

Abstract
Background
Osteoarthritis is a progressive inflammatory joint disease resulting in damage to articular cartilage. G-protein coupled estrogen receptor (GPER/GPR30) activates cell signaling in response to 17β-estradiol, which can be blocked by the GPR30 agonist, G15, an analog of G-1. The aims of this study were to investigate the effects of 17β-estradiol on the expression of G-protein coupled estrogen receptor (GPER/GPR30) on mitophagy and the PI3K/Akt signaling pathway in ATDC5 chondrocytes in vitro.
Material/Methods
Cultured ATDC5 chondrocytes were treated with increasing concentrations of 17β-estradiol with and without G15, p38 inhibitor (SB203580), JNK inhibitor (SP600125), PI3K inhibitor (LY294002, S1737), and mTOR inhibitor (S1842). Expression of GPER/GPR30 and components of the PI3K/Akt pathway in cultured ATDC5 chondrocytes were detected by immunofluorescence (IF) staining, Western blot, and real-time polymerase chain reaction (RT-PCR). Transmission electron microscopy (TEM) and IF were used to detect mitophagosomes. Expression of LC-3, LAMP2, TOM20, Hsp60, p-Akt, p-mTOR, p-p38, and p-JNK was investigated by Western blot. Proliferation and viability of the ATDC5 chondrocytes were determined using BrdU and MTT assays.
Results
In 17β-estradiol-treated ATDC5 chondrocytes, increased expression of GPER/GPR30 was found, but fewer mitophagosomes were observed, and decreased numbers of TOM20-positive granules were co-localized with decreased LAMP2 and increased expression levels of TOM20, Hsp60, p-Akt, and p-mTOR, and reduced expression of LC3-II, were found. In 17β-estradiol-treated ATDC5 chondrocytes, the proliferation and viability of the 17β-estradiol-treated ATDC5 chondrocytes were significantly elevated.
Conclusions
Treatment with 17β-estradiol protected ATDC5 chondrocytes against mitophagy via the GPER/GPR30 and the PI3K/Akt signaling pathway.
MeSH Keywords: Chondrocytes, Estradiol, Mitochondrial Degradation, Phosphatidylinositol 3-Kinases
Background
The steroid hormone, 17β-estradiol, plays an important role in mammalian physiology and pathology. Estrogen receptors (ERs) and 17β-estradiol combine to regulate many physiological functions of the reproductive, cardiovascular, nervous, muscular, skeletal, and endocrine systems [1]. Currently, three main ERs have been identified, ERα [2], ERβ [3] and the G-protein coupled estrogen receptor (GPER/GPR30) [4]. The conventional effects of estrogen are mediated by classical nuclear hormone receptors (ERα and ERβ), which act as ligand-activated transcription factors that usually take hours to days to mediate genomic signaling [5].
The G-protein coupled estrogen receptor (GPER), GPER/GPR30 is a member of the superfamily of 7-transmembrane GPERs, which has been recognized to mediate rapid cellular responses that occur in seconds to minutes, including the activation of ion channels, kinases, and signaling pathways [6,7]. Recent studies have identified GPER/GPR30 as a critical mediator of cell signaling in response to estrogen. However, these events cannot be explained by the actions of classical nuclear ERα or ERβ alone. GPER/GPR30 is widely distributed throughout the body, with the highest expression in the lymphoid system, lungs, heart, placenta, liver, skeletal muscle, kidneys, and chondrocytes of the growth plate [8–10]. ERα and ERβ are localized mainly in the cell nucleus but are also found in the mitochondria [11]. The estrogen receptor GPER/GPR30 is mainly localized in the endoplasmic reticulum but is also found in the mitochondria [12].
ERα, ERβ, and GPER/GPR30 located in mitochondria are also known as mitochondrial estrogen receptors (mtERs) [12,13]. The mtERs have been detected in many tissues and cell types and may mediate the protective effects of estrogen directly on mitochondria [12,14]. GPER/GPR30 has been shown to play a role in the protective effects of estrogen on neurons, endothelial cells, and C2C12 murine skeletal myoblasts [12,15,16]. In C2C12 murine skeletal myoblasts, ERα or ERβ may also act together with GPER/GPR30 to mediate the actions of estrogens. Currently, the role of GPER/GPR30 in the effects of estrogen on chondrocytes remains unclear.
A previously published study has shown that 17β-estradiol reduces oxidative stress and inflammatory responses by activating the PI3K/Akt signal transduction pathway in the Raw 264.7 macrophage cell line [17]. 17β-estradiol has been shown to protect osteoblasts against serum deprivation-induced apoptosis by promoting autophagy through the ER-ERK-mTOR pathway [18]. Also, 17β-estradiol has been shown to be a potential treatment for spinal cord injury due to its ability to stimulate early astroglial responses and cytokine release, which reduce the activation of calpain, a family of Ca2+-activated neutral cysteine endoproteases, and inhibition of cell apoptosis [19,20]. Recent studies have shown that estrogen has a potentially protective effect on chondrocytes and might have benefits in the treatment of osteoarthritis [21–23]. However, the molecular mechanisms of estrogen-mediated protection of chondrocytes remain unclear.
Autophagy is a process of degradation of cytoplasmic macromolecules and organelles and is a biological phenomenon found in eukaryote cells [24]. Autophagy is highly regulated to maintain the balance between the synthesis, degradation, and subsequent recycling of cell products that are essential for cell growth, survival, differentiation, development, and homeostasis. Under normal conditions, autophagy occurs at a low or basal rate in cells but can be upregulated in response to environmental stress and signaling stimuli, such as cell starvation, amino acid depletion, oxidative stress, cell proliferation, hypoxia, and by several pharmacological agents [25,26]. Furthermore, autophagy plays a significant role in many human diseases, including cancer [27], infectious diseases [28], cartilage injury and osteoarthritis [29], and pulmonary disease [30]. The regulation of the activity of autophagy is closely associated with tumor formation, progression, and response to therapy [31]. In cancer cells, chemical inhibitors of autophagy enhance apoptosis via active-site mTOR inhibitors or dual PI3K/mTOR inhibitors, which suggests that the PI3K/Akt pathway is involved in the process of autophagy [32].
