

ALCOHOLISM IS A DISEASE CHARACTERIZED BY CONTINUED USE DESPITE NEGATIVE CONSEQUENCES
Alcoholism is a major societal problem due to its negative impact on health and socioeconomics. Excessive alcohol use and abuse are among the leading causes of premature death and disability in the United States (Michaud et al., 2006), and are ranked third behind tobacco use and poor diet/obesity as a leading cause of preventable mortality (Mokdad et al., 2004). According to a large, population-based study by the National Institute on Alcohol Abuse and Alcoholism on alcohol drinking habits in the United States (the National Epidemiological Survey on Alcohol and Related Conditions: NESARC), approximately 65% of American adults report consuming alcohol (i.e., one or more drinks in the last 12-month period). Within this population, 72% of individuals reported consuming low, safe levels of alcohol (men: fewer than 5 drinks daily, 14 drinks weekly; women: fewer than 4 drinks daily, 7 drinks weekly) while the remaining 28% of individuals reported heavy drinking (frequent consumption of 5 or more drinks per day: Grant et al., 2004, 2007). Approximately three-quarters of all alcohol ingested in the United States is consumed by 10% of the population (Li, 2008). In addition to the increased risk of developing a variety of health problems, heavy-drinking patterns also increase the likelihood of developing an addiction to alcohol.
According to the Diagnostic and Statistical Manual of Mental Disorders-5 (DSM-5), a psychiatric diagnosis of substance abuse involves repeated use of alcohol or other drugs despite problems related to use of the substance (American Psychiatric Association, 2013). This is exemplified by the requirement that the individual’s drinking habit lead to clinically significant impairment or distress. Central to a medical diagnosis of alcohol dependence is loss of control over use and preoccupation with alcohol. Diminished control over alcohol use refers to escalating use, inability to reduce or control use, and continued, persistent use despite harm to the individual, family, and community due to impaired ability to alter behavior. Preoccupation refers to increased time spent obtaining, using, and recovering from alcohol as well as abandoning important social and family obligations in favor of alcohol use. The acute effects of alcohol on behavior include disinhibition and increased impulsivity, which are indicative of reduced prefrontal cortex (PFC) executive function (Weafer and Fillmore, 2008; Dick et al., 2010).
Although the acute effects of alcohol ingestion on brain and behavior have received considerable attention (Volkow et al., 1990; Varlinskaya and Spear, 2002), we are only beginning to unravel the long-term consequences of prolonged alcohol on neurobiology. In support of persistent alcohol-induced changes to the brain, alcoholics return to their heavy-drinking habits during relapse and do not initiate their drinking behaviors from baseline, earlier levels. A key component of the behavioral pathology of alcohol dependence and addiction centers on loss of cortical executive function and cognitive flexibility, and mounting limbic anxiety and impulsivity. In the chapter that follows, mechanisms that may underlie the shift from moderate-drinking practices to heavy-drinking patterns and alcoholism will be discussed in relation to alcohol-induced changes to brain structure and function.
DIMINISHED EXECUTIVE FUNCTION IN THE ALCOHOLIC IS CONSISTENT WITH A COMPROMISE OF PREFRONTAL CORTEX FUNCTION
Chronic repetitive use of alcohol culminates in changes to brain and behavior that are characterized by diminished behavioral control, difficulty avoiding negative consequences, and increased preoccupation that is associated with mounting craving and limbic negative affect. Alcohol dependence and addiction involve a disruption of the normal balance between self-control mechanisms and emotional drive. The PFC and limbic system structures are particularly vulnerable to the neurotoxic effects of alcohol, and are critically involved in the aforementioned processes. The PFC modulates decision making and other executive functions, such as motivation, planning and goal setting, and impulse inhibition. The limbic system, which consists of the amygdala, hippocampus, and diencephalon, contributes to regulation of memory, emotion, and mood. Continued use of alcohol and other drugs of abuse alters cortical and limbic system neurobiology, culminating in a loss of attention, impaired decision making, and increased impulsivity and anxiety that drive repeated cycles of binge drinking, further disrupting cognitive-limbic circuits through progressive neuroimmune activation and loss of control over alcohol use (Fig. 27.1).
Fig. 27.1.
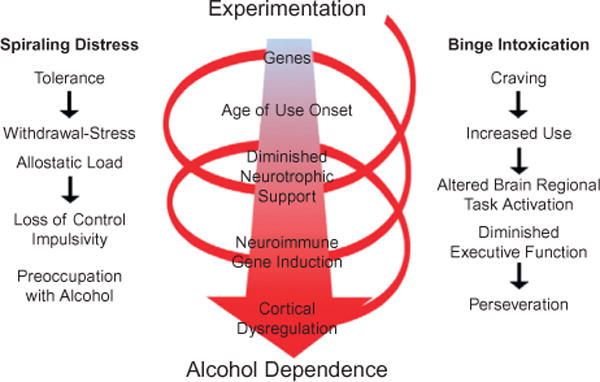
Spiral of distress modeling the progression from alcohol experimentation to alcohol dependence. Left: spiraling distress. Cycles of alcohol intoxication and withdrawal stress increase tolerance and increased allostatic load that promote craving and increased use, leading to further cycles of binge intoxication that progress to loss of control over alcohol use (Koob and Le Moal, 1997; Koob et al., 2004). Middle: Genetics and age of drinking onset are factors that influence binge drinking which, over cycles, alters brain gene expression, e.g., decreased trophic factor expression and increased neuroimmune gene expression that ultimately lead to cortical dysfunction. Right: Craving increases use and binge drinking that disrupts frontal cortical function, reduces behavioral flexibility and executive functions, increasing perseverative-compulsive actions and decreasing goal orientation. The spiral of distress links binge intoxication cycles to alcohol dependence, characterized by preoccupation with alcohol and impulsive drug taking that is likely due in part to diminished frontal cortical executive function. These mechanisms promote alcohol dependence and addiction through increased craving and other negative feelings as well as perseveration and loss of control over drinking. (Adapted from Koob and Le Moal, 1997.)
Human prefrontal cortex case studies
The PFC regulates behavior through decision-making processes and other executive functions, such as motivation, planning and goal setting, impulse inhibition, and regulation of limbic drive. Executive function encompasses abstract thinking, decision making, motivation, planning, attention to tasks, and inhibition of impulsive responses. Impulsivity, which is regulated by the PFC, refers to actions that are poorly conceived, prematurely expressed, and unduly risky or inappropriate to the situation and that often result in undesirable consequences (de Wit, 2009) that increase during intoxication and alcoholism. In addition, the PFC is involved in the retention of long-term emotional memory and modification of emotions (Bechara et al., 2000) to fit societal norms necessary for functional integration into society.
Some of the behavioral symptoms of alcoholism (e.g., loss of attention, poor decision making, and increased impulsivity) are similar to deficits observed in individuals with lesions of the PFC (Bechara et al., 1994; Bechara, 2005). The PFC consists of several structures, including the dorsolateral PFC (dlPFC), orbitofrontal cortex (OFC), ventromedial PFC (vmPFC), and anterior cingulate cortex (ACC). Although precise functional contributions of each of these intersecting structures are difficult to delineate due to considerable interconnections and overlapping functions, human case studies involving lesions and insults to these cortical regions have offered some insight into their respective contributions to executive function and addiction-like behavior. The dlPFC (Brodmann areas 9 and 46) contributes to executive function through modulation of attention as well as behavioral organization (Cummings, 1998; Stuss and Alexander, 2000). In humans, lesions of the dlPFC produce a “frontal dysexecutive syndrome” characterized by impaired planning, strategy development, and working memory as well as diminished cognitive flexibility (Clark and Manes, 2004). Similar working-memory deficits have been observed in non-human primates with lesions of this structure (Baddeley, 1992; Goldman-Rakic, 1992). The dlPFC is also associated with behavioral regulation due to its ability to maintain and integrate sensory and affective information (Carmichael and Price, 1995a, b), allowing for the representation of expected outcomes used to guide behavior (Schoenbaum et al., 2006). The OFC (Brodmann areas 11, 12, and 13) is also critically involved in the generation and use of outcome expectancies (Schoenbaum et al., 2006) as damage to this region results in profound deficits in self-regulation and control. These deficits were exemplified in the famous case of Phineas Gage, a railroad worker who survived the passage of a tamping rod through his OFC. He experienced several cognitive changes as a consequence of this insult that included behavioral disinhibition and loss of behavioral flexibility (Harlow, 1848, 1868). Other studies of OFC lesions have identified behavioral changes involving increased impulsivity and perseverative behaviors as well as deficits in emotional recognition and impaired judgment in social contexts (Rolls et al., 1994; Berlin et al., 2004; Bechara, 2005; Coutlee and Huettel, 2012; Rosenbloom et al., 2012). Studies of humans with bilateral damage of the vmPFC (Brodmann area 10) reveal severe impairments in decision making (Eslinger et al., 1984; Damasio et al., 1991; Bechara et al., 1998), defined as selecting the most advantageous response from an array of available options. Finally, the ACC (Brodmann areas 24, 32, and 33), especially the dorsal subregion, has been implicated in inhibitory control (Yucel and Lubman, 2007), whereas the anterior component plays a role in linking decision making to emotion (Cella et al., 2010). These human case studies suggest that damage to the PFC might contribute mechanistically to the development and maintenance of alcoholism. Thus, subregions of the PFC each contribute to executive functioning, and damage to these regions results in behavioral impairments that are commonly observed in the alcoholic individual (e.g., diminished cognitive flexibility and decision making, perseveration, behavioral dysregulation, and emotive dysfunction).
Alcohol-induced neurodegeneration
Human lesion studies suggest that loss of PFC function contributes to cognitive inflexibility, impulsivity, and loss of behavioral control, which are hallmark features common in alcoholism. Alcoholics are capable of ingesting large quantities of alcohol, which can result in PFC neurodegeneration (Crews and Boettiger, 2009) and might contribute to the loss of executive function associated with alcoholism. In the alcoholic brain, neuroimaging studies find reduced gray- and white-matter volumes across multiple regions, which are greatest in the PFC as well as white-matter tracts of the frontal cortex (Pfefferbaum et al., 1995; Kubota et al., 2001; Sullivan and Pfefferbaum, 2005). Although neuronal loss does not appear to account for all of the volumetric loss observed in the alcoholic brain, both the superior frontal cortex (Harper and Kril, 1989) and OFC (Miguel-Hidalgo et al., 2006; Qin and Crews, 2012b) show neuronal loss, while the temporal lobes do not show evidence of similar loss (Kril and Harper, 1989). This occurs in the absence of major nutritional deficiencies (e.g., thiamine (vitamin B1)), although they can also cause neurodegeneration and potentiate alcoholic neurodegeneration (Bowden et al., 2001; Qin and Crews, 2014). It has been proposed that progressive increases in alcohol consumption, a common feature of alcoholism, lead to PFC alterations that reduce executive functioning that promote further alcohol abuse and neurodegeneration (Crews et al., 2004). Alcohol-induced PFC degeneration leads to loss of executive function and diminished limbic system regulation, leading to increased impulsive behavior that contributes to the persistence of addiction (Bechara, 2005; Crews et al., 2005).
Alcoholic individuals display several components of impulsivity, including deficits in delay discounting and behavioral inhibition (de Wit, 2009). Connections between the vmPFC, ACC, and amygdala are thought to be critical for impulse control (Bechara, 2005). Human neuroimaging studies have also found hyperactivation in the amygdala in response to stimuli that elicit craving, which is indicative of impulsivity (Breiter et al., 1997; Childress et al., 1999; Kilts et al., 2001). In contrast, there is evidence for hypoactivation of the PFC in the addicted brain. Employing positron emission tomography in conjunction with 2-deoxy-2[18F] fluoro-D-glucose, Volkow and colleagues (1997) found reductions in PFC metabolism that were evident 2–3 months following detoxification, suggesting that cortical hypoactivity represents a persistent consequence of alcoholism. It is possible that PFC hypoactivity could lead to compulsive drug use in response to drug-related stimuli (Volkow et al., 2004). Thus, hyperactivity in the amygdala, coupled with a hypofunctional PFC, might be a mechanism underlying the development of alcoholism.
