

Abstract
Purpose
TMEM16A is a calcium-activated chloride channel that is amplified in a variety of cancers, including 30% of head and neck squamous cell carcinomas (HNSCC), raising the possibility of an anti-apoptotic role in malignant cells. The present study investigated this using a multi-modal, translational investigation.
Experimental Design
Combination of 1) in vitro HNSCC cell culture experiments assessing cell viability, apoptotic activation, and protein expression 2) in vivo studies assessing similar outcomes, and 3) molecular and staining analysis of human HNSCC samples.
Results
TMEM16A expression was found to correlate with greater tumor size, increased Erk 1/2 activity, less Bim expression, and less apoptotic activity overall in human HNSCC. These findings were corroborated in subsequent in vitro and in vivo studies and expanded to include a cisplatin-resistant phenotype with TMEM16A overexpression. A cohort of 41 patients with laryngeal cancer demonstrated that cases that recurred after chemoradiation failure were associated with a greater TMEM16A overexpression rate than HNSCC that did not recur.
Conclusions
Ultimately, this study implicates TMEM16A as a contributor to tumor progression by limiting apoptosis and as a potential biomarker of more aggressive disease.
Keywords: TMEM16A, ANO1, apoptosis, HNSCC, Bim
Introduction
Apoptosis is a highly conserved series of cellular events that leads to programmed cell death. Physiologically, apoptosis plays a central role in balancing cell growth and cell death, in processes like growth & development and in maintaining healthy tissue homeostasis [1, 2]. Mechanistically, apoptosis can be triggered by an extrinsic pathway, involving extracellular, pro-apoptotic, ligand-receptor interactions [3], or an intrinsic pathway, involving the Bcl-2 family of proteins leading to mitochondrial permeability changes [4]. Ultimately, pro-apoptotic signals converge and activate the caspase cascade, which works to dismantle key cellular machinery leading to cell death [5]. Dysregulation at any level of the apoptotic response has been implicated in a variety of pathological states, including neurodegenerative, autoimmune, and neoplastic diseases [3, 6]. The ability to bypass apoptosis is a “Hallmark of Cancer” and is required for tumorigenesis [7]. Furthermore, suppression of apoptosis is an important mechanism of acquired resistance to chemotherapy. However, the mechanisms by which cancer cells prevent apoptosis remain incompletely understood.
TMEM16A (also known as ANO1) belongs to a family of calcium-activated chloride channels and is the prototypical member of this family [8]. TMEM16A is found on the plasma membrane and is thought to contribute to maintaining homeostatic chloride fluxes in a variety of tissues, including cardiovascular endothelia and gastrointestinal epithelia [9, 10]. Clinically, TMEM16A overexpression has been demonstrated in a variety of tumors and is correlated with worse patient survival in head and neck squamous cell carcinoma (HNSCC) [11, 12]. It has been previously demonstrated that TMEM16A contributes to proliferation and tumor growth [11, 13] but its effect on apoptosis has not been explored. Therefore we sought to determine if TMEM16A/ANO1 impacts oncogenesis by inhibiting apoptosis.
Materials and Methods
Materials
Cleaved-PARP (D64E10, 5625P), cleaved-Caspase 3 (9661S), PARP (9542), Bid (2002P), Bad (9239P), Bim (2933S), Puma (4976), pERK1/2 (P-p44/42 MAPK T202/Y204, 9101S), Erk 1/2 (p44/42 MAPK, 9102S), beta actin (4970), and GAPDH (2118) antibodies were from Cell Signaling Technologies (CST). Beta tubulin (ab6046) antibody was from abcam. TMEM16A (Dog-1 sp31, MA5-16358) antibody was from Thermo Scientific.
CellTiter-Glo and CaspaseGlo were from Promega. Matrigel was from Corning Life Sciences. Cisplatin was from Sigma. Complete Mini protease inhibitor cocktail and PhosStop phosphatase inhibitor were from Roche. Bradford’s protein estimation reagent and molecular weight markers were from BioRad. Secondary antibodies were from LiCor for use with the LiCor Odyssey imaging system.
Cell lines and media
OSC19, FaDu, and UM-SCC-1 cells were obtained from ATCC and cultured in DMEM and 10% FBS and 1% Penicillin-Streptomycin. All cells were used for 10 passages and then discarded. All cell lines were authenticated using commercial SNP analysis and used within 6 months of authentication.
Control or TMEM16A overexpressing cells were engineered by transducing viral pBABE-puromycin control or TMEM16A plasmid as previously described by Shiwarski, Duvvuri et. al. [11]. Cells were selected with puromycin-containing media 48–72 hours after transduction.
