) which exhibited extremely poor survival of both ionizing radiation and CPT (Fig. S4), clearly worse than the ATM-depleted cells with no complementation, suggestive of a dominant negative effect (34).
Fig. 3. ATM deficient in activation via MRN exhibits defects in survival of DNA damage and in DSB resection.
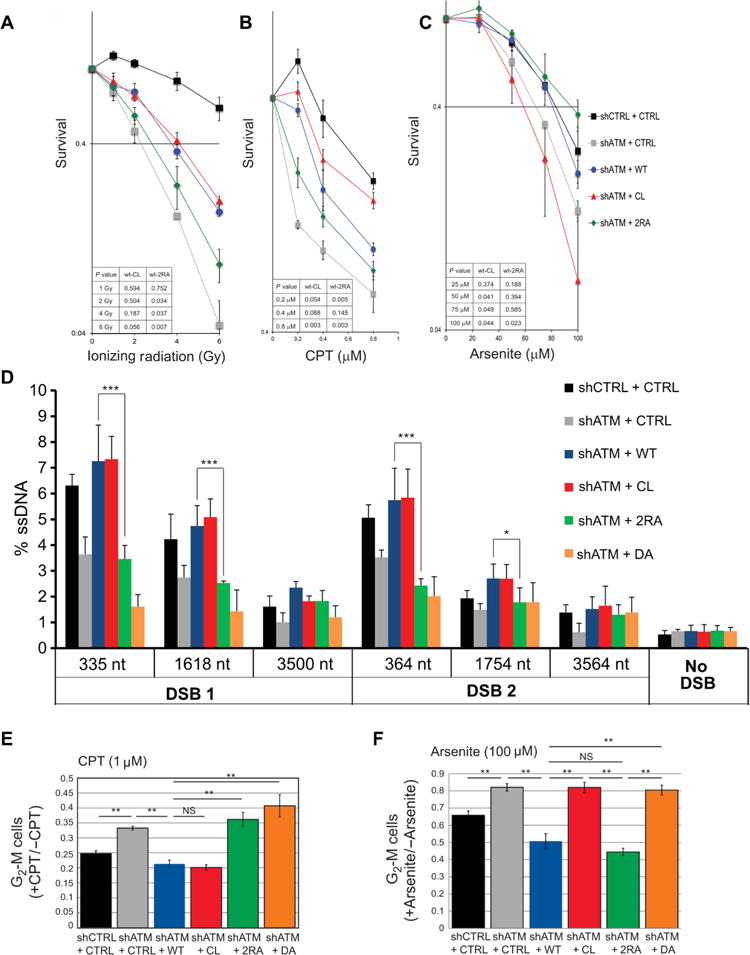
(A-C) U2OS cells depleted for endogenous ATM (shATM; Fig. 2) or treated with a control shRNA (shCTRL) were induced to express vector only (CTRL) or various ATM alleles (1 μg/ml doxycycline) as indicated, and were analyzed for cell survival after treatment with IR (A), CPT (B), or Arsenite (C) as indicated Data are means ± S.E. of 3 independent experiments. p-values (inset) assessed by Student’s t-test. (D) DNA end resection at AsiSI-induced breaks in cells expressing various ATM alleles. Endogenous ATM in U2OS-ER-AsiSI cells was depleted by shRNA treatment (KD) and cells were complemented with induced expression of ATM alleles (1 μg/ml doxycycline) as indicated, 3 days before treatment with 600 nM 4-OHT, which induces nuclear translocation of the AsiSI enzyme (98). After 4 hrs, cells were harvested, genomic DNA was prepared, and either digested with restriction enzymes to distinguish between single-stranded and double-stranded DNA, or mock-digested as described (99). Quantitation of single-stranded DNA intermediates generated by resection was performed by real-time PCR. Data are means ± S.D. of 3 experiments. * p < 0.05; *** p < 0.0005. (E and F) U2OS cells depleted of endogenous ATM with shRNA and inducibly expressing various ATM alleles (1 μg/ml doxycycline) were synchronized in G1/early S with aphidicolin (2 μg/ml, 17 hrs). The intra-S cell cycle checkpoint was analyzed by quantification of the percentage of G2/M cells 17 hr after removal of aphidicolin and treatment with CPT (1 μM) (E) or arsenite (100 μM) (F) compared with untreated group, as indicated. Data are means ± S.E. of 3 independent experiments. *p<0.05 and **p<0.005.
The cells were also treated with sodium arsenite to test for survival of oxidative stress. While ATM-depleted cells were not as sensitive to this range of arsenite treatment compared to the DNA damaging agents, it was clear that ATM-depleted cells did exhibit reduced survival compared to control shRNA expressing cells (Fig. 3C). The cells expressing CL ATM showed a marked sensitivity to arsenite compared to ATM depleted cells, a pattern opposite to that seen with IR and CPT. In contrast, both the 2RA and wild-type ATM alleles complemented the survival deficit of ATM depleted cells after exposure to arsenite (Fig. 3C). These results are generally consistent with in vitro results indicating that 2RA and CL ATM are blocked in activation after DSBs and oxidative stress, respectively (Fig. 1 and 2).
MRN-mediated ATM stimulation regulates DNA end resection
ATM activity is important for homologous recombination (35), which initiates with DNA end resection. To examine the effects of the ATM alleles on DNA end resection in human cells, we introduced an estrogen receptor fusion of AsiSI endonuclease (ER-AsiSI) to our U2OS Flp-In T-Rex cells containing wild-type or mutant ATM alleles and used a quantitative PCR (qPCR)-based method to measure the levels of single-stranded DNA produced at two sites in the genome that exhibit high efficiency AsiSI cleavage (36). Consistent with the critical role of ATM in DNA resection, depletion of ATM leads to decreased DNA resection at both DSB sites (Fig. 3D). The resection is rescued by wild-type ATM or CL ATM expression, but not by 2RA or DA kinase-deficient ATM, suggesting that MRN-dependent ATM activation is specifically required for DNA resection after DNA DSBs.
Cell cycle checkpoints are disrupted in cells expressing ATM separation of function alleles
ATM activation is required for DNA damage-induced cell cycle checkpoints (11) and A-T cells also fail to exhibit checkpoint activation in response to ROS (37). We therefore investigated whether the 2RA and CL ATM mutants efficiently arrest the cell cycle after DNA damage or oxidative stress. We monitored the intra-S phase checkpoint for these experiments, which has previously been shown to be dependent on ATM (38). To monitor S phase checkpoint activation, we blocked cells in early S phase with a low level of aphidicolin and added CPT or arsenite when cells were released into S phase. The percentage of cells in G2/M was then measured using propidium iodide staining. Using this method, we confirmed that ATM depletion generated a higher percentage of cells in G2/M after either CPT or arsenite treatment (Fig. 3E, F), suggesting that the cell cycle checkpoint is not efficiently activated. The ratio of G2/M cells after CPT or arsenite treatment dropped significantly when wild-type ATM was expressed (Fig. 3E, F). Expression of the CL ATM allele in ATM-depleted cells efficiently activated the cell cycle checkpoint after CPT treatment but not after arsenite treatment. As expected, 2RA-expressing cells showed an opposite pattern to that of CL-expressing cells, with efficient checkpoint activation after arsenite treatment but not with CPT-mediated DNA damage. Lastly, cells expressing DA ATM showed defects in cell cycle arrest after both CPT and arsenite treatment. These differences are not due to changes in the S phase populations of the cell lines expressing the various ATM alleles (Fig. S5). These results further establish that there are two distinct mechanisms to activate ATM—DNA damage and oxidative stress—and indicate that this separation of function also applies to the activation of the intra-S phase checkpoint.
Stress-specific activation of ATM controls ROS, mitochondrial function, and autophagy
ATM-deficient cells have been reported to have higher levels of reactive oxygen species and to have impaired mitochondrial function (2, 39, 40), although the underlying basis of the mitochondrial phenotype is not clear. We have also previously reported that ROS levels are higher in ATM-deficient lymphoblast cells overexpressing the CL allele of ATM compared to cells overexpressing the wild-type allele (3). Here we confirm that expression of the CL allele in U2OS cells depleted of ATM results in a higher amount of ROS compared to cells expressing the wild-type allele, as measured by the fluorescent reporter for total ROS levels, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Fig. 4A). In contrast, cells expressing the 2RA allele showed wild-type ROS levels, indicating that in untreated human cells, ATM responds to oxidative stress to control ROS levels. Because many types of reactive oxygen species exist in human cells, we sought to determine whether an excess of superoxide accounts for the increase measured by H2DCFDA. For this we used the dihydroethidium (DHE) fluorescent reporter which monitors superoxide and found that cells expressing CL ATM showed significantly lower levels of superoxide than cells expressing wild-type ATM (Fig. 4B). These results suggest that cells expressing CL ATM exhibit defects in redox homeostasis but do not accumulate superoxide, an observation generally consistent with a previous finding that overexpression of superoxide dismutase in ATM-deficient mice exacerbates rather than alleviates their radiation sensitivity and hematopoietic abnormalities (41). Results using the previously described human lymphoblast AT1ABR cell line (42) with an inducible CL allele also show this pattern of DHE staining (Fig. S6).
Fig. 4. The mechanism of ATM activation determines the functional response of ATM in cell cycle checkpoint regulation and in ROS homeostasis.
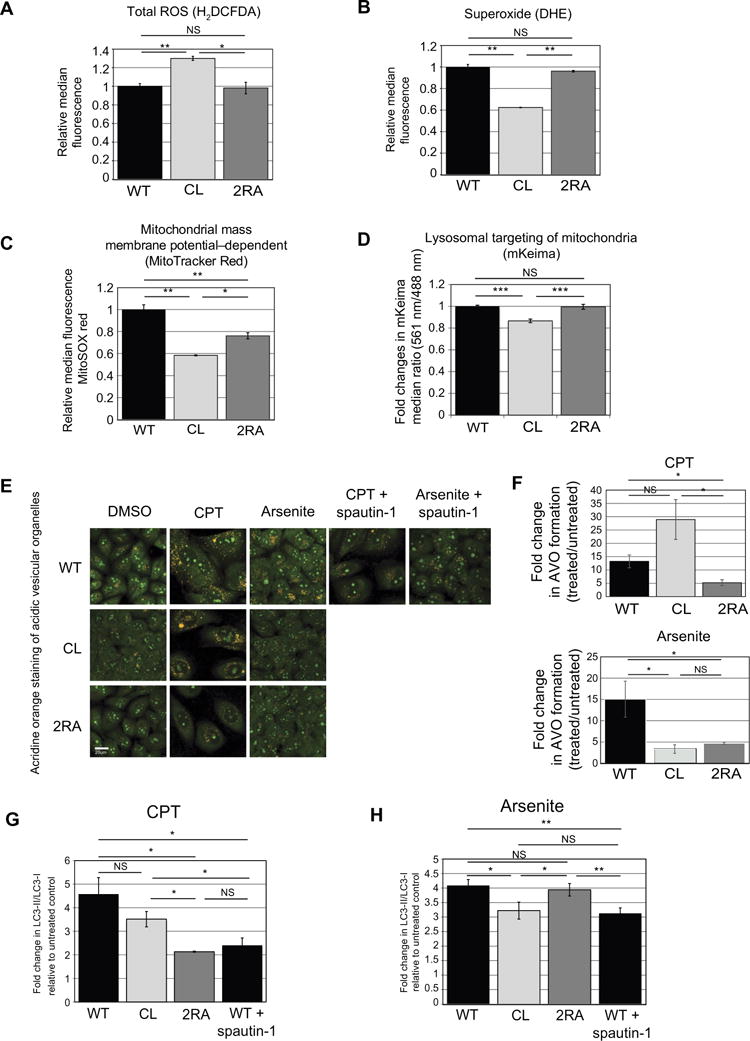
ROS levels were measured in U2OS cells depleted of endogenous ATM with shRNA (Fig. 2) and inducibly expressing various ATM alleles (1 μg/ml doxycycline) using the general ROS indicator H2DCFDA (A) or a probe for superoxide levels DHE (B). Fluorescence was analyzed by flow cytometry using 10,000 cells per cell line and normalized with data from cells expressing wild-type ATM. (C) Cells expressing various ATM alleles as indicated were analyzed for membrane potential-dependent mitochondrial mass using MitoTracker Red staining, followed by analysis using flow cytometry with 10,000 cells per cell line and normalization with data from cells expressing wild-type ATM. Data are means ± S.D. from 3 independent experiments. *p<0.05 and **p<0.005. (D) U2OS Flp-In cells expressing wild-type, CL, or 2RA alleles were infected with retrovirus containing the mKeima mitochondrial-targeted pH indicator and pH changes in the mitochondria were measured by comparison of the emission when excited at 561 nm or at 488 nm. The Y axis shows the ratio of the emission values, normalized to the cells expressing the wild-type allele. Quantitation of the mean (± S.D.) emission ratios (561 nm / 488 nM) are each from 4 independent experiments with 10,000 cells analyzed per cell line. *** p<0.0005. (E) Acridine orange was used to stain acidic vesicular organelles (AVO) in U2OS cells expressing wild-type, CL, or 2RA alleles, with either no treatment, CPT (5 μM), or arsenite treatment (100 μM) as indicated. The autophagy inhibitor Spautin-1 (10 μM) was used to confirm that acridine orange reflects autophagy-dependent vesicles. (F) Acridine orange staining in U2OS cells expressing wild-type, CL, or 2RA alleles, with either no treatment, CPT (5 μM), or arsenite treatment (100 μM) as indicated was quantified by FACS, here showing the fold increase in the percentage of cells with 585 nm emission relative to untreated cells. Data are means ± S.D. of 3 biological replicates with approximately 5,000 cells measured per replicate. * p<0.05. (G) and (H) U2OS cell lines expressing wild-type or mutant cell lines as indicated were treated with 5 μM CPT or 100 μM Arsenite for 120 min, followed by 48-hour recovery, then western blotting for LC3-II and LC3-I. Data are means ± S.D. from 3 independent biological replicates, plotted as fold increase relative to the untreated controls.