Frontal cortex dysfunction is common in the human alcoholic brain (Crews and Boettiger, 2009; Crews and Nixon, 2009). Compromised frontal cortical executive function could lead to alcohol abuse, or alcohol use might insult PFC function. Using animal models with high blood levels of ethanol induces limbic and cortical brain damage (Crews et al., 2004; Crews and Nixon, 2009). Studies investigating changes in human cortical thickness find thinning in many cortical brain regions, including entorhinal cortex, piriform cortex, and perirhinal cortex (Fortier et al., 2011) that are also found to be sensitive to binge-drinking rat models of brain damage (Fig. 27.2) (Obernier et al., 2002a, b). The brain damage in the rat binge ethanol study did not alter learning ability assessed using the Morris water maze special learning task; however, it did alter reversal learning. Reversal learning tasks assess behavioral or cognitive flexibility. In these tasks, which are applicable across species, subjects first learn a stimulus–reward association, and then the reward association is switched. This ostensibly simple process taxes multiple executive functions, including attention, working memory, and response inhibition. Alcoholic humans and binge-treated animals show a loss of flexibility. Cognitive or behavioral flexibility is a critical executive function that can be broadly defined as the ability to adapt behaviors in response to changes in the environment. Binge ethanol treatment for 4 days did not alter learning ability, but 3 weeks after the binge treatment ethanol-treated rats took more time and a greater number of trials to learn the reversal location (Fig. 27.3) (Obernier et al., 2002b). Reversal learning provides a measure of rule shifting, an executive function involving the ability to revise a previously learned behavioral response with a new response when outcomes do not match those predicted by the preceding cues (Stalnaker et al., 2009). Loss of control of drinking alcohol as a key pathology of alcoholism likely includes a loss of behavioral flexibility related to binge drinking-induced changes in brain. More recent studies have found that adolescent binge ethanol exposure of mice (Coleman et al., 2009, 2011) or adolescent exposure of rats (Vetreno et al., 2013) shows similar deficits in behavioral flexibility as adults assessed using reversal learning tasks. This behavioral paradigm models the perseveration and inability of human alcoholics to learn new, adaptive behaviors. Taken together, these human and animal studies implicate dysfunctional PFC and limbic system structures in the behavioral symptoms of addiction.
Fig. 27.2.
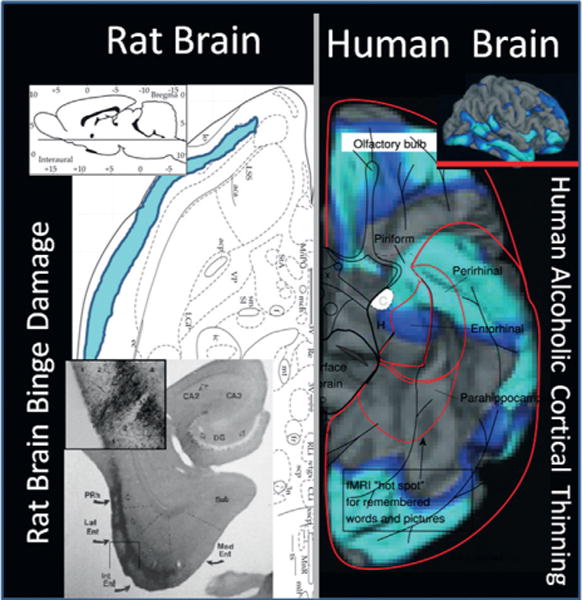
Comparison of human alcoholic cortical thinning with rat binge alcohol treatment-induced neurodegeneration. Shown are a horizontal section through the ventral rat brain and the ventral surface of the human brain. (Left side) Rat brain anatomic section in the upper left indicates location of ventral horizontal section. The bottom left shows a brain section with silver cell death stain (black areas) showing neurodegeneration from a binge-drinking model that includes association cortical areas, piriform and perirhinal cortex, as well as entorhinal cortex. Blue shading indicates that rat piriform neurodegeneration extends to frontal regions. (Adapted from Obernier et al., 2002a; Crews et al., 2004.) On the right side is a ventral view of the cortex from the human brain. Human alcoholic ventral cortical thinning is shown as a significance map of group differences in cortical thickness in abstinent alcoholics as compared to non-alcoholic controls with areas showing brain regions with significant cortical thinning in alcoholics highlighted in blue (light blue, p<0.01; dark blue, p<0.05). (Adapted from Fortier et al., 2011.) Note perirhinal, entorhinal, and piriform cortex are areas with cortical thinning in humans and areas of binge-drinking neurodegeneration in rats, suggesting binge-drinking human alcoholics are damaging association cortical areas. (Adapted from Fortier et al., 2011.)
Fig. 27.3.
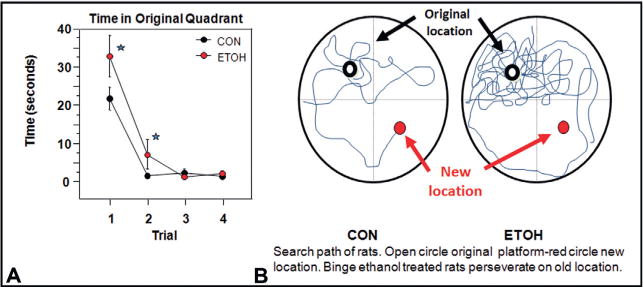
Binge drinking reduces behavioral flexibility as assessed by reversal learning. This study treated rats with a 4-day binge-drinking model and then assessed learning and reversal learning after weeks of abstinence to assess long-term changes induced by binge drinking. All animals (both control (CON) and alcohol binge-treated (EtOH)) learned the initial location without any apparent learning differences. Both CON and EtOH animals learned the location of the platform in the Morris water maze task, suggesting binge drinking did not alter learning. For reversal learning the submerged platform was moved to the opposite quadrant and animals were given four trials (21 days postbinge treatment). Binge-treated animals had difficulty. They took more time to locate the new location, spent more time in the previous location consistent with perseveration on the old location, and took longer to reach criteria in the reversal task. (A) Shown is the mean ± SEM of the time required to find the reversal location during the reversal trials. Reversal criterion was set at 2 sd from the distance to platform on the last day of the reference memory task. EtOH animals required a significantly greater number of trials to reach criterion than CON animals [t(14) = 2.376; *p<0.05]. (B) A vertical view of the track taken by a CON and an EtOH rat during the first reversal trial. The open circle represents the location of the submerged platform the animals initially learned. The red circle represents the location of the platform during the reversal learning task. The EtOH animal has numerous reentries into the original goal quadrant due to perseverative behavior and poor search strategy and fails to reach the new platform location within the 90-second time allowed. The binge-treated animals have not received ethanol for several weeks. The reversal learning deficits are consistent with a loss of behavioral flexibility due to disruption of frontal cortical circuits. (Data summarized from Obernier et al., 2002b.)
Alcohol alters the neurotrophin/neuroimmune balance
Although the cellular and molecular mechanisms underlying addiction remain to be fully elucidated, recent evidence suggests the potential involvement of a shift in neurotrophic/neuroimmune signaling (Fig. 27.4). Transcription factors, such as cyclic AMP-responsive element-binding protein (CREB) and nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) regulate expression of a diversity of central nervous system (CNS) genes, and have recently been implicated as a mechanism underlying alcoholism. CREB and its many target genes, including neuropeptide Y (NPY) and brain-derived neurotrophic factor (BDNF), are involved in promoting neuronal survival and protecting neurons from excitotoxicity and apoptosis (Lonze and Ginty, 2002). Glutamatergic N-methyl-D-aspartate (NMDA) receptors regulate synaptic plasticity and can be coupled to CREB. However, high levels of extrasynaptic NMDA receptor activation and NMDA receptor-mediated Ca2+ flux trigger excitotoxicity, leading to either rapid or delayed neuronal death, which is associated with diminished CREB activation (Hardingham and Bading, 2010). Levels of CREB-DNA binding and phosphorylated CREB, as well as the target gene BDNF, are decreased in the rat frontal cortex following a 24-hour withdrawal from chronic ethanol exposure (Pandey et al., 1999, 2001). In addition, NPY levels are reduced in the cortex following ethanol treatment, an effect that was complemented by reduced phosphorylated CREB (Bison and Crews, 2003). Using in vitro slice culture, our laboratory found that ethanol dose-dependently reduces CREB-DNA binding while simultaneously increasing NF-κB-DNA binding (Zou and Crews, 2006).
Fig. 27.4.
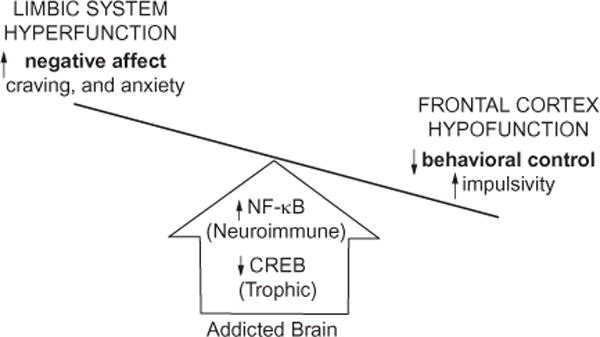
Alcohol-induced shift in neurotrophic/neuroimmune axis. A simplified schematic distinguishing the frontal cortical and limbic system behavioral changes that characterize drug-induced activation of the innate immune system. Both alcohol addiction and stress upregulate nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) signaling while reducing cyclic AMP-responsive element-binding protein (CREB) expression, which reduces glutamate transporters, leading to prefrontal cortex hyperexcitability (Crews et al., 2006a; Zou and Crews, 2006; Reissner and Kalivas, 2010) and diminished cortical behavioral control and behavioral inflexibility (Stalnaker et al., 2009; Gruber et al., 2010). Concurrently, induction of innate immune genes in limbic brain regions increases negative affect, craving, and anxiety-like behaviors, prompting further drug abuse and self-medication (Kelley and Dantzer, 2011). The long-term deleterious effects of prolonged alcohol abuse involve diminished activation of frontal cortical behavioral control circuits that lead to a progressive loss of attention and poor decision making that combine with increased negative affect and anxiety that motivate further drug-taking behaviors. Taken together, these innate immune gene-induced behavioral changes characterize the drug-addicted brain (Crews and Vetreno, 2011).
In the CNS, NF-κB is well known for its ubiquitous roll in inflammatory and neuroimmune responses (Mattson and Camandola, 2001). This transcription factor is expressed in neurons and glia (Kaltschmidt et al., 1993; Mattson and Camandola, 2001) where it regulates both physiologic processes and neurodegeneration (Grilli and Memo, 1999; Schneider et al., 1999; Pizzi et al., 2002). Nuclear activation of NF-κB is associated with induction of several downstream proinflammatory cytokines (e.g., tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, and IL-6), chemokines (e.g., monocyte chemoattractant protein-1 (MCP-1, CCL-2)), and oxidases (inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and NADPH oxidase (NOX2)) that lead to positive loops of amplification (Fig. 27.5). Activation of the proinflammatory NF-κB cascade with simultaneous reductions of neurotropic CREB signaling results in hyperexcitability, neuronal injury, and network reorganization that might also represent a mechanism that contributes to the development of alcoholism.
Fig. 27.5.
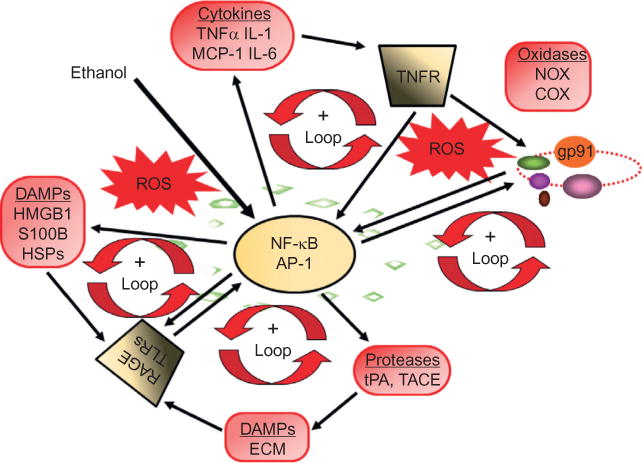
Loops of signaling converge on NF-κB, increasing the expression of chemokines, cytokines, oxidases, and proteases. The proinflammatory transcription factor NF-κB is involved in the induction of neuroimmune genes (Knapp and Crews, 1999; He and Crews, 2008; Qin et al., 2008; Alfonso-Loeches et al., 2010; Zou and Crews, 2010). Alcohol (ethanol) increases NF-κB-DNA binding and transcription. Positive loops of activation occur through induction of genes that stimulate further NF-κB activation, leading to autocrine and paracrine amplification and persistent signals. Cytokines and chemokines, such as TNF-α, IL-1β, IL-6, and MCP-1, as well as their receptors (TNFR in figure), are induced, resulting in amplification loops. Reactive oxygen species (ROS) resulting from oxidases such as NADPH oxidase or ethanol metabolism increase NF-κB transcription of NOX2phox (gp91), a key NOX catalytic subunit (Cao et al., 2005) that produces ROS (Qin et al., 2008). Toll-like receptors (TLRs) and the receptor for advanced glycation end product (RAGE) are increased by ethanol (Alfonso-Loeches et al., 2010; Crews et al., 2013; Vetreno et al., 2013), as are other neuroimmune signaling molecules, resulting in the formation of positive activation loops (Crews et al., 2013; Vetreno et al., 2013). These loops spread neuroimmune signaling across the brain, causing altered neurocircuitry and neurobiology. AP-1, activator protein-1; COX, cyclooxygenase; DAMPs, damage- (or danger-) associated molecular patterns; ECM, extracellular matrix; HMGB1, high-mobility group box 1; HSPs, heat shock proteins; IL-1, interleukin-1; MCP-1, monocyte chemoattractant protein-1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases; TACE,TNF-α converting enzyme; TNF-α, tissue necrosis factor-alpha; tPA, tissue plasminogen activator.
