

Abstract
Hydrogel delivery systems can leverage therapeutically beneficial outcomes of drug delivery and have found clinical use. Hydrogels can provide spatial and temporal control over the release of various therapeutic agents, including small-molecule drugs, macromolecular drugs and cells. Owing to their tunable physical properties, controllable degradability and capability to protect labile drugs from degradation, hydrogels serve as a platform in which various physiochemical interactions with the encapsulated drugs control their release. In this Review, we cover multiscale mechanisms underlying the design of hydrogel drug delivery systems, focusing on physical and chemical properties of the hydrogel network and the hydrogel–drug interactions across the network, mesh, and molecular (or atomistic) scales. We discuss how different mechanisms interact and can be integrated to exert fine control in time and space over the drug presentation. We also collect experimental release data from the literature, review clinical translation to date of these systems, and present quantitative comparisons between different systems to provide guidelines for the rational design of hydrogel delivery systems.
Conventional drug administration often requires high dosages or repeated administration to stimulate a therapeutic effect, which can lower overall efficacy and patient compliance, and result in severe side effects and even toxicity1–3. For example, intravenously administered Interleukin-12 (IL-12) resulted in systematic toxicities, including deaths in a clinical trial4. Oral administration, which is the most common approach for delivering pharmaceuticals, is frequently limited by poor targeting and short circulation times (<12 hours)5. Peptide and protein drugs often have short serum half-lives of only minutes to hours6. To address these issues, controlled drug delivery systems, including membranes, nanoparticles, liposomes and hydrogels have been focused on in recent decades7,8. These drug delivery systems can control how the drugs are available to cells and tissues over time and in space. They can, in principle, leverage beneficial outcomes of therapeutics by enhancing their efficacy, and reducing their toxicity and required dosage. The clinical use of drug delivery systems is appreciable7, with a global market of over $150 US billion in 2013.
Hydrogels are a particularly appealing type of drug delivery system, and have been used in many branches of medicine, including cardiology, oncology, immunology, wound healing, and pain management. Hydrogels are composed of a large amount of water and a cross-linked polymer network. The high water content (typically 70–99%) provides physical similarity to tissues, and can give the hydrogels excellent biocompatibility and the capability to easily encapsulate hydrophilic drugs. Moreover, because they are typically formed in aqueous solutions, the risk of drug denaturation and aggregation upon exposure to organic solvents is minimized. The cross-linked polymer network makes hydrogels solid-like, and they can possess a wide range of mechanical properties. For example, their stiffness can be tuneable9 from 0.5 kPa to 5 MPa, allowing their physical properties to be matched with different soft tissues in the human body10–12. The cross-linked network can impede penetration of various proteins13, and thus is believed to protect bioactive therapeutics from premature degradation by inwardly diffusing enzymes. This feature is particularly critical for highly labile macromolecular drugs (for example, recombinant proteins and monoclonal antibodies), which comprise an increasing percentage of new drugs approved, with many others under development14. Since the introduction of human insulin, more than 130 protein therapeutics have been approved by the Food and Drug Administration (FDA)15. The attributes mentioned above make hydrogels attractive material systems for the delivery of a large range of therapeutics.
Hydrogels differ in size, architecture and function, and together these features dictate how they are utilized for drug delivery. In hydrogels, there are features with length scales spanning from centimetres to sub-nanometres (FIG. 1). The macroscopic design largely determines the routes by which hydrogels can be delivered into the human body (FIG. 1a). Hydrogels can be formed into almost any overall size and shape. Micropores, if present, will dramatically affect the overall physical properties (for example, the deformability), while allowing for convective drug transport. On the several-nanometre scale, a cross-linked polymeric network surrounds the water contained in the hydrogel network. Such networks contain open spaces, the size of which is referred to as the mesh size of the network. Importantly, the mesh size governs how drugs diffuse inside the hydrogel network (FIG. 1b). Finally, at the molecular and atomistic scale, various chemical interactions may occur between the drugs and the polymer chains (FIG. 1c). The polymer chains can possess numerous sites for binding interactions with the drugs, and these can be pre-designed using a diversity of physical and chemical strategies. The features at the mesh scale and the molecular and atomistic scale are essential for controlled drug release. Because they are decoupled from the macroscopic properties of the hydrogel, desirable features at each length scale can often be designed independently of the other. This multiscale nature enables the modular design of hydrogels, which can serve as a versatile platform to meet specific application-based requirements.
Figure 1. Multiscale properties of hydrogels.
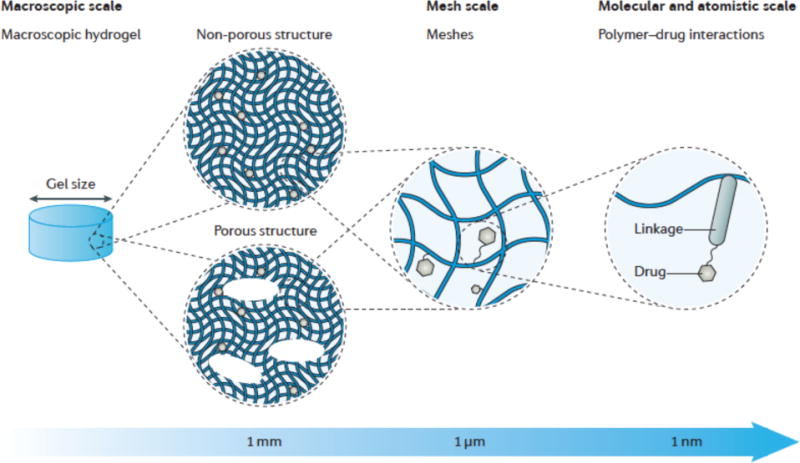
a| The macroscopic design of hydrogels includes the size and porous structure. Hydrogels can be either non-porous or contain macroscopic pores of 10–500 μm. b| The spacing between polymer molecules in the network (that is, the mesh size) is tuneable from around 5 to around 100 nm. c| At the molecular (or atomistic) scale, drugs can interact with the polymer chains via a range of mechanisms; shown here is a covalent linkage to a polymer chain.
