

Abstract
The purpose of this method is to provide a flexible, rapid, and quantitative technique to examine the kinetics of DNA-protein crosslink (DPC) repair in mammalian cell lines. Rather than globally assaying removal of xenobiotic-induced or spontaneous chromosomal DPC removal, this assay examines the repair of a homogeneous, chemically defined lesion specifically introduced at one site within a plasmid DNA substrate. Importantly, this approach avoids the use of radioactive materials and is not dependent on expensive or highly-specialized technology. Instead, it relies on standard recombinant DNA procedures and widely available real-time, quantitative polymerase chain reaction (qPCR) instrumentation. Given the inherent flexibility of the strategy utilized, the size of the crosslinked protein, as well as the nature of the chemical linkage and the precise DNA sequence context of the attachment site can be varied to address the respective contributions of these parameters to the overall efficiency of DPC repair. Using this method, plasmids containing a site-specific DPC were transfected into cells and low molecular weight DNA recovered at various times post-transfection. Recovered DNA is then subjected to strand-specific primer extension (SSPE) using a primer complementary to the damaged strand of the plasmid. Since the DPC lesion blocks Taq DNA polymerase, the ratio of repaired to un-repaired DNA can be quantitatively assessed using qPCR. Cycle threshold (CT) values are used to calculate percent repair at various time points in the respective cell lines. This SSPE-qPCR method can also be used to quantitatively assess the repair kinetics of any DNA adduct that blocks Taq polymerase.
Keywords: Genetics, Issue 133, DNA-protein crosslinks, DNA repair, quantitative polymerase chain reaction, 8-oxoguanine, human oxoguanine glycosylase, Taq polymerase
Introduction
Described herein is a PCR-based assay termed SSPE-qPCR. The purpose of this method is to quantify DPC repair on plasmid DNA transfected into repair deficient and proficient mammalian cells. This assay is rapid, quantitative, extremely flexible, and directly measures repair activity. While this report focuses on the use of this methodology to study repair of DPCs, results presented below illustrate that repair of any lesion that blocks Taq polymerase can be studied using this methodology.
The rationale behind the development of this method is to gain insight into the mechanisms through which mammalian cells repair DPCs. Unlike other types of DNA damage, DPCs are massively diverse1,2. Studies have demonstrated that hundreds of cellular proteins can become crosslinked to DNA and that for each protein there are, in principle, numerous amino acid side chains that could become covalently attached to cellular DNA3. In addition, there are numerous chemical attachment points for proteins onto the DNA backbone, including several positions on the nucleotide bases as well as on the ribose sugar4,5. This chemical diversity raises the prospect that distinct biochemical pathways may be relied upon to repair different types of DPCs. It was with this concern in mind that the SSPE-qPCR assay was developed.
Several techniques have been developed to gain insight into the molecular biology of cellular DPC repair. The following provides an overview of the major approaches that have been developed, with a summary of the major strengths and weaknesses each possess. It is worth stressing that while this summary focuses on studies of DPC repair in mammalian cell culture systems, significant contributions to the current model of DPC repair have been made using microbial and cell-free systems that are not discussed in this manuscript.
Perhaps the easiest strategy that can be taken to gain insight into the genetics of DPC repair is to assess the respective sensitivity to cell death observed in wild-type and mutant cells exposed to agents that induce DPCs6,7. This strategy is relatively fast, inexpensive, and does not require specialized expertise beyond the ability to perform basic cell culture techniques. Counterbalancing these advantages are numerous limitations to this approach including the following. First, the assay does not directly measure DNA repair. The working assumption underlying this strategy is that inactivating mutations in genes encoding relevant DNA repair proteins results in an accumulation of DNA damage that triggers programmed cell death. However, mutations in genes encoding non-DNA-repair proteins could, in principal, enhance (or reduce) cellular sensitivity to xenobiotic-induced cell death. Second, agents that create DPCs invariably induce other types of DNA damage (one exception is 5-aza-2'-deoxycytadine, but this agent also depletes cellular methyltransferase levels8). Consequently, it is conceivable that enhanced cellular hypersensitivity to the agent in question may reflect defects in repair of interstrand crosslinks or other lesions. Third, as was mentioned above, DPCs represent a vastly heterogeneous class comprised of different types of chemical crosslinks, involving different protein partners. It is possible that while repair of one or more sub-types of these lesions may be altered in a particular genetic background, this difference may not be sufficient to significantly alter cellular hypersensitivity to death induced by this agent. In summary, while this strategy represents an attractive starting point, the limitations outlined above highlight the importance of pursuing other, more direct methods to study the kinetics of DPC repair.