ALCOHOL AND INNATE IMMUNE SYSTEM ACTIVATION
Over the last decade, the innate immune system has received considerable attention for its involvement in alcoholism. Innate immunity, together with other immune-regulating nervous system components, regulates the neuroimmune response in the CNS. Alcohol exerts its deleterious effects first on the periphery through proinflammatory cascades (Crews et al., 2006b) that activate both the adaptive and innate immune system (Szabo and Mandrekar, 2009; McClain et al., 2011; Vetreno et al., 2013). The adaptive immune system consists of a class of highly specialized lymphocytic cells that recognize, remember, and target specific pathogens through the production of antibodies. It is through the formation of an immunologic memory of the pathogen that the adaptive immune system protects the organism from future insults. In contrast to adaptive immunity, the innate immune system mounts non-specific immune responses to pathogen invasion via secretion of proinflammatory cytokines and chemokines that activate neuroimmune cells (e.g., microglia: Dantzer et al., 2008).
Microglia, the resident innate immune macrophage-like cells of the CNS, along with astrocytes, modulate important metabolic, trophic, and synaptic functions in addition to responding to and amplifying neuroimmune responses in the CNS (Streit et al., 2004; Farina et al., 2007). In the healthy brain, ramified or “resting” microglia contribute to the integration of sensory systems and overall survey of the brain milieu (Raivich, 2005). In response to an endogenous or exogenous insult, microglia undergo distinct morphologic and gene expression changes associated with cellular activation (Graeber, 2010). During the initial stages of activation, microglia become enlarged and increase expression of cell matrix and cell adhesion proteins. They also begin to secrete neuroimmune signaling molecules that upregulate expression of major histocompatibility complex (MHC) proteins and Toll-like receptors (TLRs), key signaling molecules that respond to bacterial endotoxins (e.g., lipopolysaccharide (LPS)). In general, the TLR family of pattern recognition receptors consists of an extracellular domain of leucine-rich repeat motifs, a transmembrane domain that determines receptor location, and an intracellular domain (Akira et al., 2001; Takeda et al., 2003). To date, 13 TLRs have been identified (TLRs 1–13) in mammals (Medzhitov, 2001; Takeda et al., 2003) that signal through similar cascades to activate NF-κB transcription of additional proinflammatory cytokines through paracrine and autocrine amplification. Activated microglia secrete many proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6). Cytokines are signaling molecules that regulate many processes and were first discovered for their roles in orchestrating complex multicellular processes involved in defending against pathogen infection and clearing damaged cells and tissue, including signals that attract specific cells toward sites of tissue damage-initiating proinflammatory-oxidative cascades that resolve and are followed by trophic, wound-healing, cell growth cascades (Merrill and Benveniste, 1996; Pollmacher et al., 2002). Cytokine signals interact in proinflammatory signaling cascades that spread and amplify in an autocrine and paracrine fashion. As neuroimmune signaling cascades continue to intensify, microglia progress toward mitosis, proliferation, and phagocytic oxidative surges that dissolve and engulf cellular debris (Fig. 27.6). Highly activated microglia are termed “ameboid” and are characterized by a phagocytic, rounded macrophage-like morphology associated with maximal proinflammatory activation and microglial proliferation (Graeber, 2010). Microglia are further characterized as either proinflammatory (M1), or neurotrophic (M2), depending on whether they are functioning to “kill” or “repair,” respectively, although the morphologic characterization of these microglial subtypes is not entirely understood (Colton, 2009; Michelucci et al., 2009). Astrocytes are also pivotal components of the neuroimmune system as they secrete cytokines and inflammatory modulators following insult-induced activation (Aschner, 1998; Aschner et al., 2002). Indeed, S100B, which is predominantly expressed in astrocytes, is an agonist at the receptor for advanced glycation end products (RAGE), another neuroimmune receptor (Sparvero et al., 2009; Sims et al., 2010; Han et al., 2011). Together, microglia and astrocytes are the major constituents of the innate immune system in the CNS.
Fig. 27.6.
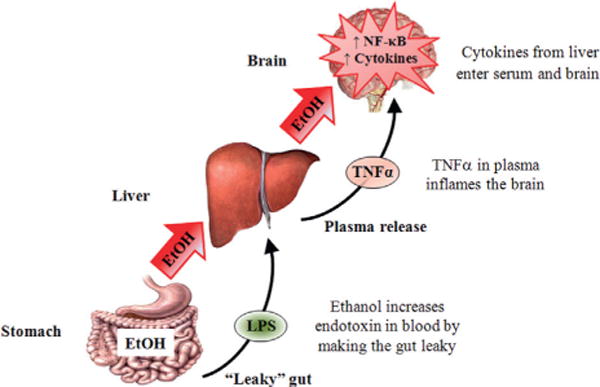
Systemic ethanol (EtOH) activation of cytokine signaling in the brain. Consumed EtOH enters the stomach and makes it “leaky,” allowing lipopolysaccharide (LPS) to enter the plasma. The circulating EtOH and LPS lead to liver proinflammatory cytokine induction, which results in the production and secretion of tumor necrosis factor-alpha (TNF-α) and other proinflammatory cytokines. TNF-α is transported into the brain from the blood and increases neuroimmune expression through increased activation and the synthesis of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) and other cytokines. See Qin et al. (2007) for details.
TLRs and RAGE recognize and respond to a variety of endogenous neuroimmune signaling molecules. Research has begun to identity several endogenous neuroimmune signaling molecules, including high-mobility group box 1 (HMGB1: Garg et al., 2010), heat shock proteins (Vabulas et al., 2002), extracellular matrix breakdown proteins (Scheibner et al., 2006), and S100 proteins (Sparvero et al., 2009). HMGB1 has received the most attention because it is an endogenous neuroimmune agonist at both TLRs and RAGE. It is a ubiquitous evolutionarily conserved nuclear protein present in most eukaryotic cells. It consists of three distinct domains: two homologous HMG boxes (box A and box B), and a C-terminal tail (Huang et al., 2010). Several studies have identified box B as the domain responsible for the proinflammatory properties of HMGB1 (Li et al., 2003; Messmer et al., 2004), whereas box A is anti-inflammatory and capable of attenuating box B-induced inflammation (Yang et al., 2004). Physiologically, HMGB1is involved in DNA bending and transcription stability (Paull et al., 1993; Thomas, 2001), but following secretion from cells, by either active release from neuroimmune cells or passive release by leaky necrotic cells, HMGB1 exerts cytokine-like activity (Sims et al., 2010). Although defense against invading pathogens is key to the survival of all mammals, timely resolution of the neuroimmune response is critical to prevent inflammation-induced tissue damage. Indeed, persistent activation of this system leads to chronic inflammation, which may contribute to the development and progression of multiple diseases, including Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis (Glass et al., 2010), and possibly alcoholism and addiction.
Alcohol activates the neuroimmune signaling system
Following ingestion, alcohol causes the stomach to become permeable or “leaky” (Ferrier et al., 2006). Recent studies report that intragastric administration of ethanol at doses >2 g/kg (Ferrier et al., 2006) potentiates gut innate immune signaling, disrupting gut tight junctions and opening sites that allow the gut biome bacteria and their fragments to enter portal circulation (Sims et al., 2010). Biome leakage contains endotoxins including LPS, which potentiates alcohol-induced liver inflammation and secretion of proinflammatory cytokines, including the proinflammatory cytokine TNF-α, which is released into the blood (Qin et al., 2007). Proinflammatory cytokines in the blood are then either transported across the blood–brain barrier (e.g., TNF receptor (TNFR): Qin et al., 2007; Banks and Erickson, 2010) or activate endothelial cells to release cytokines (Watkins et al., 1995) that, combined with ethanol, enter the brain where they induce neuroimmune activation (Fig. 27.7). The precise mechanism for the movement of proinflammatory cytokines from the periphery to the CNS remains to be fully elucidated, but does not appear to involve a compromise of the blood–brain barrier (BBB). Employing a 4-day binge rat model of alcoholic brain damage, Marshall and colleagues (2013) assessed BBB integrity using immunohistochemistry for albumin, a 140-kDa protein that does not cross an intact BBB, and found no evidence of BBB dysfunction. The resolution of liver and blood cytokine increases in the periphery occurs over time, whereas marked cytokine induction within the brain, once activated, remains active and primed for additional activation that persists for months. The enduring and progressive induction of the neuroimmune response in brain is consistent with the cumulative progression and persistence of addiction, and might represent a mechanism contributing to the development of alcoholism.
Fig. 27.7.
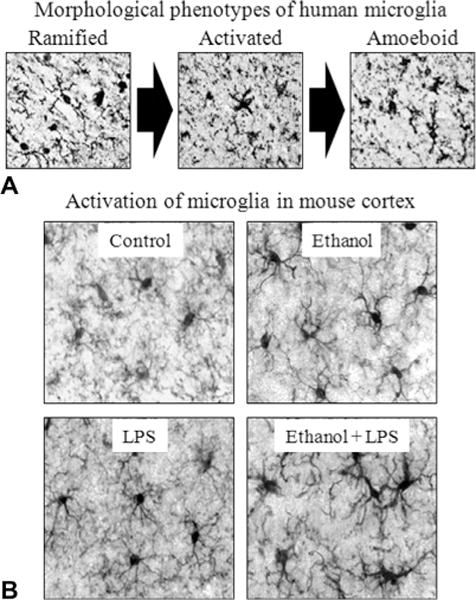
Microglial morphology, lipopolysaccharide (LPS) and alcohol in the brain. (A) Representative figures depicting characteristic stages of microglial activation. Ramified or “resting” microglia are characterized by long, highly ramified processes with comparatively small cell bodies. Activated microglia are characterized by swollen, truncated processes and enlarged cell bodies. Ameboid or “phagocytic” microglia are characterized by large, ameba-like cell body with no or few small processes (Kreutzberg, 1996; Raivich et al., 1999). Photomicrographs depict stages of microglial activation in postmortem human brain tissue (He and Crews, 2008). (B) Representative figures depicting microglial activation in the mouse cortex following ethanol and/or LPS treatment. Male C57BL/6 mice were treated with saline or ethanol (5 g/kg, i.g.) for 10 days. Half of the subjects received a dose of LPS (3 mg/kg, i.p.) 24 hours after ethanol/saline treatment, and were sacrificed 1 hour later. Ten daily doses of ethanol exposure potentiated LPS-induced microglial activation. (Adapted from Qin et al., 2008.)
Although there is an increasing body of literature supporting the hypothesis that alcohol is a potent immunomodulatory drug, it has opposing effects depending on whether administration is acute or chronic (Szabo and Mandrekar, 2009). Indeed, acute ethanol was found to suppress the innate immune response in both in vivo and in vitro models. For instance, LPS-induced TNF-α and IL-1β production was blunted in blood monocytes obtained from healthy human volunteers 16 hours after acute alcohol exposure (2 mL vodka/kg body weight: Crews et al., 2006b). In animal models, acute ethanol exposure attenuated the TNF-α, IL-1β, and IL-6 immune response to a pathogen challenge (Pruett et al., 2004). Similarly, Szabo’s group reported that acute ethanol (25 mM) inhibited LPS-induced TNF-α expression, but not TLR2-induced upregulation of the TNF-α in the presence of ethanol (Crews et al., 2006b). In contrast, chronic ethanol exposure is associated with increased neuroimmune activation. Through a series of elegant animal studies, Guerri’s laboratory demonstrated both in vivo and in vitro that chronic ethanol treatment induces astrocytic and microglial activation (Valles et al., 2004; Blanco et al., 2005; Fernandez-Lizarbe et al., 2009; Alfonso-Loeches et al., 2010; Pascual et al., 2011). For instance, in vitro administration of ethanol at physiologically relevant levels activates Toll-IL-1 receptor (TIR) signaling pathways in cultured astrocytes in a time-dependent fashion similar to what occurs following LPS and IL-1β administration, although at a much smaller response. Similarly, our laboratory found increased TLR2, TLR3, and TLR4 expression in the OFC of adult mice 24 hours after exposure to 10 days of 5 g/kg ethanol (Crews et al., 2013). Although ethanol and stress activate microglia, they do not produce the phagocytic oxidative bursts associated with maximal microglial activation by LPS or severe neurodegeneration. Thus, ethanol generally suppresses neuroimmune responses when it is present, but causes adaptations, particularly with chronic treatment, that sensitize responses during abstinence.