Because high amounts of ROS are often associated with mitochondrial dysfunction and loss of mitochondrial integrity has been reported in ATM-deficient cells, we asked whether a failure in oxidative activation of ATM is responsible for this phenotype. To do this we measured mitochondrial mass (dependent on membrane potential) with Mito Tracker Red CMXRos (43). The mitochondrial membrane potential in cells expressing CL ATM was reduced in ATM-deficient cells compared to that in cells expressing wild-type ATM (Fig. 4C), and the cells expressing the 2RA showed an intermediate level. These results indicate that both activation pathways affect mitochondrial function, although the CL mutation blocking ATM activation via oxidative stress exhibits a more striking effect. Similar to the assays for ROS levels, the mitochondrial assays also showed a strong effect of the CL allele in the lymphoblast AT1ABR cell line (Fig. S6). We also quantitated the amounts of carnitine and its derivatives, given that acyl carnitine metabolites are reportedly biomarkers of mitochondrial stress (44), and a recent study also showed that disruption of carnitine homeostasis resulted in mitochondrial dysfunction (45). Overall levels of carnitine and its derivatives were significantly higher in A-T patient lymphoblast cells expressing CL ATM than in cells expressing wild-type ATM (Fig. S6), consistent with the hypothesis that mitochondrial function in cells expressing CL ATM is impaired.
Lastly, we investigated whether turnover of mitochondria and other organelles is affected by the ATM separation of function alleles by measuring mitophagy and autophagy, respectively. We examined mitophagy in the cell lines by expressing a mitochondrial targeted pH indicator protein that responds to different wavelengths of light in a manner that depends on the pH of the environment (46, 47). The amount of lysosome-dependent mitochondrial turnover can thus be measured by the pH of the mitochondria-targeted probe. We tested the mKeima probe using the mTOR inhibitor rapamycin, which induces mitophagy (48), in comparison to wortmannin, which blocks mitophagy through inhibition of PI3 kinase (49, 50). In wild-type U2OS cells, rapamycin increased the fold change in mKeima emission whereas wortmannin reduced the ratio, as expected (Fig. S7). To test the ATM alleles for effects on mitophagy, we expressed the mKeima sensor in cells treated with shRNA against ATM and expressing either the wild-type or CL alleles. We found that the ratio of emission with 561 nm excitation relative to emission with 488 nm excitation was significantly lower in cells expressing the CL allele compared to wild-type or 2RA-expressing cells (Fig. 4D), indicating less efficient delivery of mitochondria to the lysosome.
In addition to mitophagy, ATM has also been implicated generally in the control of macroautophagy, the process by which proteins and organelles are degraded and recycled (51–54). To determine if the either pathway of ATM activation affects control over macroautophagy we used acridine orange to quantify acidic lysosomal vesicles in the U2OS cell lines expressing ATM alleles. This technique has previously been used to identify a role for ATM in adriamycin-induced autophagy (54), which also confirmed the specificity of this technique for ATM-dependent autophagic flux using p62 and LC3 quantitation. Here we found that both CPT and arsenite treatments increased the number of acidic vesicles in U2OS cells, and that this increase was largely blocked by the macroautophagy inhibitor Spautin-1 (55)(Fig. 4E). While all the cell lines exhibited an increase in acridine orange staining with stress, the cells expressing the 2RA ATM allele showed significantly less change in comparison to untreated cells, in response to either treatment (Fig. 4, E and F). In contrast, cells expressing the CL ATM allele were more proficient than cells expressing wild-type ATM in their response to CPT but were deficient in their response to arsenite (Fig. 4E, F). In addition, we analyzed cells depleted for ATM in the absence of mutant allele expression and also observed a deficiency in CPT and arsenite-induced acidic vesicles (Fig. S8).
To confirm this result using a different method, we also measured he amount of lipidated, membrane-bound LC3 (LC3-II) relative to soluble LC3 (LC3-I) which is a commonly used marker for autophagosome formation (56). Quantitation of these ratios from 3 experiments showed that expression of the 2RA allele of ATM during CPT treatment reduced the amount of LC3-II to a similar level as seen in wild-type cells treated with Spautin-1, an inhibitor of macroautophagy (55), whereas expression of the CL allele had an intermediate effect with this treatment (Fig. 4G). In contrast, the amount of LC3-II in response to treatment with arsenite was only reduced with expression of the CL allele, which was equivalent to the effect of Spautin-1in wild-type cells (Fig. 4H). Thus, the oxidative response of ATM is critical for an increase in macroautophagy after oxidation.
ATM activation via oxidative stress is required for CK2 function
Our studies using ATM protein that is deficient in oxidative activation suggested there are global effects on redox homeostasis and metabolism. To examine these effects in detail and also to determine whether there are specific phosphorylation targets downstream of ATM for each activation pathway, we performed phosphoproteomic analysis using ATM-deficient AT1ABR patient lymphoblasts expressing wild-type, CL, 2RA, or 2RA+CL ATM alleles. Through this analysis, we identified 2,694 phosphopeptides with a false discovery rate of 1% (table S1). A histogram of the raw intensities from each cell line shows the phosphoproteomes are grossly similar, as expected (Fig. 5A), and comparisons of the nonparametric Spearman’s rank correlation coefficient, ρ, also suggest the phosphopeptide quantifications are grossly similar across all the cell lines (Fig. 5B). However, examination of the ρ values reveals that the cells expressing 2RA and CL ATM were the most dissimilar in phosphopeptide quantitation while cells expressing CL and 2RA+CL ATM were the most similar to each other and to the uncomplemented AT1ABR cell line. Specifically, comparison of the CL/AT1ABR Spearman rank correlation coefficient with the 2RA/AT1ABR coefficient after Fisher z transformation yields a p-value < 0.00001, as did comparisons of the CL/WT with the 2RA/WT data. Thus, cells expressing the wild-type or 2RA alleles were statistically very different from the parental A-T cell line compared to cells expressing the CL ATM alleles, and the expression of ATM C2991L results in fewer changes in the phosphoproteome compared to the ATM-deficient cell line.
Fig. 5. Phosphoproteomic analysis of A-T patient lymphoblast cells expressing an oxidation-deficient ATM mutant shows defects in global phosphorylation patterns.
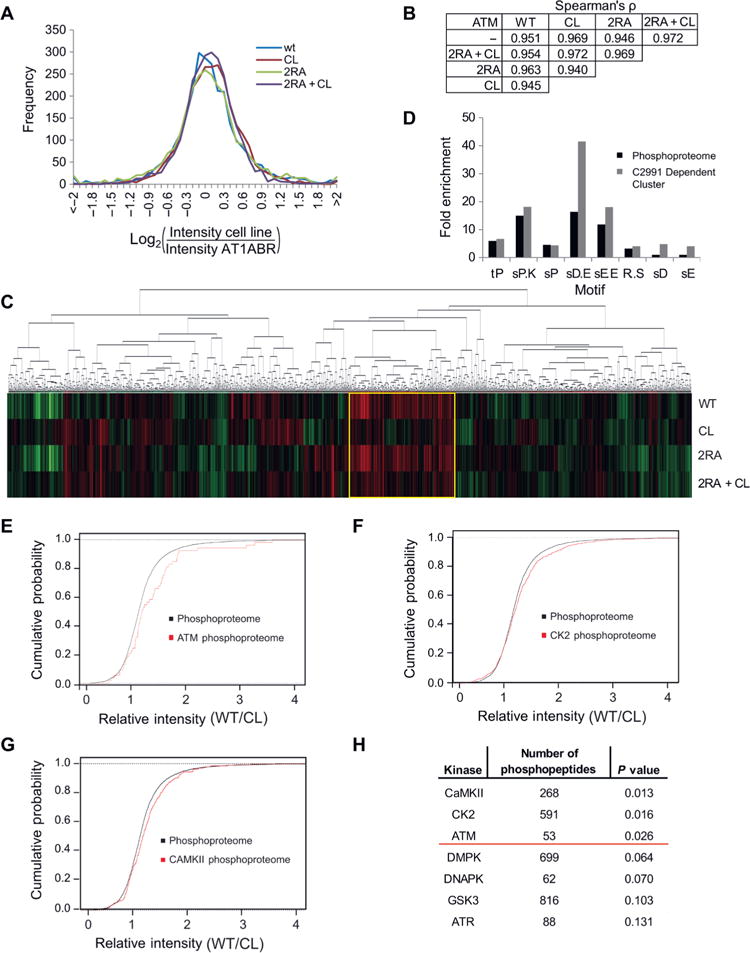
(A) AT1ABR lymphoblast cells deficient in wild-type ATM were complemented with inducible expression of wild-type, CL, 2RA, or 2RA+CL ATM alleles. Phosphopeptides were enriched and analyzed by mass spectrometry. A histogram of total phosphopeptide counts (log2 values) in each cell line is shown after normalizing raw values for each cell line to the parental AT1ABR cell line. (B) Pairwise comparisons of the phosphoproteomic raw data from AT1ABR cells with or without expression of ATM. The raw intensity of each phosphopeptide was analyzed in pairwise comparisons of each cell line, and Spearman’s rank correlation coefficient, ρ, was calculated for each pairwise comparison. (C) Hierarchical clustering of all the phosphopeptides. Phosphopeptide levels were normalized to the parental AT1ABR cell line. Each vertical line within a cell line is a phosphopeptide and vary from green to black to red to represent decreased, equal, or increased levels of the phosphopeptides compared to the parental AT1ABR cell line, respectively. (D) Phosphopeptides from the phosphoproteome or the C2991 Dependent Cluster [indicated by the yellow box in (C)] were analyzed with motif-x. Sequences contained 6 residues N-terminal and C-terminal of each phosphorylation event. Fold enrichment is shown relative to the abundance of motifs in the proteome. (E and F) Empirical cumulative distribution functions of the ratio of phosphopeptide intensities of cells expressing wild-type ATM or cells expressing the CL allele,comparing the phosphoproteome and the predicted ATM phosphoproteome (E) or the predicted CK2 phosphoproteome (F). (G) Results from Kolmogorov-Smirnov tests showing the predicted number of phosphopeptides in the dataset for each kinase and the p-value of the observed changes in the CL and 2RA+CL expressing cell lines relative to cells expressing the wild-type allele. The red line marks p = 0.05.
Analysis of the total phosphoproteome dataset by hierarchical clustering revealed a group of 314 peptides significantly less phosphorylated in the parental AT1ABR cells and cells expressing CL ATM (both CL ATM and 2RA+CL ATM), hereafter referred to as the C2991 Dependent Cluster (Fig. 5C; in the heat map, all values were normalized to the uncomplemented AT1ABR cell line phosphopeptide signal). Examination of the sequences surrounding these phosphorylation sites revealed that few of these phosphopeptides contained an S/TQ motif, the canonical sequence for ATM targets, whereas the remaining sites showed sequences distinct from this motif. Because ATM was not the predominant kinase responsible for phosphorylating the proteins in this group, we performed motif-x analysis (57, 58) to extract motifs from the phosphoproteome and the C2991 Dependent Cluster to identify which kinase(s) might be responsible for phosphorylating each phosphosite. Nine motifs were extracted from the C2991 Dependent Cluster – tP, sPXK, sP, sDXE, RXXs, sD, and sE – and the results were compared to the motifs from the entire phosphoproteome. The four motifs in which acidic residues followed the phosphorylated residue, which is typical of substrates for the protein kinase CK2, were significantly enriched in the C2991 Dependent Cluster (Fig. 5D).