A number of related approaches have been developed to achieve this objective. For instance, investigators have developed methods to distinguish between 'free' DNA and 'protein-bound' DNA9,10,11. Using these approaches, it is possible to compare steady-state levels of DPCs or following exposure to a DPC-producing agent in different genetic backgrounds. The two strategies that have been most widely used involve separating DPC-containing DNA from free DNA using either a nitrocellulose membrane binding strategy or KCl/SDS precipitation12,13. In the former approach cells are lysed and passed through a nitrocellulose filter. Because nitrocellulose binds protein, the filter retains protein-linked DNA, permitting free DNA to pass through. In the latter strategy protein-bound DNA is separated from free DNA based on the fact that SDS binds to protein but not DNA, and can be precipitated by the addition of KCl. Consequently, protein-linked DNA becomes insoluble while unbound DNA remains in solution. DPC-containing DNA can then be quantitated using radiolabeled thymidine (if cells were initially metabolically labeled) or by a DNA-selective fluorescent dye like Hoechst 33258. These methods are reproducible and require a small number of steps. However, they do not provide information regarding the nature of the chemical crosslink through which protein is attached to DNA. Furthermore, it is important to note, these assays may over-estimate DPC repair by falsely scoring incomplete repair, i.e., proteolytic processing to smaller DNA-peptide crosslinks that may not be as easily trapped or precipitated, as bona fide DNA repair.
Comet assays can be used to visualize DPC formation in cells14. In these experiments, the presence of DPCs decrease DNA migration which can then be reversed by pretreating with proteinase K. Therefore, the length of the tail can be used to estimate DPC formation. However, as mentioned above, DPC-forming drugs create other types of DNA damage which could alter tail length. This protocol is also highly technical and requires expertise and training in confocal imaging.
Mass spectrometry can be used to study DPC repair kinetics following treatment with crosslinking agents15,16,17. These experiments treat cells with DPC-forming agents and isolate DPCs via biotin capture or phenol:chloroform (1:1) extraction. Mass spectrometry can then be used to identify the crosslinked proteins or quantitate the amount of DPCs formed over time. The major advantage of this approach is the nature of the data produced. It is possible to precisely catalog the types of proteins that become crosslinked following exposure to a xenobiotic, however, this protocol is expensive, time consuming, and is limited by the type of crosslink that can be detected.
Maizels et al. developed a sensitive 'RADAR' (rapid approach to DNA adduct recovery) assay to quantitate immunodetection of DNA-protein adducts as well as an ELISA-based RADAR assay18,19. These assays are especially useful for trapping DNA-protein intermediates that transiently form in cells and generate samples suitable for mass spectroscopy to identify new protein adducts. This immunodetection assay relies on the availability of antibodies to capture the DNA-protein crosslink and, therefore, may not be capable of detecting degraded DNA-peptide adducts that form during repair. Recently, a specific DPC repair pathway linked to DNA replication and a DNA-dependent metalloprotease Spartan was discovered in which DPCs are proteolyzed to smaller peptides during repair20,21. Inherited mutations in this gene are associated with Ruijs-Aalfs syndrome in humans, a disease characterized by genomic instability, premature ageing, and liver cancer22. Mice with genetically engineered Spartan gene defects display similar phenotypes23.
Host-cell reactivation of transcriptional activity has been used to study the repair of defined lesions present on transfected plasmid DNA substrates24,25. In these experiments, plasmid containing DPCs (or other types of DNA lesions) that block the transcription of a reporter, such as luciferase, are transfected into cells. Luminescence measurements taken 24 - 72 h later are then correlated with DPC repair. However, these indirect repair assays are incapable of detecting repair events earlier than 24 h post-transfection and cannot distinguish between RNA polymerase bypass of partially repaired substrates and complete repair.
Each of the methods described above has advantages and has contributed to the current model of DPC repair. However, the SSPE-qPCR assay circumvents several of the limitations associated with these other approaches and consequently can provide more specific insight into DPC repair mechanisms. For example, the SSPE-qPCR assay can directly measure repair of site-specific DPCs on DNA in intact mammalian cells. This method is versatile and has been used to obtain repair results following transfection in hamster and human cell lines. Transfection of the plasmid can be performed using lipofection or electroporation in cultured mammalian cell lines. It also ensures that only repair of defined DPCs is measured, and not other types of DNA damage induced by most DPC-forming agents. The SSPE-qPCR is easy to perform, inexpensive, and rapid. Results obtained using this assay have detected repair events as early as 2 h post-transfection. Using this method, variables that may influence DPC repair outcomes can be studied in a manner that is sensitive and efficient. For example, the role of transcription in DPC repair has yet to be rigorously evaluated. Due to the flexibility of the SSPE-qPCR assay, the crosslinking site of the DPC can be manipulated to address this question. In addition, introduction of an origin of replication into the DPC-bearing plasmid can be used to address the influence of replication on DPC repair. Additionally, multiple crosslinks can be created on the plasmid to examine differences in repair of a single DPC versus multiple crosslinks. These are questions that would be difficult to answer using chromosomal DNA but can easily be addressed using the SSPE-qPCR assay. Overall, the SSPE-qPCR assay requires purified, plasmid DNA in microgram quantities containing a lesion of a known location. Adducts besides DPC can be used in this assay, however, the lesion must be capable of blocking extension by Taq polymerase.