Involvement of the neuroimmune signaling system in alcoholism is supported by findings in the postmortem human alcoholic brain. Our laboratory recently discovered microglial activation as well as upregulated levels of MCP-1 (CCR-2) in the ventral tegmental area, substantia nigra, hippocampus, and amygdala of postmortem human alcoholic brain tissue, relative to moderate drinking controls (He and Crews, 2008). Our laboratory recently found upregulated expression of TLR2, TLR3, TLR4, and RAGE in the postmortem OFC of human alcoholics, which correlated with lifetime alcohol consumption (Crews et al., 2013). Although the mechanisms underlying ethanol-induced upregulation of neuroimmune signaling remains unknown, a potential signaling molecule that likely plays a role is HMGB1. Upon upregulation of HMGB1, it is released from the nucleus to the extracellular space where it exerts its cytokine-like effects (Muller et al., 2004; Hock et al., 2007; Maroso et al., 2010). Following its release, HMGB1 activates TLRs and RAGE, which facilitate proinflammatory responses in a cyclic fashion (Fig. 27.8) (Park et al., 2004; Yang et al., 2010; Rauvala and Rouhiainen, 2010; Volz et al., 2010). We discovered that alcohol causes increased expression and extracellular release of HMGB1 in brain slice cultures and in animal models of chronic ethanol exposure (Fig. 27.9) (Crews et al., 2013). Activation of this pathway leads to induction of NF-κB and activator protein-1, which is accompanied by an upregulation of iNOS and COX-2 expression in astrocytes that, in turn, leads to further downstream activation of the NF-κB signaling (Blanco et al., 2005). In support of the role of neuroimmunity in alcoholism, treatment with indomethacin, an anti-inflammatory drug, reduces chronic intermittent ethanol induction of brain innate immune genes (iNOS and COX-2) in astrocytes and reduces markers of cell death and behavioral dysfunction (Pascual et al., 2007). In addition, administration of minocycline, an antibiotic that blocks microglial activation (Plane et al., 2010; Qin and Crews, 2012a), reduces ethanol-induced neuroimmune activation and neural cell death (Qin and Crews, 2012a). These data provide evidence that ethanol-induced induction of neuroimmune signaling contributes to the development and maintenance of alcoholism.
Fig. 27.8.
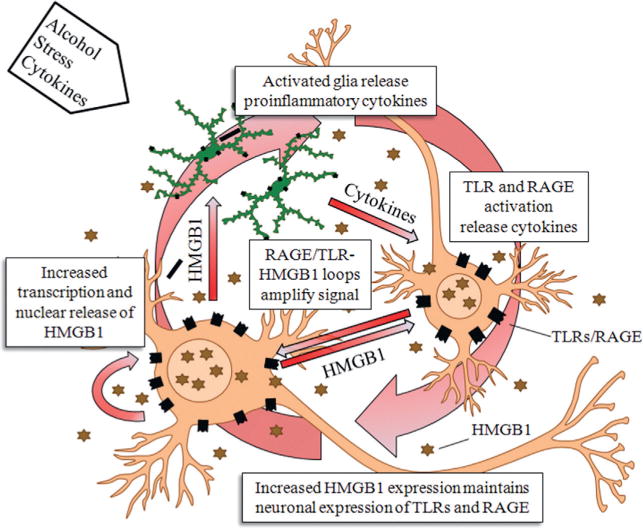
Neuroimmune activation through HMGB1-TLR signaling loops. A single injection of lipopolysaccharide (LPS: 5 mg/kg, i.p.) in C57BL/6 mice caused a long-lasting increase in protein expression of tumor necrosis factor-alpha (TNF-α). (See Qin et al., 2007.)
The persistent priming of neuroimmune activation lasts for long periods due to positive loops of signaling between neurons and glia. HMGB1 released from neurons stimulates glial activation and glial synthesis of proinflammatory cytokines that further induce additional neuroimmune genes consistent with the long-lasting impact of alcoholism and the persistence, e.g., the folklore that once an alcoholic always an alcoholic (e.g., the neuroimmune priming persists and does not go away, like the risk of relapse to alcoholism). (Adapted from Vetreno et al., 2013.)
Fig. 27.9.
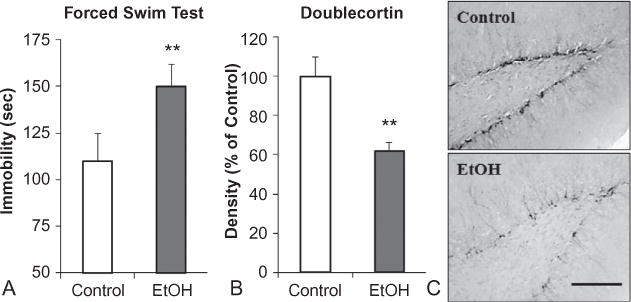
Chronic ethanol (EtOH) self-administration induces depression-like behavior and inhibits hippocampal neurogenesis. C57BL/6J mice self-administered either ethanol (10% v/v) or water for 28 days. (A) Increased immobility (seconds) on the forced-swim test provides an index of depression-like behavior. Following a period of abstinence from chronic ethanol consumption, mice evidenced increased immobility time relative to controls. (B) Doublecortin (DCX) expression, a marker of neurogenesis, was reduced in the dentate gyrus of mice exposed to chronic ethanol self-administration. (C) Representative photomicrographs depicting the reduced DCX expression. Alcohol-induced reduced neurogenesis and neuroprogenitor cell proliferation are associated with increased depression-like behavior. Antidepressants reverse both (Stevenson et al., 2009). Scale bar = 200 mm. **p<0.01, relative to control. (Adapted from Stevenson et al., 2009.)
Induction of neuroimmune cascades contributes to addiction-like behaviors
Executive functioning is mediated by neural circuitry, including the PFC and interconnected subcortical regions, which regulate mood and cognition through reciprocal glutamatergic connections with multiple limbic system brain regions. In the frontal cortex, positive loops of neuroimmune activation likely lead to hyperexcitability (Crews et al., 2006a). Ethanol-induced activation of microglial NF-κB cascades is relatively small compared to the bursts of innate immune genes responding to cell death in brain associated with stroke, traumatic brain injury, and other diseases involving major neurodegeneration (Qin et al., 2008). In astrocytes, ethanol exposure induces NF-κB transcription leading to increased expression of proinflammatory innate immune genes (Zou and Crews, 2006, 2010; Pascual et al., 2007) and reduced astrocyte glutamate transport (Zou and Crews, 2005). This increase in extracellular glutamate leads to increased neuronal excitation, microglial activation, and excitotoxicity (Zou and Crews, 2006; (Ward et al., 2009). The increase in glutamate excitotoxicity might also contribute to ethanol-induced increases of caspase-3 and COX-2 in the frontal cortex (Knapp and Crews, 1999; Alfonso-Loeches et al., 2010).
Innate immune genes also play a critical role in modulating neural plasticity by affecting long-term potentiation (LTP) and long-term depression (LTD), both of which are neural correlates of learning and memory (Stanton, 1996; Dantzer et al., 2008; Boulanger, 2009). During periods of health, the delicate neuronal–glial communication balance maintains neural plasticity through secretion of several neuroimmune mediators, including IL-1β, IL-6, and IL-18, which are induced during hippocampal LTP and related to synaptic strengthening (Schneider et al., 1998; Balschun et al., 2004; del Rey et al., 2013). However, innate immune system activation by endogenous or exogenous stimuli results in a neuroimmune imbalance, resulting in a shift in the neuronal–glial communication balance (Besedovsky and del Rey, 2011), and might impair LTP and LTD, leading to deficits in plasticity. Indeed, Maggio et al. (2013) found that a brief exposure to systemic LPS (1 mg/kg, i.p. twice a week for 1 week) transiently impaired hippocampal LTP, which was recovered to control levels 7 days following the conclusion of inflammagen exposure. In contrast, longer exposure to LPS (1 mg/kg, i.p. twice a week for 1 month) persistently altered LTP, which was evident 2 months following the conclusion of inflammagen exposure. Thus, ethanol-induced disruption of neural plasticity, particularly following binge drinking, might contribute to the long-term deficits in learning and memory associated with alcoholism.
Disruption of frontal cortex and limbic system circuits due to ethanol-induced induction of neuroimmune signaling might contribute to the loss of cognitive flexibility and other addiction-like behaviors. Neurogenesis, a process whereby newborn neurons are generated and functionally integrated into the existing hippocampal circuitry (Altman and Das, 1965; Zhao et al., 2006), is critically involved in learning and emotive function. This process is also particularly vulnerable to ethanol-induced neuroimmune activation. Using an ex vivo model of organotypic hippocampal-entorhinal cortex brain slice culture, (Zou and Crews, 2012) found that 4 days of 100 mM ethanol reduced cellular proliferation and neurogenesis. This effect was accompanied by a shift in the neuroimmune/neurotrophin balance, wherein mRNA levels of the proinflammatory cytokine IL-1β were increased while expression of CREB and BDNF was reduced. Interestingly, ethanol-induced reductions of neurogenesis were reversed by administration of a neutralizing anti-IL-1β antibody as well as blockade of IL-1 surface receptors. Further, administration of rolipram, a phosphodiesterase 4 inhibitor that increases cAMP leading to increased PKA activation and inhibition of NF-κB induction (Takahashi et al., 2002), and BHT, an antioxidant that blocks NF-κB induction of neuroimmune genes (Crews et al., 2006a; Zou and Crews, 2010), reversed ethanol-induced inhibition of neurogenesis. Similarly, using an in vivo 4-day chronic binge model of ethanol exposure, Nixon and Crews (2002) found diminished hippocampal neural stem cell proliferation and neurogenesis. Diminished neurogenesis in this structure likely contributes to deficits in learning and alterations in mood. A study following chronic alcohol administration to mice found reduced neurogenesis and increased immobility in the forced-swim test, a finding consistent with negative affect – depression and hopelessness behavior (Stevenson et al., 2009) (Fig. 27.9). Interestingly, antidepressant treatment reversed both the lost neurogenesis and the depression-like behavior. Thus, components of the negative affect and bad feelings in alcoholism may be related to alcohol-induced changes in neurogenesis.
The involvement of innate immune genes in the reduction of glutamate transporters and subsequent hyperexcitability inactivates the frontal cortex and might also contribute to the neurobiology of addiction (Crews et al., 2006a), as evidenced by decreased ability to adapt behavior to successful outcomes when there are changes in the situation. Alcohol activates microglia and astrocytes (Lee et al., 2004; Qin et al., 2007; Alfonso-Loeches et al., 2010), which might lead to glial adaptive conditioning (Shpargel et al., 2008). Adaptive conditioning occurs when the brain is exposed to persistent low levels of noxious stimuli that culminate in neuroprotection by priming of the brain for future insults (i.e., increased expression of anti-inflammatory molecules). However, repeated bouts of exposure to these stimuli (e.g., intermittent) leads to persistent activation of microglia and astrocytes (McClain et al., 2011) that secrete proinflammatory cytokines that disrupt neurocircuits, and promote neuronal damage and cell death (Valles et al., 2004). In support of a glial contribution to addiction, administration of prednisone, a synthetic glucocorticoid, increases anxiety-like behavior on the elevated plus-maze as well as microglial populations in the frontal cortex and hippocampus (Gonzalez-Perez et al., 2001). In human postmortem frontal cortical tissue of depressed individuals that committed suicide, astrocytes were found to exhibit enlarger cell bodies and more ramified processes, which is indicative of activated astrocytes (Torres-Platas et al., 2011). Taken together, there is mounting evidence to support a role for neuroimmune signaling in the development and maintenance of alcoholism (Fig. 27.10). Further, the neuroimmune signaling system might offer a novel target in the treatment of alcoholism and other addictions.
Fig. 27.10.