Mitochondria drive the major metabolic pathways of the cell. Mitophagy is a form of selective autophagy that is involved in the removal of dysfunctional mitochondria through degradation in the autophagy-lysosomal system [33]. Microtubule-associated protein-1 light chain-3 (LC3) is the mammalian ortholog of the yeast autophagy-associated protein, ATG8. During the process of autophagy, cytosolic LC3 (LC3-I) binds phosphatidylethanolamine to form an LC3-phosphatidylethanolamine conjugate (LC3-II), which is a good index of the process of autophagy [34]. TOM 20 is a subunit of the translocase of the outer membrane (TOM) complex that is responsible for recognizing early mitochondrial sequences and is considered to be a potential marker of mitophagy [35]. Also, some specific antioxidant enzymes that are located in the mitochondria, including heat shock protein 60 (HSP60), peroxiredoxin 3 (Prx3), and thioredoxin 2 (Trx2) protect mitochondria against reactive oxygen species (ROS)-induced damage by catalyzing the reduction of H2O2 to H2O [36–38].
Recent studies have shown that 17β-estradiol is not only associated with oxidative stress, the inflammatory response, and apoptosis but is also associated with autophagy. The neuroprotective effects of 17β-estradiol have been shown to be related to the suppression of excessive autophagy in a rat spinal cord injury model, indicating that the protective effect of estrogen against spinal cord injury occurs through GPER/GPR30 and not through nuclear ERs [39]. G15 is an analog of G-1 that binds to GPR30 with high affinity but has no affinity for ERα and ERβ. It has been shown that when G15 was administrated, the effects of GPER/GPR30 of estrogen on glomerular endothelial cells were reversed [40]. Estradiol has been shown to modify the PI3K/Akt-mTOR signaling pathway and to regulate autophagy, reducing the extent of damage following spinal cord injury [41]. LY294002 is a PI3K inhibitor, which is a morpholino-derivative of quercetin. Rapamycin is a specific mTOR antagonist. LY294002 and rapamycin have been used in several studies to provide evidence for the involvement of the PI3K-mTOR pathway in various biological systems [42,43]. SB203580 is a p38 mitogen-activated protein kinase inhibitor, and SP600125 is a broad-spectrum JNK inhibitor for JNK1, JNK2, and JNK3, which have been used to block the downstream signaling elements of p38 and JNK in the PI3K-mTOR pathway [44].
Currently, the molecular mechanisms by which estrogen protects chondrocytes and might alleviate osteoarthritis remain unclear. The aims of this study were to investigate the effects of 17β-estradiol on the expression of GPER/GPR30 on mitophagy and the PI3K/Akt signaling pathway in ATDC5 chondrocytes in vitro. The findings of this study might provide insight into estradiol as a potential treatment for osteoarthritis.
Material and Methods
Reagents and antibodies
The 17β-estradiol was purchased from Sigma-Aldrich (Sigma, St. Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 containing L-glutamine and HEPES, and fetal bovine serum (FBS) were obtained from HyClone (Logan, UT, USA). The selective the G-protein coupled estrogen receptor (GPER/GPR30) antagonist, G15, was purchased from Tocris Bioscience (Minneapolis, MA, USA). The p38 inhibitor (SB203580), JNK inhibitor (SP600125), PI3K inhibitor (LY294002, S1737), mTOR inhibitor (S1842) and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime Biotechnology (Jiangsu, China). The GPER/GPR30 polyclonal antibody (sc-48525-R) used at a dilution of 1: 200 for Western Blot (WB) and at a dilution of 1: 50 for immunofluorescence (IF), the Lamp2 polyclonal antibody (sc-8100) (IF 1: 50), the TOM20 polyclonal antibody (sc-11415) (WB 1: 200, IF 1: 50), the LC3 monoclonal antibody (sc-376404) (WB 1: 100), and the Hsp60 polyclonal antibody (sc-1052) (WB 1: 200) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The total/phosphor-Akt polyclonal antibody and total/phosphor-p38 antibody were purchased from Abcam (Cambridge, MA, USA). The total/phosphor-mTOR antibody was purchased from Sigma-Aldrich (Sigma, St. Louis, MO, USA). The total/phosphor-JNK antibody was purchased from ImmunoWay (Newark, DE, USA). The β-actin monoclonal antibody (AF0003) (1: 1000) was purchased from Biosynthesis Biotechnology (Beijing, China). The IF staining kit with Alexa Fluor 555-labeled donkey anti-rabbit IgG (P0179) was purchased from Beyotime Biotechnology (Jiangsu, China). IFKine® green-conjugated donkey anti-goat IgG (A24231) was obtained from Abbkine Scientific Co., Ltd. (California, USA). The Cell Proliferation ELISA bromodeoxyuridine (BrdU) colorimetric kit was purchased from Roche Diagnostics (Penzberg, Germany).
Cell culture
The mouse chondroprogenitor cell line ATDC5 was purchased from Kebai (Nanjing, China). The ATDC5 cells were cultured in DMEM/F12 medium supplemented with 5% FBS. The cells were incubated in a humidified atmosphere containing 5% CO2 at 37°C. The cell culture medium was replaced every two days. Before the addition of 17β-estradiol, the cells were transferred into serum-free medium in Petri dishes and incubated for 24 hours, and then treated with increasing concentrations of 17β-estradiol (0 μM, 10−3 μM, 10−2 μM and 10−1 μM) with or without the addition of 15 μM G15 or 20 μM of LY294002, and incubated for a further 24 hours.
Real-time polymerase chain reaction (RT-PCR)
For the analysis of the expression levels of mRNA coding for GPER/GPR30, total RNA was extracted from the ATDC5 chondrocytes using TRIzol reagent (Life Technologies Co., Carlsbad, CA, USA). Real-time polymerase chain reaction (RT-PCR) was performed to detect the expression of GPER/GPR30 mRNA using TaqMan reagents (Takara, Otsu, Japan). The following specific primers were used: GPER/GPR30 forward, 5′-AACAGAGCAGCGATCTGGAC-3′ and GPER/GPR30 reverse, 5′-GCAGAGTCCTTGGATGGCTT-3′.
The data were normalized based on the expression of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, which is an internal control for reverse transcription (RT) and reaction efficiency. The following specific primers were used: GAPDH forward, 5′-GGAAGGGCTCATGACCACAGT-3′, and GAPDH reverse, 5′-GGAAGGCCATGCCAGTGA-3′. The reactions were amplified at 95°C for 30 s followed by 40 cycles of 95°C for 5 s followed by 60°C for 30 s. All primers and TaqMan probes specific to GPER/GPR30 and GAPDH were designed using the Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA).