FaDu cells were engineered to express control scrambled shRNA or TMEM16A-targeting shRNA in doxycycline-inducible manner as previously described by Britschgi, Bill et al. [14, 15]. These cells were cultured in DMEM containing 10% tetracycline-free serum. For experimental use, cells were cultured in 10ng/ml doxycycline containing media for 72 hours, in addition to experimental conditions as noted, to achieve induction of shRNA. Cells expressing Bcl-2 and Bim shRNA were created using lentiviral infection of the TMEM16A-shRNA expressing FaDu cells. Lentiviral particles were generated using a three-plasmid system in HEK-293T cells. Infection was performed as described in the TRC Library Production and Performance Protocols, RNAi Consortium, Broad Institute. shRNA constructs were obtained from the Broad RNAi Consortium and clone IDs as are follows: BIM: TRCN0000355973 (shBim-1). The pLKO.1-shRNA scramble vector was obtained from Dr. David M. Sabatini through Addgene (Addgene plasmid 1864).
Cell viability and apoptotic activity assays
For viability assays and apoptosis assays, 5 × 103 cells were plated in 96 well plates in triplicates per condition and left untreated as controls or treated as indicated. 100uL of CellTiter-Glo reagent or CaspaseGlo reagent was added 24–72 hours after treatments, allowed to incubate for one hour on an orbital shaker, and then fluorescence was read. Viability or apoptotic activity was assessed by normalizing readings from treated wells to readings from untreated control wells. CaspaseGlo reagent generates a fluorescent signal proportional to the amount of the cleaved effector caspases 3 and 7.
Western blot assay
Cells were cultured and treated as indicated followed by lysis in ice cold lysis buffer containing protease and phosphatase inhibitors. Insoluble material was removed by centrifugation at 13,000 RPM for 10 min at 4C; protein concentration was subsequently estimated by Bradford’s method. For analysis in western blots, equal amounts of protein was denatured, separated in 8–12% SDS-PAGE gels, and transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies followed by secondary antibody and imaging in the LiCor Odyssey system or standard camera luminescence. Densitometry analysis was conducted using signal quantification software provided in the LiCor Odyssey system. Cropped images are presented for conciseness and were cropped using Microsoft PowerPoint (Redmond, Washington, USA). All changes to images (adjusting brightness or contrast) were applied uniformly to the entire image using the software provided with LiCor Odyssey system. Full length images of all presented blots are available in supplemental materials.
Tumor Xenografts
1.5 × 106 control or TMEM16A overexpressing OSC19 or UM-SCC-1 cells were implanted in 100μL of Matrigel subcutaneously in nude mice. In OSC19 xenograft, each mouse had control or TMEM16A overexpressing tumors on either flank and were treated with vehicle control (60μL sterile water; n = 11) or cisplatin (3mg/kg in sterile water; n = 10). At the end point mice were euthanized in accordance with University of Pittsburgh guidelines for animal use. Tumors were removed and weights were recorded. Lysates of tumors were prepared and protein was estimated. Equal amount of protein was separated in 8% gels and detected in western blots analyses with the indicated antibodies in LiCor imaging system (LiCor Odyssey Classic). All experiments performed in mice were conducted with approval from the University of Pittsburgh Institutional Animal Care and Use Committee.
Tissue Analysis
Tumor xenografts were formalin fixed, paraffin embedded, and subsequently stained for Ki67, Terminal deoxynucleotidyl dUTP nick end labeling (TUNEL), or TMEM16A (using anti-TMEM16A antisera; clone SP31, Thermo Scientific) at core facilities at our institution. TUNEL scoring was performed by counting the number of positive nuclei per high powered field by two independent raters (authors RS and UD).
Biochemical Analysis of Primary Human Tumor Samples
Human tumor samples were obtained from the University of Pittsburgh Medical Center in accordance with established University of Pittsburgh IRB guidelines. All tumors were removed from the primary site of malignancy and histologically confirmed. Tumors lysates were prepared for Western blot by using a sonic dismembrator followed by chemical lysis with lysis buffer with phosphatase and protease inhibitors. Tissue expression of TMEM16A was assessed using immunohistochemical analysis
IHC Analysis of Recurrent Laryngeal Cancers
Tumors samples were identified in accordance with University of Pittsburgh Medical Center IRB guidelines. A cohort of patients were identified retrospectively that were treated initially with non-surgical therapy with curative intent (see Fig. 6A for demographics). This cohort was then stratified by whether disease recurred, defined as re-detection of disease at the primary site after initial management, or whether the disease did not recur. Sections of the primary tumor tissue were prepared, stained for TMEM16A, and analyzed for the TMEM16A H-score (H-score = percentage of cells expressing protein x staining intensity). Author RS performed analysis of the tissue sections and assigned H-scores across all sections. The maximum tumor H-score across all stained tumor sections was used for analysis. The threshold for overexpression was defined as greater than the average H-score for tumors that did not recur. TMEM16A scoring was compared between non-recurrent primary tumors (PrimaryNR) vs. primary tumors that went on to recur (PrimaryR). Representative H&E stain and anti-TMEM16A images for PrimaryNR with average H-score and PrimaryR with high H-score are available in Supplemental Figure 4.