Protocol
1. Generation of DPC-containing Plasmid
- Combine 80 pmol of oligonucleotide containing an 8-oxoguanine residue (20 µL) with 10 units of T4 polynucleotide kinase (1 µL) in 10x ligase buffer (5 µL). Add water to reach a total volume of 50 µL and incubate at 37 °C for 30 min in a heated water bath.
- Combine the phosphorylated oligonucleotide (50 µL), 80 pmol of single-stranded DNA (80 μL), 100 units Taq polymerase (20 µL) in 10x Taq reaction buffer (25 µL), 100 mM ATP (20 µL), 10 mM dNTPs (20 µL), NEB buffer 2 (25 µL), and 8 µg BSA (20 µL) to total 260 µL. Incubate the sample in a thermocycler set at 75 °C for 15 min followed by 37 °C for 5 min. Next, add 60 U of T4 polymerase (20 µL), 8,000 U T4 ligase (20 µL), 100 mM ATP (20 µL), 10 mM dNTPs (20 µL), NEB buffer 2 (15 µL), and 8 µg BSA (20 µL) to total 375 µL. Incubate the reaction overnight at 37 °C (Figure 1A).
- Create a 50:50 buffer-saturated phenol:chloroform mixture. Add equal volumes of each component, mix, and spin in a table-top centrifuge at 21,130 x g for 5 min. Chloroform drives water out of the buffer-saturated phenol to form two layers: an upper, aqueous layer and a bottom, organic layer. Add 375 µL of the lower, organic layer to the primer extension reaction, mix, and spin in a table-top centrifuge at 21,130 x g for 5 min. CAUTION: Phenol and chloroform are hazardous substances and precautions should be taken to avoid contact with the eyes or skin.
- Following centrifugation, carefully extract the top layer and mix with ammonium acetate added to a final concentration of 0.3 M followed by 2 volumes of 100% ethanol. Store the solution at -20 °C for a minimum of 30 min or overnight.
- Spin the sample in a table-top centrifuge at 15,000 x rpm at 4 °C for 10 min. Remove the supernatant and wash the pellet in 1 mL of 70% ethanol. Spin the sample in a table-top centrifuge at 15,000 x rpm at 4 °C for 5 min. Remove the supernatant and resuspend the pellet in 100 µL of water.
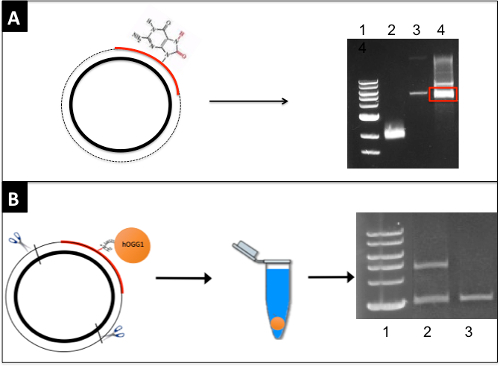
- Combine 50 µL of DNA from the previous step with 16 µL 6x gel-loading dye, and 34 µL water and subject to electrophoresis on a 0.8% low-melt agarose gel containing 0.5 µg/mL ethidium bromide. Run the gel at 2 V/cm on a 10-cm gel for 6 h in 1x TAE buffer. Excise the supercoiled band using a razor blade and weigh the gel slice (Figure 1A). Run a positive control to serve as a marker for where the covalently closed, supercoiled DNA migrates. CAUTION: Ethidium bromide is a mutagen and precautions should be taken to avoid contact with the skin. Also, it is important to minimize the time that the plasmid sample is exposed to UV in order to reduce the formation of UV photoproducts.
- Digest the gel slice by adding 10% β-agarase reaction buffer for every mg of gel weight. Incubate at 65 °C for 10 min and cool to 42 °C. Add 10 U of β-agarase and incubate at 42 °C for 1 h.
- Following incubation, measure the volume and add ammonium acetate to a final concentration of 0.3 M and chill on ice for 15 min. Centrifuge at 15,000 x g for 15 min at room temperature, collect the supernatant, and add 2 volumes of isopropanol. Chill at -20 °C overnight.
- Centrifuge the purified, supercoiled DNA for 10 min at 15,000 x rpm in a table-top centrifuge at 4 °C and resuspend the pellet in 40 µL of water.
Crosslink 12 pmol (15 µL) of DNA to 36 pmol of oxoguanine glycosylase (1 µL) in buffer containing 100 mM NaCl (3 µL), 1 mM MgCl2 (3 µL), 20 mM Tris-HCl pH 7.0 (6 µL), and 10 mM sodium cyanoborohydride (2 µL) to reach a final volume of 30 µL at 37 °C for 30 min26. Remove 1 µL of crosslinked sample and dilute in 500 µL of water to serve as a 0 h data point and analyze on PCR in step 3.