To further characterize the kinases that could be phosphorylating the phosphopeptides identified in the C2991 Dependent Cluster, the ratios of intensities of phosphopeptides from cells expressing wild-type or CL ATM in the phosphoproteome and each kinase-specific predicted phosphoproteome were analyzed using the two-sample Kolmogorov-Smirnov (K-S) test, a nonparametric test to determine whether two empirical cumulative distribution functions (ECDFs) are from the same or separate distributions. The K-S statistic is the largest difference between two ECDFs. If the K-S statistic is above a critical value, the null hypothesis that the samples are drawn from the same distribution is rejected, suggesting predicted substrates of the kinase are either over- or under-represented in the C2991 Dependent Cluster. We used Group-based Prediction system (59) to predict the substrates for each kinase based on known substrates. These putative substrates were entered as the phosphoproteomes of each kinase, the ratios of the phosphopeptides in wild-type vs. CL ATM expressing cells were calculated, and the ECDFs of the kinase-specific phosphoproteomes and the full phosphoproteome were compared using the K-S test. From this analysis, the phosphorylation of substrates by the kinase CaMKII, CK2, and ATM were predicted to depend on oxidation-induced ATM activation, given that the ECDFs of the phosphoproteome and the kinase-specific phosphoproteomes were significantly different (Fig. 5E, F, G, H). As the WT/CL ratio of these phosphopeptides is shifted to the right, these phosphopeptides had lower abundances in cells expressing CL ATM. In contrast, the ECDF of CK1 and several other cellular kinases tightly overlaps the ECDF of the full phosphoproteome, suggesting that there is no difference between the CK1 phosphoproteome and the entire phosphoproteome as measured by the K-S test (Fig. 5H Fig. S9).
Functional ATM is required to maintain cellular ROS amounts and CK2 activity
Because a reduction in CK2 activity in AT1ABR cells expressing CL ATM was suggested by the phosphoproteomic analysis, we investigated the causes of this phenomenon. First, we compared the abundances of CK2 subunits - CK2α, CK2α′, and CK2β - in AT1ABR cells expressing wild-type and CL ATM. Cells expressing CL ATM had similar amounts of all three subunits compared to cells expressing wild-type ATM, suggesting that the reduction in predicted phosphorylation targets of CK2 is not due to loss of protein (Fig. S10). We then examined whether there is a change in CK2 activity in the absence of ATM function by immunoprecipitating CK2 complexes expressed in HEK-293T cells in the presence or absence of 10 μM KU-55933, an ATM inhibitor. We tested the immunoprecipitated CK2 in an in vitro assay and observed a reduction in CK2 activity with the enzyme isolated from ATM inhibitor-treated cells compared to the untreated cells (Fig. 6A). However, there was little difference in the specific activity of the kinase with or without ATM inhibitor when the kinase activity was normalized to the level of CK2β-V5 protein immunoprecipitated. We found overall immunoprecipitated CK2β levels approximately 2-fold lower in HEK-293T cells in the presence of ATM inhibitor than in cells without treatment, although there were similar amounts of CK2β-V5 in the input lysates of the untreated and KU-55933 treated cells (Fig. 6B). This suggests that ATM inhibition reduces the efficiency of CK2β immunoprecipitation and corresponding levels of CK2 activity in the in vitro assay.
Fig. 6. Loss of ATM oxidative stress activation causes aggregation of CK2β.
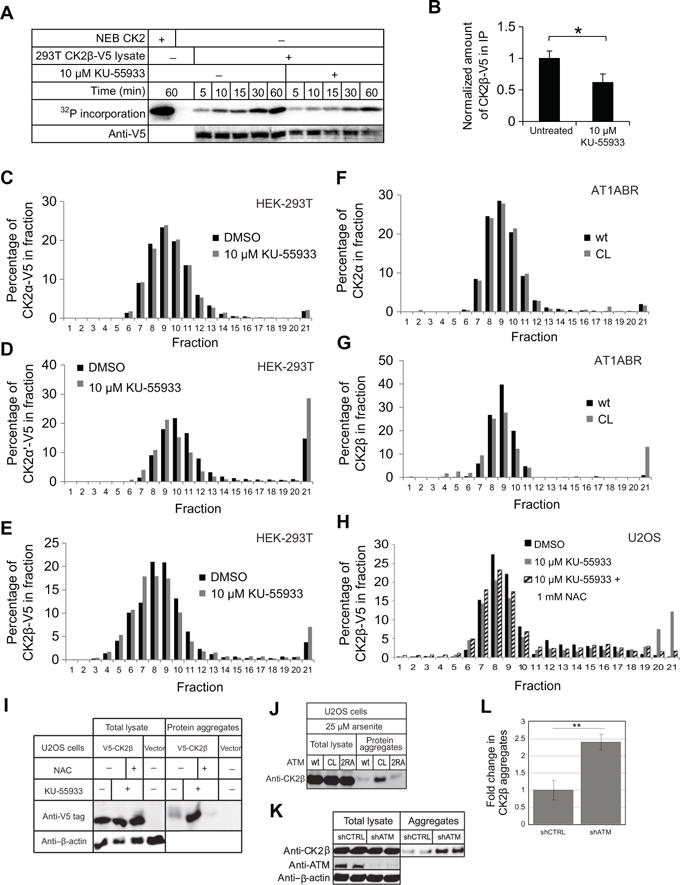
(A) In vitro kinase assay with immunoprecipitated CK2. HEK-293T cells stably expressing V5-tagged CK2β were treated with 10 μM ATM inhibitor (KU-55933) or an equivalent amount of DMSO for 16 hours and CK2β was immunoprecipitated with magnetic anti-V5 beads. CK2 was incubated with 1.67 mM ATP, 5 fCi ATP-γ-32P, and 1.15 μg of GST-CK2 substrate for 1 hour, and 32P-labeled substrate was analyzed by phosphorimager. (B) Normalized amount of immunoprecipitated CK2β-V5 from (A) with the levels of CK2β-V5 normalized to levels in mock-treated cells. Quantitation was performed on the Licor system using Image Studio Ver 4.0. P-values: * < 0.05 (C-E) Distribution profile of CK2 subunits CK2α (C), CK2α′ (D), and CK2β (E), in HEK-293T cells after sucrose gradient sedimentation. HEK-293T cells expressing CK2α-V5, CK2α′-V5, or CK2β-V5 were treated with 10 μM KU-55933 or an equivalent amount of DMSO for 16 hours, harvested, and lysed in the absence of detergent. 1 mg of lysate in 500 μl lysis buffer total was added to the top of a sucrose gradient made with 1 ml layers of 50% to 5% sucrose in 5% increments. After ultracentrifugation, 500 μl fractions were collected and analyzed by western blot. (F and G) the distribution profile of CK2α (F) and CK2β (G) from the AT1ABR cells expressing wild-type or CL ATM alleles, analyzed by sucrose gradient sedimentation. Lysates were analyzed as in (C-E) except with induced AT1ABR cells expressing wild-type or CL alleles of ATM as indicated. (H) Sucrose gradient sedimentation pattern of V5-tagged CK2β stably expressed in U2OS cells treated with 10 μM KU-55933 or an equivalent amount of DMSO for 16 hours, or treated with a combination of KU-55933 and 1 mM NAC, as indicated. (I) Analysis of detergent-resistant aggregates in U2OS cells treated with 10 μM KU-55933 or DMSO, or a combination of KU-55933 and 1 mM NAC, as indicated. Aggregate fractions were isolated and compared with total lysate using western blot analysis for stably expressed V5-tagged CK2β. (J) Analysis of detergent-resistant aggregates in U2OS Flp-In cells expressing wt, CL, or 2RA ATM treated with 25 μM arsenite as shown in (I). (K) Aggregate fractions were isolated from U2OS cells expressing shCTRL or ATM shRNA and probed for CK2β, ATM, and β-actin as indicated from two independent cultures. (L) Means ± S.D. of CK2β abundance in aggregates were quantified from 3 independent experiments [including the replicates shown in (K)]. ** p< 0.005.
CK2 is composed of two catalytic alpha subunits (CK2α or CK2α′) bound to two beta subunits (CK2β) in a heterotetramer, with larger oligomeric forms also observed (60). The CK2 enzyme is known to aggregate in response to oxidative stress (61–64), with the CK2α′ specifically shown to be prone to aggregation (65). Based on this evidence, we hypothesized that CK2 aggregation may be the cause of reduced activity and reduced efficiency of immunoprecipitation from ATM inhibitor-treated cells. To test this possibility, we treated cells expressing V5-tagged CK2α, CK2α′, or CK2β with KU-55933 or DMSO as a control and separated protein in the lysates using sucrose gradient sedimentation, followed by Western blotting. CK2α-V5 showed a similar distribution pattern in ATM inhibitor treated cells and untreated cells (Fig. 6C). However, a subset of CK2α′-V5 and CK2β-V5 protein showed a shift to the denser fractions, and ATM inhibitor-treated cells showed a 2-fold increase in the level of protein observed in these fractions (Fig. 6, D and E). To examine whether this ATM inhibitor-triggered aggregation is related to the reduction of endogenous CK2 activity in AT1ABR cells and to use an inhibitor-independent approach, we compared the distribution profile of CK2α or CK2β from the AT1ABR cells expressing wild-type or CL ATM after sucrose gradient analysis. Although CK2α showed similar distributions in both cell lines (Fig. 6F), CK2β again showed a large (~10-fold) increase in the aggregated fraction in the cell line expressing the CL allele compared to the wild-type expressing line (Fig. 6G). This is similar to aggregation previously reported for CK2 and may provide an explanation for the reduction in overall CK2 activity in AT1ABR cells expressing the CL ATM allele.
Because the C2991L ATM mutant is impaired in oxidative activation by H2O2 (3) and human cells expressing CL ATM exhibit higher levels of ROS (Fig. 4A and fig. S1), it is possible that the increase in ROS caused by ATM inhibition may cause CK2β aggregation. To test this hypothesis, we compared distribution profiles of CK2β expressed in U2OS cells treated with ATM inhibitor by itself or in the presence of the reducing agent N-acetylcysteine (NAC). As expected, CK2β showed a dramatic shift to the denser fraction with 20% of the protein appearing in the aggregated form from cells treated with ATM inhibitor. However, in the presence of both ATM inhibitor and 1 mM NAC, the levels of CK2β aggregate were significantly reduced and similar to the levels in untreated cells (Fig. 6H).
To confirm this, we isolated aggregated proteins from cellular lysates by sequential detergent extractions and sedimentation analysis (66). The level of CK2β-V5 in the insoluble fraction containing protein aggregates was increased 4-fold in the ATM inhibitor-treated cells in comparison to the untreated cells and was dramatically reduced in cells treated with both ATM inhibitor and NAC (1 mM), although CK2β levels were identical in the total lysates (Fig. 6I). Similarly, expression of the CL mutant allele in U2OS cells also promoted aggregation of CK2β as measured by the increased presence of CK2β in the aggregate fraction (Fig. 6J). These results suggest that functional ATM is required for maintaining protein homeostasis and that the loss of this activity of ATM alters the solubility of the CK2 kinase and thus the subset of the phosphoproteome dependent on CK2 (Fig. 5).