Figure 6. Recurrence of laryngeal cancer is associated with greater rates of TMEM16A overexpression.
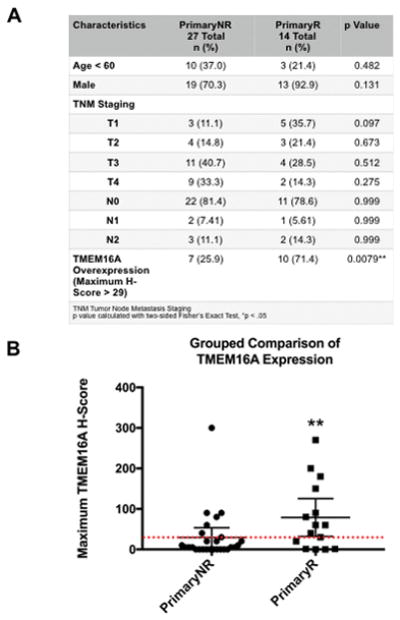
A: Clinical and demographic data of n = 41 cases of laryngeal cancer; cases were separated by the absence (n = 27) or presence (n = 14) of disease recurrence. P-values calculated by two-sided Fisher’s Exact Test. B: Primary tumors that did not recur (PrimaryNR) had a significantly smaller proportion of tumors overexpressing TMEM16A than primary tumors that did recur (PrimaryR). Threshold of overexpression (red dotted line) was defined as the mean H-Score of PrimaryNR. Lines represent mean −/+ 95% CI. *p < .05 calculated by Fisher’s Exact Test.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 6 or Microsoft Excel. All data are reported as mean ± SEM unless stated otherwise. A paired t test or ANOVA was used to test significance as appropriate. P < 0.05 was considered statistically significant. Linear regression and Fisher exact tests were performed in GraphPad Prism 6 for human tumor data.
Results
TMEM16A expression is correlated with tumor size and decreased apoptosis in 11 human HNSCC tumors
To begin investigating the role that TMEM16A plays in apoptosis and cancer, a panel of n = 11 human HNSCC tumors (Sup. Fig. 1 A) was assembled and assessed for expression of TMEM16A by western blot (Fig. 1A). The TMEM16A signal intensity was quantified to investigate whether TMEM16A expression correlated with clinical factors or other proteins. First, TMEM16A expression had a strong, positive correlation with tumor size at time of resection (R2 = 0.71), while TMEM16A was modestly, negatively correlated with cleaved PARP (cl-PARP) (R2 = 0.35), a molecular marker of apoptotic activity; in both cases, the slope of the linear regression was significantly non-zero (p = .0005 and p = .041, resp.) (Fig. 1B).
Figure 1. TMEM16A expression correlates with tumor size and apoptosis resistance in human HNSCC tumor samples.
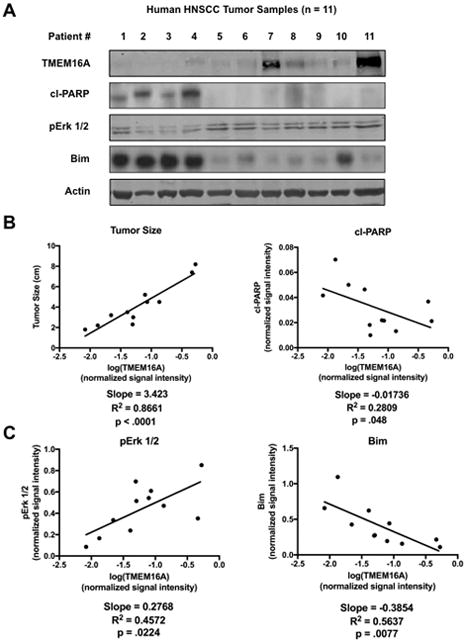
A: n = 11 human head and neck squamous cell carcinoma (HNSCC) samples were assessed for TMEM16A, cl-PARP, pErk 1/2, and Bim expression by western blot. B: TMEM16A expression had a strong, positive correlation with tumor size and a modest, negative correlation with cl-PARP in human HNSCC samples. C: TMEM16A expression had a strong, positive correlation with pErk 1/2 expression and a strong, negative correlation with Bim. p < .05 denotes a significantly non-zero slope of the trend line. Blots cropped for conciseness; clinical demographic information and full length blots available in supplemental materials.