2. KCl/SDS Precipitation
- To visualize crosslinking efficiency, restriction digest 1 µg of the crosslinked plasmid with 1 μL BspDI in 10x buffer at 37 °C for 1 h to generate two different sized DNA fragments. NOTE: One fragment will be crosslinked to protein (4.4 kb) and the other will not (2.8 kB) (Figure 1B).
- Divide the samples in half, add SDS to both to a final concentration of 0.5%, and incubate at 65 °C for 10 min.
- Add KCl (final concentration 100 mM) to one of the samples and incubate on ice for 5 min.
- Centrifuge both samples at 12,000 x g for 5 min at 4 °C and run the supernatants on an agarose gel to estimate the percent conjugation of protein to DNA12. NOTE: KCl/SDS precipitation is used for quality control, not substrate preparation.
3. Transfection into Mammalian Cells
1 day prior to transfection, plate 0.5 x 106 cells/well in a 6-well plate. The next day, mix 1.5 µg (30 µL) of the DPC-containing plasmid (from step 1.6) with 300 µL of serum-free culture media. In another tube, mix 12 µL of transfection reagent and 300 µL of serum-free culture media. Combine 300 µL of diluted DNA with 300 µL of diluted transfection reagent and incubate for 5 min at room temperature. Add 250 µL of the complexes to each of two wells and incubate for a minimum of 1 h at 37 °C.
Following incubation, remove media, add 1 mL of 0.6% SDS/0.01 M EDTA, and incubate at room temperature for 10 - 15 min. Scrape the cells using a rubber policeman and transfer to a 1.5 mL microfuge tube. Add 200 µL of 5 M NaCl (final concentration of 1 M), invert gently 5 times, and incubate at 4 °C overnight27.
Centrifuge the samples at 21,130 x g in a table-top centrifuge at 4 °C for 30 min, collect the supernatant, ethanol precipitate as described above, and resuspend in 50 µL of water.
4. SSPE-qPCR
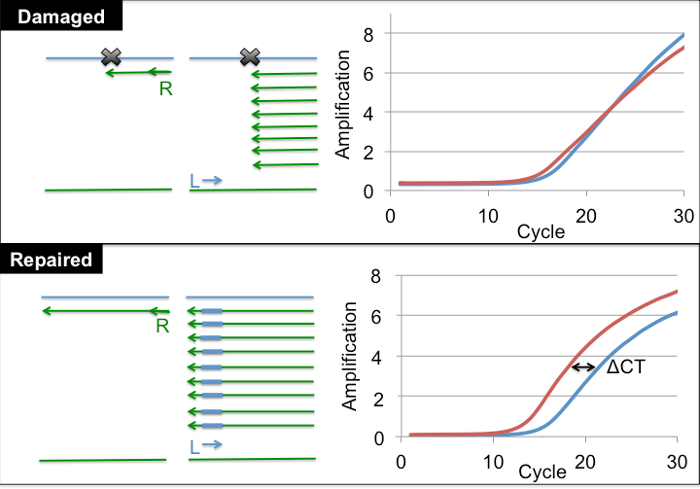
Mix together 1 µL of recovered DNA from each time point (including 0 h), 2x SYBR green PCR master mix (30 µL), 27 µL of water, and 1 µL (100 pMol) of primer complementary to the damage strand of the plasmid (Primer R, Figure 2). Perform PCR using the following conditions: Initial pre-melt for 10 min at 90 °C, followed by 8 cycles of: 90 °C, 15 s, 65 °C, 1 min. At the completion of cycle 8 add 1 µL (100 pMol) of the second primer to total 60 µL (Primer L, Figure 2)28 (see Table of Materials for reagent details).
Mix 1 µL of unamplified recovered DNA from each time point (including 0 h), 2x master mix (30 µL), 27 µL of water, and 1 µL of each primer (100 pMol) to total 60 µL.
Load a 96-well PCR plate with the samples from steps 4.1 and 4.2 (20 µL/well, in triplicate) and perform qPCR for 30 cycles using the conditions described above.
Average the CT values from each set of triplicate samples. Subtract the CT values generated in step 4.1 from those in step 4.2 to obtain the delta CT value for each sample. Subtract the 0 h time point from the delta CT value to remove any background (See Representative Results section for a detailed description).