Immunofluorescence (IF) analysis
The ATDC5 chondrocytes were cultured in 24-well plates. After incubation, the cells were fixed with 4% paraformaldehyde buffered with 0.1 M phosphate (pH 7.3) for 30 min. The fixed samples were then washed with phosphate-buffered saline (PBS) for 10 min. The cells were treated with 0.1% TritonX-100 for 30 min to permeabilize the cell membrane and then washed with PBS for 10 min. The samples were blocked with 5% bovine serum albumin (BSA) in TBST for 30 min and then incubated overnight at 4°C with the following specific primary antibodies: goat anti-Lamp2 (1: 50), rabbit anti-TOM20 (1: 50), and rabbit anti-GPER/GPR30 antibody (1: 50). Then, the ATDC5 chondrocytes were washed with PBS for 10 min and incubated for 30 min with the following secondary antibodies: donkey anti-goat IgG (1: 1000) and donkey anti-rabbit IgG (1: 1000). The cells were stained with DAPI for 5 min, washed with PBS for 15 min and observed with an Olympus FV1000 confocal laser-scanning microscope with the peak emission wavelengths of 518 nm (green) and 565 nm (red).
Protein preparation and Western blot analysis
The cultured ATDC5 chondrocytes were washed with PBS, and the total protein was harvested in radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China) supplemented with the protease inhibitor phenylmethanesulfonyl fluoride (PMSF) (Beijing, Jiangsu, China) and phosphatase inhibitors (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). The protein concentration was determined with a Bicinchoninic Acid (BCA) Protein Assay Kit, according to the manufacturer’s instructions (Beyotime, China). The samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes using a transfer buffer at 70 V for 1.5 hours. To block nonspecific binding, the membranes were incubated in dried skimmed milk powder for 120 min at room temperature and were washed three times with Tris-buffered saline (TBS) containing Tween 20 (TBST) for 30 min. The membranes were incubated overnight at 4°C with the primary antibodies and then washed with TBST for 30 min and incubated with horseradish peroxidase (HRP)-conjugated anti-species secondary antibody (Beyotime, China) for 2 hours at room temperature. After the membranes were washed three times with TBST for 30 min, the proteins were visualized with the BeyoECL plus kit (Beyotime, China).
Transmission electron microscopy (TEM) analysis
The cells were trypsinized and collected by centrifugation at 1,000×g for 5 min. The cells were fixed in 2.5% phosphate-buffered glutaraldehyde and post-fixed in 1% osmium tetroxide in water. After being subjected to gradient acetone dehydration, the cells were embedded and sectioned. The samples were double-stained with uranyl acetate and lead citrate, and the ultrastructure of the cells was observed with a JEM-1200EX transmission electron microscope (TEM) (Tokyo, Japan).
Cell proliferation assay
ATDC5 chondrocytes were seeded in 96-well plates (2×103 cells/well) and cultured for 24 hours. The cells were serum-deprived for 24 hours and then treated with 17β-estradiol (0 or 10−1 μM), with or without G15 and PI3Ki for a further 24 hours. Then, the BrdU solution (1 μM) was added, and the cells were incubated for 2.5 hours. BrdU incorporation into the DNA was measured using the Cell Proliferation ELISA BrdU colorimetric kit (Roche Diagnostics).
Cell viability
ATDC5 chondrocytes were seeded in 96-well plates (2×103 cells/well) and cultured for 24 hours. After incubation in serum-free DMEM/F12 medium supplemented with 17β-estradiol (10−1 μM) at 37°C for 24 hours, 10 μl of MTT reagent (5 mg/ml) was added, and the cells were incubated at 37°C for 4 hours. Then, the culture medium was removed, and formazan crystals were dissolved with 100 μl of dimethyl sulfoxide (DMSO). All experiments were performed in triplicate. The optical density (OD) was measured using a Spectra Max Plus 384 Microplate Reader (Molecular Devices, Ismaning, Germany) at 570 nm (reference wavelength).
Statistical analysis
The data were presented as the mean ± standard deviations (SD). Normality was assessed with the Shapiro–Wilk test, and the homoscedastic sequence of random variables was assessed using Bartlett’s method (also known as the method of averaged periodograms). The data that passed the homogeneity test were analyzed with the Student’s t-test or one-way analyses of variance (ANOVA), followed by the least significant difference (LSD) test. The statistical analysis was performed using SPSS version 15.0 (SPSS, Inc., IL, USA). P<0.05 was considered statistically significant.
Results
G-protein coupled estrogen receptor (GPER/GPR30) expression in ATDC5 chondrocytes was stimulated by 17β-estradiol
Positive expression of GPER/GPR30 in the ATDC5 cells was detected by real-time polymerase chain reaction (RT-PCR) (Figure 1A), Western blot (Figure 1B, 1C), and immunofluorescence (IF) staining (Figure 1D).
Figure 1.

Expression of the G-protein coupled estrogen receptor (GPER/GPR30) stimulated by 17β-estradiol in serum-starved ATDC5 chondrocytes. (A) Expression of GPER/GPR30 mRNA in serum-starved ATDC5 chondrocytes treated with increasing concentrations of 17β-estradiol (0 μM, 10−3 μM, 10−2 μM, and 10−1 μM), as assessed with quantitative real-time polymerase chain reaction (RT-PCR). (B) Expression of GPER/GPR30 protein in ATDC5 chondrocytes treated with 17β-estradiol (0 μM, 10 1 μM) for 0, 5, 30, 60, or 120 min, as assessed with Western blot. (C) Expressions of GPER/GPR30 protein in serum-starved ATDC5 chondrocytes treated with increasing concentrations of 17β-estradiol (0 μM, 10−3 μM, 10−2 μM, and 10−1 μM) with or without 15 μM of G15, as assessed by Western blot. (D) Confocal immunofluorescence (IF) staining of ATDC5 chondrocytes for GPER/GPR30 (red). The cell nuclei are stained with 4′, 6-diamidino-2-phenylindole (DAPI) (blue). The intensity of the GPER/GPR30 staining is increased in the serum-starved ATDC5 cells that were cultured in serum-free medium with 17β-estradiol (P <0.05). The figure data represent experiments performed in triplicate. The asterisks indicate significant differences compared with the control (* P<0.05).