Earlier studies implicate Erk 1/2 as an intracellular mediator of the oncogenic capacity of TMEM16A [11]. Therefore, the activity of Erk 1/2, as measured by the phosphorylated Erk 1/2 (pErk 1/2) fraction, was assessed in the tumor panel. TMEM16A strongly correlated with pErk 1/2 expression (R2 = 0.51) (Fig. 1C) corroborating previous studies. Next, the expression of a number of apoptotic proteins was measured to see if TMEM16A affected protein mediators of apoptosis. TMEM16A expression had a strong, negative correlation with the pro-apoptotic protein Bim (R2 = 0.56) (Fig. 1C); this effect was specific to Bim, as similar correlations were not found with a variety of other apoptotic proteins, including Bad and Puma (Sup. Fig. 1 B). Additionally, the slope of the linear regression line was significantly non-zero for both pErk 1/2 and Bim (p = .0083 and p = .005, resp.).
This data identified an anti-apoptotic clinical phenotype in a subset of human tumors with greater TMEM16A expression and implicated a specific protein change, reduced Bim, that could mediate that phenotype.
TMEM16A overexpression inhibits in vivo apoptotic activation
Next, UM-SCC-1 control and TMEM16A overexpressing (TMEM16A) xenografts (Sup. Fig. 2) were created as preclinical models of TMEM16A overexpression and to investigate any effects on apoptosis. TMEM16A overexpressing tumors demonstrated significantly greater tumor volumes than controls tumors (Fig. 2A). Subsequent IHC analysis of tumor samples confirmed in vivo TMEME16A overexpression in the overexpression group (Fig. 2B) and, furthermore, TMEM16A overexpressing tumors demonstrated significantly less TUNEL staining, a marker of apoptosis, than control tumors (Fig. 2C; representative IHC images Sup. Fig. 3). Finally, analysis of tumor lysates demonstrated that TMEM16A overexpressing tumors contained significantly greater pErk 1/2 and significantly less Bim (Fig. 2D) than control tumors.
Figure 2. TMEM16A correlates with in vivo Erk 1/2 activation and suppression of Bim.
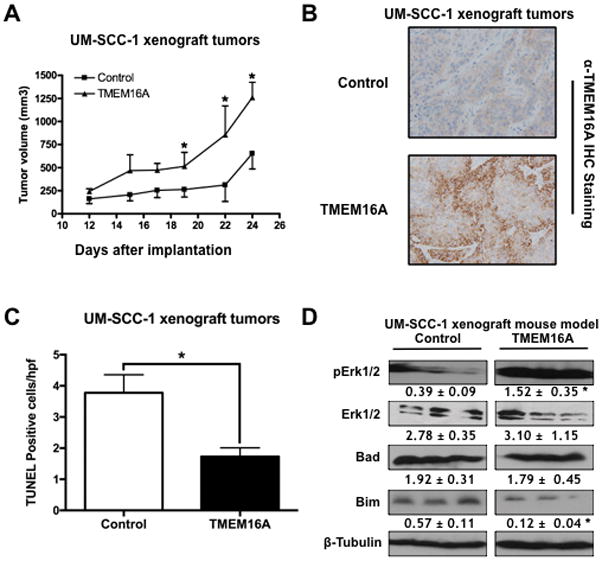
A & B: UM-SCC1 TMEM16A overexpressing (TMEM16A) tumor xenografts were significantly larger than control tumor xenografts. C: UM-SCC-1 TMEM16A tumors demonstrated significantly less TUNEL staining than control tumors. D: UM-SCC1 TMEM16A overexpressing tumors expressed significantly greater pErk 1/2 with selective suppression of Bim. All data expressed as mean ± SEM relative to control conditions; *p < .05. Blots cropped for conciseness; full length blots available in supplemental materials.
These results corroborated the initial clinical findings and tied TMEM16A overexpression to an in vitro reduction in apoptotic activation.