Although some design requirements are common to all hydrogel delivery systems, others are specific to the desired therapeutic application. In general, the fabrication of hydrogel delivery systems needs to maintain the drug bioactivity, and through packaging, transport and storage, both the drug and hydrogel must be chemically and physically stable. Hydrogels can be delivered in a variety of manners, such as surgical implantation, local needle injection or systemic delivery via intravenous infusion. The choice of delivery method for a given application is based on maximizing the overall efficacy and patient compliance. How the hydrogel releases the drug is often essential to achieve desirable therapeutic outcomes, and the required duration of drug availability (short term versus long term) and its release profile (continuous versus pulsatile) depend on the specific application. When the drug is exhausted, the hydrogel should be designed to either degrade to avoid surgical removal, or to be re-used by drug refilling. The degradation of the hydrogel may also need to be tailored to coordinate with tissue regeneration. Besides the general requirements, there exist other application-based requirements. For example, in the treatment of skin wounds, hydrogels are placed on dynamic surfaces to which they need to be adhesive and conform, while being tough enough to tolerate the surface movement (for example, strain of knee bending up to 50%) and deformation derived from the environment (for example, compression and scratching).16–18
The multiscale properties of hydrogels are essential for their functions to protect, target and locally deliver drugs in a controllable manner. This Review will first illustrate the multiscale structural properties of hydrogels, and how they affect encapsulation, delivery and release of therapeutic agents. The discussion will start from the macroscopic scale, in which the key design parameters include architectural factors (such as hydrogel size and porous structure). The Review will proceed to the mesh scale, in which drug diffusion is regulated by the mesh size and its temporal or stimuli-responsive evolution. The Review will then focus on the molecular and atomistic scale. Any affinity or binding between the drugs and the polymer chains will have a crucial role in the sustained or on-demand release of the drugs. Various mechanisms to bind drugs with the polymer network, ranging from covalent conjugation to secondary bonds such as electrostatic interactions and hydrophobic associations, will be discussed. After a detailed discussion of the three length scales, this Review will provide quantitative comparisons of the release kinetics between systems utilizing one or a hybridization of different mechanisms. The use of hydrogels for the delivery of cells that secrete therapeutic agents will also be discussed. The goal of this Review is to link a fundamental understanding of hydrogels and their interactions with drugs to the rational and practical design of hydrogel drug delivery systems.
Macroscopic design and delivery routes
The size of a hydrogel matters. Hydrogels can be cast or formed into practically any shape and size, according to the requirements of the delivery route into the human body. Hydrogel delivery systems can be classified into three main categories based on their size: macroscopic hydrogels, microgels and nanogels (FIG. 2). Microgels and nanogels are particulate hydrogels with dimensions on the order of micrometres and nanometres, respectively.
Figure 2. Macroscopic design determines the delivery route.
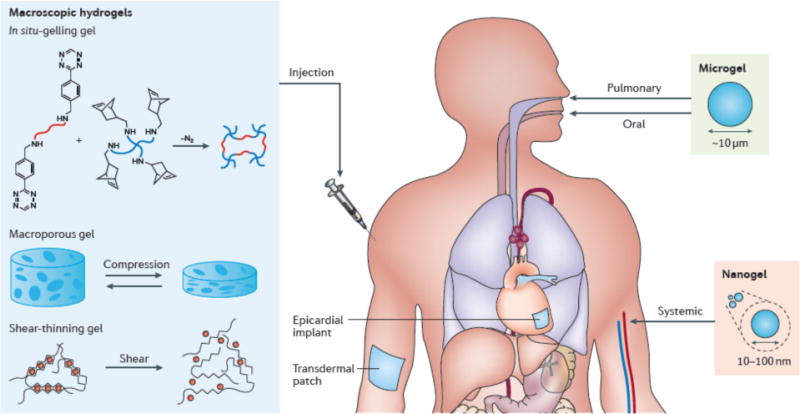
a| Macroscopic hydrogels are used for transepithelial delivery and placement inside the body. Injectable macroscopic hydrogels that can be delivered via syringe-needle injection include (b) in situ-gelling hydrogels such as a hydrogel formed with tetrazine-norbornene chemistry, (c) shear-thinning hydrogels such as alginate hydrogels cross-linked with multivalent ions and (d) macroporous hydrogels that can undergo reversible dramatic volumetric change. In addition to transepithelial and local injection, microgels (e) are suitable for oral, pulmonary and intrabony delivery and nanogels (f) are suitable for systemic administration of drugs.
Macroscopic hydrogels
The size of macroscopic hydrogels is typically on the order of millimetres to centimetres. Correspondingly, they are usually either implanted surgically into the body or are placed in contact with the body for transepithelial drug delivery (FIG. 2a)7. Success has been achieved with surgically implanted hydrogels for drug delivery in the clinic, as exemplified by INFUSE, a type I collagen gel that releases recombinant human bone morphogenetic protein-2 (BMP-2), which is implanted surgically into the body for the treatment of long bone fracture and spinal fusion19.
Epithelial barriers that have been exploited for drug delivery include the skin, intestinal epithelium and mucosa. Although these are impenetrable to macroscopic hydrogels, they can be permeable to drugs released from the hydrogels. Hydrogels, including those fabricated from synthetic polymers such as poly(vinyl alcohol) and poly(hydroxyl alkyl methacrylate)20, and biopolymers such as alginate21, collagen22 and chitosan23, are widely used as wound dressings. Adaptation of these materials for transdermal drug delivery is highly appealing, and these hydrogels have been used to deliver proteins such as insulin and calcitonin7. Alginate hydrogels, similar to those in use for decades to treat wounds24, have been shown to controllably deliver potential therapeutics like substance P to promote wound healing25. Some of these materials are currently being evaluated in clinical trials. Indeed, a recent clinical trial showed the efficacy and safety of a hydrogel delivering recombinant human granulocyte-macrophage colony-stimulating factor (rhGMC-SF) for the healing of deep, second-degree burn wounds26.