In this study, serum-starved ATDC5 chondrocytes were treated with different concentrations of 17β-estradiol (0 μM, 10−3 μM, 10−2 μM and 10−1 μM, with or without 15 μM G15) for 24 hours. G15 is a high-affinity and selective GPER antagonist. The data from this study indicated that 17β-estradiol could increase GPER/GPR30 mRNA and protein levels in a dose-dependent manner up to the highest dose of 10−1 μM (P<0.05) (Figure 1A, 1C). Also, the level of expression of GPER/GPR30 protein increased in a time-dependent manner.
Western blot analysis showed that estradiol significantly increased the protein expression over time (as assessed at 0 min, 5 min, 30 min, 1 hour, and 2 hours) (Figure 1B). Western blot showed that the GPER protein was expressed at a higher level in the serum-deprived 17β-estradiol-treated ATDC5 chondrocytes compared with cells that were treated with 17β-estradiol plus the GPER/GPR30-specific antagonist G15 (15 μM) (Figure 1C). Also, immunofluorescence (IF) staining showed that, compared with the control group, 17β-estradiol increased the expression of GPER/GPR30 in the cell membrane and the cytoplasm (P<0.05) (Figure 1D).
Inhibition of mitophagy in ATDC5 cells was involved in the function of 17β-estradiol via GPER/GPR30
Mitophagy was be visualized based on mitophagosomes or autophagosomes seen on transmission electron microscopy (TEM) and the co-localization of mitochondria with mitophagic proteins and lysosomes on IF staining. In this study, the prominent features of the mitophagosomes and autophagosomes were analyzed by TEM (Figure 2). Compared with the control group, fewer mitophagosomes were observed in the 17β-estradiol-treated ATDC5 cells, which indicated that 17β-estradiol inhibited mitophagy in the ATDC5 cells. Also, a significantly increased number of double membrane vacuoles were seen in the ATDC5 cells that were treated with 17β-estradiol and G15 (which inhibits binding to estrogen receptors) compared with the cells that were treated with 17β-estradiol alone (Figure 2), which indicated that GPER was involved in mitophagy.
Figure 2.

Transmission electron microscopy (TEM) showing mitophagosomes in the ATDC5 cells. Double membrane vacuoles (mitophagosomes) in the ATDC5 cells that were treated with 17β-estradiol (0 μM and 10−1 μM) with or without 15 μM of G15 seen by transmission electron microscopy (TEM). N represents the nucleus. The vacuoles are indicated with arrows. Black bar=5 μm.
The co-localization of TOM20 with Lamp2 was measured with IF staining. The results indicated that decreased numbers of TOM20-positive granules were co-localized with the decreased Lamp2 in the ATDC5 cells that were exposed to 17β-estradiol, which indicates that the number of mitophagosomes or autophagosomes decreased (Figure 3). However, these effects of 17β-estradiol were abolished by the presence of the inhibitors G15 or LY294002 (Figure 3).
Figure 3.

Confocal immunofluorescence (IF) staining for TOM20 and Lamp2 of ATDC5 cells. Confocal immunofluorescence staining for TOM20 (red) and Lamp2 (green). The cell nuclei are stained blue with 4,6-diamidino-2-phenylindole (DAPI). Serum-free ATDC5 cells were treated with 17β-estradiol (0 μM and 10−1 μM) with or without 15 μM of G15 and 20 μM of PI3K inhibitor. The staining intensity is lower in the serum-free 17β-estradiol-treated ATDC5 cell group compared with the other groups (P<0.05). White bar=50 μm.
17β-Estradiol inhibited mitophagy by regulating the phosphorylation of LC3, TOM20, and Hsp60 in ATDC5 cells
To understand the effect of 17β-estradiol on mitophagy protein activity, the expression of LC3 (LC3-I and LC3-II), which are typical markers of completed autophagy, TOM20, and Hsp60 proteins were measured by Western blot. The expression level of LC3-II was significantly decreased in the 17β-estradiol-treated ATDC5 cells compared with the control group, and this effect was abolished in the cells that were treated with 17β-estradiol and G15 or LY294002 (Figure 4). The levels of TOM20 and Hsp60 in the group of ATDC5 cells that were exposed to 17β-estradiol were greater compared with those in the control group. Also, these effects were diminished by G15 or LY294002 treatment (Figure 4).
Figure 4.

Western blot showing expression of the LC3, TOM20, and Hsp60 proteins in ATDC5 cells treated with 17β-estradiol with or without G15 or the PI3K inhibitor. (A) Western blot shows expression of the LC3 protein in ATDC5 chondrocytes that were treated with 17β-estradiol (0 μM and 10−1 μM) with or without 15 μM of G15 and 20 μM of PI3K inhibitor. (B) Western blot shows expression of the TOM20 protein in ATDC5 chondrocytes that were treated with 17β-estradiol (0 μM and 10−1 μM) with or without 15 μM of G15 and 20 μM of PI3K inhibitor. (C) Western blot shows expression of the Hsp60 protein in ATDC5 chondrocytes that were treated with 17β-estradiol (0 μM and 10−1 μM) with or without 15 μM of G15 and 20 μM of PI3K inhibitor. The figure data represent experiments performed in triplicate. The asterisks indicate significant differences compared with the control (* P<0.05).
17β-Estradiol protected ATDC5 chondrocytes through the GPER/GPR30/PI3K/Akt pathway
The GPER/GPR30/PI3K/Akt pathway plays important roles in cell proliferation, apoptosis, and autophagy. Akt phosphorylation has been widely used as an indicator of PI3K/Akt pathway activation. In this study, Western blot showed increased phosphor-Akt protein levels in the 17β-estradiol-treated ATDC5 cells, while the level of total Akt expression remained comparable. The opposite results were observed when the cells were treated with G15 (15 μM) or the PI3K inhibitor, LY294002 (20 μM) (Figure 5A), which indicated that 17β-estradiol (10−1 μM) activated the PI3K/Akt pathway via GPER/GPR30 in the ATDC5 cells. Also, the inhibition of phospho-mTOR (p-mTOR) was selected as an indicator of autophagy pathway activation. The data showed that 17β-estradiol (10−1 μM) increased the p-mTOR protein levels in the ATDC5 cells, and the opposite results were found following treatment of the cells with G15 or the mTOR inhibitor, rapamycin (10−1 μM) (Figure 5B), which indicated that 17β-estradiol (10−1 μM) increased the phosphorylation of mTOR via GPER/GPR30 in the ATDC5 cells and inhibited mitophagy.