Convert the delta CT value into percent repair using the formula:
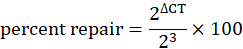
Representative Results
In order to utilize the SSPE-qPCR assay to assess cellular DPC repair, a sufficient quantity (µg amounts) of high-quality DPC repair substrate must be prepared. To obtain this product, an oligonucleotide containing an 8-oxoguanine residue was annealed to complementary single-stranded DNA, primer extended, and gel purified to ensure that only covalently, closed circular product was used for transfections (Figure 1A). This report focuses on substrates in which borohydride-trapping is used to create a covalent crosslink between recombinant human oxoguanine glycosylase and a ribose unit on a double-stranded circular plasmid molecule, although analogous approaches can be used to obtain chemically diverse DPC substrates. The oxoguanine glycosylase/borohydride trapping strategy is especially attractive given the extremely high efficiency of the crosslinking reaction. As shown in Figure 1B, KCl/SDS precipitation performed on a DPC substrate that had been digested into two fragments selectively and nearly quantitatively depleted the DNA fragment harboring the DPC. These results support the conclusion that essentially 100% of the plasmid substrate molecules contain a protein crosslink.
The SSPE-qPCR assay described in the Protocol section utilizes CT values generated by qPCR to calculate the percent of DPC repair following transfection in mammalian cells. As Figure 2 illustrates, the protein crosslink blocks Taq polymerase from extending the 'R' primer annealed to the DPC-containing strand. A second, critical feature of this assay is the incorporation of eight rounds of SSPE reactions (using the R primer) prior to performing the qPCR assay. While undamaged (or repaired) DNA will be extended during these SSPE reactions, creating a binding site for the downstream 'L' primer, damaged DNA (or incompletely repaired DNA) will not be extended. Consequently, SSPE increases the abundance of each undamaged (or repaired) strand by eight-fold. This means that repaired samples in which the SSPE step has been performed prior to qPCR will display a CT value that is three units lower than that obtained for an identical sample in which SSPE was not performed prior to qPCR (Figure 2). The three-unit difference in CT values observed for the two treatments, referred to as ΔCT, reflects the fact that 8 = 23. This delta CT value can be used to calculate cellular DNA repair activity by using the formula: percent DNA repair = (2ΔCT/23) x 100. Control experiments confirmed that a delta CT value of approximately 3 was consistently observed when undamaged substrate plasmids were subjected to SSPE-qPCR (data not shown).
Table 1 depicts example CT values that were generated from DPC-containing plasmid substrates that were recovered from Chinese hamster lung fibroblasts 3 h and 8 h post-transfection (Table 1A), as well as from time zero samples, i.e., samples that were prepared as described in the Protocol section, but not transfected into cells (Table1B). The table also provides the ΔCT, and percent repair values (calculated using the formula 2ΔCT/23 x 100) obtained from these samples. As illustrated in Table 1A, the percent repair calculated from samples harvested 3 h and 8 h post-transfection was 66% and 93%, respectively. These values are then subtracted from that obtained from analysis of the 0 h sample to determine the percent repair. Table 1B depicts CT values for two different 0 h samples in which efficient crosslinking was or was not achieved prior to transfection. Efficiently crosslinked sample resulted in a delta CT value of 0.8 whereas a poorly crosslinked sample resulted in a delta CT value of 2.5. To date, it has not been possible to precisely determine the source of the 0.8 background signal in the efficiently crosslinked 0 h sample. It is unlikely that this background reflects a low level of either spontaneous DPC removal, or is due to a low level of Taq polymerase 'bypass' synthesis through the DPC lesion: extensive experimentation performed on highly purified single-stranded M13 substrate consistently yielded a delta CT value of approximately 0.8, i.e., essentially identical to this 'background' extension seen in DPC-containing substrates. Consequently, it is likely that there is a small degree of mis-priming of the 'R' primer during SSPE that results in the generation of a small amount of product that can subsequently be amplified when the 'L' primer is added, and qPCR performed. Efforts to eliminate this background by manipulating PCR conditions have, to date, failed to remedy this defect. However, the subtraction strategy described above permits accurate estimation of DPC repair activity. It is worth noting that inefficient crosslinking of the protein onto the plasmid will result in an elevated background value. This is depicted in the sample data provided in Table 1B where inefficient protein-DNA crosslinking resulted in a higher delta CT value for time 0. Therefore, control experiments are invariably performed prior to initiating transfections and substrates with delta CT values elevated above the 0.8 threshold level are discarded. As mentioned in the qPCR protocol, each sample is divided into 3 wells to ensure pipetting consistency. These replicates are then averaged to obtain the CT value for that sample. Replicates that deviate more than ± 0.1 are eliminated from the data set. If all three replicates deviate more than ± 0.1 from each other, the SSPE-qPCR assay is redone until consistent values are obtained. Experience shows that at least 3 independent transfections must be performed to obtain reliable average values that can then be subjected to statistical analysis to determine the influence of various parameters on DPC repair efficiency.