Placement of the hydrogel in the body may be necessary when the target site for drug delivery is located deep within the tissue or when the biological barriers have low permeability to the drug of interests. Placement of hydrogels within the body directly bypasses the epithelial barriers and concentrates drug release at the target site27,28. However, surgical implantation is invasive and potentially leads to patient discomfort and the risks associated with surgery29. To address this issue, there has been great emphasis placed on the development of injectable macroscopic hydrogels. These are largely designed on the principles of gelation in the body (in situ-gelling hydrogels), gelation outside of the body but with a transition to a flowable state upon application of sufficient shear stress (shear-thinning hydrogels) to allow injection, or gelation outside of the body to a physical form that can be readily collapsed for minimally invasive delivery followed by shape recovery in vivo (shape-memory hydrogels).
In situ-gelling hydrogels
These systems can be injected in liquid form, followed by a sol-gel transition inside the human body. The resulting hydrogels will take the shape of the available space at the injection site and the sol–gel transition can be achieved with different strategies. The simplest strategy is to use slow-gelling systems that allow gelation to be initiated outside the body. Because this process occurs so slowly, the solution can be injected before solidification occurs. Ideal kinetics are slow enough to prevent needle clotting, but fast enough to prevent dilution of the pre-gel solution by body fluids once in the body. This strategy has been applied with numerous gelation mechanisms, including charge interaction30,31, stereocomplexation32 and Michael addition33. As an example of a system exploiting charge interactions, elastin-like polypeptides have been cross-linked via electrostatic interactions between their cationic lysine residues and anionic organophosphorus cross-linkers34. Non-covalent interactions between heparin and heparin-binding peptides and proteins can be used to form hydrogels for growth factor delivery35,36. Another in situ-gelling hydrogel was formed with a polyelectrolyte complex, which showed a sustained release of proteins (for example, insulin and Avidin) over two weeks37. Certain pre-gel solutions may require monomers or catalysts that are harmful to cells and tissues, which provides a strong impetus to develop bio-orthogonal cross-linking reactions such as copper-free click reactions for gelation (FIG. 2b)38–40. Click chemistry provides high specificity, quantitative conversion and modest gelation kinetics that are tuneable from a few minutes to one hour, with no side reactions with biomolecules in the body.
Temperature-responsive systems that gel at body temperature have also been explored for in situ gelation. Most natural polymers like gelatin form a gel upon lowering of temperature, which would require their introduction to the body at supraphysiologic temperatures. By contrast, certain synthetic polymers such as poly(N-isopropylacrylamide) (PNIPAm) and poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO) undergo reverse thermogelation (that is, gelation at elevated temperature)41,42,43 and remain in a flowable state at room temperature, thus providing a significant practical advantage. Temperature-responsive polymers often contain hydrophobic domains that allow for the inclusion of hydrophobic drugs (for example, doxorubicin)43. However, challenges exist for these systems, such as their relatively low mechanical strength, poor physical stability, and synthetic polymers such as PNIPAm may be non-biodegradable44. The stability of temperature-responsive systems may be improved by adding additional covalent cross-links after initial gelation45,46.
Shear-thinning hydrogels
Certain hydrogels can be pre-gelled outside of the body, and then injected by application of shear stress. These shear-thinning hydrogels flow like low-viscosity fluids under shear stress during injection, but quickly recover their initial stiffness after removal of shear stress in the body. The shear-thinning behaviour is a result of reversible physical cross-links (FIG. 2c). In contrast to covalent bonding, physical cross-links are reversible, resulting from a dynamic competition between pro-assembly forces (for example, hydrophobic interactions, electrostatic interactions and hydrogen bonding) and anti-assembly forces (for example, solvation and electrostatic repulsion)47. Self-assembling peptides have been extensively explored to make shear-thinning hydrogels owing to the diversity of amino acids and the ease of sequence-specific peptide modification48–50. A family of β-hairpin peptides (namely, MAX peptides) has been developed to make injectable hydrogels for drug delivery51,52. These peptides contain two blocks of alternating hydrophobic and charged amino acids in the form of long fibrils (up to 200 nm) that can undergo a sol–gel transition. Peptide self-assembly can also be realized by taking advantage of interactions between metal cations and amino acid residues of the peptides. This was demonstrated with gelation of a β-sheet-rich fibrillar hydrogel with zinc ions53. Alginate hydrogels are also shear-thinning and have been studied extensively. These hydrogels are formed via electrostatic interactions between alginate and multivalent cations (for example, calcium and zinc) (FIG. 2c); they can be readily injected via a needle after gelation in a syringe and have been used to achieve sustained local delivery of bioactive vascular endothelial growth factor (VEGF) in ischemic murine hindlimbs for 15 days, in contrast to complete VEGF deprivation after 72 hours with bolus injection30,31.
Another approach to make shear-thinning hydrogels is to use dynamic covalent bonds54,55. Such bonds are reversible and the hydrogels can behave similarly to self-assembling peptides. The most widely used reactions are the dynamic exchange of C=N bonds in imines, hydrazones and oximes. Other dynamic covalent reactions include reversible Diels–Alder reactions (for example, furan and maleimide), boronic acid condensations and disulfide exchange56. For example, complexation of boronic acids and diol compounds was used to form a shear-thinning hydrogel57. Although the variety of dynamic covalent reactions is rapidly expanding, many dynamic covalent reactions suffer from harsh reaction conditions (for example, elevated temperature), and it would be of great interest to develop dynamic reactions and catalysts that are biocompatible and suitable for aqueous solutions and biomedical applications.