Figure 5.

Western blot showing expression of p-Akt, p-mTOR, p-p38, and p-JNK in ATDC5 cells treated with 17β-estradiol with or without G15, the PI3K inhibitor, the mTOR inhibitor, the p38i inhibitor, and the JNK inhibitor. (A) Western blot shows the expression of p-Akt in ATDC5 cells treated with 17β-estradiol (0 μM or 10−1 μM), with or without G15, the PI3K inhibitor, the mTOR inhibitor, the p38i inhibitor, and the JNK inhibitor. (B) Western blot shows the expression of p-mTOR in ATDC5 cells treated with 17β-estradiol estradiol (0 μM or 10−1 μM), with or without G15, the PI3K inhibitor, the mTOR inhibitor, the p38i inhibitor, and the JNK inhibitor. (C) Western blot shows the expression of p-p38 in ATDC5 cells treated with 17β-estradiol estradiol (0 μM or 10−1 μM), with or without G15, the PI3K inhibitor, the mTOR inhibitor, the p38i inhibitor, and the JNK inhibitor. (D) Western blot shows the expression of p-JNK in ATDC5 cells treated with 17β-estradiol estradiol (0 μM or 10−1 μM), with or without G15, the PI3K inhibitor, the mTOR inhibitor, the p38i inhibitor, and the JNK inhibitor. The figure data represent experiments performed in triplicate. The asterisks indicate significant differences compared with the control (* P<0.05).
Also, the effect of 17β-estradiol on the ATDC5 cells was abolished by the GPER/GPR30 inhibitor G15 (Figures 2–6), the PI3K inhibitor LY294002 (Figures 3–6), and by the mTOR inhibitor (Figure 5B), which suggests that 17β-estradiol inhibited mitophagy through the PI3K/Akt pathway via GPER/GPR30.
Figure 6.
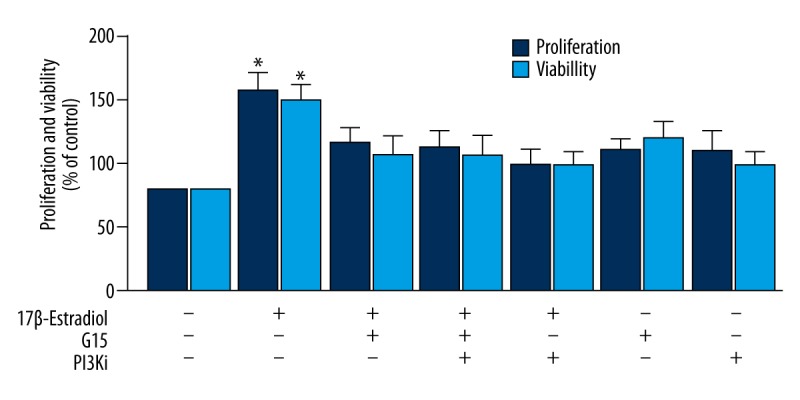
BrdU and MTT assays for proliferation and viability of ATDC5 chondrocytes that were treated with 17β-estradiol with or without G15 and PI3K inhibitor. Analysis of the proliferation and viability of ATDC5 chondrocytes that were treated with 17β-estradiol (0 μM and 10−1 μM) with or without G15 and PI3K inhibitor as assessed with the BrdU assays and the MTT assay. The figure data represent experiments performed in triplicate. The asterisks indicate significant differences compared with the control (* P<0.05).
Mitogen-activated protein kinases (MAPKs), including those of the JNK and p38 signaling pathways, have also been linked to the regulation of autophagy [45]. To further determine whether estradiol could protect ATDC5 chondrocytes through the GPER/GPR30/JNK or GPER/GPR30/p38 pathways, serum-free ATDC5 cells were treated with 10−1 μM of 17β-estradiol with or without the JNK inhibitor, SP600125 (10 μM), the p38 inhibitor, SB203580 (10 μM), G15 (15 μM), or with the combination of inhibitors for 24 hours. The data indicated no changes in p38 or JNK phosphorylation in the 17β-estradiol-treated ATDC5 cells, and no reversal of effects was observed with G15, the p38 inhibitor, or the JNK inhibitor (Figure 5C, 5D).
Finally, a BrdU assay and an MTT assay showed that the proliferation and viability of the 17β-estradiol-treated ATDC5 chondrocytes were increased by 1.5-fold compared with the control group (P<0.05), and the effects were reduced by treatment with G15 and the PI3K inhibitor, LY294002 (Figure 6). These findings indicated that 17β-estradiol promoted the proliferation and viability of the ATDC5 chondrocytes under serum-starved conditions through the GPER/GPR30/PI3K/Akt pathway.
Discussion
Osteoarthritis is a degenerative joint disease that is characterized by articular cartilage damage. Chondrocytes are the main cell type that form the articular cartilage, and the gradual loss of these cells is the main cause of osteoarthritis [46]. There has been increasing published evidence that estrogen can protect chondrocytes and alleviate osteoarthritis in vivo and in vitro [21–23]. In this study, the proliferation and viability of 17β-estradiol-treated ATDC5 chondrocytes were significantly increased compared with the control group, which confirmed the protective effect of 17β-estradiol on chondrocytes. However, the molecular mechanism of the estrogen-mediated protection of chondrocytes from damage remains unclear.
In the present study, fewer mitophagosomes were observed using transmission electron microscopy (TEM), and decreased numbers of TOM20-positive granules were co-localized with decreased Lamp2 shown by immunofluorescence (IF) analysis in the 17β-estradiol-treated ATDC5 cells, which showed that mitophagy was suppressed by 17β-estradiol. These findings are consistent with those of a recent study that showed that the protective effect of estradiol was due to the inhibition of autophagy in neurocytes [41]. There is increasing evidence to indicate that mitochondria are essential for cell survival and cell death because they can be considered to be the metabolic ‘powerhouse’ of the cell [47].