TMEM16A overexpression suppresses in vitro apoptotic activation in response to cisplatin
To further test the identified link between TMEM16A and blunted apoptotic activation, scrambled control shRNA (shScram) or shRNA against TMEM16A (shTMEM16A) was introduced into FaDu cells, which have high endogenous expression of TMEM16A. TMEM16A knockdown led to a significant decrease in cell viability (p = .0001); furthermore, this reduction in viability was due to apoptotic activation, as the addition of the pan-caspase inhibitor, Q-VD-OPh, prevented the reduction in cell viability (Fig. 3A). This finding was corroborated by demonstrating that cl-PARP was eliminated with Q-VD-OPh treatment (Fig. 3A).
Figure 3. TMEM16A inhibits apoptosis induced by cisplatin.
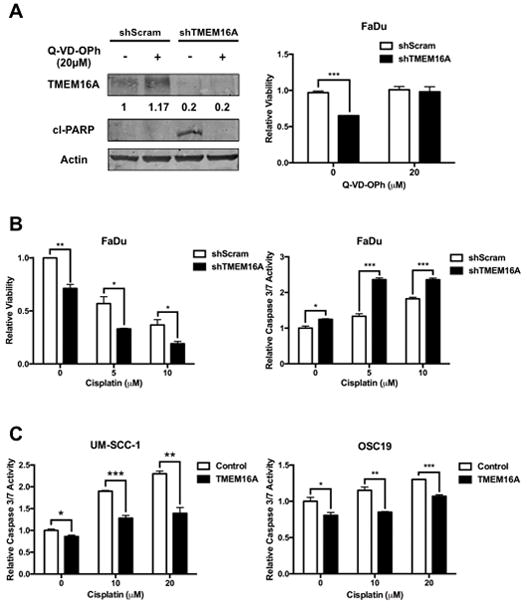
A: Pan-caspase inhibition with Q-VD-OPh increased FaDu cell viability following TMEM16A knockdown and prevented PARP cleavage. B: FaDu cells treated with shTMEM16A and cisplatin demonstrated significantly less viability and significantly greater caspase 3/7 activity at all concentrations of cisplatin. C: UM-SCC-1 and OSC19 TMEM16A overexpressing cells demonstrate significantly less caspase 3/7 activation at all concentrations of cisplatin treatment relative to control cells. All data expressed as mean ± SEM relative to control conditions; *p < .05, ***p< .001. Blots presented with tubulin-normalized densitometric quantification and cropped for conciseness; full length blots available in supplemental materials.
Next, FaDu cells with either shScram or shTMEM16A were treated with cisplatin, a potent apoptotic stimulus, and assessed for cell viability (Fig. 3B). FaDu cells treated with both shTMEM16A and cisplatin had significantly less viability than shScram treated FaDu cells (p = .03 at 10μM cisplatin). To clarify whether the observed effect was purely anti-proliferative or pro-apoptotic, these cells were also assessed for caspase 3 and caspase 7 activity. Again, shTMEM16A FaDu cells treated with cisplatin demonstrated significantly greater caspase 3/7 activity at all concentrations of cisplatin treatment (p = .0009 at 10μM cisplatin). To further corroborate this, FaDu cells were treated with single-agent or combination of cisplatin and CaCCInh-AO1, a specific inhibitor of TMEM16A and assessed for cell viability (Sup. Fig. 4). Here again, TMEM16A inhibition along with cisplatin treatment resulted in significantly less cell viability (p < .0001 for CaCCInh-AO1 or cisplatin alone vs. combination) then with either agent alone.
Next, UM-SCC-1 and OSC19 cells, both with low endogenous TMEM16A, were transfected with either a control or TMEM16A overexpressing plasmid (Sup. Fig. 2). Both control and TMEM16A cells were treated with cisplatin and assessed for caspase 3 and caspase 7 activation. In both UM-SCC-1 and OSC19 cell lines, TMEM16A cells demonstrated significantly less caspase 3/7 activity relative to control at all cisplatin concentrations (p = .003 and p = .0004 for UM-SCC-1 and OSC19, respectively, at 20μM cisplatin treatment) (Fig. 3C).
Furthermore, inhibition of caspase 9, a key mediator of the intrinsic apoptotic pathway, but not caspase 8 inhibition, reduced the magnitude of apoptotic activity in UM-SCC-1 control and TMEM16A cells treated with cisplatin (Sup. Fig. 5). However, there was still significant differential apoptotic activity between control and TMEM16A cells.
These results demonstrate that TMEM16A expression is able to modulate apoptosis, both at baseline and, notably, in response to cisplatin. Additionally, this effect was demonstrated in cell lines with differing endogenous levels of TMEM16A, suggesting that the effect is generalizable to TMEM16A expression and is not entirely cell line specific.