Macroporous hydrogels
An alternative strategy for making injectable hydrogels is to preform large hydrogels with interconnected pores that can mechanically collapse and recover reversibly (FIG. 2d). Such hydrogels can be fabricated with cryogelation58, gas foaming59, microemulsion formation60, freeze drying61, and porogen leaching62. When the hydrogel is delivered via injection with a needle and syringe, water is squeezed out from the pores, which causes the hydrogel to collapse, allowing it to pass through the needle. Once the hydrogel has left the needle and the mechanical constraint imposed by the needle walls is removed, the hydrogel can recover its original shape almost immediately in the body. These hydrogels behave like a foam and can be reversibly compressed at up to 90% strain without any permanent damage to the network63. Compared with slow-gelling and shear-thinning systems, macroporous systems allow researchers to create pre-defined geometries and highly defined volumes for drug delivery vehicles. The macropores can allow cell infiltration and incorporation of payloads after gelation. The potential for this approach has been demonstrated with alginate and gelatin cryogels that were injected subcutaneously into mice to deliver immunomodulatory factors in a controlled manner64, which led to regression of established tumors. Despite having many advantages, interconnected pores may significantly reduce the diffusion length for drug release and the volume fraction of polymer, potentially leading to release that is too rapid and limited drug loading capacity.
Microgels and Nanogels
An alternative solution for minimally invasive delivery of hydrogels is to use small hydrogel particles. Nanogels and microgels have some advantages over their macroscopic analogs. First, their size is much smaller than the inner diameter of most needles (~1 mm). The small size not only makes them needle-injectable, but also leads to a large surface area for bioconjugation, facile natural clearance and can enhance penetration through tissue barriers.
The size of hydrogels determines how they transport and adhere if introduced into blood vessels, airways or the gastro-intestinal tract. In addition to transepithelial and local injection (such as intraperitoneal and intrabony injection), microgels and nanogels also enable other routes for drug delivery. Microgels smaller than 5 μm are used in oral or pulmonary delivery, but are generally not considered suitable for intravascular injection owing to their rapid circulation clearance (FIG. 2e). Nanogels of size 10–100 nm are suitable for systemic drug administration, because they can leave small blood vessels through fenestrations in the endothelial lining, allowing for extravasation into tissues (FIG. 2f). Hydrogels below 10 nm in diameter can be cleared by kidney filtration, while those of 0.5–10 μm can be phagocytized by macrophages65. Along with the size and size distribution, deformability, shape and surface chemistry are other factors to consider in designing drug delivery systems66,67. It has been revealed that cellular internalization is faster for nanogels of positive zeta potential or with high aspect ratios (for example, those with a rod-like shape)68, and that size and deformability determine the biodistribution and circulation persistence of microgels in mice69.
Nanogels are particularly appealing for delivery of nucleotide-based drugs such as plasmid DNA, which is used for gene therapy. Gene therapy holds promise for the treatment of cancers, hemophilia and viral infections70. DNA delivery using nanogels can improve cellular uptake and prolong circulation time, as compared to non-encapsulated DNA. They are particularly useful to target drugs to tumours, as the leaky tumour vasculature enhances nanoparticle accumulation, while ineffective lymphatic drainage limits nanoparticle clearance (that is, the enhanced permeability and retention effect)71. Cationic nanogels consisting of PEO and poly(ethylenimine) were found to increase the transport of oligonucleotides across the gastrointestinal epithelium and even the blood–brain barrier72. Nanogels consisting of polymer–protein conjugates were shown to prolong the plasma half-life and enhance protein stability73. DNA nanogels can integrate multiple modular elements, including oligonucleotides for inhibiting cell proliferation, DNAzymes for inhibiting cell migration and aptamers for targeting specific cancer cells74.
Many fabrication techniques have been developed to produce hydrogel particles of different sizes. The dimensions can be controlled through the gelation conditions, such as the polymer and surfactant concentrations, or the fabrication parameters, such as the nozzle size and flow rate (the reader is referred to a previous review for further details75). Microgels can be produced with microfluidics and micromolding methods, and nanogels can be produced with emulsion and nanomolding techniques75. As an example, a molding technique consisting of particle replication in nonwetting templates (PRINT) produced monodisperse hydrogel particles76–78 ranging from below 200 nm to 10 μm.
Bioadhesion and toughness
In addition to overall size, the bioadhesive properties of hydrogels are important factors in the selection of their delivery routes. Biological barriers such as intestinal epithelium and mucosa are often wet, dynamic and slippery, limiting the ability of many hydrogels to adhere. A hydrogel that can adhere well to the epithelium can prolong the retention of the system at a target site, and thus provide sufficient drug dose for the desired therapeutic effect; this is particularly important for nasal and oral delivery79. For example, nasal mucosa was found to limit the residence time of nanogels, which caused dose loss into other parts of the respiratory tract80.
Extensive efforts have been made to develop bioadhesive hydrogels to enable improved drug delivery79,81. Some polymers such as chitosan and poly(acrylic acid) are found to be mucoadhesive82–84. Poly(acrylic acid) is capable of forming hydrogen bonds with the mucosa, whereas positively charged chitosan can form electrostatic interactions with negatively charged surfaces of tissues and cells84. Hydrogels consisting of these mucoadhesive polymers were shown to have prolonged retention for oral, nasal and vaginal drug delivery85–88. Another strategy is inspired by marine mussels, incorporating a catechol moiety (3,4-dihydroxy-L-phenylalanine, DOPA) in hydrogels to promote bioadhesion. Its high efficacy is exemplified with a poly(ethylene glycol) hydrogel (PEG) containing a DOPA moiety that was shown to adhere on epididymal fat pad and external liver surfaces for up to one year89,90,91.