Widespread protein aggregation occurs in cells lacking oxidative activation of ATM
Our observation of CK2β aggregation in cells expressing the CL allele of ATM suggested that other polypeptides may also be prone to aggregation in these cells. To identify these, we isolated the detergent-insoluble fraction as described above in U2OS cells expressing the wild-type, CL, or 2RA alleles of ATM and analyzed the proteins by mass spectrometry. We also did this analysis with concurrent arsenite treatment to model conditions of oxidative stress. The results show that a small number of proteins are enriched in both CL- and 2RA-expressing cells compared to wild-type (approximately 40 per cell line; see also Table S2) but that a large number of proteins (approximately 500) appear in the aggregate fraction of CL-expressing cells exposed to arsenite (Fig. 7A). The fold enrichment of these proteins relative to their levels in the cells expressing the wild-type allele is over 100-fold for some polypeptides (> 1.5-fold shown in Fig. 7B). The levels of CL/WT enrichment for CK2β and CAMKIIδ kinases in the aggregate fraction are 9-fold and 150-fold, respectively (Table S2). Gene ontology analysis of the proteins that appear in the aggregate fraction of the cells expressing the CL allele show a strong enrichment for nuclear proteins including the RFC complex, DNA repair proteins (including Mre11 and Rad50), and RNA processing enyzmes (Fig. 7C). The presence of Rad50 in the aggregate fraction was validated by western blot (Fig. S11). The aggregated proteins isolated from the cells expressing the CL allele of ATM are also predicted to have a higher risk of aggregation, based on the TANGO and WALTZ algorithms that estimate aggregation propensity and amyloid-like structures (67, 68) and are significantly longer than average polypeptides in human cells (Fig. 7D). Overall, this analysis shows that expression of mutant ATM that cannot be activated by oxidative stress results in a global increase in protein aggregation, particularly in cells that are also exposed to low levels of ROS.
Fig. 7. Loss of ATM oxidative stress activation causes global protein aggregation.
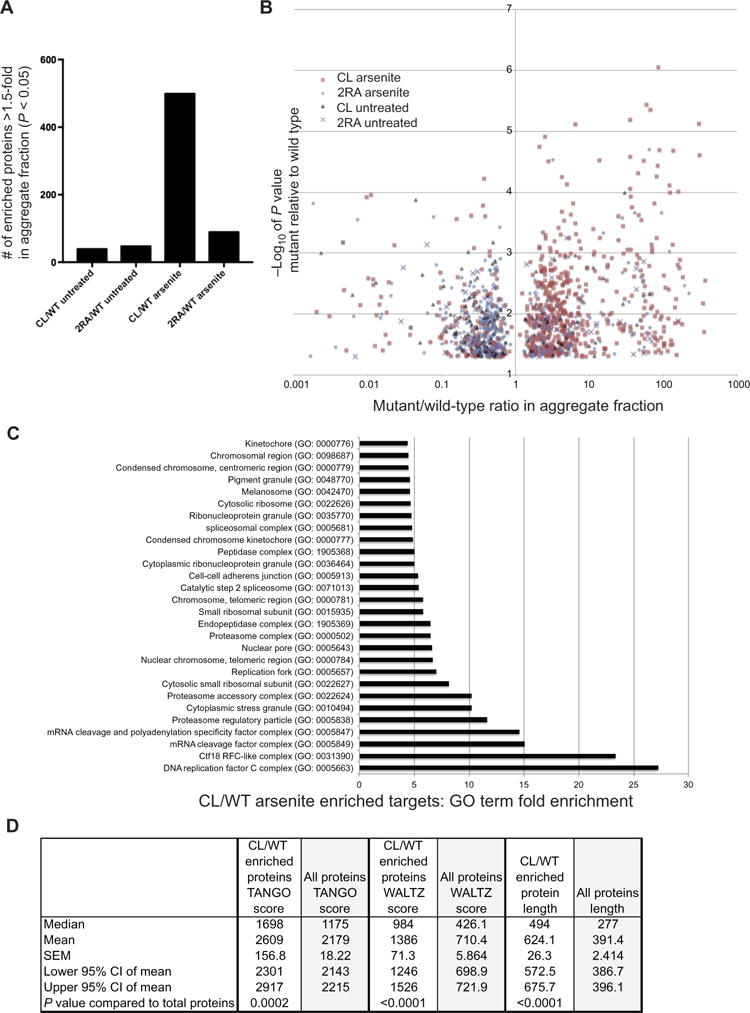
(A) U2OS cells depleted for endogenous ATM and inducibly expressing wild-type, CL, or 2RA alleles with or without concurrent arsenite treatment (25 μM) were lysed, and detergent-resistant aggregate fractions were isolated and analyzed by mass spectrometry using label-free quantification. 3 biological replicates were analyzed for each cell line, and proteins enriched by ≥ 1.5-fold in cells expressing the CL or 2RA alleles compared to the level in cells expressing wild-type ATM are shown (only those with p< 0.05 by a t-test). (B) The fold-enrichment over wild-type for proteins identified in (A) is shown for each target. (C) Gene ontology analysis of proteins enriched in aggregate fraction of cells expressing the CL allele with arsenite treatment (Panther Gene Ontology database). The analysis included Bonferroni correction for multiple testing; all results shown had p < 0.05. (D) Comparison of TANGO and WALTZ scores for 497 polypeptides isolated from U2OS cells expressing the CL allele with arsenite treatment in (A) compared to the entire proteome. Polypeptide length is shown in amino acids. N = 3 biological replicates per sample; p-values by two-tailed t-test.
Discussion
The ATM protein kinase responds to both DNA double-strand breaks as well as to oxidative stress. In this work we identified two adjacent arginine residues, R2579/R2580 (2RA), at the end of the FAT domain in ATM that are essential for ATM activation by MRN and DNA in vitro. These mutations do not affect H2O2-dependent ATM activation, indicating that 2RA ATM is only blocked in MRN-dependent ATM activation, likely due to the deficiency in binding to Mre11/Rad50 that we have observed here. However, it is currently unknown if the R2579/R2580 region of ATM directly interacts with Mre11/Rad50. Heat repeats 17/18 and 21/22 in S. pombe Tel1, which are N-terminal to the location of R2579/2580 in human ATM, were identified as Nbs1-binding regions previously (69), but a detailed understanding of this interaction will likely require structural analysis.
Most of our functional characterization of the 2RA ATM allele was performed with a tet-inducible system in human U2OS cells combined with shRNA-mediated depletion of the endogenous protein. This conditional system is preferable to analysis of A-T patient cells expressing mutant alleles because ATM-deficient cells are extremely sensitive to transfection and because ATM deficiency induces adaptive compensatory responses over time that are not fully understood and can confound analysis of mutant alleles (2). Using this inducible system, we found that survival of CPT-induced DNA damage, checkpoint activation, and resection of DNA ends were specifically impaired with expression of the 2RA ATM allele, suggesting that the activation of ATM via the MRN/DSB pathway is specifically responsible for these DNA damage-related outcomes.
In contrast to cells expressing the 2RA allele, cells expressing the C2991L allele of ATM (2, 3) fail to phosphorylate downstream substrates in response to low levels of peroxide or arsenite stress, a defect most apparent with phosphorylation of Chk2. Similarly, these cells failed to activate the intra-S phase checkpoint in response to arsenite exposure and showed poor survival in clonogenic assays after this treatment. Our results with arsenite in this work as well as our previous observations with hydrogen peroxide indicate that the oxidative pathway of ATM activation is specifically required for ATM responses to ROS in human cells.
With expression of the C2991L of ATM, we observe higher overall ROS levels compared to cells expressing the wild-type allele, while levels of superoxide are clearly lower with expression of the mutant. This is generally consistent with our previous observations as well as a number of other groups indicating high levels of ROS when ATM activity is absent (2, 3). Our previous results with hydrogen peroxide in cells and in vitro are consistent with a higher level of this compound in ATM-deficient cells, which is also supported by a report that overexpression of mitochondrial-targeted catalase (which removes hydrogen peroxide) reduces or even rescues hematopoiesis deficiencies, thymic lymphomas, and immune system function in mice lacking ATM (70). Catalase activity is lower in ATM-deficient human cells compared to normal controls (71) and in ATM−/− mice where the deficit was reported specifically in the cerebellum (72), an important detail given the Purkinje cell specificity of the A-T disorder in humans. The ATM protein has also been reported to be associated with peroxisomes (52, 73), the location of catalase activity in human cells, which is perhaps relevant to the overall deficit in catalase function in cells lacking ATM.
Mitochondrial dysfunction has previously been observed in ATM-deficient mouse thymocytes and human cells; specifically, an increase in total mitochondrial mass, a decrease in mitochondrial complex I activity, a loss of mitochondrial membrane potential, and a decrease in mitophagy were measured in the absence of ATM function (21, 39, 40). Our experiments with the C2991L ATM mutant indicate that a loss of oxidative activation of ATM does lead to lower levels of mitochondria with normal membrane potential as well as a deficiency in mitophagy. Lastly, levels of carnitine derivatives are significantly higher in cells expressing the ATM C2991L allele, indicating aberrant regulation or flux in fatty acid oxidation pathways, perhaps as a compensatory mechanism in response to oxidative stress. Carnitine derivatives are important for transport of long-chain fatty acids into the mitochondria and have been observed to act as neuroprotective agents (74). Treatment of lymphoblasts from A-T patients with L-carnitine reduced levels of chromosomal abnormalities after exposure to oxidative DNA damage (75). Taken together, it is clear that there are changes in mitochondrial function that occur with ATM inhibition or deletion, and that the C2991L allele that lacks oxidative stress activation promotes these changes.
Consistent with the mitophagy observation, we also observed that cells expressing the ATM C2991L allele are specifically deficient in arsenite-induced macroautophagy, a process that recycles proteins and organelles. ATM has previously been shown to regulate autophagy through a pathway that is induced by ROS and controls mTOR activity through LKB1, AMPK, and TSC2 (51). We found that cells expressing the 2RA ATM allele are deficient in CPT-induced macroautophagy, reminiscent of the role for ATM in adriamycin induction of autophagy reported recently (54), and also exhibits a deficit in response to arsenite. It is not clear why the 2RA-expressing cells show this sensitivity while most of the other readouts of ATM function in oxidative stress are normal in these cells, although it is clear that the 2RA cells also show a modest increase in protein aggregation in response to arsenite as well.
Our analysis of global phosphorylation patterns in cells expressing the ATM separation of function alleles in the absence of exogenous stress pointed toward effects of ATM on other kinases: CAMKII and CK2. We focused on CK2 in this work, an enzyme that is often considered to be constitutively active although cellular localization and protein-protein interactions appear to be critical for modulating its effects and substrates (76). Importantly for this study, CK2 is known to aggregate in vivo and in vitro and this property has been shown to negatively affect its kinase activity (60–64, 77, 78). Our results indicate aggregation of a subset of CK2 in cells expressing the C2991L allele of ATM that is specific to the CK2α′ subunit and CK2β. This aggregation is relieved by treatment with NAC, thus it appears to be a result of increased ROS, although we cannot rule out the possibility that downstream effects of ATM deficiency may be alleviated by antioxidants indirectly. This collaborative relationship between ATM function and CK2 activity is consistent with previous observations that a significant fraction of DNA damage-induced phosphorylation events are predicted to be catalyzed by CK2 and that these are reduced upon inhibition of ATM (79). Since CK2 is an important enzyme in the DNA damage response, phosphorylating Mdc1 and promoting Nbs1 association with DSBs as well as many other critical interactions between signaling and repair proteins (80), the partial loss of CK2 function in the absence of ATM oxidative activation is likely to also have effects on the DNA damage response.
Our analyses of protein aggregates formed with expression of the C2991L allele of ATM during arsenite exposure identified approximately 500 polypeptides that form detergent-resistant precipitates more efficiently than in the cell line expressing wild-type ATM. These proteins only appear in the detergent-resistant aggregate fraction with low-level arsenite treatment and expression of the CL allele, suggesting that the combined effects of these treatments result in large-scale protein aggregation. It is interesting that the most over-represented groups of polypeptides among these are those that function in the DNA metabolism and gene expression. ATM has been implicated in the control of RNA splicing (81) and many groups have noted the relationships between efficient RNA processing and genomic stability (82). It is not yet clear why these specific proteins show aggregation with loss of ATM function, but the predicted aggregation propensity of the proteins we identified is significantly higher than the total proteome. The aggregation of proteins, particularly in neurons, is very likely relevant to the loss of Purkinje cell function that is the hallmark of the A-T disorder as protein aggregation is a common link between many if not all neurodegenerative diseases (83).
Loss of normal proteostasis in A-T patients, particularly if it is centered on proteins relevant to DNA metabolism, may play a critical role in the pathogenesis of A-T. Further investigation will clearly be required to determine how this phenomenon relates to the A-T phenotype and also how this relates to ATLD patients who have deficiencies in Mre11 or Rad50 (84–87); these patients are hypomorphic for MRN function yet also experience progressive neurodegeneration. Lastly, it is important to investigate the basis of the extreme dominant-negative effect of kinase-deficient ATM on cells. This is almost certainly related to the embryonic lethal phenotype of this allele in the mouse (33, 34), but it is currently unknown whether this is caused by stable binding of the mutant ATM to certain substrates and if so, which substrates are the cause of the cellular toxicity during stress responses and during development.