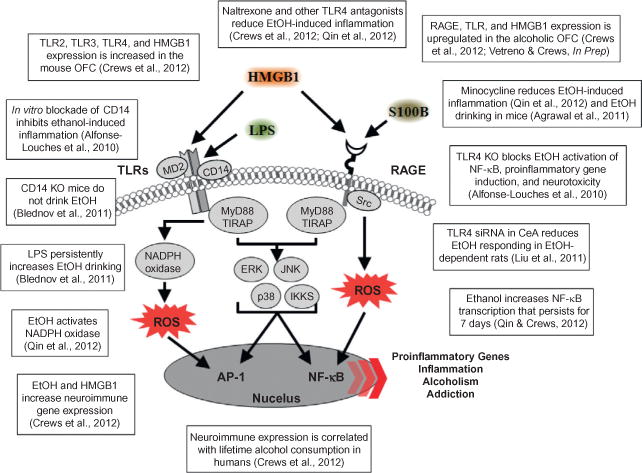
Neuroimmune signaling cascade activation and evidence for involvement in alcohol-induced neurodegeneration. A simplified schematic of the TLR and RAGE receptor signaling cascades. Stimulation of TLRs leads to the generation of ROS and downstream activation of NF-κB. Similarly, activation of the RAGE receptor leads to caspase-3 induction and downstream activation of NF-κB. The production of NF-κB leads to the secretion of proinflammatory gene expression, neuroimmune induction, and cell death. AP-1, activator protein-1; CD14, cluster of differentiation 14; ERK, extracellular signal-regulated kinase; HMGB1, high-mobility group box-1; IKK, inhibitor of nuclear factor kappa-B; JNK, c-Jun N-terminal kinases; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response gene 88; NADPH oxidase, nicotinamide adenine dinucleotide phosphate-oxidase; NF-κB, nuclear factor kappa-light-chain enhancer of activated B cells; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; S100B, S100 calcium-binding protein B; Src, proto-oncogene tyrosine-protein kinase; TIRAP, Toll/interleukin-1 receptor domain-containing adaptor protein; TLRs, Toll-like receptors.
RISK FACTORS THAT CONTRIBUTE TO THE PROGRESSION OF ALCOHOL DEPENDENCE
Genetic contribution to alcohol dependence
Similar to other psychopathologies (e.g., depression and schizophrenia), alcohol dependence stems from a combination of precipitating environmental factors and inherent biologic/genetic predispositions (Sander et al., 1999). Specific variants of multiple neuroimmune genes are associated with the genetic risk for alcohol dependence in humans (Crews, 2012) which further supports the connection between the innate immune system of the CNS and alcoholism. Approximately 50% of the risk for alcohol dependence stems from genetic factors (Schuckit, 2009). Variations on chromosomes at specific locations on DNA (i.e., polymorphisms) result in gene variants (i.e., alleles) that may diminish or increase the risk for alcohol dependence. The enzyme CYP2E1 is involved in alcohol metabolism, and polymorphisms of the gene that encodes for this enzyme are associated with increased risk for alcoholism (Webb et al., 2011). In the periphery, CYP2E1 is localized to monocytes (immune cells), and ethanol metabolism by this enzyme leads to activation and consequent production of reactive oxygen species and activation of NF-κB cascades in response to LPS (Cao et al., 2005). Within the CNS, ethanol upregulates CYP2E1 expression in astrocytes (Montoliu et al., 1994, 1995), which likely contribute to astrocytic activation and induction of NF-κB cascades. Polymorphisms of the gene that encodes NF-κB have also been implicated in alcohol dependence (Flatscher-Bader et al., 2005; Okvist et al., 2007). Alleles of many other genes linked to innate immune factors are associated with alcoholism (Crews, 2012). The interactions are complex due to combinations of alleles that promote proinflammatory signals with alleles that blunt anti-inflammatory signals (Crews, 2012). Thus, human genetic factors related to alcoholism might include innate immune genes.
Gene expression studies of genetically paired rats and mice that differ primarily in their preference for ethanol consumption reveal that NF-κB, its regulatory proteins, and many innate immune genes are central to high-ethanol-drinking behaviors (Mulligan et al., 2006). Beta-2 microglobulin, an NF-κB target gene involved in MHC immune signaling (Pahl, 1999), and other transcription regulation transcriptomes (i.e., RNA molecules), were significantly overexpressed in high-ethanol-preferring mice which might induce a predisposition for excessive ethanol consumption (Mulligan et al., 2006). In addition, work from Blednov and colleagues provided interesting data supporting the hypothesis that innate immune genes regulate ethanol-drinking behavior (Blednov et al., 2005, 2011, 2012). In null mutant mice for specific genes associated with TLR and NF-κB signaling, including β2-macroglobulin, cathepsin S, cathepsin F, IL-1 receptor antagonist (Il1rn), CD14, and IL-6, these animals universally evidenced altered ethanol preference and intake relative to wild-type controls across multiple ethanol-drinking paradigms. However, across measures, the male and female Il1rn mutant mice evidenced reduced ethanol preference and intake, supporting a role for neuroimmune involvement in ethanol-drinking behavior (Blednov et al., 2012). Taken together, these findings are consistent with a modulatory role of innate immune genes in alcohol preference and consumption.
Polymorphisms of several genes related to the dopaminergic system, which is involved in frontal cortical and limbic system regulation of behavior, have been implicated in the development of impulsive behaviors (Kreek et al., 2005). For instance, genetic variation at the Val158Met locus of the catechol-O-methyltransferase (COMT) gene influences decision-making behavior and underlying activity in brain activity associated with impulsive choice (Boettiger et al., 2007). The COMT enzyme plays a significant role in regulating frontal cortex dopaminergic function (Chen et al., 2004), and individuals homozygous for the enzymatically more active 158Val allele show an increased tendency to choose immediate over delayed rewards. Such genetic variation in COMT function may also contribute to other forms of impulsive behavior (Congdon and Canli, 2005; Kreek et al., 2005), and might contribute mechanistically to alcoholism (Ishiguro et al., 1999; Samochowiec et al., 2006). Thus, genes that alter impulsivity likely contribute to risk for alcoholism and other mental diseases that have overlapping psychopathology.
Adolescent onset of alcohol consumption
Adolescence is a highly conserved critical developmental period that encompasses the transition from childhood to adulthood. It is defined by a constellation of behaviors that encompass high levels of risk taking, increased exploration, novelty and sensation seeking, and social interaction that promote the acquisition of skills necessary for maturation and independence. In addition, it is a period when the adolescent brain, particularly the frontal cortex and limbic system, undergoes both progressive and regressive changes involving synaptogenesis, finetuning of projections and inputs, and maturation of neurotransmitter systems (Spear, 2000; Ernst et al., 2009). Indeed, absolute PFC gray-matter volumes decline in humans (Sowell et al., 1999, 2001) as well as in rats (van Eden et al., 1990) during adolescence. In addition, there is considerable synaptic pruning during adolescence, especially of excitatory glutamatergic inputs to the PFC in humans and non-human primates (Huttenlocher, 1984; Zecevic et al., 1989). By contrast, dopaminergic, serotonergic, and cholinergic inputs to the PFC increase, ultimately reaching mature levels (Kalsbeek et al., 1988; Kostovic, 1990; Gould et al., 1991; Rosenberg and Lewis, 1994). Greater incidence of novelty/sensation seeking is also a strong predictor of alcohol and drug use (Baumrind, 1987; Wills et al., 1994) that might alter cortical maturation, leading to addiction due to the vulnerability of the developing PFC (Crews et al., 2007).
In humans, initiation of alcohol use typically occurs during adolescence and is highly prevalent as 8% of eighth-grade, 16% of 10th-grade, and 25% of 12th-grade adolescents report heavy episodic drinking (i.e., >5 consecutive alcohol drinks per episode) over the past 2 weeks (Johnston et al., 2009). This heavy-drinking pattern persists into college, as 44% of students report binge drinking every 2 weeks and 19% report >3 binge-drinking episodes per week (Wechsler et al., 1995; O’Malley et al., 1998). The effects of alcohol on the adolescent brain are different from those observed in adulthood. For instance, adolescents are less sensitive to the sedative effects of alcohol (Silveri and Spear, 1998), which allows them to binge drink. In contrast, they are more vulnerable to alcohol-induced neurotoxicity (Monti et al., 2005; Crews et al., 2007). Heavy drinking among human adolescent males was found to increase impulsivity the following year in those individuals predisposed to adolescent-typical impulsivity (White et al., 2011). Furthermore, early onset of alcohol use (<13 years of age) is associated with increased drinking frequency and physical violence (Gruber et al., 1996), which is consistent with diminished impulse inhibition. In addition, adolescents and adults with alcohol use disorders evidence deficits in executive functioning (Brown et al., 2000; Tapert and Brown, 2000; Hanson et al., 2011). Using animal models of adolescent binge drinking, we found impaired reversal learning (Coleman et al., 2011; Vetreno and Crews, 2012), The increased sensitivity of the adolescent brain to alcohol-induced toxicity, coupled with the dynamic synaptic remodeling that characterizes this stage of development, might strengthen the learning components of heavy-drinking behaviors and perpetuate the loss of important self-control and goal-setting components of the maturing brain’s executive centers. Indeed, an early age of drinking onset is highly predictive of later development of alcoholism, regardless of family history (Grant, 1998).
Neuroimmune gene activation in the brain remains elevated for long periods (Qin et al., 2007, 2008), consistent with the enduring nature of addiction. The persistence of neuroimmune activation is likely amplified in the adolescent brain (Spear, 2000; Vetreno and Crews, 2012; Vetreno et al., 2013) because of their increased ethanol consumption (Silveri and Spear, 1998). Similar to our adult findings, our laboratory recently discovered that adolescent binge ethanol exposure increases TLRs, RAGE, and HMGB1 neuroimmune expression in the adolescent brain, an effect that persists into adulthood. These increases were accompanied by persistent upregulation of proinflammatory cytokines (TNF-α and MCP-1), oxidases (COX-2 and NOX2), and neuroimmune signaling molecules (HMGB1 and S100β: Vetreno and Crews, 2012; Vetreno et al., 2013). The induction of neuroimmune signaling includes neuronal-glial signaling that leads to hyperexcitability in neuronal networks that contribute to addiction (Fig. 27.11). Consistent with the increased excitability and excitotoxic vulnerability of the PFC during adolescence, an earlier age of drinking onset correlated with increased expression of TLRs, RAGE, and HMGB1 in the postmortem human alcoholic OFC (Fig. 27.12). Although the mechanism underlying the persistence of neuroimmune activation remains to be elucidated, it does appear to involve positive loops of activation (Crews et al., 2011). This hypothesis would predict a relationship between TLR/RAGE and HMGB1 expression. Indeed, expression of HMGB1 is positively correlated with RAGE and TLR immunoreactivity in the postmortem human alcoholic OFC (Vetreno et al., 2013) (Fig. 27.13) and in the rat PFC following adolescent binge ethanol exposure (Vetreno and Crews, 2012; Vetreno et al., 2013).
Fig. 27.11.
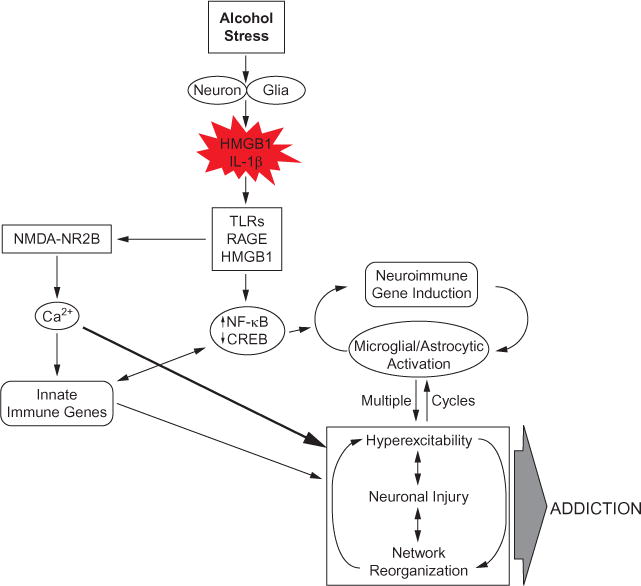
Hyperexcitability contributes to the neurobiology of addiction. A simplified schematic depicting how neuroimmune signaling leads to hyperexcitability and the neurobiology of addiction. Alcohol and stress activate neurons and glia in the central nervous system, resulting in the release of various neuroimmune signals (e.g., high-mobility group box 1 (HMGB1) and interleukin-1β (IL-1β)) that activate neuroimmune receptors (i.e., Toll-like receptors (TLRs) and receptor for advanced glycation end products (RAGE)). Neuroimmune receptor stimulation leads to activation of glutamatergic N-methyl-D-aspartate (NMDA) receptors, e.g., NR2B (Maroso et al., 2010; Iori et al., 2013), which increases Ca2+ flux, triggering induction of neuroimmune genes. In addition, TLR/RAGE activation leads to downstream transcription of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) signaling that might be accompanied by diminished cyclic AMP-responsive element-binding protein (CREB) expression, which contributes to neuroimmune gene induction. These two pathways converge, leading to cycles of neuroimmune gene induction that lead to hyperexcitability, neuronal cell death, and network reorganization that culminates in addiction.
Fig. 27.12.