TMEM16A protects against cisplatin-induced apoptosis in vivo
To extend the in vitro findings, mice were inoculated with OSC19 control and TMEM16A cells to explore whether TMEM16A expression led to in vivo resistance to cisplatin. Mice bearing control and TMEM16A overexpressing tumors were treated with either vehicle (n = 11) or cisplatin (n = 10). Control tumors treated with vehicle were significantly smaller than TMEM16A overexpressing tumors treated with vehicle (p < .0001 at 28 days). Furthermore, the addition of cisplatin appeared to prevent the growth of control tumors (p = .0001 vehicle treated control vs. cisplatin treated control at 28 days), whereas it had no effect on the TMEM16A overexpressing tumors (Fig. 4A). As expected, OSC19 control tumors demonstrated significantly greater IHC TUNEL staining (p = .006), a marker of apoptotic activity, with cisplatin treatment than the TMEM16A overexpressing tumors treated with cisplatin (Fig. 4B & C).
Figure 4. TMEM16A contributes to in vivo cisplatin resistance.
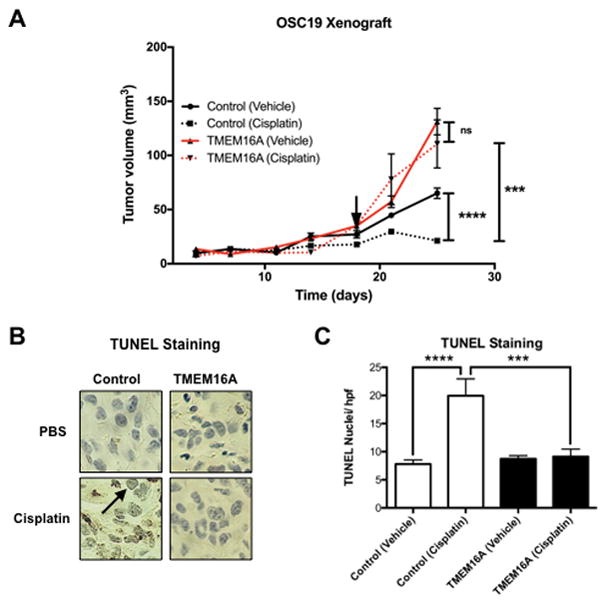
A: OSC19 TMEM16A overexpressing (TMEM16A) tumor xenografts (n = 10) were significantly larger than control tumor xenografts (n = 11) and were not affected by treatment with cisplatin. Cisplatin treatment initiated at arrowhead. B & C: OSC19 TMEM16A overexpressing tumors experienced significantly less apoptosis as shown by reduced TUNEL staining on IHC section. All data expressed as mean ± SEM relative to control conditions, unless otherwise noted; *p < .05, **p < .01, ****p< .0001.
TMEM16A knockdown-induced apoptosis requires Bim expression
To clarify the observed inverse relation between TMEM16A and Bim, Bcl-2, a protein that opposes the action of Bim, was overexpressed in FaDu cells treated with shTMEM16A. As before, shTMEM16A led to a decrease in cell viability and an increase in cleaved PARP, indicating apoptotic cell death. However, overexpression of Bcl-2 was able to prevent reduction in cell viability and PARP cleavage (Fig. 5A & C). To demonstrate the role that Bim plays in TMEM16A knockdown-induced apoptosis more directly, shRNA against Bim (shBim) was introduced in the FaDu cells. Control, scrambled shRNA (shScram) had no effect in preventing TMEM16A knockdown-induced apoptosis, while shBim prevented PARP cleavage and ameliorated the reduction in cell viability following TMEM16A knockdown (Fig. 5B & D).
Figure 5. Bim is a key mediator of TMEM16A knockdown-induced apoptosis.
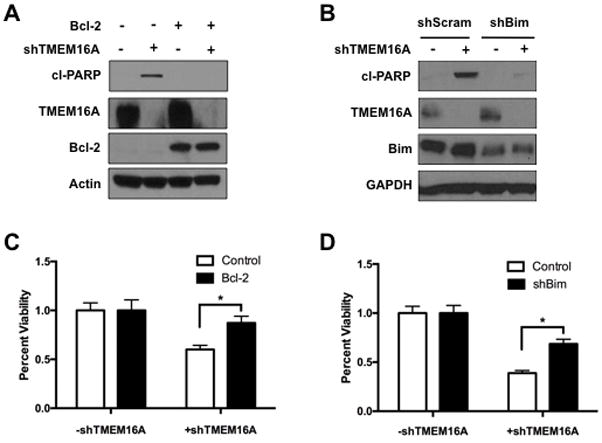
A & C: Overexpression of Bcl-2 prevented PARP cleavage (cl-PARP) and viability loss in FaDu cells following TMEM16A knockdown. B & D: shRNA knockdown of Bim (shBim) prevented PARP cleavage and viability loss in FaDu cells following TMEM16A knockdown. All data expressed as mean ± SEM relative to control conditions; *p < .05. Blots cropped for conciseness; full length blots available in supplemental materials.