Figure 1. Generation of a DPC-containing plasmid. (A) An oligonucleotide containing an 8-oxo-guanine residue (red) is annealed to single-stranded DNA (bold black circle) and extended to create double-stranded plasmid (primer extension product indicated by dashed line). Following electrophoresis in ethidium bromide, the band corresponding to supercoiled DNA (red box) is excised from a low-melt agarose gel and digested with β-agarase. Lanes: (1) Molecular weight marker, (2) single-stranded DNA, (3) double-stranded DNA, (4) primer extended sample. (B) Oxoguanine glycosylase (abbreviated hOGG1, depicted as an orange circle) is crosslinked to the 8-oxo-guanine residue via sodium cyanoborohydride and the resulting DPC substrate digested to generate a 2,800 base-pair, free DNA fragment and a 4,400 base-pair fragment attached to the protein. SDS is added to the sample which is then divided into two portions, one of which is treated with KCl and centrifuged to sediment DPC-containing DNA (depicted as an orange pellet). The supernatant from this latter sample (depicted as blue fluid in centrifuge tube) and the sample not exposed to KCl are subjected to gel electrophoresis. Lanes: (1) Molecular weight marker, (2) -KCl, (3) +KCl. Please click here to view a larger version of this figure.
Figure 2: Quantification of damaged plasmid via SSPE-qPCR. Plasmid DNA is denatured and a primer complementary to the damaged strand (R) is annealed and extended. This process is repeated for a total of 8 cycles. With a damaged substrate (top), Taq polymerase is blocked by the DPC lesion and will not produce full length product strands. In contrast, with a repaired substrate (bottom), Taq polymerase will produce full-length product strands containing the binding site for primer L. After 8 cycles, primer L is added, and cycle threshold values are determined using qPCR. Example amplification results for damaged and undamaged substrates are illustrated on the right with the red line representing DNA that underwent primer extension prior to qPCR and blue representing DNA that was not primer extended prior to qPCR. Please click here to view a larger version of this figure.
| Table 1A | ||||
| Time | - Primer extension | + Primer extension | Δ CT | Percent Repair |
| (2ΔCT/23x 100) | ||||
| 3 h | 23.5 | 21.1 | 2.4 | 66 |
| 8 h | 29.4 | 26.5 | 2.9 | 93 |
| Table 1B | ||||
| Time | - Primer extension | + Primer extension | Δ CT | Percent Background |
| (2ΔCT/23x 100) | ||||
| 0 h | 15.4 | 14.6 | 0.8 | 22 |
| 0 h | 15.4 | 12.9 | 2.5 | 71 |
Table 1: Calculating percent repair using CT values generated from SSPE-qPCR. (A) Repair of a DPC-containing substrate transfected in V79 Chinese hamster lung fibroblasts 3 h and 8 h post-transfection. (B) SSPE-qPCR of 0 h samples not transfected into cells. In one sample the efficiency of the crosslinking reaction between DNA and oxoguanine glycosylase was high (this sample yielded a low delta CT value) while in the other sample the efficiency of protein crosslinking was low (this sample yielded a relatively high delta CT value). See text of Representative Results for details.
Discussion
The SSPE-qPCR method offers numerous advantages over other approaches by examining the repair of a homogenous population containing a single, defined DPC lesion. It is noteworthy that in addition to controlling the identity of the protein and the type of chemical crosslink used to connect the protein to the DNA, it is possible to easily manipulate the sequence context into which the DPC lesion is introduced. We have explored the influence on DPC repair of introducing the lesion on either the template or coding strand of a plasmid downstream of an active promotor locus. Similarly, we are in the process of investigating the influence on DPC repair of replication using a M13 plasmid containing an SV40 origin of replication transfected into HEK293T cells. The assay described herein directly measures DPC repair, as opposed to other strategies such as host cell reactivation that indirectly estimates repair activity24,25. In addition, the system is robust, sensitive, and quantitative. Unlike other systems, which measure DPC removal, this assay only detects complete repair events, i.e., it requires not only that the DPC lesion be removed but that the integrity of the duplex DNA be fully restored17. This is because abasic sites and nicks or breaks in the phosphodiester backbone block the assay as effectively as the original DPC lesion29.
While this report focuses on a particular type of DPC, which was created by borohydride trapping of an enzyme reaction intermediate, we are currently developing approaches to study repair of DPCs involving other proteins and lesions in which the protein linkage to the DNA occurs through the nucleoside base, rather than the ribose position. Using reductive amination, we have created protein and peptide crosslinks attached to the guanine or cytosine base of a DNA primer30. These oligonucleotides were purified to homogeneity and used to generate supercoiled plasmids containing DNA-protein and DNA-peptide crosslinks. While the efficiency of these reactions is somewhat reduced relative to that of the oxoguanine glycosylase crosslink approach described in detail above, they were nevertheless successful and permit the examination of the repair of these substrates in wild-type and nucleotide excision repair-deficient mammalian cell lines. We have also used the SSPE-qPCR assay to study the repair of oxoguanine lesions and a synthetic ribose-cholesterol conjugate, which was previously shown to be repaired via the cellular nucleotide excision repair machinery31. As Figure 2 graphically depicts, repair of any lesion that blocks primer extension by Taq polymerase can, in principle, be measured using the SSPE-qPCR assay.