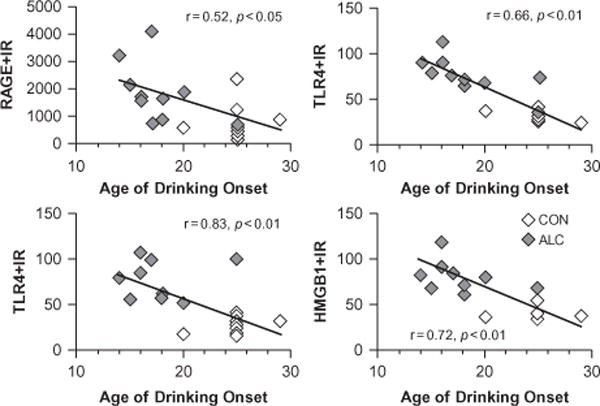
Receptor for advanced glycation end product (RAGE), Toll-like receptor (TLR) 3 and 4, and high-mobility group box 1 (HMGB1) expression in the human alcoholic postmortem orbitofrontal cortex correlates with age of drinking onset. The younger the age of drinking onset, the greater the risks of developing alcohol dependence (Grant, 1998). Neuroimmune signaling shows a similar correlation consistent with alcohol-induced increased neuroimmune signaling, increasing the risk for alcoholism. Depicted are individual self and family reports of age of drinking onset vs RAGE (×10 000 pixels/mm2), TLR3 (cells/mm3), TLR4 (cells/mm3), and HMGB1 (cells/mm3) immunoreactivity. Across subjects, age of drinking onset negatively correlated with neuroimmune signal immunoreactivity (r = −0.42, p<0.001). Moderate alcohol drinking controls (CON) tended to self-report a later age of drinking onset (25 ± 1 years of age) in comparison to individuals that met criteria for alcoholism (18 ± 1 years of age). (Adapted from Vetreno et al., 2013).
Fig. 27.13.
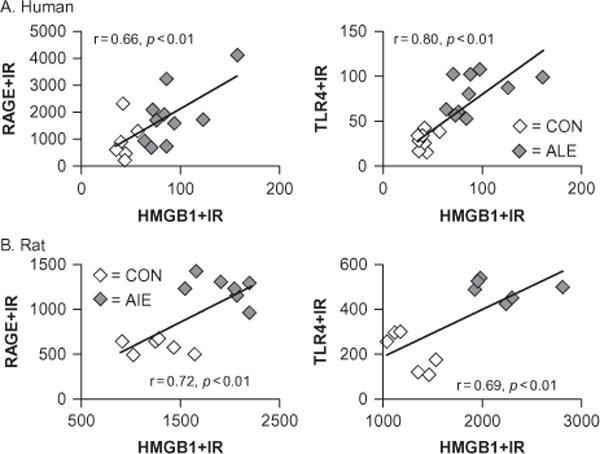
Receptor for advanced glycation end product (RAGE) and Toll-like receptor 4 (TLR4) expression correlates with high-mobility group box 1 (HMGB1) expression in the prefrontal cortex. The persistent nature of neuroimmune gene induction is likely due to upregulation of both the agonist, HMGB1, and receptors, e.g., TLR receptors that progressively increase in concert with chronic alcohol abuse. (A) Depicted are correlations of individual human RAGE (×10 000 pixels/mm2) and TLR4 (cells/mm3) immunoreactivity versus HMGB1 (cells/mm3) expression in the orbitofrontal cortex (OFC) of alcoholics and moderate-drinking controls. Across subjects, RAGE and TLR4 expression correlated with HMGB1. The correlation of HMGB1 with RAGE/TLR4 is consistent with neuroimmune loops of amplification. (B) Depicted are correlations of RAGE (×10 000 pixels/mm2) and TLR4 (cells/mm2) immunoreactivity versus HMGB1 (cells/mm2) expression in the OFC of adult rats exposed to adolescent binge ethanol. Across subjects, RAGE and TLR4 expression correlated with HMGB1. The correlation of RAGE/TLR4 with HMGB1 indicates the persistence of neuroimmune loops of activation. (Adapted from Vetreno and Crews, 2012; Crews et al., 2013; Vetreno et al., 2013).
Disorders of PFC function
Innate PFC dysfunction represents another risk factor that could contribute to the development of alcohol and other addictions. Loss of PFC control over impulsivity is a cardinal feature of both attention-deficit hyperactivity disorder (ADHD) and addiction. ADHD is a highly prevalent psychiatric disorder (NIH, 2000) characterized by symptoms of inattention, hyperactivity, and impulsivity, as well as diminished executive function (Biederman, 1998; APA, 2000; Vaurio et al., 2008). Neuroimaging studies have revealed abnormal prefrontal cortical and striatal activation in ADHD individuals (Herrmann et al., 2010) as well as significant gray-matter reductions in the frontal gyrus and cingulate gyrus (Overmeyer et al., 2001). Animal models of ADHD implicate serotonergic and dopaminergic function in impulsivity and attention (Puumala and Sirvio, 1998), suggesting that dysfunctional catecholaminergic signaling might underlie some of the impulsivity symptoms. A number of studies have been conducted to assess the potential relationship between ADHD and the development of alcoholism and other drug addictions (Wilson and Marcotte, 1996; Biederman et al., 1997; Murphy et al., 2002). Although there is a lack of agreement regarding the association with ADHD and addiction, diminished PFC function is likely a contributing factor to heavy drinking (Span and Earleywine, 1999). This seems especially true given the involvement of the PFC in executive functioning and its dysregulation in the alcoholic brain. Indeed, perhaps due to increased impulsivity, college students with ADHD are more likely than others to abuse alcohol and other drugs (Murphy et al., 2002).
CONCLUSIONS
Over time, continued alcohol use leads to the development of alcoholism-promoting behaviors. Diminished executive functioning due to hypoactive PFC executive control and hyperactive limbic system anxiety and negative emotion might contribute mechanistically to the shift from experimental use to alcoholism and dependence. Human neuroimaging studies and animal models find that chronic alcohol exposure causes neurodegeneration that is most prominent in the PFC, which might contribute to cortical hypofunction and diminished executive function. Further, alcohol diminishes CREB/neurotrophin support while increasing transcription of NF-κB, which in turn leads to downstream induction of proinflammatory chemokines, cytokines, oxidases, proteases, and other neuroimmune genes in a cyclic fashion that spreads across the CNS. These findings, coupled with the neuroimmune genetic finds of Blednov and colleagues (2012), support the role for the involvement of innate immune gene expression in the development of alcoholism. Furthermore, hyperexcitability and behavioral inflexibility are associated with innate immune gene expression in the frontal cortex (Vetreno et al., 2013). The emerging view that innate immunity underlies addiction has important ramifications for the interaction of early onset of drug and alcohol abuse with genetics. Furthermore, it suggests that therapies aimed at suppressing innate immune activation in the CNS might represent a novel therapeutic approach to prevent the development, and perhaps treat addiction and/or anxiety disorders.
References
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, et al. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J, DAS GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- APA. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. Washington, D.C: American Psychiatric Association; 2000. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. American Psychiatric Publishing; Arlington, VA: 2013. [Google Scholar]
- Aschner M. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology. 1998;19:269–281. [PubMed] [Google Scholar]
- Aschner M, Sonnewald U, Tan KH. Astrocyte modulation of neurotoxic injury. Brain Pathol. 2002;12:475–481. doi: 10.1111/j.1750-3639.2002.tb00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A. Working memory. Science. 1992;255:556–559. doi: 10.1126/science.1736359. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wetzel W, Del Rey A, et al. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- Banks WA, Erickson MA. The blood–brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Baumrind D. A developmental perspective on adolescent risk taking in contemporary America. New Dir Child Adolesc Dev. 1987:93–125. doi: 10.1002/cd.23219873706. [DOI] [PubMed] [Google Scholar]
- Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8:1458–1463. doi: 10.1038/nn1584. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio AR, Damasio H, et al. Insensitivity to future consequences following damage to human prefrontal cortex. Cognition. 1994;50:7–15. doi: 10.1016/0010-0277(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Tranel D, et al. Dissociation of working memory from decision making within the human prefrontal cortex. J Neurosci. 1998;18:428–437. doi: 10.1523/JNEUROSCI.18-01-00428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR. Emotion, decision making and the orbitofrontal cortex. Cereb Cortex. 2000;10:295–307. doi: 10.1093/cercor/10.3.295. [DOI] [PubMed] [Google Scholar]
- Berlin HA, Rolls ET, Kischka U. Impulsivity, time perception, emotion and reinforcement sensitivity in patients with orbitofrontal cortex lesions. Brain. 2004;127:1108–1126. doi: 10.1093/brain/awh135. [DOI] [PubMed] [Google Scholar]
- Besedovsky HO, Del Rey A. Central and peripheral cytokines mediate immune-brain connectivity. Neurochem Res. 2011;36:1–6. doi: 10.1007/s11064-010-0252-x. [DOI] [PubMed] [Google Scholar]
- Biederman J. Attention-deficit/hyperactivity disorder: a life-span perspective. J Clin Psychiatry. 1998;59(Suppl 7):4–16. [PubMed] [Google Scholar]
- Biederman J, Wilens T, Mick E, et al. Is ADHD a risk factor for psychoactive substance use disorders? Findings from a four-year prospective follow-up study. J Am Acad Child Adolesc Psychiatry. 1997;36:21–29. doi: 10.1097/00004583-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Bison S, Crews F. Alcohol withdrawal increases neuropeptide Y immunoreactivity in rat brain. Alcohol Clin Exp Res. 2003;27:1173–1183. doi: 10.1097/01.ALC.0000075827.74538.FE. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M, et al. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol. 2005;175:6893–6899. doi: 10.4049/jimmunol.175.10.6893. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Bergeson SE, Walker D, et al. Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res. 2005;165:110–125. doi: 10.1016/j.bbr.2005.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, et al. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011;25(Suppl 1):S92–S105. doi: 10.1016/j.bbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, et al. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol. 2012;17:108–120. doi: 10.1111/j.1369-1600.2010.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger CA, Mitchell JM, Tavares VC, et al. Immediate reward bias in humans: fronto-parietal networks and a role for the catechol-O-methyltransferase 158(Val/Val) genotype. J Neurosci. 2007;27:14383–14391. doi: 10.1523/JNEUROSCI.2551-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Bowden SC, Crews FT, Bates ME, et al. Neurotoxicity and neurocognitive impairments with alcohol and drug-use disorders: potential roles in addiction and recovery. Alcohol Clin Exp Res. 2001;25:317–321. [PubMed] [Google Scholar]
- Breiter HC, Gollub RL, Weisskoff RM, et al. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- Brown SA, Tapert SF, Granholm E, et al. Neurocognitive functioning of adolescents: effects of protracted alcohol use. Alcohol Clin Exp Res. 2000;24:164–171. [PubMed] [Google Scholar]
- Cao Q, Mak KM, Lieber CS. Cytochrome P4502E1 primes macrophages to increase TNF-alpha production in response to lipopolysaccharide. Am J Physiol. 2005;289:G95–G107. doi: 10.1152/ajpgi.00383.2004. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Price JL. Limbic connections of the orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol. 1995a;363:615–641. doi: 10.1002/cne.903630408. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Price JL. Sensory and premotor connections of the orbital and medial prefrontal cortex of macaque monkeys. J Comp Neurol. 1995b;363:642–664. doi: 10.1002/cne.903630409. [DOI] [PubMed] [Google Scholar]
- Cella M, Dymond S, Cooper A. Impaired flexible decision-making in major depressive disorder. J Affect Disord. 2010;124:207–210. doi: 10.1016/j.jad.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Halim N, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress AR, Mozley PD, Mcelgin W, et al. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–18. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L, Manes F. Social and emotional decision-making following frontal lobe injury. Neurocase. 2004;10:398–403. doi: 10.1080/13554790490882799. [DOI] [PubMed] [Google Scholar]
- Coleman LG, Jr, Jarskog LF, Moy SS, et al. Deficits in adult prefrontal cortex neurons and behavior following early post-natal NMDA antagonist treatment. Pharmacol Biochem Behav. 2009;93:322–330. doi: 10.1016/j.pbb.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Jr, He J, Lee J, et al. Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res. 2011;35:671–688. doi: 10.1111/j.1530-0277.2010.01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon E, Canli T. The endophenotype of impulsivity: reaching consilience through behavioral, genetic, and neuroimaging approaches. Behav Cogn Neurosci Rev. 2005;4:262–281. doi: 10.1177/1534582305285980. [DOI] [PubMed] [Google Scholar]
- Coutlee CG, Huettel SA. The functional neuroanatomy of decision making: prefrontal control of thought and action. Brain Res. 2012;1428:3–12. doi: 10.1016/j.brainres.2011.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT. Immune function genes, genetics, and the neurobiology of addiction. Alcohol Res Curr Rev. 2012;34:355–361. doi: 10.35946/arcr.v34.3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Boettiger CA. Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav. 2009;93:237–247. doi: 10.1016/j.pbb.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP. Addiction, adolescence, and innate immune gene induction. Frontiers Psychiatry. 2011;2:19. doi: 10.3389/fpsyt.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Collins MA, Dlugos C, et al. Alcohol-induced neurodegeneration: when, where and why? Alcohol Clin Exp Res. 2004;28:350–364. doi: 10.1097/01.alc.0000113416.65546.01. [DOI] [PubMed] [Google Scholar]
- Crews FT, Buckley T, Dodd PR, et al. Alcoholic neurobiology: changes in dependence and recovery. Alcohol Clin Exp Res. 2005;29:1504–1513. doi: 10.1097/01.alc.0000175013.50644.61. [DOI] [PubMed] [Google Scholar]