Mitochondria are essential for the intermediary metabolism of protein, carbohydrate, and fat. Mitochondria also play a key role in adenosine triphosphate (ATP) production and provide more than 90% of the energy of the cell. It has been established that the activity of mitochondria modulate cell survival, cycle differentiation, cell proliferation, apoptosis, and the generation of reactive oxygen species (ROS) [48,49]. Oxidative stress can lead to mitochondrial damage, and if that damage surpasses the membrane potential across the inner mitochondrial membrane, the entire population of dysfunctional mitochondria is removed via the autophagy pathway, a process known as mitophagy.
Autophagy has been proposed to be a ‘double-edged sword’; while mitophagy can protect cells from apoptosis by removing mitochondria that have been damaged by oxidative stress, excessive mitophagy can reduce essential cellular components through the degradation of the bulk cytoplasm and thus cause cell death [50]. In ATDC5 cells in vitro, excess autophagy can be induced by glucocorticoids (GCs), which results in a significant reduction in cell viability [51]. Previous studies have shown that autophagy could reduce the growth rate of growing cells through the elimination of growth-promoting molecules and organelles, such as the protein p62 (sequestosome-1). When p62 is depleted or knocked out, mTOR activation fails, which leads to an impairment of cell growth [52]. In response to excessive mitophagy, mitochondria employ other mechanisms to repair or remove damaged cell components, such as products arising during cycles of fusion and fission, and act a an organelle quality control mechanism known as the mitochondrial unfolded protein response (UPRmt). These mechanisms ensure that the mitochondria maintain their normal functions [53]. The cell can activate the UPRmt pathway to protect the mitochondria from dysfunction by increasing mitochondrial chaperone levels, including Hsp60 and Hsp70, and increasing the expression of proteases [54,55]. TOM20 is a protein marker in the mitochondrial outer membrane. Serum starvation of HeLa cells can induce mitophagy along with an increased expression of LC3-II and can decrease the expression of TOM20 [56]. Similarly, in this study, the expression level of the protein LC3-II was reduced, while the expressions of the mitochondrial outer membrane protein TOM20 and Hsp60 were increased. Therefore, it can be concluded that 17β-estradiol might protect ATDC5 chondrocytes by inhibiting mitophagy and may help to repair the mitochondrial damage.
In this study, mitophagy was suppressed by 17β-estradiol in the ATDC5 cells via the G-protein coupled estrogen receptor (GPER/GPR30) and the PI3K/Akt-mTOR signaling pathway, which suggests that the PI3K/Akt-mTOR signaling pathway might be a target for osteoarthritis therapy. Previous studies have shown that 17β-estradiol can bind to GPER/GPR30 receptors that are expressed in cardiomyocytes and cancer cells to affect their physiological functions [57,58]. In this study, GPER/GPR30 was expressed in ATDC5 chondrocytes, and treatment with 17β-estradiol at increasing doses (0 μM, 10−3 μM, 10−2 μM, and 10−1 μM) increased the GPER/GPR30 protein levels in a dose-dependent manner, with strong increase at the concentration of 10−1 μM, which indicates that 17β-estradiol might act on GPER/GPR30 to exert its physiological regulatory function in chondrocytes.
Also, in this study, Western blot analysis also showed a time-dependent induction of the levels of the GPER/GPR30 protein. Immunofluorescence (IF) staining was performed and showed that GPER/GPR30 was present in the regions outside the nucleus in ATDC5 chondrocytes, which implies that the GPER/GPR30 receptor may not be an intra-nuclear receptor and that 17β-estradiol may act on ATDC5 chondrocytes through GPER/GPR30 in the membrane, cytoplasm and mitochondrial. More studies are needed to elucidate the distribution of GPER/GPR30 in ATDC5 chondrocytes, to clarify the mechanism of action of GPER/GPR30 in ATDC5 cells. The effects of 17β-estradiol were abolished the GPER/GPR30 antagonist G15, which caused an increase in the expression of LC3-II and reductions in the expressions of TOM20 and Hsp60. Therefore, 17β-estradiol may inhibit mitophagy via GPER/GPR30. The role of ERα and ERβ in mediating 17β-estradiol actions on ATDC5 chondrocytes remain to be studied, to further evaluate the possible interactions among ERα, ERβ, and GPER/GPR30 in 17β-estradiol actions on chondrocytes.
The PI3K/Akt signaling pathway is involved in almost every aspect of physiological and pathological cell functions, including growth, tumorigenesis, apoptosis, and autophagy [59,60]. In this study, the phospho-Akt protein level was increased in the ATDC5 chondrocytes that were cultured with 17β-estradiol for 24 hours, while the expression levels of the total Akt protein remained unchanged. It has previously been reported that mTOR, which is positively regulated by PI3K, is a negative regulator of autophagy. In the present study, the expression level of p-mTOR was significantly increased in the 17β-estradiol-treated ATDC5 cells compared with the control cells. In addition to incubation with 17β-estradiol, ATDC5 cells were incubated with an inhibitor of GPER/GPR30 (G15), an inhibitor of PI3Ki or an inhibitor of mTOR. The results showed that the inhibition of GPER, PI3K or mTOR abolished the effect of 17β-estradiol on mitophagy, which indicated that 17β-estradiol inhibited mitophagy through the PI3K/Akt-mTOR signaling pathway via GPER/GPR30. Similar findings have also been in previous studies. For example, 17β-estradiol has been shown to suppress lipopolysaccharide-induced acute lung injury through PI3K/Akt signaling [61]. Moreover, 17β-estradiol decreases the rate of apoptosis in nucleus pulposus cells in rats via the PI3K/Akt/caspase-3 pathway [62]. Furthermore, PI3K/Akt signaling is inversely associated with mitochondrial oxidative stress, and the activation of PI3K/Akt signaling reduces ROS production [63,64]. Other MAPK signaling pathways, including the JNK and p38 pathways, might be linked to the regulation of autophagy. To verify whether 17β-estradiol also modulated mitophagy through JNK and p38, in this study, cells were pretreated with a p38i and a JNKi for 30 min. The P-p38 and P-JNK levels exhibited non significant differences between the experimental groups. These results indicated that 17β-estradiol did not regulate mitophagy through the JNK or p38 signaling pathways.
Future studies will focus on the function of estradiol in chondrocytes and the relevant molecular mechanisms in animal models and clinical trials with the goals of identifying the clinical effects of estradiol on chondrocytes and potential therapeutic targets for osteoarthritis.