Materials and Methods
Cell culture and ATM induction
U2OS T-Rex FLP-in cells containing control vector, wild-type, R2579A/R2580A, C2991L, or D2889A ATM alleles were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) containing 15 μg/ml Blastocidin (A1113903, Life Technology), 100 units/ml penicillin-streptomycin (15140-122, Life Technology), and 200 μg/ml Hygromycin (400052-50ml, Life Technology). Depletion of endogenous ATM was performed by incubating cells with lentivirus containing shRNA toward ATM (sc-29761-SH, Santa Cruz Biotechnology) overnight and selecting with media containing 1 μg/ml puromycin (Invitrogen) for 5-7 days. To induce wild-type ATM or mutant ATM, doxycycline (1 μg/ml or as indicated) was added to the media as final concentration 3 days before treatment with DNA damage or ROS-inducing agent. For experiments with H2O2 (H325-100, Fisher) or arsenite (S7400-100G, Sigma), media was changed to serum-free media when cells were treated and changed back to media containing serum and appropriate antibiotics after treatment.
HEK-293T cells (ATCC, CRL-11268) were grown in DMEM supplemented with 10% FBS. AT1ABR cells and AT1ABR cells expressing the various pMAT1-ATM constructs were a gift from M. Lavin. AT1ABR cells were grown in RPMI-1640 medium (Sigma, R8758) supplemented with 15% FBS. The media for AT1ABR cells with pMAT1-ATM constructs was supplemented with 200 μg/ml hygromycin B (EMD Millipore, 400052) to select for pMAT1-ATM retention. ATM expression was induced from the metallothionein II promoter for 16 hours as described previously using 2 μM CdCl2 (42).
Virus production and transduction
HEK-293T cells were plated in 10 cm dishes and allowed to grow to near confluence. A solution of OptiMEM and plasmids was made using 20 μg pLX304 vector containing the gene of interest, 12 μg pCMV-dR8.91 (Delta 8.9), and 8 μg VSV-G and brought to 500 μl with OptiMEM. In another tube, 60 μl Lipofectamine 2000 (Invitrogen, 11668-027) was mixed 440 μl OptiMEM. The plasmid and Lipofectamine 2000 solutions were combined, incubated for 5 minutes at room temperature, and then added to the HEK-293T cells. The media was changed the next day and left for 48 hours. The media was harvested after 48 hours and replaced and then harvested again 24 hours later. The media with virus was combined, filtered with 0.45 μm filters, aliquoted in 500 μl aliquots, and stored at −20 °C for transduction. HEK-293T or U2OS cells were plated in 24-well plates and allowed to reach confluency. The media was removed and replaced with 500 μl of the viral aliquots. Polybrene (EMD Millipore, TR-1003-G) was added at a final concentration of 10 μg/ml to increase viral transduction. The cells were grown overnight and the media was replaced the next day. The following day selection agent was added - for pLX304 plasmids cells were treated with 10 μg/ml blasticidin (InvivoGen, ant-bl-1), and cells were allowed to grow for approximately one week until the control untransduced cells had died. The stable cell lines were checked for expression of V5-tagged protein and used in subsequent assays.
V5 immunoprecipitation and Western blotting
HEK-293T cells stably expressing V5-tagged CK2 subunits were lysed with 1X Cell Lysis Buffer (Cell Signaling Technology, 9803) according to the manufacturer’s instructions. Protein quantitation was performed by Bradford assay with Coomassie Plus Protein Assay Reagent (Pierce, 23236). For each immunoprecipitation, 2.5 μg lysate was combined with 4 μl mouse anti-V5 magnetic beads (Medical and Biological Laboratories, M167-9) and brought to a final volume of 50 μl with 1X Cell Lysis Buffer. Tubes were rotated for 30 minutes at 4 °C and then spun briefly. A magnetic stand was used to wash the beads 3 times with 500 μl 1× Cell Lysis Buffer. Beads were immediately used for the CK2 kinase reaction. The following antibodies were used for western blotting: mouse anti-V5 (Invitrogen, R960-25), rabbit anti-CK2α (Pierce, PA1-86381), mouse anti-CK2β (EMD Millipore, 218712), goat anti-mouse IgG IRDye 800 Conjugated (Rockland Immunochemicals, RL-610-132-121), and goat anti-rabbit IgG IRDye 700 Conjugated (Rockland Immunochemicals, RL605-430-003).
AT1ABR tissue culture for staining and qPCR
AT1ABR cells expressing wild-type-ATM or C2991L-ATM were seeded at a density of 500,000 cells/ml in 15 ml. The following day, 4 aliquots of 1.5 ml of each cell line were harvested for the uninduced controls. The aliquots were pelleted for 5 minutes at 100 g. The supernatant was removed and the cell pellets were washed with 1 ml PBS and pelleted again. The pellets were immediately stained or frozen in liquid nitrogen for subsequent RNA or protein extraction. The remaining cells were induced for 16 hours with 2 μM CdCl2. After induction, the cells were gently spun down for 5 minutes at 100 g and resuspended in fresh media without CdCl2. The cells were allowed to recover for 48 hours and 4 aliquots of 1.5 ml of each cell line were harvested and immediately stained or pelleted and frozen in liquid nitrogen for subsequent protein extraction.
Analysis of carnitine derivatives in AT1ABR cells
AT1ABR cells expressing wild-type-ATM or C2991L-ATM were grown and induced for ATM expression as described above. 6 biological replicates with 5 × 10^6 cells per sample were analyzed. Metabolic profiling was performed by Metabolon Inc. and identification of compounds was done by comparison to known purified standards.
Sucrose gradient sedimentation
Cell lysates were prepared in buffer containing 25 mM Tris, pH 8, 100 mM NaCl, and 10% glycerol supplemented immediately before use with 1 mM DTT, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM β-glycerophosphate, and 2.5 mM sodium pyrophosphate. Lysis was performed by douncing 50 times on ice. For both methods, the protein concentration was quantitated by Bradford assay with Coomassie Plus Protein Assay Reagent (Pierce, 23236). The different concentrations of sucrose were made in 20 mM Tris, pH 7.4 and 150 mM NaCl and layered in ThinWall Ultra-Clear Tubes (Beckman-Coulter, 344059) using gravity flow from a pipet bulb. The sucrose percentages from the bottom were 50%, 45%, 40%, 35%, 30%, 25%, 20%, 15%, 10%, and 5% (W/V) and each layer was 1 ml. Between 10 μg and 2 mg of lysate in 500 μl of the appropriate cell lysis buffer was added to the top of the sucrose gradient and then spun for 16 hours at 4 °C and 180,000 g in a swinging bucket SW 41 Ti rotor (Beckman-Coulter, 331362). Fractions of 500 μl were collected by pipet. Western blots were performed with 16 or 120 μl each fraction combined with 4 or 30 μl 5× SDS and loading controls of 2.5 or 100 μg lysates were run on each blot. The blots were probed as before and the percentage of probed protein was quantitated using the Li-Cor Odyssey system.
An identical sucrose gradient was performed with molecular weight markers to determine the fractions corresponding to different size complexes. The fractions were run on a NuPage 4-12% Bis-Tris Gel (Invitrogen, NP0336BOX) and the colloidal Coomassie-stained gels. Aldolase (40 kDa monomer) appeared mostly in fractions 6 and 7. Catalase (60 kDa monomer, 240 kDa tetramer) ran in fractions 8-10. Ferritin (21 kDa monomer, 500 kDa 24-mer) and thyroglobulin (330 kDa monomer, 660 kDa dimer) appeared in the sucrose gradient starting in fraction 12. However, some amount of ferritin and thyroglobulin are present through the last fraction suggesting there are larger complexes or aggregates in the sucrose gradient.
Expression constructs and protein expression
Wild-type MRN complex was expressed in Sf21 insect cells by coexpression with baculovirus for wild-type Rad50, wild-type Mre11, and wild-type Nbs1 as described previously (88). To make biotinylated MRN or MR, Nbs1 or Mre11 were modified at its C-terminus with a biotinylation epitope for the BirA enzyme and coexpressed with other component(s) as described previously (89) as well as baculovirus expressing BirA. Expression constructs for Flag-tagged wild-type and HA-tagged ATM were gifts from M. Kastan and R. Abraham, respectively. 2RA and CL ATM were generated using Quikchange XL site-directed mutagenesis (Stratagene) from wild-type ATM pcDNA3 expression plasmid (sequences available upon request). Cesium-purified plasmid DNA was used to transfect human HEK-293T cells and express recombinant ATM as described previously (24). The E.coli expression constructs for GST-p53 were described previously (88, 90). To generate U2OS T-Rex FLP-in cells expressing wt, 2RA, CL, or DA ATM, ATM gene was digested from ATM pcDNA3 expression plasmid with Not1/Apa1 and inserted into pcDNA5-FRT/TO-intron vector, which was gift from B. Xhemalce, and co-transfected with pOG44 Flp-Recombinase expression vector into U2OS T-Rex Flp-In cells (J. Parvin). Transfected U2OS cells containing inducible ATM genes were selected with 200 μg/ml hygromycin in DMEM (10% Tet System Approved FBS (Clontech, Cat No. 631106), 15 μg/ml blasticidin, and 100 units/ml penicillin-streptomycin (15140-122, Life Technology)).
ATM expression and purification
Flag- and HA-tagged wild-type ATM were generous gifts from M. Kastan and R. Abraham, respectively. R2579A/R2580A and C2991L ATM were generated using QuickChange XL site-directed mutagenesis kit (Stratagene) from wild-type ATM pcDNA3 expression plasmid. Plasmid DNA prepared with QIAFilter plasmid Maxi kit (Qiagen) was used to transfect human HEK-293T cells and express recombinant ATM. To generate ATM heterodimer, plasmid containing Flag-ATM (R2579A/R2580A) was co-transfected with plasmid containing HA-wild-type ATM or HA-ATM (C2991L) in HEK-293T cells and purified through Flag and HA columns, sequentially as described previously (24).
In vitro kinase assay
ATM kinase assays were performed in kinase buffer (50 mM HEPES, pH 7.5, 50 mM potassium chloride, 5 mM magnesium chloride, 10% glycerol, 1 mM ATP, and 1 mM DTT) for 90 min at 30°C in a volume of 40 μl as previously described (24). To test for MRN and DNA-dependent ATM activation, 9.6 nM MRN and 10 ng linear DNA (~140 nM) were used in kinase assays with 6.25 nM GST-p53. For H2O2-dependent ATM activation, 12.5 nM GST-p53 was incubated with various concentrations of H2O2 as indicated without additional DTT. Phosphorylated p53 (Ser-15) was detected as described previously (90) using phospho-specific antibody from Calbiochem (PC461).
In vitro binding assay
Biotinylated proteins (20 nM MRN or MR) were incubated with 50 nM wild-type or 2RA ATM protein in buffer A (100 mM NaCl, 25 mM Tris pH 8, 1 mM DTT, and 10% Glycerol) for 30 min at 30°C in a final volume of 100 μl and then incubated with streptavidin‐coated magnetic beads (Dynal) and 0.1% CHAPS (Sigma), rotating at 4°C for 15 min. Beads with associated proteins were washed three times with buffer A containing 0.1% CHAPS and bound proteins were eluted by boiling the beads in SDS loading buffer, and analyzed by SDS–PAGE and western blotting using antibodies directed against ATM (sc-135663, Santa Cruz), Rad50 (GTX70228, Genetex), and Nbs1 (MSNBS10PX1, Genetex).
ATM-dependent phosphorylation in cells
For the experiment with IR, cells were irradiated with 10 Gy and incubated for 1hr before harvesting. For the experiments with CPT, H2O2, and arsenite, cells were incubated with media containing 10 μM CPT, 100 μM H2O2, or arsenite for 1hr before harvesting. Cells were lysed in 10× cell lysis buffer (9803, Cell Signaling) and lysate (20 μg) was separated by 8% SDS-PAGE gel and analyzed by western blotting. Proteins were transferred to PVDF-FL membrane (Millipore) and probed with antibodies directed against ATM (sc-135663, Santa Cruz), phospho-ATM Ser-1981 (AF-1655, R&D Systems), KAP1 (ab22553, Abcam), phospho-KAP1 Ser-824 (A300-767A, Bethyl Laboratories), Chk2 (GTX70295, Genetex), and phospho-Chk2 Thr-68 (2661S, Cell Signaling) followed by detection with IRdye 800 anti-mouse (RL-610-132-121, Rockland) or Alexa Fluor 680 anti-rabbit (A21076, Invitrogen) secondary antibodies. Western blots were analyzed and quantitated using a Licor Odyssey system.