- Crews F, Nixon K, Kim D, et al. BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res. 2006a;30:1938–1949. doi: 10.1111/j.1530-0277.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Bechara R, Brown LA, et al. Cytokines and alcohol. Alcohol Clin Exp Res. 2006b;30:720–730. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav. 2007;86:189–199. doi: 10.1016/j.pbb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011;25(Suppl 1):S4–S12. doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, et al. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry. 2013;73:602–612. doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL. Frontal-subcortical circuits and human behavior. J Psychosom Res. 1998;44:627–628. doi: 10.1016/s0022-3999(98)00034-8. [DOI] [PubMed] [Google Scholar]
- Damasio H, Kuljis RO, Yuh W, et al. Magnetic resonance imaging of human intracortical structure in vivo. Cereb Cortex. 1991;1:374–379. doi: 10.1093/cercor/1.5.374. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit H. Impulsivity as a determinant and consequence of drug use: a review of underlying processes. Addict Biol. 2009;14:22–31. doi: 10.1111/j.1369-1600.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rey A, Balschun D, Wetzel W, et al. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav Immun. 2013;33:15–23. doi: 10.1016/j.bbi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Dick DM, Smith G, Olausson P, et al. Understanding the construct of impulsivity and its relationship to alcohol use disorders. Addict Biol. 2010;15:217–226. doi: 10.1111/j.1369-1600.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Romeo RD, Andersen SL. Neurobiology of the development of motivated behaviors in adolescence: a window into a neural systems model. Pharmacol Biochem Behav. 2009;93:199–211. doi: 10.1016/j.pbb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Eslinger PJ, Damasio H, Graff-Radford N, et al. Examining the relationship between computed tomography and neuropsychological measures in normal and demented elderly. J Neurol Neurosurg Psychiatry. 1984;47:1319–1325. doi: 10.1136/jnnp.47.12.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol. 2009;183:4733–4744. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- Ferrier L, Berard F, Debrauwer L, et al. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatscher-Bader T, Van Der Brug M, Hwang JW, et al. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. J Neurochem. 2005;93:359–370. doi: 10.1111/j.1471-4159.2004.03021.x. [DOI] [PubMed] [Google Scholar]
- Fortier CB, Leritz EC, Salat DH, et al. Reduced cortical thickness in abstinent alcoholics and association with alcoholic behavior. Alcohol Clin Exp Res. 2011;35:2193–2201. doi: 10.1111/j.1530-0277.2011.01576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Nowis D, Golab J, et al. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. doi: 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Working memory and the mind. Sci Am. 1992;267:110–117. doi: 10.1038/scientificamerican0992-110. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez O, Ramos-Remus C, Garcia-Estrada J, et al. Prednisone induces anxiety and glial cerebral changes in rats. J Rheumatol. 2001;28:2529–2534. [PubMed] [Google Scholar]
- Gould E, Woolf NJ, Butcher LL. Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res Bull. 1991;27:767–789. doi: 10.1016/0361-9230(91)90209-3. [DOI] [PubMed] [Google Scholar]
- Graeber MB. Changing face of microglia. Science. 2010;330:783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- Grant BF. The impact of a family history of alcoholism on the relationship between age at onset of alcohol use and DSM-IV alcohol dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. Alcohol Health Res World. 1998;22:144–147. [PMC free article] [PubMed] [Google Scholar]
- Grant B, Moore TC, Shepard J, et al. Source and Accuracy Statement: Wave 1 National Epidemiologic Survey on Alcohol and Related Conditions NESARC. National Institute on Alcohol Abuse and Alcoholism; Bethesda, MD: 2004. Available online at www.niaaa.nih.gov. [Google Scholar]
- Grant BF, Harford TC, Muthen BO, et al. DSM-IV alcohol dependence and abuse: further evidence of validity in the general population. Drug Alcohol Depend. 2007;86:154–166. doi: 10.1016/j.drugalcdep.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Grilli M, Memo M. Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ. 1999;6:22–27. doi: 10.1038/sj.cdd.4400463. [DOI] [PubMed] [Google Scholar]
- Gruber E, Diclemente RJ, Anderson MM, et al. Early drinking onset and its association with alcohol use and problem behavior in late adolescence. Prev Med. 1996;25:293–300. doi: 10.1006/pmed.1996.0059. [DOI] [PubMed] [Google Scholar]
- Gruber AJ, Calhoon GG, Shusterman I, et al. More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J Neurosci. 2010;30:17102–17110. doi: 10.1523/JNEUROSCI.4623-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Kim YH, Mook-Jung I. RAGE: the beneficial and deleterious effects by diverse mechanisms of actions. Mol Cells. 2011;31:91–97. doi: 10.1007/s10059-011-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson KL, Medina KL, Padula CB, et al. Impact of adolescent alcohol and drug use on neuropsychological functioning in young adulthood: 10-year outcomes. J Child Adolesc Substance Abuse. 2011;20:135–154. doi: 10.1080/1067828X.2011.555272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow JM. Passage of an iron rod through the head. Boston Med Surg J. 1848;39:389–393. [PMC free article] [PubMed] [Google Scholar]
- Harlow JM. Recovery from the passage of an iron rod through the head. Massachus Med Soc. 1868;2:327–346. [Google Scholar]
- Harper C, Kril J. Patterns of neuronal loss in the cerebral cortex in chronic alcoholic patients. J Neurol Sci. 1989;92:81–89. doi: 10.1016/0022-510x(89)90177-9. [DOI] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann MJ, Biehl SC, Jacob C, et al. Neurobiological and psychophysiological correlates of emotional dysregulation in ADHD patients. Attention Deficit Hyperactivity Disord. 2010;2:233–239. doi: 10.1007/s12402-010-0047-6. [DOI] [PubMed] [Google Scholar]
- Hock R, Furusawa T, Ueda T, et al. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007;17:72–79. doi: 10.1016/j.tcb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine. 2010;51:119–126. doi: 10.1016/j.cyto.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Synapse elimination and plasticity in developing human cerebral cortex. Am J Ment Defic. 1984;88:488–496. [PubMed] [Google Scholar]
- Iori V, Maroso M, Rizzi M, et al. Receptor for advanced glycation endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis. 2013 doi: 10.1016/j.nbd.2013.03.006. http://dx.doi.org/10.1016/j.nbd.2013.03.006. [DOI] [PubMed]
- Ishiguro H, Haruo Shibuya T, Toru M, et al. Association study between high and low activity polymorphism of catechol-O-methyltransferase gene and alcoholism. Psychiatr Genet. 1999;9:135–138. doi: 10.1097/00041444-199909000-00004. [DOI] [PubMed] [Google Scholar]
- Johnston L, O’Malley P, Bachman J, et al. Seconday school students, 2008. Vol. 1. National Institute on Drug Abuse; Bethesda, MD: 2009. Monitoring the future: National survey results on drug use, 1975–2008. [Google Scholar]
- Kalsbeek A, Voorn P, Buijs RM, et al. Development of the dopaminergic innervation in the prefrontal cortex of the rat. J Comp Neurol. 1988;269:58–72. doi: 10.1002/cne.902690105. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Baeuerle PA, Kaltschmidt C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol Aspects Med. 1993;14:171–190. doi: 10.1016/0098-2997(93)90004-w. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun. 2011;25(Suppl 1):S13–S20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, et al. Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry. 2001;58:334–341. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Crews FT. Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol Clin Exp Res. 1999;23:633–643. [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Ahmed SH, Boutrel B, et al. Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2004;27:739–749. doi: 10.1016/j.neubiorev.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Kostovic I. Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life. Prog Brain Res. 1990;85:223–239. doi: 10.1016/s0079-6123(08)62682-5. discussion 239–240. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Nielsen DA, Butelman ER, et al. Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nat Neurosci. 2005;8:1450–1457. doi: 10.1038/nn1583. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Harper CG. Neuronal counts from four cortical regions of alcoholic brains. Acta Neuropathol. 1989;79:200–204. doi: 10.1007/BF00294379. [DOI] [PubMed] [Google Scholar]
- Kubota M, Nakazaki S, Hirai S, et al. Alcohol consumption and frontal lobe shrinkage: study of 1432 non-alcoholic subjects. J Neurol Neurosurg Psychiatry. 2001;71:104–106. doi: 10.1136/jnnp.71.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Jeong J, Son E, et al. Ethanol selectively modulates inflammatory activation signaling of brain microglia. J Neuroimmunol. 2004;156:88–95. doi: 10.1016/j.jneuroim.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Li TK. Quantifying the risk for alcohol-use and alcohol-attributable health disorders: present findings and future research needs. J Gastroenterol Hepatol. 2008;23(Suppl 1):S2–S8. doi: 10.1111/j.1440-1746.2007.05298.x. [DOI] [PubMed] [Google Scholar]
- Li J, Kokkola R, Tabibzadeh S, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45. [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Maggio N, Shavit-Stein E, Dori A, et al. Prolonged systemic inflammation persistently modifies synaptic plasticity in the hippocampus: modulation by the stress hormones. Frontiers Mol Neurosci. 2013;6:46. doi: 10.3389/fnmol.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Marshall SA, Mcclain JA, Kelso ML, et al. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol Dis. 2013;54:239–251. doi: 10.1016/j.nbd.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcclain JA, Morris SA, Deeny MA, et al. Adolescent binge alcohol exposure induces long-lasting partialactivation of microglia. Brain Behav Immun. 2011;25(Suppl 1):S120–S128. doi: 10.1016/j.bbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Messmer D, Yang H, Telusma G, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- Michaud CM, Mckenna MT, Begg S, et al. The burden of disease and injury in the United States 1996. Population Hlth Metrics. 2006;4:11. doi: 10.1186/1478-7954-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, et al. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Overholser JC, Meltzer HY, et al. Reduced glial and neuronal packing density in the orbitofrontal cortex in alcohol dependence and its relationship with suicide and duration of alcohol dependence. Alcohol Clin Exp Res. 2006;30:1845–1855. doi: 10.1111/j.1530-0277.2006.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup DF, et al. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- Monti PM, Miranda R, Jr, Nixon K, et al. Adolescence: booze, brains, and behavior. Alcohol Clin Exp Res. 2005;29:207–220. doi: 10.1097/01.alc.0000153551.11000.f3. [DOI] [PubMed] [Google Scholar]
- Montoliu C, Valles S, Renau-Piqueras J, et al. Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: effect of chronic alcohol consumption. J Neurochem. 1994;63:1855–1862. doi: 10.1046/j.1471-4159.1994.63051855.x. [DOI] [PubMed] [Google Scholar]
- Montoliu C, Sancho-Tello M, Azorin I, et al. Ethanol increases cytochrome P4502E1 and induces oxidative stress in astrocytes. J Neurochem. 1995;65:2561–2570. doi: 10.1046/j.1471-4159.1995.65062561.x. [DOI] [PubMed] [Google Scholar]
- Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KR, Barkley RA, Bush T. Young adults with attention deficit hyperactivity disorder: subtype differences in comorbidity, educational, and clinical history. J Nerv Ment Dis. 2002;190:147–157. doi: 10.1097/00005053-200203000-00003. [DOI] [PubMed] [Google Scholar]
- NIH. National Institutes of Health Consensus Development Conference Statement: diagnosis and treatment of attention-deficit/hyperactivity disorder (ADHD) J Am Acad Child Adolesc Psychiatry. 2000;39:182–193. doi: 10.1097/00004583-200002000-00018. [DOI] [PubMed] [Google Scholar]
- Nixon K, Crews FT. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem. 2002;83:1087–1093. doi: 10.1046/j.1471-4159.2002.01214.x. [DOI] [PubMed] [Google Scholar]
- O’Malley PM, Johnston LD, Bachman JG. Alcohol use among adolescents. Alcohol Health Res World. 1998;22:85–93. [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, Crews FT. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res. 2002a;26:547–557. [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, et al. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav. 2002b;72:521–532. doi: 10.1016/s0091-3057(02)00715-3. [DOI] [PubMed] [Google Scholar]
- Okvist A, Johansson S, Kuzmin A, et al. Neuroadaptations in human chronic alcoholics: dysregulation of the NF-kappaB system. PLoS One. 2007;2:e930. doi: 10.1371/journal.pone.0000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer S, Bullmore ET, Suckling J, et al. Distributed grey and white matter deficits in hyperkinetic disorder: MRI evidence for anatomical abnormality in an attentional network. Psychol Med. 2001;31:1425–1435. doi: 10.1017/s0033291701004706. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Zhang D, Mittal N, et al. Potential role of the gene transcription factor cyclic AMP-responsive element binding protein in ethanol withdrawal-related anxiety. J Pharmacol Exp Ther. 1999;288:866–878. [PubMed] [Google Scholar]