The toughness of a hydrogel is key to its ability to maintain its structure and avoid fracture during use and after tissue adhesion. Especially when using hydrogels as immunoisolating membranes, one needs to ensure the hydrogel matrix is tough enough to resist rupture and to prevent cell escape and the associated risks92. To increase the resistance to rupture (that is, the toughness), one can modulate the cross-link density93 or use interpenetrating networks to form hydrogels.94 For example, an alginate–polyacrylamide interpenetrating network hydrogel achieved extremely high toughness95,96, and mechanically resembled soft tissues such as cartilage and tendon11.
Mesh size controls diffusion and release
Hydrogels consist of a cross-linked polymer network, and open spaces (that is, meshes) between polymer chains; the meshes allow for liquid and small solute diffusion. Typical mesh sizes97 reported for hydrogels range from 5 to 100 nm, and a number of approaches exist to determine the mesh size (BOX 1). To put these mesh sizes into context, the widely used model proteins lysozyme and immunoglobulin G (IgG) have hydrodynamic diameters of 4.1 and 10.7 nm, respectively98. The mesh size depends on polymer and cross-linker concentrations, as well as external stimuli such as temperature and pH. Because of network heterogeneity and polymer polydispersity, most hydrogels have a wide distribution of mesh sizes. This is particularly common when the hydrogels possess non-ideal network structures (for example, dangling chains, and closed loops) resulted from certain gelation mechanisms like free radical polymerization99. Notably, a network with homogeneous mesh size can be obtained with gelation of symmetrical tetrahedron-like macromeres of the same size100. The cross-linking principles and basic material characteristics of hydrogel materials are reviewed elsewhere101,102.
Box 1. Experimental characterization of hydrogel mesh sizes.
The mesh size of a hydrogel can be determined with various mechanical tests and characterization techniques. First, theoretical and experimental methods have been developed to calculate mesh size with mechanical tests. The classical theory of rubber elasticity relates the shear modulus G to the mesh size rmesh by227,228:
where R is the gas constant, T is the absolute temperature, and Nav is Avogadro’s number. Experimentally, this relation allows one to estimate the mesh size based on the measurement of shear modulus of a hydrogel229. Relevant mechanical tests include rheology228, swelling tests230, indentation231, compression and tensile tests232. From the data from these tests, additional structural properties can also be extracted229. For instance, along with the mesh size, force-relaxation indentation can be used to determine diffusivity and permeability of water and solute in a hydrogel, as the relaxation rate is closely related to liquid migration inside the network231. The mechanical tests are widely used due to ease of application, but they provide indirect characterization of the hydrogel network.
To directly characterize the mesh size, there exist techniques such as confocal microscopy233, electron microscopy72, atomic force microscopy234, small-angle X-ray scattering (SAXS)235, and small-angle neutron scattering (SANS)236. Electron microscopy enables extremely high spatial resolution, but specimen preparation may alter the morphology of hydrogels. The application of confocal microscopy is typically limited by the spatial resolution (>1 μm), but by combining this technique with measurements of the transport of molecular probes (for example, fluorescently labeled proteins, fluorescent beads) across the hydrogels, one can determine the mesh size of hydrogels237·238.
The mesh size determines how drugs diffuse through a hydrogel, as it will control steric interactions between the drugs and the polymer network. When the mesh is larger than the drug (rmesh/rdrug >1) (FIG. 3a), the drug release process is dominated by diffusion. Small drug molecules migrate freely through the network, and diffusion is largely independent of the mesh size. The diffusivity, D, in this situation depends on the radius of the drug molecule (rdrug) and the viscosity of the solution (η) via the Stokes–Einstein equation103:
where R is the gas constant and T is the absolute temperature. The radius of the drug molecule typically scales with its molecular weight. Macromolecular drugs have large radii and thus slower diffusion. For proteins, the measured diffusivity103 is typically between 10−11 and 10−9 m2 s−1.
Figure 3. Mesh size mediates drug diffusion.
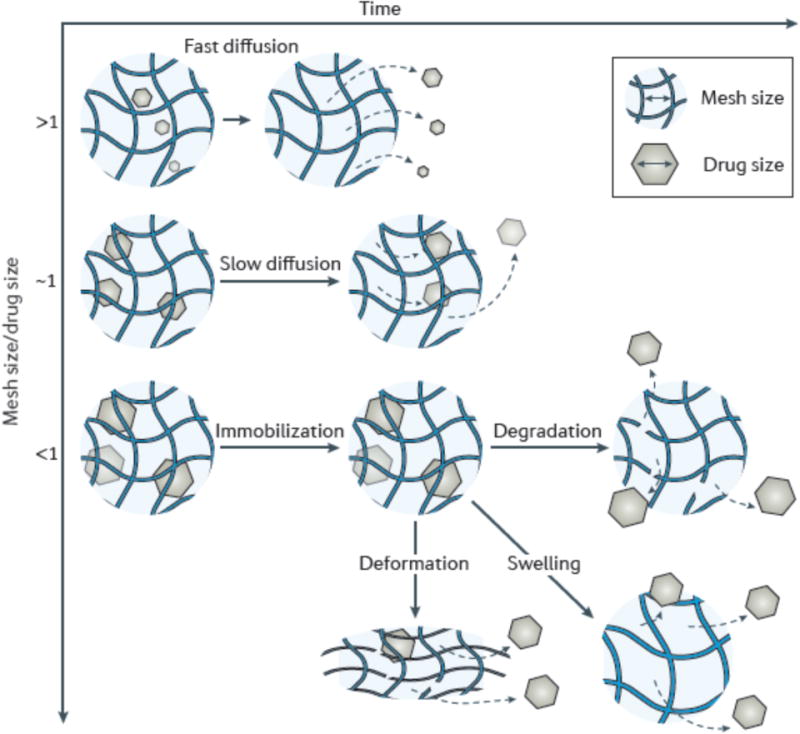
a| A small drug relative to the mesh size diffuses rapidly through the hydrogel, resulting in a short release duration. b| When the size of drug approaches the mesh size (rmesh/rdrug ≈ 1), drug release is dramatically slowed. c| When the drug is larger than the mesh size (rmesh/rdrug <1), drugs are physically entrapped inside the network. To release the originally immobilized drugs, the mesh size can be enlarged through network degradation (d), swelling (e), or via applying deformation to disrupt the network (f). The gray dashed lines refer to the diffusion pathway of drugs.