Clonogenic cell survival assay
U2OS cells were treated with various dose of IR, CPT, or arsenite as indicated. CPT and arsenite were added to media for 1hr and removed by washing cells with PBS followed by changing to complete media without drugs. Cells were incubated for 10-14 days as described (91). Colonies were stained with staining solution (0.5% crystal violet and 20% ethanol) for 30 min followed by washing with dH2O and were counted with Image J. The percentage of cell viability was calculated with untreated group and error bars show standard deviation from 3 independent experiments.
Quantitation of DNA end resection
U2OS Flp-In cells with wild-type or mutant ATM alleles were infected with ER-AsiSI retro-virus and selected with 1 μg/ml of puromycin for two weeks. Cells were further infected with lentivirus expressing control shRNA (shCTRL) or shRNA against ATM (shATM) and expression of exogenous ATM alleles was induced with doxycycline treatment (1 μg/ml). DNA end resection in cells was measured using a method developed previously (36). Briefly, after 4 h treatment with 600 nM 4-Hydroxytamoxifen (4-OHT), cells were trypsinized and resuspended in 0.6% low-gelling-temperature agarose at a final concentration of 2×106 cells/ml. The agar balls with cells were used for genomic DNA extraction and DNA end resection at selected DSB sites were quantitated by quantitative PCR (qPCR) as detailed previously (92).
Cell cycle checkpoint assay
U2OS FLP-in cells after knocking down endogenous ATM and induction of wild-type or mutant ATM were synchronized at G1 with 2 μg/ml aphidicolin (14007, Cayman Chemical) for 17 hr and treated with 1 μM CPT in serum-containing media or 100 μM Arsenite in serum-free media for 1hr. Cells were washed with PBS and incubated in media containing 400 ng/ml nocodazole for 17 hr followed by harvesting with low spin (100 g). For PI staining, cell pellets were washed with 2 times with PBS and resuspended with cold PBS followed by mixing with 100 % ethanol to generate 70 % final concentration and were stored at 4°C overnight. Cells were washed with PBS 2 times and incubated with PI staining solution (3.8 mM sodium citrate, 40 μg/ml propidium iodide, and 0.5 μg/ml RNase A) overnight followed by analysis of the percentage of G2/M cells by flow cytometry.
H2DCFDA, DHE, Mitotracker Green, and MitoTracker Red staining
For staining, all centrifugation occurred for 5 minutes at 100 g unless otherwise stated. AT1ABR pellets were resuspended in PBS and stained with H2DCFDA (ThermoFisher Scientific, D399), DHE (ThermoFisher Scientific, D1168), MitoTracker Green FM (ThermoFisher Scientific, M7514), or MitoTracker Red CMXRos(ThermoFisher Scientific, M7512) as follows. For analysis of ROS levels, washed cells were incubated for 30 minutes with 1 μM H2DCFDA, then centrifuged, and the supernatant removed. The pellets were washed 3 times with PBS and then transferred to 5 ml polystyrene round bottom tubes (VWR, 60818-496) in 0.3 – 1 ml PBS for analysis by flow cytometry. For analysis of superoxide levels, washed cells were incubated for 30 minutes with 5 μM DHE, then centrifuged, and the supernatant removed. The pellets were washed 3 times with PBS and then transferred to 5 ml polystyrene round bottom tubes (VWR, 60818-496) in 0.3 – 1 ml PBS for analysis by flow cytometry. For mitochondrial mass staining, washed cells were incubated with 0.2 μM MitoTracker Green FM for 20 minutes, then centrifuged, and the supernatant removed. The pellets were washed 3 times with PBS and then transferred to 5 ml polystyrene round bottom tubes in 0.3 – 1 ml PBS for analysis by flow cytometry. For mitochondrial staining based on membrane potential, washed cells were incubated with 0.2 μM MitoTracker Red CMXRos for 20 minutes, then centrifuged, and the supernatant removed. The pellets were washed 3 times with PBS and then transferred to 5 ml polystyrene round bottom tubes in 0.3 – 1 ml PBS for analysis by flow cytometry.
Flow cytometry
Stained samples were analyzed on a BD LSRFortessa cell analyzer using the FITC setting (excitation 488 nm/emission 515-545 nm) for samples stained with H2DCFDA, PE setting (excitation 561 nm/emission 578-590 nm) for samples stained with DHE, and the PE-Texas Red setting (excitation 488 nm/emission 600-620 nm) for samples stained with MitoTracker Red CMXRos. Live cells were gated according to the forward scatter and side scatter before analysis of fluorescence. For each sample 10,000 cells were analyzed. The median values from 4-6 biological replicates were used to calculate the means and standard deviations.
mKeima mitophagy assay
pCHAC-mt-mKeima was a gift from Richard Youle (Addgene plasmid # 72342)(47). The plasmid was transfected with retroviral helper plasmids in HEK-293T cells to make recombinant virus. U2OS T-Rex Flp-In cells inducibly expressing wild-type, CL, or 2RA ATM were seeded into 6 well plate with DMEM media containing 1 μg/ml of doxycycline one day before infection. After infection with mKeima retrovirus overnight, media were changed and cells incubated 2 more days. Cells were harvested with Trypsin and cell pellets were washed with PBS one time. For analysis with flow cytometry, cells were resuspended with 1 ml of PBS. To measure mKeima, 488 nm and 561 nm lasers with 610/20 nm emission filters were used. For each sample, 20,000 events were collected for each of 4 biological replicates and analyzed with BD FACSDiva™ software.
Acridine orange staining of acidic lysosomal vesicles
U2OS cell lines expressing inducible ATM alleles were stained for acidic vesicular organelles using acridine orange as previously described (54). Cells incubated with 1 μg/ml of doxycycline was added to the media as final concentration 3 days before treatment with 5 μM CPT or 100 μM Arsenite for 2 hr and cells were washed with PBS and incubated with fresh media without CPT or Arsenite for 48 hr for FACS or 72 hr for confocal microscopy. For FACS anaysis, cells were trypsinized, washed with PBS 2 times, and stained with 1 μg/ml Acridine orange in PBS for 15 min, followed by washing 2 times with PBS. Cells were fixed with 4% paraformaldehyde for 5 min and washed with PBS one time. For confocal microscopy, cells, which were seeded on a glass bottom dish (Willco Wells, HBST-3522) before adding doxycycline, were washed with PBS 2 times and stained with 1 μg/ml Acridine orange in the media for 20 min, followed by replacing with PBS for microscopy. FACS analysis of the acridine orange signal was performed as previously described (54), and the ratio of cells with high 585 nm signal in treated vs untreated cells was calculated for each of 3 biological replicates.
In vitro CK2 kinase reactions
CK2 kinase assays were performed with either 1 μl CK2 (NEB, P6010L) or with immunoprecipitated CK2 on beads. The reactions were performed in 40 μl total with 1× CK2 Reaction Buffer (NEB) (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 10 mM MgCl2) supplemented with 1 mM ATP, 1 μl ATP-γ-32P (NEN Radiochemicals, BLU002H), and 1.15 μg GST-CK2 substrate. For the time course, a master mix of immunoprecipitated CK2β-V5 in the kinase reaction was made and split into 5 tubes. The tubes were incubated at 30 °C for various times and stopped by the addition of 6 μl 5× SDS loading buffer. Quantitation was performed using GelQuant and normalized to protein levels quantified through western blots. Proteins were run on SDS-PAGE gels and transferred to PVDF membrane following standard protocols. V5-tagged CK2 levels were probed using rabbit anti-V5 (Novus Biologicals, NB600-381) and Alexa Fluor 680 goat anti-rabbit (Invitrogen, A21076) and scanned and quantitated on the Li-Cor Odyssey system. Then the membrane was exposed to a PhosphorImager screen for at least 16 hours. The screen was scanned using Typhoon Imager to analyze 32P incorporation.
Protein aggregate isolation and analysis
Cell pellets from 150 mm dishes were frozen in liquid N2. In order to prepare cell lysates, the pellets were resuspended in lysis buffer (20mM Na-phosphate, pH6.8, 10mM DTT, 1mM EDTA, 0.1% Tween, 1mM PMSF, protease inhibitor mini tablets, EDTA free [Pierce]) and rotated at 4°C for 30 min. Cells were lysed in a 4°C water bath-based sonicator (Bioruptor®; eight times, level 4.5, 50% duty cycle) and centrifuged for 20 min at 200 xg at 4°C. Supernatants were adjusted to similar concentration and protein aggregates were pelleted at 16,000 xg for 20 min at 4°C. After removing supernatants, protein aggregates were washed twice with 2% NP-40 (in 20mM Na-phosphate, pH 6.8, 1 mM PMSF and protease inhibitor mini tablets, EDTA free [Pierce]), sonicated (six times at level 4.5 and duty cycle 50%), and centrifuged at 16,000 ×g for 20 min at 4°C. Aggregated proteins were washed in buffer without NP-40 (sonication, four times at level 3 and duty cycle 50%), boiled in 2.5× SDS sample buffer, runned in 4-12% Bis-Tris protein gels (NuPAGE™ Novex™), and analyzed by western blotting.
Aggregate samples were prepared in triplicate and each was resuspended in 15 μL 10% SDS with 50 mM beta-mercaptoethanol and boiled at 100°C for 5 minutes. The samples were diluted with 200 μL of UA buffer (8M Urea, 0.1M Tris pH 8.8). Microcon®-30 centrifugal filter units (Millipore, MRCF0R030) were equilibrated with 20% ACN, 2% formic acid solution (14,000×g for 10 min) prior to use. Diluted samples were loaded on the filters then washed with 400 μL UA buffer (8M Urea, 0.1M Tris pH 8.8) 3 times. After washing, samples were reduced with 400 μL 50 mM DTT in UA buffer by addition of buffer to filter, incubation for 5 minutes at room temperature, and centrifugation. Samples were alkylated with 400 μL 50 mM iodoacetamide in UA buffer with incubation for 5 minutes at room temperature and centrifugation. Samples were de-salted with 400 μL 40 mM ammonium bicarbonate (ABC) 3 times. 100 μL 40 mM ABC with 0.5 μL of trypsin gold (Promega, V528A) in PBS was added to sample and were incubated overnight (37°C). The next morning peptides were eluted by centrifugation; filters were washed with 20% ACN, 2% formic acid solution and filtrate was combined with eluted peptides in ABC buffer. Collected samples were lyophilized at room temperature. Dried samples were resuspended in 10 μL 0.1% formic acid with 0.1% trifluoroacetic acid then de-salted with C18 tips (Pierce, QK224796) according to manufacturer’s protocol. Finally samples were resuspended in 80% ACN 2% formic acid for LC-MS analysis. All centrifugations were done at 14,000×g for 20 min at room temperature unless otherwise noted. Protein identification by LC-MS/MS was provided by the University of Texas at Austin Proteomics Facility on an Orbitrap Fusion following previously published procedures (93). Raw files were analyzed using label-free quantification with Proteome Discoverer 2.15. Proteins with at least two peptides identified by MS/MS were used for quantification and results show only proteins with p values ≤ 0.05 using the Students t-Test with a 2-tailed distribution.
Mass spectrometry for phosphopeptide analysis
Cell lysis and protein quantitation
AT1ABR cell pellets were lysed in 4 volumes of cell lysis buffer (8 M urea, 50 mM Tris, pH 8, 5 mM CaCl2, 30 mM NaCl, 50 mM NaF, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1× mini EDTA-free protease inhibitor (Roche Diagnostics) and 1× phosSTOP phosphatase inhibitor (Roche Diagnostics). The lysate was sonicated until clear and centrifuged for 10 minutes at 20,000 g. The supernatant was collected and the protein concentration was quantitated by BCA assay (Pierce, 23227).
Protein digestion
Equal amounts of lysate from each cell line (1 mg) were reduced with 5 mM dithiothreitol (DTT) for 30 minutes at 37 °C. Following reduction, the cysteine residues were alkylated by the addition of 15 mM iodoacetic acid for 30 minutes at room temperature protected from light. The lysates were then reduced again with 5 mM DTT for 15 minutes at room temperature. The protein sample was diluted with 50 mM Tris, pH 8 and 5 mM CaCl2 to a final concentration of 1.5 M urea. To digest the proteins, LysC (10 μg) (Wako Chemicals, 129-02541) was added and the samples were digested for 2 hours at 37 °C. Trypsin (20 μg) (Promega, V5113) was added and the samples were digested overnight at room temperature.