Conclusions
The findings of the present study have shown that 17β-estradiol protected ATDC5 chondrocytes cultured in vitro from mitophagy by activating the PI3K/Akt-mTOR signaling pathway via the G-protein coupled estrogen receptor (GPER/GPR30). The findings of this study also suggest that estradiol might be a promising treatment for osteoarthritis and that the PI3K/Akt-mTOR signaling pathway might be a potential target for future therapy for patients with osteoarthritis.
Footnotes
Conflicts of interest
None.
Source of support: This study was supported by the National Natural Science Foundation of China (Grant Nos. 81470998, 81071460, and 81271996) and the Natural Science Foundation of Liaoning Province (Grant No. 20170541033)
References
- 1.Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7:715–26. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green S, Walter P, Greene G, et al. Cloning of the human oestrogen receptor cDNA. J Steroid Biochem. 1986;24:77–83. doi: 10.1016/0022-4731(86)90035-x. [DOI] [PubMed] [Google Scholar]
- 3.Kuiper GG, Enmark E, Pelto-Huikko M, et al. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmeci C, Thompson DA, Ring HZ, et al. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–17. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 5.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–76. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 6.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–63. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. 2010;204:105–14. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- 8.Chagin AS, Savendahl L. GPR30 estrogen receptor expression in the growth plate declines as puberty progresses. J Clin Endocrinol Metab. 2007;92:4873–77. doi: 10.1210/jc.2007-0814. [DOI] [PubMed] [Google Scholar]
- 9.Kvingedal AM, Smeland EB. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997;407:59–62. doi: 10.1016/s0014-5793(97)00278-0. [DOI] [PubMed] [Google Scholar]
- 10.Takada Y, Kato C, Kondo S, et al. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–41. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 11.Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab. 2015;29:557–68. doi: 10.1016/j.beem.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Ronda AC, Boland RL. Intracellular distribution and involvement of GPR30 in the actions of E2 on C2C12 cells. J Cell Biochem. 2016;117:793–805. doi: 10.1002/jcb.25369. [DOI] [PubMed] [Google Scholar]
- 13.Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–73. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- 14.Liao TL, Tzeng CR, Yu CL, et al. Estrogen receptor-beta in mitochondria: Implications for mitochondrial bioenergetics and tumorigenesis. Ann N Y Acad Sci. 2015;1350:52–60. doi: 10.1111/nyas.12872. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Hu R, Ge H, et al. G-protein-coupled receptor 30-mediated antiapoptotic effect of estrogen on spinal motor neurons following injury and its underlying mechanisms. Mol Med Rep. 2015;12:1733–40. doi: 10.3892/mmr.2015.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Q, Hussain Y, Chorazyczewski J, et al. GPER-independent effects of estrogen in rat aortic vascular endothelial cells. Mol Cell Endocrinol. 2015;399:60–68. doi: 10.1016/j.mce.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, He Y, Zong Y, et al. 17beta-estradiol attenuates homocysteine-induced oxidative stress and inflammatory response as well as MAPKs cascade via activating PI3-K/Akt signal transduction pathway in Raw 264.7 cells. Acta Biochim Biophys Sin (Shanghai) 2015;47:65–72. doi: 10.1093/abbs/gmu124. [DOI] [PubMed] [Google Scholar]
- 18.Yang YH, Chen K, Li B, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18:1363–75. doi: 10.1007/s10495-013-0867-x. [DOI] [PubMed] [Google Scholar]
- 19.Ritz MF, Hausmann ON. Effect of 17beta-estradiol on functional outcome, release of cytokines, astrocyte reactivity and inflammatory spreading after spinal cord injury in male rats. Brain Res. 2008;1203:177–88. doi: 10.1016/j.brainres.2008.01.091. [DOI] [PubMed] [Google Scholar]
- 20.Sribnick EA, Matzelle DD, Ray SK, Banik NL. Estrogen treatment of spinal cord injury attenuates calpain activation and apoptosis. J Neurosci Res. 2006;84:1064–75. doi: 10.1002/jnr.21016. [DOI] [PubMed] [Google Scholar]
- 21.Funck-Brentano T, Lin H, Hay E, et al. Targeting bone alleviates osteoarthritis in osteopenic mice and modulates cartilage catabolism. PLoS One. 2012;7:e33543. doi: 10.1371/journal.pone.0033543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maneix L, Servent A, Poree B, et al. Up-regulation of type II collagen gene by 17beta-estradiol in articular chondrocytes involves Sp1/3, Sox-9, and estrogen receptor alpha. J Mol Med (Berl) 2014;92:1179–200. doi: 10.1007/s00109-014-1195-5. [DOI] [PubMed] [Google Scholar]
- 23.Tao Y, Sun H, Sun H, et al. 17beta-estradiol activates mTOR in chondrocytes by AKT-dependent and AKT-independent signaling pathways. Int J Clin Exp Pathol. 2015;8:15911–18. [PMC free article] [PubMed] [Google Scholar]
- 24.Xiaowei Z, Cuicui L, Ning H, et al. Progress on the autophagic regulators and receptors in plants. Yi Chuan. 2016;38:644–50. doi: 10.16288/j.yczz.15-525. [DOI] [PubMed] [Google Scholar]
- 25.Xie K, Tian L, Guo X, et al. BmATG5 and BmATG6 mediate apoptosis following autophagy induced by 20-hydroxyecdysone or starvation. Autophagy. 2016;12:381–96. doi: 10.1080/15548627.2015.1134079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitter SK, Song C, Qi X, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989–2005. doi: 10.4161/auto.36184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orvedahl A, MacPherson S, Sumpter R, Jr, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–27. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lotz M, Carames B. Autophagy: A new therapeutic target in cartilage injury and osteoarthritis. J Am Acad Orthop Surg. 2012;20:261–62. doi: 10.5435/JAAOS-20-04-261. [DOI] [PubMed] [Google Scholar]
- 30.Mizumura K, Choi AM, Ryter SW. Emerging role of selective autophagy in human diseases. Front Pharmacol. 2014;5:244. doi: 10.3389/fphar.2014.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–39. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 32.Fan QW, Cheng C, Hackett C, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palikaras K, Lionaki E, Tavernarakis N. Mitophagy: In sickness and in health. Mol Cell Oncol. 2016;3:e1056332. doi: 10.1080/23723556.2015.1056332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1–12. doi: 10.1016/S0076-6879(08)03601-X. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Li S, Ren R, Li J, Cui X. Recombinant buckwheat trypsin inhibitor induces mitophagy by directly targeting mitochondria and causes mitochondrial dysfunction in Hep G2 Cells. J Agric Food Chem. 2015;63:7795–804. doi: 10.1021/acs.jafc.5b02644. [DOI] [PubMed] [Google Scholar]