Taken together these data indicate that TMEM16A knockdown induced apoptosis proceeds in a Bim-dependent manner.
Recurrence of laryngeal cancer is associated with greater rates of TMEM16A overexpression
Finally, a panel of 41 primary laryngeal cancer tumors was assembled to further test the clinical impact of TMEM16A overexpression. This cohort was then separated by the clinical progression of the disease: whether the tumor did not (n = 27) or did recur (n = 14). The clinical and demographic background of the panel was found to be proportional between the two groups (Fig. 6A).
TMEM16A has been shown to be overexpressed in about 30% of HNSCC, according to data presented in The Cancer Genome Atlas (TCGA) [16, 17]. Analysis of the maximum TMEM16A H-score across the two clinical subsets revealed that a significantly greater proportion of tumors that went on to recur overexpressed TMEM16A (n = 10/14, 71%) than tumors that did not recur (n = 7/27, 25.9% consistent with TCGA overexpression rate) (*p = .023) (Fig. 6B).
Ultimately, this data underscores the important connection between TMEM16A and clinical disease burden, possibly mediated through the described Bim mediated anti-apoptotic phenotype.
Discussion
Ion channels, specifically chloride channels, have been postulated to be involved in the control of cell growth and cell cycle regulation [9, 10, 18]. The calcium-activated chloride channel, TMEM16A/ANO1 is frequently overexpressed in epithelial malignancies and contributes to tumor growth and proliferation [11]. Additionally, it has been shown that patients with HNSCC malignancies overexpressing TMEM16A have worse survival outcomes [11, 12]. However, the role of TMEM16A in mediating poor outcomes and the control of apoptosis and cell death remains unclear.
The data presented here demonstrate a reduction in apoptotic activity, including in response to cisplatin, in multiple models of TMEM16A overexpression. Furthermore, it identifies a specific reduction in the pro-apoptotic protein, Bim, as a key event in mediating this effect. This molecular phenotype was found to also exist in a panel 11 human HNSCC tumors. Finally, analysis of 41 human HNSCC samples found a greater proportion of TMEM16A overexpression in primary tumors that went on to recur (i.e., more aggressive clinical disease) than tumors that did not recur. In conjunction with the pre-clinical data, these clinical observations provide strong evidence for the translational importance of TMEM16A and a possible mechanism underlying the clinical phenomena of more aggressive disease.
The present findings suggest that TMEM16A inhibition or knockdown results in apoptotic cell death. Furthermore, recent data using the RIP-kinase inhibitor, necrostatin, suggest that TMEM16A knockdown induced cell death does not proceed in a necroptotic manner [19]. While this does not preclude the involvement of other cell death pathways (e.g., autophagy), it does suggest that apoptotic cell death is a key mediator.
Recent investigations into recurrent and metastatic HNSCC found a 10.4% amplification rate of chromosomal locus 11q13, which houses TMEM16A [20] - a much lower rate of amplification than the ~30% amplification rate reported by the TCGA in primary HNSCC samples [17]. Consistent with this data, analysis of the recurrent tumor tissue of 10 of the 14 cases of laryngeal cancer that recurred (data not shown), demonstrated a non-significant reduction in maximum H-score as compared to the primary tumor tissue. This data corroborates the analysis of recurrent and metastatic HNSCC and, in the context of this study, could support the idea that different subpopulations of the cancer expressing different protein profiles are preferentially selected for during different times of cancer progression.
Beyond strongly supporting TMEM16A as a protein that contributes to oncogenicity, this study has important translational implications. First, the human tumor analysis strongly suggests that TMEM16A expression is associated with larger tumors, likely by avoiding apoptosis through the mechanisms presented. Furthermore, the data suggests that TMEM16A overexpression may be linked to disease recurrence, a major cause of morbidity and mortality in the HNSCC population. Accordingly, TMEM16A has the potential to be a useful clinical biomarker to predict specific facets of tumor progression (i.e., chemotherapy resistance and disease recurrence), beyond poor survival outcomes.