Peptide desalting
Sep-Pak Accell Plus CM Vac cartridges (Waters, WAT023625) were prepared under light vacuum using the following washes (all percentages as V/V): 3 ml 100% acetonitrile (ACN); 1 ml 70% ACN, 0.25% acetic acid (AA); 1 ml 40% ACN, 0.5% AA; 1 ml 20% ACN, 0.5% AA; and 3 ml 0.1% trifluoroacetic acid (TFA). The peptide samples were brought to pH 2 with TFA and loaded onto the pre-washed cartridge. The bound peptides were washed with 3 ml 0.1% TFA followed by 300 μl 0.5% AA. The peptides were eluted in sequential applications of 1 ml 40% ACN, 0.25% AA and by 750 μl 70% ACN, 0.25% AA and the samples were dried in a speed vacuum concentrator.
Peptide TMT labeling
The TMTsixplex Isobaric Label Reagent Set (Pierce, 90061) was used to individually tag the samples with 6 unique mass tags. The dried peptide samples were resuspended in 100 μl 200 mM tetraethylammonium bromide. TMT Label Reagents (0.8 mg/sample) were brought to room temperature, spun down, and resuspended in 50 μl ACN. The peptide samples and TMT Label Reagents were combined and incubated at room temperature for 2 hours. The labeling reactions were quenched with 0.8 μl 50% hydroxylamine (V/V) and then shaken for 15 minutes. Aliquots (5 μl) of the samples were combined with 50 μl water and dried down and the remaining samples were immediately frozen. The aliquots of labeled peptides were resuspended in 0.2% formic acid then run on the mass spectrometer to determine the ratios of the samples to load for equal protein abundance. The frozen samples were thawed, mixed according to the determined ratios, dried down, and desalted as before.
Strong cation exchange fractionation
Strong cation exchange fractionation was performed at a flow rate of 3.0 mL/min using a polySULFOETHYL A column (9.4 × 200 mM, PolyLC, 209-SE05) on a Surveyor LC quaternary pump. Tagged samples were resuspended in buffer A (30% ACN, 5 mM KH2PO4, pH 2.6) and separated using the following gradient: 0-2 min, 100% buffer A; 2-5 min, 0-15% buffer B (30% ACN, 350 mM KCl, 5 mM KH2PO4, pH 2.6); 5-35 min, 15-100% buffer B. Buffer B was then held at 100% for 10 min. The column was washed with buffer C (500 mM KCl, 50 mM KH2PO4, pH 7.5) and water and re-equilibrated with buffer A. All fractions were collected by hand, frozen, lyophilized and de-salted over a Sep-Pak Accell Plus CM Vac cartridges (Waters, WAT023625).
Phosphopeptide enrichment
Phosphopeptide enrichment was performed using immobilized metal affinity chromatography with metal beads (Qiagen). Before loading the peptides, the beads were equilibrated with water and then incubated with 40 mM EDTA, pH 8.0 for 30 minutes with shaking. EDTA was removed by three successive water washes, and then the beads were incubated with 100 mM FeCl3 for 30 minutes while shaking. For a final wash, the beads were washed with four rounds of 80% ACN, 0.1% TFA. Peptides were resuspended in 80% ACN, 0.1% TFA and incubated with the beads for one hour with shaking. Non-phosphorylated peptides were removed by washing the beads with 80% ACN, 0.1% TFA and these were used for the proteomic analysis. Phosphorylated peptides were eluted from the beads with 50% ACN, 0.7% NH4OH and immediately acidified with 4% formic acid.
LC-MS/MS
Tandem mass spectrometry was performed using a NanoAcquity UPLC system (Waters) coupled to an OrbiTrap Elite (ThermoFisher). Peptides were loaded onto a 75 μm inner diameter and 360 μm outer diameter bare fused silica capillary packed with 10 cm of Magic C18 particles (Bruker-Michrom) for twelve minutes at a flow rate of 1 μL/min. Peptides were then eluted onto a 50 μm inner diameter and 360 μm outer diameter analytical column packed with 17 cm of Magic C18 particles. Peptides were separated over either a 90-minute or 120-minute gradient by ramping from 2% to 35% ACN, 0.2% formic acid at a flow rate of 0.3 μL/min. Peptides were fragmented by HCD and analyzed using the Coon OMSSA Proteomics Software Suite (COMPASS) (94). The mass tolerance was set to 20 ppm for precursors and 0.01 Th for fragment ions. The carbamidomethylation of cysteines and the 6 unique mass tags of TMTsixplex reagents were searched as fixed modification. Oxidation of methionine, TMTsixplex modification of tyrosine, phosphorylation of tyrosine, and phosphorylation with neutral loss on serine and threonine were searched as variable modifications. COMPASS software was used to filter peptides to a 1% FDR which were then combined and used to filter proteins at a 1% FDR. Comparisons of quantitative values were performed by converting all values to log2 and substracting the value for uncomplemented AT1ABR (see Table S1).
Hierarchical clustering
Protein and phosphopeptides levels were clustered in Cluster 3.0 using Hierarchical Clustering with Centered Correlation and Average Linkage (95). Results were visualized and gene lists extracted using Java TreeView (96).
Pairwise comparisons
Pairwise comparison graphs were made in Microsoft Excel 2010. The proteins or phosphopeptides were ranked from lowest intensity to highest intensity separately in each cell line and the Spearman’s coefficients were calculated as , where di = xi − yi is the difference in ranks for one protein between two cell lines. The t statistic for each Spearman’s coefficient was calculated as and the significance at the 5 × 10−16 level was calculated using the qt() function in R version 3.0.3.
Sequence analysis
Peptide sequences 6 amino acids N-terminal and C-terminal to each phosphorylated residue were extracted and formatted for submission to motif-x v1.2 10.05.06 (57, 58). The following settings were used for analysis of the phosphoproteome: foreground format = MS/MS; extend from = IPI Human Proteome; central character = s, t, or y in three separate runs; width = 13, occurrences = 20, significance = 0.000001; background = IPI human proteome. For the C2991 Dependent Cluster, the occurrences were reduced to 10 to reflect the smaller number of phosphopeptides. The analysis was performed using the default significance value of 0.000001 which corresponds to a maximal p-value of <0.0002.
Kinase motif prediction
Peptide sequences 6 amino acids N-terminal and C-terminal to each phosphorylated residue were extracted and formatted for submission to Group-based Prediction System ver 3.0 (GPS) (97). The program was run with the threshold set to high and the results for multiple kinases were combined in one table for further analysis.
Kolmogorov-Smirnov (K-S) tests
The empirical distribution functions (EDFs) were generated and K-S tests were performed in R version 3.0.3. Briefly, the ratios of intensities from AT1ABR cells expressing wild-type-ATM and C2991L-ATM for the phosphopeptides from the entire phosphoproteome or those predicted to be phosphorylated by a specific kinase using GPS were calculated. These ratios were entered into R as a table and converted to numeric vectors. The empirical cumulative distribution function for the phosphoproteome and the phosphopeptides predicted to be substrates of a specific kinase were plotted with the plot() and ecdf() functions and the K-S test was performed using the ks.test() function.
Supplementary Material
Figure S1. Recombinant ATM protein expression.
Figure S2. Arsenite and peroxide treatments do not induce double-strand breaks in ATM-inducible U2OS cell lines.
Figure S3. The 2RA ATM mutant is deficient in CPT-induced KAP1 phosphorylation.
Figure S4. Expression of kinase-deficient ATM sensitizes cells to exogenous stress.
Figure S5. U2OS cells expressing wild-type and mutant alleles of ATM have equivalent ratios of cells in S phase.
Figure S6. Mitochondrial dysfunction in AT1ABR cells expressing CL ATM.
Figure S7. The mKeima mitochondria-targeted pH probe responds to induction or repression of mitophagy.
Figure S8. ATM depletion reduces macroautophagy responses of human cells in response to DNA damage or oxidative stress.
Figure S9. ATM mutant expression does not alter the predicted phosphorylation targets of CK1 or CDK.
Figure S10. CK2 subunit levels are not reduced in A-T cells expressing the C2991L ATM allele.
Figure S11. Expression of C2991L ATM causes Rad50 accumulation in protein aggregates.
Table S1: Phosphorylated peptides observed in AT1ABR cells with inducibly expressed wild-type, CL, 2RA, or 2RA/CL ATM alleles.
Table S2: Proteins detected in aggregate fractions.
Acknowledgments
We thank Blerta Xhelmace for gifts of cell lines and expression constructs, Edward Marcotte for helpful advice with statistical analysis, Anthony Harvey for assistance with analysis of mass spectrometry data, Joost Schymkowitz for assistance with TANGO/WALTZ analysis, and Paull laboratory members for helpful suggestions. Funding: C-H. K. was supported by The Ministry of Education Technologies Incubation Scholarship grant of Taiwan. Studies in the Coon laboratory were supported by NIH (P41 GM108538). Studies in the Paull laboratory were supported in part by CPRIT grant RP100670.
Footnotes
Author contributions: J-H. L. and M.M. conducted and analyzed experiments and contributed to the writing of the manuscript. C-H. K., S.R., Y.Z., and A.R conducted and analyzed experiments. J.C. analyzed experiments and helped to edit the manuscript. T.T.P analyzed experiments and wrote and edited the manuscript.
The authors declare that they have no competing interests.
Data and materials availability: The mass spectrometry and phosphoproteomics data are deposited to CHORUS.