- 36.Hwang IK, Yoo KY, Kim DW, et al. Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radic Biol Med. 2010;48:1242–51. doi: 10.1016/j.freeradbiomed.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Lee CH, Park JH, Cho JH, et al. Differences in the protein expression levels of Trx2 and Prx3 in the hippocampal CA1 region between adult and aged gerbils following transient global cerebral ischemia. Mol Med Rep. 2015;12:2555–62. doi: 10.3892/mmr.2015.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall L, Martinus RD. Hyperglycaemia and oxidative stress upregulate HSP60 and HSP70 expression in HeLa cells. Springerplus. 2013;2:431. doi: 10.1186/2193-1801-2-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu R, Sun H, Zhang Q, et al. G-protein coupled estrogen receptor 1 mediated estrogenic neuroprotection against spinal cord injury. Crit Care Med. 2012;40:3230–37. doi: 10.1097/CCM.0b013e3182657560. [DOI] [PubMed] [Google Scholar]
- 40.Hutchens MP, Fujiyoshi T, Komers R, et al. Estrogen protects renal endothelial barrier function from ischemia-reperfusion in vitro and in vivo. Am J Physiol Renal Physiol. 2012;303:F377–85. doi: 10.1152/ajprenal.00354.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin CW, Chen B, Huang KL, et al. Inhibition of autophagy by estradiol promotes locomotor recovery after spinal cord injury in rats. Neurosci Bull. 2016;32:137–44. doi: 10.1007/s12264-016-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maira SM, Stauffer F, Schnell C, Garcia-Echeverria C. PI3K inhibitors for cancer treatment: where do we stand? Biochem Soc Trans. 2009;37:265–72. doi: 10.1042/BST0370265. [DOI] [PubMed] [Google Scholar]
- 43.Mosley JD, Poirier JT, Seachrist DD, et al. Rapamycin inhibits multiple stages of c-Neu/ErbB2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol Cancer Ther. 2007;6:2188–97. doi: 10.1158/1535-7163.MCT-07-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Y, Jiang Y, Shi L, et al. 7-O-Geranylquercetin induces apoptosis in gastric cancer cells via ROS-MAPK mediated mitochondrial signaling pathway activation. Biomed Pharmacother. 2017;87:527–38. doi: 10.1016/j.biopha.2016.12.095. [DOI] [PubMed] [Google Scholar]
- 45.Periyasamy-Thandavan S, Jiang M, et al. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297:F244–56. doi: 10.1152/ajprenal.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hosseinzadeh A, Kamrava SK, Joghataei MT, et al. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J Pineal Res. 2016;61:411–25. doi: 10.1111/jpi.12362. [DOI] [PubMed] [Google Scholar]
- 47.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–14. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao L, Laude K, Cai H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet Clin North Am Small Anim Pract. 2008;38:137–55. vi. doi: 10.1016/j.cvsm.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: A hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 50.Tsujimoto Y, Shimizu S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–34. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Y, Zuo Y, Huo H, et al. Dexamethasone reduces ATDC5 chondrocyte cell viability by inducing autophagy. Mol Med Rep. 2014;9:923–27. doi: 10.3892/mmr.2014.1915. [DOI] [PubMed] [Google Scholar]
- 52.Neufeld TP. Autophagy and cell growth – the yin and yang of nutrient responses. J Cell Sci. 2012;125:2359–68. doi: 10.1242/jcs.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roberts RF, Tang MY, Fon EA, Durcan TM. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int J Biochem Cell Biol. 2016;79:427–36. doi: 10.1016/j.biocel.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 54.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao Q, Wang J, Levichkin IV, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–19. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukhopadhyay S, Naik PP, Panda PK, et al. Serum starvation induces anti-apoptotic cIAP1 to promote mitophagy through ubiquitination. Biochem Biophys Res Commun. 2016;479:940–46. doi: 10.1016/j.bbrc.2016.09.143. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, Liao Y, Fan S, et al. G protein-coupled estrogen receptor (GPER) mediates NSCLC progression induced by 17beta-estradiol (E2) and selective agonist G1. Med Oncol. 2015;32:104. doi: 10.1007/s12032-015-0558-2. [DOI] [PubMed] [Google Scholar]
- 58.Sbert-Roig M, Bauza-Thorbrugge M, Galmes-Pascual BM, et al. GPER mediates the effects of 17beta-estradiol in cardiac mitochondrial biogenesis and function. Mol Cell Endocrinol. 2016;420:116–24. doi: 10.1016/j.mce.2015.11.027. [DOI] [PubMed] [Google Scholar]
- 59.Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: An emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40:980–89. doi: 10.1016/j.ctrv.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Li T, Wang G. Computer-aided targeting of the PI3K/Akt/mTOR pathway: Toxicity reduction and therapeutic opportunities. Int J Mol Sci. 2014;15:18856–91. doi: 10.3390/ijms151018856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qi D, He J, Wang D, et al. 17beta-estradiol suppresses lipopolysaccharide-induced acute lung injury through PI3K/Akt/SGK1 mediated up-regulation of epithelial sodium channel (ENaC) in vivo and in vitro. Respir Res. 2014;15:159. doi: 10.1186/s12931-014-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang SD, Ma L, Yang DL, Ding WY. Combined effect of 17beta-estradiol and resveratrol against apoptosis induced by interleukin-1beta in rat nucleus pulposus cells via PI3K/Akt/caspase-3 pathway. Peer J. 2016;4:e1640. doi: 10.7717/peerj.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dai H, Song D, Xu J, Li B, et al. Ammonia-induced Na,K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem Int. 2013;63:610–25. doi: 10.1016/j.neuint.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 64.Zhu Z, Li R, Stricker R, Reiser G. Extracellular alpha-crystallin protects astrocytes from cell death through activation of MAPK, PI3K/Akt signaling pathway and blockade of ROS release from mitochondria. Brain Res. 2015;1620:17–28. doi: 10.1016/j.brainres.2015.05.011. [DOI] [PubMed] [Google Scholar]