Cisplatin based chemotherapy is a cornerstone of treatment for a variety of human malignancies, including HNSCC. Clinically, resistance to cisplatin therapy is a major problem and contributes to poor patient outcomes [21]. Multiple mechanisms of resistance have been proposed, including: reduced intracellular accumulation of drug, drug inactivation, and alterations in DNA damage-sensing mechanisms [22]. Of particular interest to the current discussion, is the role that Erk 1/2 may play in mediating resistance. Evidence is mixed regarding the effect that Erk 1/2 has – some work suggests that Erk 1/2 sensitizes cells to cisplatin therapy [23] while others suggest that Erk 1/2 activity is associated with resistance to cisplatin therapy [23, 24]. Knowing that TMEM16A drives Erk 1/2 activity and is correlated with reduced apoptosis in response to stimuli like cisplatin, supports the latter possibility. Ultimately, this study provides strong evidence that, in a portion of cisplatin-resistant malignancies, overexpression of TMEM16A may lead to inhibition of apoptosis and subsequent cisplatin-resistance. Furthermore, this raises the possibility that pharmacologic inhibition of either TMEM16A or its relevant downstream effectors (i.e., ERK 1/2) could be strategically used to overcome cisplatin resistant malignancies in TMEM16A expressing malignancies.
Supplementary Material
Translational Relevance.
The presented study investigates the role that TMEM16A overexpression plays in head and neck squamous cell carcinoma (HNSCC) and ties it to a distinct, clinical anti-apoptotic and cisplatin-resistant phenotype, possibly through an identified molecular mediator: Bim. Ultimately, 30% of HNSCC demonstrate TMEM16A overexpression and cisplatin resistance is a common clinical occurrence – accordingly, the potential translational benefit of understanding how TMEM16A is involved and, in the future, could be targeted, would be an invaluable resource for a large population of patients with HNSCC.
Acknowledgments
Financial Support: Head and Neck Cancer SPORE grant (U. Duvvuri, P50-CA097190), Career Development Award and Pilot Project Grant from the Department of Veterans Affairs Biomedical Laboratory Science Research and Development (U. Duvvuri), PNC Foundation (U. Duvvuri), and the University of Pittsburgh Cancer Institute Cancer Center Support Grant (U. Duvvuri, P30CA047904).
The authors would like to acknowledge A. Bill and L. Alex Gaither for generously providing cell lines and reagents.
References
- 1.Basu A, Haldar S. The relationship between BcI2, Bax and p53: consequences for cell cycle progression and cell death. Mol Hum Reprod. 1998;4(12):1099–109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 2.Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23(12):620–33. doi: 10.1016/j.tcb.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JC, et al. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2(3):156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 5.Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5(6) doi: 10.1101/cshperspect.a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 2015;16(2):2942–55. doi: 10.3390/ijms16022942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Brunner JD, et al. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516(7530):207–12. doi: 10.1038/nature13984. [DOI] [PubMed] [Google Scholar]
- 9.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–58. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 10.Yu K, et al. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res. 2012;110(7):990–9. doi: 10.1161/CIRCRESAHA.112.264440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duvvuri U, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13):3270–81. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz C, et al. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One. 2012;7(8):e43265. doi: 10.1371/journal.pone.0043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiwarski DJ, et al. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin Cancer Res. 2014;20(17):4673–88. doi: 10.1158/1078-0432.CCR-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bill A, et al. Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J Biol Chem. 2014;289(16):11029–41. doi: 10.1074/jbc.M114.549188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Britschgi A, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110(11):E1026–34. doi: 10.1073/pnas.1217072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada Y, et al. Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch. 2004;448(3):287–95. doi: 10.1007/s00424-004-1276-3. [DOI] [PubMed] [Google Scholar]
- 19.Kulkarni S, et al. TMEM16A/ANO1 suppression improves response to antibody-mediated targeted therapy of EGFR and HER2/ERBB2. Genes Chromosomes Cancer. 2017;56(6):460–471. doi: 10.1002/gcc.22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris LG, et al. The Molecular Landscape of Recurrent and Metastatic Head and Neck Cancers: Insights From a Precision Oncology Sequencing Platform. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2016.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozols RF. Ovarian cancer: new clinical approaches. Cancer Treat Rev. 1991;18(Suppl A):77–83. doi: 10.1016/0305-7372(91)90027-w. [DOI] [PubMed] [Google Scholar]
- 22.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 23.Mandic A, et al. The MEK1 inhibitor PD98059 sensitizes C8161 melanoma cells to cisplatin-induced apoptosis. Melanoma Res. 2001;11(1):11–9. doi: 10.1097/00008390-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Yeh PY, et al. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol. 2002;63(8):1423–30. doi: 10.1016/s0006-2952(02)00908-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.