The time required for a drug to diffuse through the hydrogel (diffusion time, tdiff) also depends on the diffusion length (for example, the hydrogel thickness, H), and can be estimated104 by H2/D. For a hydrogel of thickness H = 1 mm, the diffusion time of a protein out of the hydrogel is typically around 0.3–30 hours. This approximation is consistent with many experimental observations; for example, the release of bovine serum albumin (BSA) from alginate/chitosan beads of 3 mm diameter was completed within 6 hours105. In general, diffusion-dominated drug release cannot achieve extended release beyond hours to a day.
When the mesh size approaches the drug size (rmesh/rdrug ≈ 1), the effect of steric hindrance on drug diffusion becomes prominent. One can reduce the mesh size of a hydrogel by increasing the concentrations of the polymer or the cross-linker. In this situation, the polymer chains induce significant frictional drag on diffusing drugs, and because the meshes in some parts of the network are smaller than the drugs, the path length for drug transport increases106. This complex scenario calls for sophisticated theoretical approaches to estimate the diffusivity106. The overall effect is slow drug diffusion, which allows for slow and extended release (FIG. 3b). The potential utility of this situation is exemplified with a triblock copolymer hydrogel, in which the release of proteins (BSA or IgG) was prolonged from 6 to 14 days as the polymer concentration was increased from 20–35%45.
The upper bound of the mesh distribution defines the molecular weight cut-off for drugs that can diffuse inside the hydrogel. For an extremely small mesh size and/or a very large drug molecules (that is, for rmesh/rdrug <1), strong steric hindrance effectively immobilizes the drugs (FIG. 3c). Drugs remain physically entrapped inside the network, unless the network degrades or the mesh size is otherwise enlarged. A number of strategies have been developed to change the mesh size over time or in response to external stimuli, as reviewed in the following sections, to regulate the kinetics of drug release from these types of systems.
Controlled release through network degradation
One strategy to control the release of drug molecules initially entrapped in a hydrogel is to regulate network degradation. The mesh size increases as the network degrades (FIG. 3d), allowing drugs to diffuse out of the hydrogel. Degradation can occur in the polymer backbone or at the cross-links, and is typically mediated by hydrolysis107–109 or enzyme activity110–112. Ester bonds that can hydrolyse slowly have been used to form a family of biodegradable PEG hydrogels that can achieve extremely slow protein release with half-lives up to 17 days109. Oligo-peptide bonds are cleavable with matrix metalloproteinases (MMPs)111. One example of the use of this is the incorporation of an MMP-cleavable peptide (GGRMSMPV) into a hyaluronic acid hydrogel that was used to deliver a recombinant tissue inhibitor of MMPs in a porcine model of myocardial infarction113. In addition to MMP-responsive hydrogels, there exist other cleavage systems that respond to biomolecules present in the body, such as glucose-responsive hydrogels for insulin delivery114,115 and thrombin-responsive hydrogels to regulate blood coagulation116,117. Degradation can also be triggered in real time with externally provided cues. For example, acidic conditions typically accelerate hydrolysis118. High-energy ultra-violet (UV) light can trigger degradation of microgels containing o-nitrobenzyl ether moieties (NBE) due to cleavage of the NBE, accompanied by release of encapsulated transforming growth factor beta 1 (TGF-β1)119. Even low-energy near-infrared (NIR) light can trigger degradation of a hydrogel by using upconversion nanoparticles to convert two or more NIR photons into a UV photon.120
The loss of polymer mass through hydrogel degradation, also known as erosion, can proceed simultaneously in the bulk or preferentially on the surface of the hydrogel; bulk and surface erosion can be used to differentially control drug release. Many hydrogels undergo bulk erosion, as the network is permeable to water or enzymes that mediate degradation; if the rate of diffusion of these agents is rapid compared to the rate of bond degradation, degradation will occur simultaneously throughout the bulk of the gel121. Hydrogels consisting of oxidized polysaccharides (for example, alginate and chitosan that are oxidized with sodium periodate) typically undergo bulk erosion, and the degradation rate can be mediated by the degree of oxidation108,122. Hydrophobic polyesters such as poly(caprolactone) (PCL) and poly(lactide) (PLA) that degrade via hydrolysis are often copolymerized with hydrophilic polyethylene glycol (PEG), leading to bulk eroding hydrogels123. These copolymers are often used in high polymer contents (20–30 wt.%), which allows degradation-controlled release; for example, a triblock copolymer of PCL–PEG–PCL led to the extended release of BSA for two weeks124.
Surface erosion, in contrast, results when the rate of bond breakage is more rapid than the rate of enzyme or water diffusion from the exterior into the bulk of the gel121. Cross-linking hydrogels using hydrophobic associations (for example, between β-cyclodextrin and cholesterol) can inhibit water entry, leading to a largely surface eroding hydrogel. Mass loss can be fairly linear with respect to time for surface-eroding gels, leading to near zero-order release of encapsulated drugs125. For a variety of hydrogels, one can tune the degradation reaction and erosion mechanism to obtain desirable release kinetics ranging from weeks to months, thereby allowing for long-term release. However, one must consider that degradation products should be nontoxic and small enough for natural clearance.