References and Notes
- 1.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013;14:197–210. [PubMed] [Google Scholar]
- 2.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair. 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 3.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 4.Bencokova Z, et al. ATM activation and signaling under hypoxic conditions. Mol Cell Biol. 2009;29:526–537. doi: 10.1128/MCB.01301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt CR, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer research. 2007;67:3010–3017. doi: 10.1158/0008-5472.CAN-06-4328. [DOI] [PubMed] [Google Scholar]
- 6.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 7.Uziel T, et al. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paull TT. Mechanisms of ATM Activation. Annual review of biochemistry. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- 9.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 10.Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair. 2004;3:1219–1225. doi: 10.1016/j.dnarep.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 12.Reichenbach J, et al. Elevated oxidative stress in patients with ataxia telangiectasia. Antioxidants & redox signaling. 2002;4:465–469. doi: 10.1089/15230860260196254. [DOI] [PubMed] [Google Scholar]
- 13.Reichenbach J, et al. Anti-oxidative capacity in patients with ataxia telangiectasia. Clinical and experimental immunology. 1999;117:535–539. doi: 10.1046/j.1365-2249.1999.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yi M, Rosin MP, Anderson CK. Response of fibroblast cultures from ataxia-telanglectasia patients to oxidative stress. Cancer letters. 1990;54:43–50. doi: 10.1016/0304-3835(90)90089-g. [DOI] [PubMed] [Google Scholar]
- 15.Green MH, et al. Hypersensitivity of ataxia-telangiectasia fibroblasts to a nitric oxide donor. Free radical biology & medicine. 1997;22:343–347. doi: 10.1016/s0891-5849(96)00336-x. [DOI] [PubMed] [Google Scholar]
- 16.Ito K, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 17.Reliene R, Schiestl RH. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair. 2006;5:852–859. doi: 10.1016/j.dnarep.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Gueven N, et al. Dramatic extension of tumor latency and correction of neurobehavioral phenotype in Atm-mutant mice with a nitroxide antioxidant. Free radical biology & medicine. 2006;41:992–1000. doi: 10.1016/j.freeradbiomed.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Reliene R, Fleming SM, Chesselet MF, Schiestl RH. Effects of antioxidants on cancer prevention and neuromotor performance in Atm deficient mice. Food Chem Toxicol. 2008;46:1371–1377. doi: 10.1016/j.fct.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reliene R, Schiestl RH. Antioxidants suppress lymphoma and increase longevity in Atm-deficient mice. J Nutr. 2007;137:229S–232S. doi: 10.1093/jn/137.1.229S. [DOI] [PubMed] [Google Scholar]
- 21.Schubert R, et al. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Human molecular genetics. 2004;13:1793–1802. doi: 10.1093/hmg/ddh189. [DOI] [PubMed] [Google Scholar]
- 22.Yang C, et al. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol Cell. 2011;44:597–608. doi: 10.1016/j.molcel.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Paull TT. Purification and biochemical characterization of ataxia-telangiectasia mutated and Mre11/Rad50/Nbs1. Methods Enzymol. 2006;408:529–539. doi: 10.1016/S0076-6879(06)08033-5. [DOI] [PubMed] [Google Scholar]
- 25.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 26.Tsou TC, Tsai FY, Yeh SC, Chang LW. ATM/ATR-related checkpoint signals mediate arsenite-induced G2/M arrest in primary aortic endothelial cells. Archives of toxicology. 2006;80:804–810. doi: 10.1007/s00204-006-0110-4. [DOI] [PubMed] [Google Scholar]
- 27.Mei N, et al. Genetic predisposition to the cytotoxicity of arsenic: the role of DNA damage and ATM. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:2310–2312. doi: 10.1096/fj.02-0093fje. [DOI] [PubMed] [Google Scholar]
- 28.Cooper KL, Liu KJ, Hudson LG. Enhanced ROS production and redox signaling with combined arsenite and UVA exposure: contribution of NADPH oxidase. Free radical biology & medicine. 2009;47:381–388. doi: 10.1016/j.freeradbiomed.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellinsworth DC. Arsenic, reactive oxygen, and endothelial dysfunction. The Journal of pharmacology and experimental therapeutics. 2015;353:458–464. doi: 10.1124/jpet.115.223289. [DOI] [PubMed] [Google Scholar]
- 30.Zelenik Pevec A, Slejkovec Z, van Elteren JT, Falnoga I. As(2)O (3) oxidation by vitamin C: cell culture studies. Biometals. 2012;25:103–113. doi: 10.1007/s10534-011-9486-6. [DOI] [PubMed] [Google Scholar]
- 31.Gulden M, Jess A, Kammann J, Maser E, Seibert H. Cytotoxic potency of H2O2 in cell cultures: impact of cell concentration and exposure time. Free radical biology & medicine. 2010;49:1298–1305. doi: 10.1016/j.freeradbiomed.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Li X, et al. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007;282:36177–36189. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
- 33.Daniel JA, et al. Loss of ATM kinase activity leads to embryonic lethality in mice. The Journal of cell biology. 2012;198:295–304. doi: 10.1083/jcb.201204035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto K, et al. Kinase-dead ATM protein is highly oncogenic and can be preferentially targeted by Topo-isomerase I inhibitors. eLife. 2016;5 doi: 10.7554/eLife.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison C, et al. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Caron P, Legube G, Paull TT. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic acids research. 2014;42:e19. doi: 10.1093/nar/gkt1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shackelford RE, et al. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem. 2001;276:21951–21959. doi: 10.1074/jbc.M011303200. [DOI] [PubMed] [Google Scholar]
- 38.Siu WY, et al. Topoisomerase poisons differentially activate DNA damage checkpoints through ataxia-telangiectasia mutated-dependent and -independent mechanisms. Molecular cancer therapeutics. 2004;3:621–632. [PubMed] [Google Scholar]
- 39.Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Human molecular genetics. 2007;16:2154–2164. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- 40.Valentin-Vega YA, et al. Mitochondrial dysfunction in ataxia telangiectasia. Blood. 2011 doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peter Y, et al. Elevated Cu/Zn-SOD exacerbates radiation sensitivity and hematopoietic abnormalities of Atm-deficient mice. EMBO J. 2001;20:1538–1546. doi: 10.1093/emboj/20.7.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang N, et al. Isolation of full-length ATM cDNA and correction of the ataxia-telangiectasia cellular phenotype. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8021–8026. doi: 10.1073/pnas.94.15.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puleston D. Detection of Mitochondrial Mass, Damage, and Reactive Oxygen Species by Flow Cytometry. Cold Spring Harb Protoc. 2015;2015 doi: 10.1101/pdb.prot086298. pdb prot086298. [DOI] [PubMed] [Google Scholar]
- 44.McGill MR, et al. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch Toxicol. 2014;88:391–401. doi: 10.1007/s00204-013-1118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma S, et al. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;294:L46–56. doi: 10.1152/ajplung.00247.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chemistry & biology. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 47.Lazarou M, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Groenewoud MJ, Zwartkruis FJ. Rheb and mammalian target of rapamycin in mitochondrial homoeostasis. Open biology. 2013;3:130185. doi: 10.1098/rsob.130185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stoyanova S, et al. Lipid kinase and protein kinase activities of G-protein-coupled phosphoinositide 3-kinase gamma: structure-activity analysis and interactions with wortmannin. The Biochemical journal. 1997;324(Pt 2):489–495. doi: 10.1042/bj3240489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3) Redox biology. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander A, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Assante R, et al. Abnormal cell-clearance and accumulation of autophagic vesicles in lymphocytes from patients affected with Ataxia-Teleangiectasia. Clinical immunology. 2016;175:16–25. doi: 10.1016/j.clim.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 54.Goehe RW, et al. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? The Journal of pharmacology and experimental therapeutics. 2012;343:763–778. doi: 10.1124/jpet.112.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slobodkin MR, Elazar Z. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013;55:51–64. doi: 10.1042/bse0550051. [DOI] [PubMed] [Google Scholar]
- 57.Chou MF, Schwartz D. Biological sequence motif discovery using motif-x. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis .. [et al.] 2011 doi: 10.1002/0471250953.bi1315s35. Chapter 13, Unit 13.15-24. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 59.Xue Y, et al. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics : MCP. 2008;7:1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valero E, et al. Quaternary structure of casein kinase 2. Characterization of multiple oligomeric states and relation with its catalytic activity. J Biol Chem. 1995;270:8345–8352. doi: 10.1074/jbc.270.14.8345. [DOI] [PubMed] [Google Scholar]
- 61.Hubner GM, et al. Evidence for aggregation of protein kinase CK2 in the cell: a novel strategy for studying CK2 holoenzyme interaction by BRET(2) Molecular and cellular biochemistry. 2014;397:285–293. doi: 10.1007/s11010-014-2196-y. [DOI] [PubMed] [Google Scholar]
- 62.Glover CV. A filamentous form of Drosophila casein kinase II. J Biol Chem. 1986;261:14349–14354. [PubMed] [Google Scholar]
- 63.Lolli G, Pinna LA, Battistutta R. Structural determinants of protein kinase CK2 regulation by autoinhibitory polymerization. ACS chemical biology. 2012;7:1158–1163. doi: 10.1021/cb300054n. [DOI] [PubMed] [Google Scholar]
- 64.Schnitzler A, Olsen BB, Issinger OG, Niefind K. The protein kinase CK2(Andante) holoenzyme structure supports proposed models of autoregulation and trans-autophosphorylation. J Mol Biol. 2014;426:1871–1882. doi: 10.1016/j.jmb.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 65.Olsen BB, Boldyreff B, Niefind K, Issinger OG. Purification and characterization of the CK2alpha’-based holoenzyme, an isozyme of CK2alpha: a comparative analysis. Protein Expr Purif. 2006;47:651–661. doi: 10.1016/j.pep.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Koplin A, et al. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J Cell Biol. 2010;189:57–68. doi: 10.1083/jcb.200910074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 68.Maurer-Stroh S, et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nature methods. 2010;7:237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 69.You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.D’Souza AD, Parish IA, Krause DS, Kaech SM, Shadel GS. Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol Ther. 2013;21:42–48. doi: 10.1038/mt.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watters DJ. Oxidative stress in ataxia telangiectasia. Redox Report. 2003;8:23–29. doi: 10.1179/135100003125001206. [DOI] [PubMed] [Google Scholar]
- 72.Kamsler A, et al. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer research. 2001;61:1849–1854. [PubMed] [Google Scholar]
- 73.Watters D, et al. Localization of a portion of extranuclear ATM to peroxisomes. J Biol Chem. 1999;274:34277–34282. doi: 10.1074/jbc.274.48.34277. [DOI] [PubMed] [Google Scholar]
- 74.Ribas GS, Vargas CR, Wajner M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene. 2014;533:469–476. doi: 10.1016/j.gene.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 75.Berni A, et al. L-carnitine enhances resistance to oxidative stress by reducing DNA damage in Ataxia telangiectasia cells. Mutation research. 2008;650:165–174. doi: 10.1016/j.mrgentox.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Olsten ME, Litchfield DW. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2004;82:681–693. doi: 10.1139/o04-116. [DOI] [PubMed] [Google Scholar]
- 77.Niefind K, Issinger OG. Primary and secondary interactions between CK2alpha and CK2beta lead to ring-like structures in the crystals of the CK2 holoenzyme. Molecular and cellular biochemistry. 2005;274:3–14. doi: 10.1007/s11010-005-3114-0. [DOI] [PubMed] [Google Scholar]
- 78.Filhol O, et al. Live-cell fluorescence imaging reveals the dynamics of protein kinase CK2 individual subunits. Mol Cell Biol. 2003;23:975–987. doi: 10.1128/MCB.23.3.975-987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bensimon A, et al. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Science signaling. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 80.Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair. 2009;8:1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tresini M, et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature. 2015;523:53–58. doi: 10.1038/nature14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wickramasinghe VO, Venkitaraman AR. RNA Processing and Genome Stability: Cause and Consequence. Mol Cell. 2016;61:496–505. doi: 10.1016/j.molcel.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Experimental & molecular medicine. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Human molecular genetics. 2013;22:5146–5159. doi: 10.1093/hmg/ddt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stewart GS, et al. The DNA double-strand break repair gene hMre11 is mutated in individuals with an Ataxia-Telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 86.Delia D, et al. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Human molecular genetics. 2004;13:2155–2163. doi: 10.1093/hmg/ddh221. [DOI] [PubMed] [Google Scholar]
- 87.Fernet M, et al. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Human molecular genetics. 2005;14:307–318. doi: 10.1093/hmg/ddi027. [DOI] [PubMed] [Google Scholar]
- 88.Lee JH, et al. Ataxia Telangiectasia-Mutated (ATM) Kinase Activity Is Regulated by ATP-driven Conformational Changes in the Mre11/Rad50/Nbs1 (MRN) Complex. J Biol Chem. 2013;288:12840–12851. doi: 10.1074/jbc.M113.460378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee JH, Goodarzi AA, Jeggo PA, Paull TT. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 2010;29:574–585. doi: 10.1038/emboj.2009.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 91.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou Y, Paull TT. Direct measurement of single-stranded DNA intermediates in mammalian cells by quantitative polymerase chain reaction. Analytical biochemistry. 2015;479:48–50. doi: 10.1016/j.ab.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 93.Lee CF, Paull TT, Person MD. Proteome-wide Detection and Quantitative Analysis of Irreversible Cysteine Oxidation Using Long Column UPLC-pSRM. Journal of proteome research. 2013;12:4302–4315. doi: 10.1021/pr400201d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 96.Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 97.Xue Y, et al. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein engineering, design & selection : PEDS. 2011;24:255–260. doi: 10.1093/protein/gzq094. [DOI] [PubMed] [Google Scholar]
- 98.Iacovoni JS, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou Y, Paull TT. DNA-dependent Protein Kinase Regulates DNA End Resection in Concert with Mre11-Rad50-Nbs1 (MRN) and Ataxia Telangiectasia-mutated (ATM) J Biol Chem. 2013;288:37112–37125. doi: 10.1074/jbc.M113.514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Recombinant ATM protein expression.
Figure S2. Arsenite and peroxide treatments do not induce double-strand breaks in ATM-inducible U2OS cell lines.
Figure S3. The 2RA ATM mutant is deficient in CPT-induced KAP1 phosphorylation.
Figure S4. Expression of kinase-deficient ATM sensitizes cells to exogenous stress.
Figure S5. U2OS cells expressing wild-type and mutant alleles of ATM have equivalent ratios of cells in S phase.
Figure S6. Mitochondrial dysfunction in AT1ABR cells expressing CL ATM.
Figure S7. The mKeima mitochondria-targeted pH probe responds to induction or repression of mitophagy.
Figure S8. ATM depletion reduces macroautophagy responses of human cells in response to DNA damage or oxidative stress.
Figure S9. ATM mutant expression does not alter the predicted phosphorylation targets of CK1 or CDK.
Figure S10. CK2 subunit levels are not reduced in A-T cells expressing the C2991L ATM allele.
Figure S11. Expression of C2991L ATM causes Rad50 accumulation in protein aggregates.
Table S1: Phosphorylated peptides observed in AT1ABR cells with inducibly expressed wild-type, CL, 2RA, or 2RA/CL ATM alleles.
Table S2: Proteins detected in aggregate fractions.