- Pandey SC, Roy A, Mittal N. Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the creb gene transcription factor in rat cortex. J Pharmacol Exp Ther. 2001;296:857–868. [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, et al. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci. 2007;25:541–550. doi: 10.1111/j.1460-9568.2006.05298.x. [DOI] [PubMed] [Google Scholar]
- Pascual M, Balino P, Alfonso-Loeches S, et al. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun. 2011;25(Suppl 1):S80–S91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Paull TT, Haykinson MJ, Johnson RC. The nonspecific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev. 1993;7:1521–1534. doi: 10.1101/gad.7.8.1521. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, et al. Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res. 1995;19:1177–1191. doi: 10.1111/j.1530-0277.1995.tb01598.x. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Goffi F, Boroni F, et al. Opposing roles for NF-kappa B/Rel factors p65 and c-Rel in the modulation of neuron survival elicited by glutamate and interleukin-1beta. J Biol Chem. 2002;277:20717–20723. doi: 10.1074/jbc.M201014200. [DOI] [PubMed] [Google Scholar]
- Plane JM, Shen Y, Pleasure DE, et al. Prospects for minocycline neuroprotection. Arch Neurol. 2010;67:1442–1448. doi: 10.1001/archneurol.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollmacher T, Haack M, Schuld A, et al. Low levels of circulating inflammatory cytokines–do they affect human brain functions? Brain Behav Immun. 2002;16:525–532. doi: 10.1016/s0889-1591(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Zheng Q, Fan R, et al. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol. 2004;33:147–155. doi: 10.1016/j.alcohol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Puumala T, Sirvio J. Changes in activities of dopamine and serotonin systems in the frontal cortex underlie poor choice accuracy and impulsivity of rats in an attention task. Neuroscience. 1998;83:489–499. doi: 10.1016/s0306-4522(97)00392-8. [DOI] [PubMed] [Google Scholar]
- Qin L, Crews FT. Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J Neuroinflamm. 2012a;9:130. doi: 10.1186/1742-2094-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflamm. 2012b;9:5. doi: 10.1186/1742-2094-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT. Focal thalamic degeneration from ethanol and thiamine deficiency is associated with neuroimmune gene induction, microglial activation, and lack of monocarboxylic acid transporters. Alcohol Clin Exp Res. 2014;38:657–671. doi: 10.1111/acer.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, et al. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflamm. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, et al. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Rauvala H, Rouhiainen A. Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors. Biochim Biophys Acta. 2010;1799:164–170. doi: 10.1016/j.bbagrm.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Reissner KJ, Kalivas PW. Using glutamate homeostasis as a target for treating addictive disorders. Behav Pharmacol. 2010;21:514–522. doi: 10.1097/FBP.0b013e32833d41b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls ET, Hornak J, Wade D, et al. Emotion-related learning in patients with social and emotional changes associated with frontal lobe damage. J Neurol Neurosurg Psychiatry. 1994;57:1518–1524. doi: 10.1136/jnnp.57.12.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg DR, Lewis DA. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study. Biol Psychiatry. 1994;36:272–277. doi: 10.1016/0006-3223(94)90610-6. [DOI] [PubMed] [Google Scholar]
- Rosenbloom MH, Schmahmann JD, PRICE BH. The functional neuroanatomy of decision-making. J Neuropsychiatry Clin Neurosci. 2012;24:266–277. doi: 10.1176/appi.neuropsych.11060139. [DOI] [PubMed] [Google Scholar]
- Samochowiec J, Kucharska-Mazur J, Grzywacz A, et al. Family-based and case-control study of DRD2, DAT, 5HTT, COMT genes polymorphisms in alcohol dependence. Neurosci Lett. 2006;410:1–5. doi: 10.1016/j.neulet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Sander T, Ball D, Murray R, et al. Association analysis of sequence variants of GABA(A) alpha6, beta2, and gamma2 gene cluster and alcohol dependence. Alcohol Clin Exp Res. 1999;23:427–431. [PubMed] [Google Scholar]
- Scheibner KA, Lutz MA, Boodoo S, et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- Schneider H, Pitossi F, Balschun D, et al. A neuromodulatory role of interleukin-1beta in the hippo-campus. Proc Natl Acad Sci U S A. 1998;95:7778–7783. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, et al. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Roesch MR, Stalnaker TA. Orbitofrontal cortex, decision-making and drug addiction. Trends Neurosci. 2006;29:116–124. doi: 10.1016/j.tins.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA. An overview of genetic influences in alcoholism. J Subst Abuse Treat. 2009;36:S5–S14. [PubMed] [Google Scholar]
- Shpargel KB, Jalabi W, Jin Y, et al. Preconditioning paradigms and pathways in the brain. Cleve Clin J Med. 2008;75(Suppl 2):S77–S82. doi: 10.3949/ccjm.75.suppl_2.s77. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. Decreased sensitivity to the hypnotic effects of ethanol early in ontogeny. Alcohol Clin Exp Res. 1998;22:670–676. doi: 10.1111/j.1530-0277.1998.tb04310.x. [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Holmes CJ, et al. Localizing age-related changes in brain structure between childhood and adolescence using statistical parametric mapping. Neuroimage. 1999;9:587–597. doi: 10.1006/nimg.1999.0436. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Delis D, Stiles J, et al. Improved memory functioning and frontal lobe maturation between childhood and adolescence: a structural MRI study. J Int Neuropsychol Soc. 2001;7:312–322. doi: 10.1017/s135561770173305x. [DOI] [PubMed] [Google Scholar]
- Span SA, Earleywine M. Cognitive functioning moderates the relation between hyperactivity and drinking habits. Alcohol Clin Exp Res. 1999;23:224–229. [PubMed] [Google Scholar]
- Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Translat Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Takahashi Y, Roesch MR, et al. Neural substrates of cognitive inflexibility after chronic cocaine exposure. Neuropharmacology. 2009;56(Suppl 1):63–72. doi: 10.1016/j.neuropharm.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK. LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus. 1996;6:35–42. doi: 10.1002/(SICI)1098-1063(1996)6:1<35::AID-HIPO7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Stevenson JR, Schroeder JP, Nixon K, et al. Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology. 2009;34:1209–1222. doi: 10.1038/npp.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflamm. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuss DT, Alexander MP. Executive functions and the frontal lobes: a conceptual view. Psychol Res. 2000;63:289–298. doi: 10.1007/s004269900007. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berl) 2005;180:583–594. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220–232. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Tetsuka T, Uranishi H, et al. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem. 2002;269:4559–4565. doi: 10.1046/j.1432-1033.2002.03157.x. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tapert SF, Brown SA. Substance dependence, family history of alcohol dependence and neuropsychological functioning in adolescence. Addiction. 2000;95:1043–1053. doi: 10.1046/j.1360-0443.2000.95710436.x. [DOI] [PubMed] [Google Scholar]
- Thomas JO. HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans. 2001;29:395–401. doi: 10.1042/bst0290395. [DOI] [PubMed] [Google Scholar]
- Torres-Platas SG, Hercher C, Davoli MA, et al. Astrocytic hypertrophy in anterior cingulate white matter of depressed suicides. Neuropsychopharmacology. 2011;36:2650–2658. doi: 10.1038/npp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Braedel S, Hilf N, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–20853. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- Valles SL, Blanco AM, Pascual M, et al. Chronic ethanol treatment enhances inflammatorymediators and cell deathin the brain and in astrocytes. Brain Pathol. 2004;14:365–371. doi: 10.1111/j.1750-3639.2004.tb00079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eden CG, Kros JM, Uylings HB. The development of the rat prefrontal cortex. Its size and development of connections with thalamus, spinal cord and other cortical areas. Prog Brain Res. 1990;85:169–183. doi: 10.1016/s0079-6123(08)62680-1. [DOI] [PubMed] [Google Scholar]
- Varlinskaya EI, Spear LP. Acute effects of ethanol on social behavior of adolescent and adult rats: role of familiarity of the test situation. Alcohol Clin Exp Res. 2002;26:1502–1511. doi: 10.1097/01.ALC.0000034033.95701.E3. [DOI] [PubMed] [Google Scholar]
- Vaurio L, Riley EP, Mattson SN. Differences in executive functioning in children with heavy prenatal alcohol exposure or attention-deficit/hyperactivity disorder. J Int Neuropsychol Soc. 2008;14:119–129. doi: 10.1017/S1355617708080144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience. 2012;226:475–488. doi: 10.1016/j.neuroscience.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol Dis. 2013;59:52–62. doi: 10.1016/j.nbd.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Hitzemann R, Wolf AP, et al. Acute effects of ethanol on regional brain glucose metabolism and transport. Psychiatry Res. 1990;35:39–48. doi: 10.1016/0925-4927(90)90007-s. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Overall JE, et al. Regional brain metabolic response to lorazepam in alcoholics during early and late alcohol detoxification. Alcohol Clin Exp Res. 1997;21:1278–1284. [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, et al. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9:557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- Volz HC, Kaya Z, Katus HA, et al. The role of HMGB1/RAGE in inflammatory cardiomyopathy. Semin Thromb Hemost. 2010;36:185–194. doi: 10.1055/s-0030-1251503. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Colivicchi MA, Allen R, et al. Neuroinflammation induced in the hippocampus of ‘binge drinking’ rats may be mediated by elevated extracellular glutamate content. J Neurochem. 2009;111:1119–1128. doi: 10.1111/j.1471-4159.2009.06389.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci. 1995;57:1011–1026. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]
- Weafer J, Fillmore MT. Individual differences in acute alcohol impairment of inhibitory control predict ad libitum alcohol consumption. Psychopharmacology (Berl) 2008;201:315–324. doi: 10.1007/s00213-008-1284-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Lind PA, Kalmijn J, et al. The investigation into CYP2E1 in relation to the level of response to alcohol through a combination of linkage and association analysis. Alcohol Clin Exp Res. 2011;35:10–18. doi: 10.1111/j.1530-0277.2010.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler H, Dowdall GW, Davenport A, et al. Correlates of college student binge drinking. Am J Public Health. 1995;85:921–926. doi: 10.2105/ajph.85.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HR, Marmorstein NR, Crews FT, et al. Associations between heavy drinking and changes in impulsive behavior among adolescent boys. Alcohol Clin Exp Res. 2011;35:295–303. doi: 10.1111/j.1530-0277.2010.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TA, Vaccaro D, Mcnamara G. Novelty seeking, risk taking, and related constructs as predictors of adolescent substance use: an application of Cloninger’s theory. J Subst Abuse. 1994;6:1–20. doi: 10.1016/s0899-3289(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Marcotte AC. Psychosocial adjustment and educational outcome in adolescents with a childhood diagnosis of attention deficit disorder. J Am Acad Child Adolesc Psychiatry. 1996;35:579–587. doi: 10.1097/00004583-199605000-00012. [DOI] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Seifert A, Wu D, et al. Role of phospholipase D2/phosphatidic acid signal transduction in micro- and delta-opioid receptor endocytosis. Mol Pharmacol. 2010;78:105–113. doi: 10.1124/mol.109.063107. [DOI] [PubMed] [Google Scholar]
- Yucel M, Lubman DI. Neurocognitive and neuroimaging evidence of behavioural dysregulation in human drug addiction: implications for diagnosis, treatment and prevention. Drug Alcohol Rev. 2007;26:33–39. doi: 10.1080/09595230601036978. [DOI] [PubMed] [Google Scholar]
- Zecevic N, Bourgeois JP, Rakic P. Changes in synaptic density in motor cortex of rhesus monkey during fetal and postnatal life. Brain Res Dev Brain Res. 1989;50:11–32. doi: 10.1016/0165-3806(89)90124-7. [DOI] [PubMed] [Google Scholar]
- Zhao C, Teng EM, Summers RG, Jr, et al. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci. 2006;26:3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol. 2006;26:385–405. doi: 10.1007/s10571-006-9045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Crews F. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol Clin Exp Res. 2010;34:777–789. doi: 10.1111/j.1530-0277.2010.01150.x. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews FT. Inflammasome-IL-1beta signaling mediates ethanol inhibition of hippocampal neurogenesis. FrontiersNeurosci. 2012;6:77. doi: 10.3389/fnins.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]