Controlled release through swelling
A second strategy to release entrapped drugs is the controlled swelling of hydrogels. As a hydrogel swells, the mesh size increases (FIG. 3e). The extent of swelling of a hydrogel is a balance between forces that constrain network deformation and the osmosis that leads to water absorption126,127. The swelling behaviour can be sensitive to various external conditions, including temperature128, glucose129,130, pH131, ionic strength132, light120, and electric fields133. These cues have been widely exploited in drug delivery.
pH-responsive swelling is particularly important for oral and cancer delivery systems. In this application, the swelling of the hydrogels in the acidic stomach is typically minimal, and thus the drug is protected and entrapped physically. As the hydrogels pass down the intestinal tract where the pH is neutral, the network can be designed to swell dramatically, allowing for rapid drug diffusion. Such hydrogels have been developed using a variety of polymers with acidic or basic groups; among them, alginate is one of the more commonly used. In acidic conditions, the alginate hydrogel is so condensed that it holds the drugs tightly; as the pH becomes neutral, the carboxylic acid groups on the alginate deprotonate and generate large osmosis, leading to network swelling and drug release134. The pH-triggered release can also target drug release in solid tumours where the extra- and intra-cellular environments are more acidic than in normal tissues135. Other stimuli-responsive swelling mechanisms have also been exploited for on-demand drug release. For example, a temperature responsive nanogel was used to deliver a chemotherapy drug cisplatin to breast cancer cells in which the temperature was slightly higher than normal body temperature136.
A limitation of swelling-controlled systems is that the response is relatively slow for macroscopic hydrogels owing to the slow diffusion of water. For hydrogels of 1 mm, changes in swelling and drug release would be expected to require tens of minutes. To achieve a faster response, one can reduce the diffusion length by reducing the hydrogel size, or by constructing interconnected macropores inside the hydrogel. As an alternative to altering the bulk structure of a hydrogel, superficial layers that can quickly swell to control drug diffusion have been explored137. For example, temperature-sensitive PNIPAm has been grafted as a thin layer to the surface of a hydrogel, in essence serving as a valve to control the diffusion of drug molecules out of the hydrogel.
Controlled release through mechanical deformation
A final approach to release entrapped drug molecules is to mechanically deform the network, as this can both increase the mesh size by changing the network structure and trigger convective flow within the network (FIG. 3f)138. This strategy can generate pulsatile release patterns with fine control over the magnitude of the instantaneous release rate138. Pulsatile release may mimic some natural patterns of biological signalling, for example, in the delivery of insulin following eating139. Deformation of the network can be achieved with various approaches, including purely mechanical deformation, or using ultrasound and magnetic field-induced deformations. Direct mechanical deformation has been demonstrated to upregulate the release of a growth factor to enhance tissue vascularization140. A magnetic field can deform a hydrogel network containing magnetic nanoparticles141,142, and the inclusion of macropores results in large and rapid deformation of the scaffold that dramatically enhances the release of drug molecules143. Ultrasound can transiently disrupt the hydrogel structure, and is potentially advantageous owing to its high spatiotemporal resolution and deep penetration within tissues144. The efficacy of ultrasound to provide pulsatile drug delivery has been demonstrated with a number of drugs, including insulin and interferon gamma145. In all of these approaches, a potential concern with mechanical deformation is cumulative damage of the hydrogels, which ultimately results in mechanical failure. This problem can potentially be addressed with self-healing hydrogels. For example, alginate hydrogels that are reversibly cross-linked with divalent cations can heal under physiological conditions following ultrasound disruption, enabling repeated, near digital release of small molecules, proteins and condensed oligonucleotides138.
Drug–polymer interactions
Another approach to control drug release from hydrogels is to design chemical bonds between the drug and the polymer chains. In this strategy, moderate-to-high-affinity interactions effectively slow down, and in some cases, terminate drug diffusion through the network. This strategy is particularly important when small drugs (that is, below the tuneable range of the mesh size) are to be delivered, because they would otherwise be released within a very short period (FIG. 3a). To achieve high-affinity interactions, there exist a variety of chemical and physical interactions from which one can choose, ranging from covalent conjugation to secondary interactions, such as electrostatics and hydrophobic associations.
Covalent conjugation
Covalent linkages between the drug and polymer immobilize the drug, and can be either highly stable or cleavable. Highly stable covalent linkages retain the drug until the network degrades. Cleavable covalent linkages, much like cleavable cross-links in degradable networks, can be programmed to cleave over time, or in response to environmental cues. The drugs are released at a rate dictated by the cleavage of linkers.
A variety of covalent linkages have been explored. Those of high stability over time include amide bonds formed through carbodiimide chemistry, thiol-ene bonds and other covalent bonds formed through metal-free click chemistry (FIG. 4a). Amide bonds have been used to conjugate TGF-β1 to a PEG hydrogel via an amine–hydroxysuccinimide reaction146. A long-chain linkage like a heterobifunctional PEG was proposed to reduce steric hindrance on the tethered drugs by the hydrogel matrix146,147. Covalent conjugation was applied to bind an engineered variant of VEGF, α2PI1–8-VEGF121 to a fibrin hydrogel, which was shown to protect the growth factor from rapid clearance, and induce local and controlled blood vessel growth148,149.
Figure 4. Chemical interactions mediate drug release.