Current models of DPC repair suggest that larger DPCs (>10 kDa) are subjected to proteolytic processing to smaller peptide lesion prior to removal32,33,34. Most likely candidates responsible for this proteolysis are the proteasome or a specific protease in human cells named Spartan20,21,35,36,37,38. Further investigations into the roles of these proteases can be conducted using the SSPE-qPCR assay. Proteasome inhibitors could be used to pretreat cells prior to transfection with DPC-containing substrates. Alternatively, Spartan knockdown cell lines could be transfected with damaged plasmids to elucidate its role in proteolysis of larger DPCs.
Irrespective of the method used to create the crosslink or of the nature of the DNA lesion, it is worth stressing that the SSPE-qPCR methodology is critically dependent on the ability to generate substantial amounts of homogeneous DPC-plasmid substrate. Steps essential for this analysis include purification of covalently, closed-circular plasmid following primer extension to eliminate any nicked or linear plasmid molecules. These contaminants must be eliminated to ensure that any subsequent DPC repair observed is not due to nick-directed or double-strand break-directed repair processes. Failure to obtain sufficient quantities of supercoiled DNA following primer extension reactions may be overcome by varying the ratio of oligonucleotide to single-stranded DNA. Another important step for the SSPE-qPCR method, is the crosslinking efficiency of the protein to the plasmid. In this study, an efficient, enzymatic reaction was used to crosslink the oxoguanine glycosylase protein to an 8-oxo-guanine lesion. Sub-optimum crosslinking can be improved by varying the amount of protein added to the reaction.
While the results described in this report relied on lipofection to introduce DPC repair substrates into recipient cells, there is no reason, a priori, other transfections methods could not be employed. We have performed preliminary studies and observed that electroporation39 can also be used. However, it is worth noting that in our experience, electroporation transfection efficiency is reduced compared to lipofection, and we found it necessary to use carrier DNA (up to 5 µg) in addition to the 1.5 µg of DPC substrate to obtain repair data. Overall, the SSPE-qPCR method described above provides an innovative way to exclusively examine DPC repair on plasmid DNA and generate new insight into the DNA damage response.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was funded by the National Institutes of Health (ES023350). Lisa Chesner is supported by Training Grant 5T32HL007741. We thank Natalia Tretyakova (University of Minnesota) and Ashis Basu (University of Connecticut) and their lab members for support and technical advice during the early and intermediate stages of this work.
References
- Tretyakova NY, Groehler A, Ji S. DNA-Protein Cross-links: Formation, Structural Identities, and Biological Outcomes. Accounts of chemical research. 2015;48(6):1631–1644. doi: 10.1021/acs.accounts.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat Res. 2005;589(2):111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- At Groehler At, Degner A, Tretyakova NY. Mass Spectrometry-Based Tools to Characterize DNA-Protein Cross-Linking by Bis-Electrophiles. Basic Clin Pharmacol Toxicol. 2016;121:63–77. doi: 10.1111/bcpt.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker S, Weinfeld M, Zheng J, Li L, Murray D. Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J Biol Chem. 2005;280(40):33826–33838. doi: 10.1074/jbc.M502477200. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M, Gajewski E. Structure and mechanism of hydroxyl radical-induced formation of a DNA-protein cross-link involving thymine and lysine in nucleohistone. Cancer Res. 1989;49(13):3463–3467. [PubMed] [Google Scholar]
- Chvalova K, Brabec V, Kasparkova J. Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res. 2007;35(6):1812–1821. doi: 10.1093/nar/gkm032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speit G, Schutz P, Merk O. Induction and repair of formaldehyde-induced DNA-protein crosslinks in repair-deficient human cell lines. Mutagenesis. 2000;15(1):85–90. doi: 10.1093/mutage/15.1.85. [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Boulton SJ. Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol. 2017. [DOI] [PubMed]
- Kohn KW, Erickson LC, Ewig RA, Friedman CA. Fractionation of DNA from mammalian cells by alkaline elution. Biochemistry. 1976;19(15):4629–4637. doi: 10.1021/bi00666a013. [DOI] [PubMed] [Google Scholar]
- Ewig RA, Kohn KW. DNA damage and repair in mouse leukemia L1210 cells treated with nitrogen mustard, 1,3-bis(2-chloroethyl)-1-nitrosourea, and other nitrosoureas. Cancer Res. 1977;37:2114–2122. [PubMed] [Google Scholar]
- Ewig RA, Kohn KW. DNA-protein cross-linking and DNA interstrand cross-linking by haloethylnitrosoureas in L1210 cells. Cancer Res. 1978;38:3197–3203. [PubMed] [Google Scholar]
- Zhitkovich A, Costa M. A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo. Carcinogenesis. 1992;13(8):1485–1489. doi: 10.1093/carcin/13.8.1485. [DOI] [PubMed] [Google Scholar]
- Chiu SM, Sokany NM, Friedman LR, Oleinick NL. Differential processing of ultraviolet or ionizing radiation-induced DNA-protein cross-links in Chinese hamster cells. Int J Radiat Biol Relat Stud Phys Chem Med. 1984;46(6):681–690. doi: 10.1080/09553008414551921. [DOI] [PubMed] [Google Scholar]
- Merk O, Reiser K, Speit G. Analysis of chromate-induced DNA-protein crosslinks with the comet assay. Mutat Res. 2000;471(1-2):71–80. doi: 10.1016/s1383-5718(00)00110-8. [DOI] [PubMed] [Google Scholar]
- Loeber RL, et al. Proteomic analysis of DNA-protein cross-linking by antitumor nitrogen mustards. Chem Res Toxicol. 2009;22(6):1151–1162. doi: 10.1021/tx900078y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherezghiher TB, Ming X, Villalta PW, Campbell C, Tretyakova NY. 1,2,3,4-Diepoxybutane-induced DNA-protein cross-linking in human fibrosarcoma (HT1080) cells. J Proteome Res. 2013;12(5):2151–2164. doi: 10.1021/pr3011974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groehler At, Villalta PW, Campbell C, Tretyakova N. Covalent DNA-Protein Cross-Linking by Phosphoramide Mustard and Nornitrogen Mustard in Human Cells. Chem Res Toxicol. 2016;29(2):190–202. doi: 10.1021/acs.chemrestox.5b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, Maizels N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Research. 2013;41(9):e104. doi: 10.1093/nar/gkt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, Maizels N. Ultrasensitive isolation, identification and quantification of DNA-protein adducts by ELISA-based RADAR assay. Nucleic Acids Research. 2014;42(13):e108. doi: 10.1093/nar/gku490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, et al. Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Molecular Cell. 2016;64(4):688–703. doi: 10.1016/j.molcel.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, et al. Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol Cell. 2016;64:704–719. doi: 10.1016/j.molcel.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, et al. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 2017;45(8):4564–4576. doi: 10.1093/nar/gkx107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nature Genetics. 2014;46(11):1239–1244. doi: 10.1038/ng.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, et al. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J Biol Chem. 2007;282(31):22592–22604. doi: 10.1074/jbc.M702856200. [DOI] [PubMed] [Google Scholar]
- Ahn B, Kang D, Kim H, Wei Q. Repair of mitomycin C cross-linked DNA in mammalian cells measured by a host cell reactivation assay. Mol Cells. 2004;18(2):249–255. [PubMed] [Google Scholar]
- Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nat Struct Biol. 2003;10(3):204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Lee HW, et al. Monitoring repair of DNA damage in cell lines and human peripheral blood mononuclear cells. Anal Biochem. 2007;365(2):246–259. doi: 10.1016/j.ab.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorsky JA, Primerano DA, Fenger TW, Denvir J. DNA damage reduces Taq DNA polymerase fidelity and PCR amplification efficiency. Biochemical and biophysical research communications. 2007;355(2):431–437. doi: 10.1016/j.bbrc.2007.01.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramaratne S, Mukherjee S, Villalta PW, Scharer OD, Tretyakova NY. Synthesis of sequence-specific DNA-protein conjugates via a reductive amination strategy. Bioconjug Chem. 2013;24(9):1496–1506. doi: 10.1021/bc400018u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JW, et al. Restoration of nucleotide excision repair in a helicase-deficient XPD mutant from intragenic suppression by a trichothiodystrophy mutation. Mol Cell Biol. 2001;21(21):7355–7365. doi: 10.1128/MCB.21.21.7355-7365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko IG, et al. Initiation of Repair of DNA-Polypeptide Cross-Links by the UvrABC Nuclease. Biochemistry. 2005;44(8):3000–3009. doi: 10.1021/bi0478805. [DOI] [PubMed] [Google Scholar]
- Minko IG, Zou Y, Lloyd RS. Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc Natl Acad Sci U S A. 2002;99 doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A. Repair of DNA-polypeptide crosslinks by human excision nuclease. PNAS. 2006;103:4056–4061. doi: 10.1073/pnas.0600538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 2016;5:e21491. doi: 10.7554/eLife.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014;158(2):327–338. doi: 10.1016/j.cell.2014.04.053. [DOI] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, Walter JC. Repair of a DNA-Protein Crosslink by Replication-Coupled Proteolysis. Cell. 2014;159(2):346–357. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Habermann B, Jentsch S. DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends in Biochemical Sciences. 2015;40(2):67–71. doi: 10.1016/j.tibs.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Potter H, Heller R, et al. Ausubel FM, et al., editors. Transfection by Electroporation. Current protocols in molecular biology. 2003. CHAPTER Unit-9.3. [DOI] [PubMed]