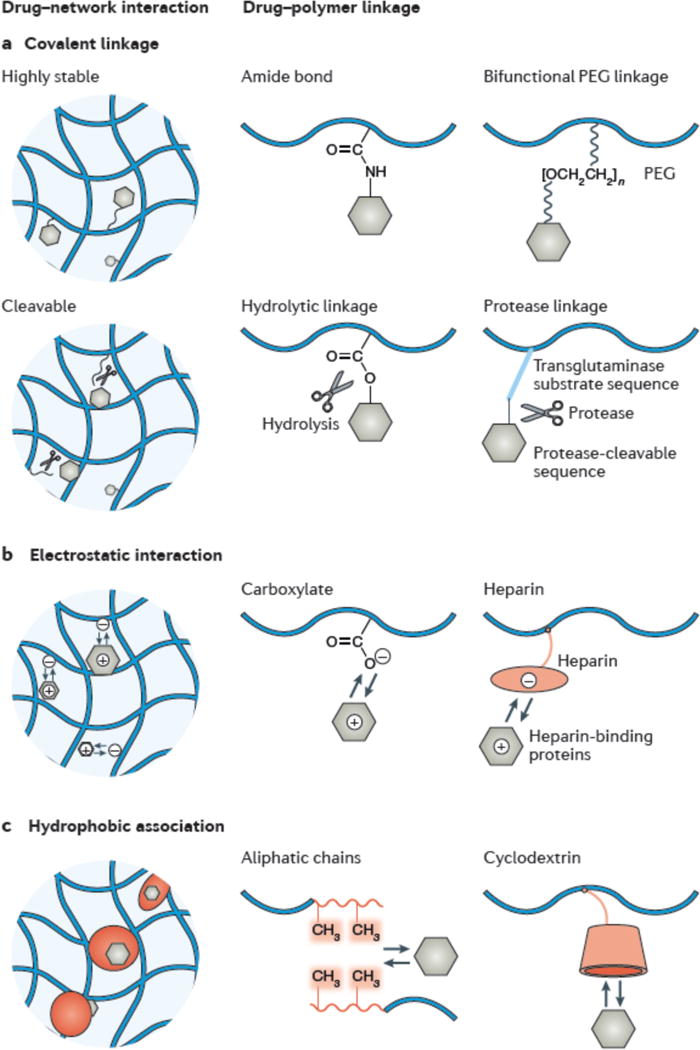
Representative chemical interactions between a drug and polymer chains. a| Highly stable covalent linkages immobilize the drugs inside the hydrogel. Strategies include the formation of amide bonds and the use of long-chain poly(ethylene glycol) (PEG) linkages. b| Cleavable covalent linkages release the drug as a result of hydrolysis or the activity of enzymes like proteases. c| Electrostatic interactions between a charged drug and the polymer chain can slow release. This can be exploited using polymers carrying charges, such as carboxylate groups and those mimicking heparin-binding groups. d| Hydrophobic drugs associate with hydrophobic domains such as aliphatic chains and cyclodextrin.
Cleavable covalent linkages range from small-molecule linkages including ester bonds, disulfide bonds, and β-elimination cleavable linkages to macromolecular linkages like peptide sequences (FIG. 4b). Peptide-based drugs flanked by enzymatically degradable peptides have also be used to form a hydrogel and release the drug upon degradation mediated by proteases like matrix metalloproteinases (MMPs)150. A family of covalent linkages that are cleavable via β-elimination reaction has been developed to achieve a wide range of degradation rates151,152. These linkages have been used to tether a 39-amino-acid peptide, exenatide, to a PEG hydrogel and provide extended release (about 7 days) in rodents6,152.
Electrostatic interactions
This mechanism has been widely exploited to form a strong affinity between drugs and the polymer chains. Because the charge-based interaction is nonspecific, it can enable controlled and simultaneously delivery of multiple drugs from a single system153,154. The drugs are released when the hydrogel is degraded or when the electrostatic interaction is screened by mobile ions from the environment. This strategy is applicable to many drugs and polymers that carry charges (FIG. 4c). Alginate hydrogels, which carry negative charges, have been used to deliver cationic, heparin-binding growth factors like VEGF to promote tissue regeneration155,156. If direct polymer–drug electrostatic interactions do not exist or are not sufficiently strong to control release, one can incorporate a third agent that will provide the desired interactions.157 Heparin, for example, has been incorporated into hydrogels to controllably deliver heparin-binding proteins such as VEGF and basic fibroblast growth factor (bFGF)158. Another example is sulfonate functional groups, which are used to increase electrostatic interactions between alginate and protein drugs to extend the release duration159. In recent approaches, the sulfation pattern of heparin has been tailored to further adjust the affinity of heparin to growth factors and thus control their release kinetics116,160.
Hydrophobic association
Hydrogels by definition possess a large amount of water, and their hydrophilic nature renders the encapsulation and release of hydrophobic drugs problematic. Phase separation between encapsulated hydrophobic drugs and the hydrogel may also deteriorate the stability and strength of the hydrogel. These issues have motivated the development of hydrogels that contain hydrophobic polymers or components to serve as binding sites for hydrophobic drugs161.
Typical approaches to provide hydrophobic domains in hydrogels include copolymerizing hydrophobic monomers and the incorporation of hydrophobic molecules (for example, cyclodextrin and cholesterol) (FIG. 4d). Hydrophobic aliphatic chains were incorporated into peptides that self-assembled to form a hydrogel for the delivery of hydrophobic anticancer drugs162,163. Thiocholesterol was incorporated into polyvinyl alcohol hydrogels for delivery of hydrophobic drugs164. However, the incorporation of hydrophobic domains may reduce the water content of the hydrogels significantly, and potentially alter their biochemical and physical properties. To this end, inclusion of cyclodextrins into hydrogels is advantageous because it does not change the overall hydrophilicity of the hydrogels. These macrocyclic oligosaccharides comprise both an external hydrophilic character and internal hydrophobic pockets to which hydrophobic drugs can associate. In such systems, the drug release is primarily controlled by the relative partitioning of solubilized drugs between the hydrogel and the release medium, and is independent of the mesh size of the hydrogel165.
It is important to recognize that hydrogels often provide numerous sites for constructing interactions with drugs, which also potentially allows one to hybridize multiple interactions into a single hydrogel166. Manipulating drug release via chemical interactions often will provide one more degree of freedom in design, but this potential advantage must be weighed against potential concerns related to the impact of chemical modifications on the drug or hydrogel biocompatibility. Chemical modifications of a drug near an active site or a region of a protein that cause a conformation change can reduce drug activity167,168. For example, some enzymes lose bioactivity when PEGylated via the ε-amino group of lysine resides169.
Designing across length scales
Each of the strategies discussed above has pros and cons, and combining mechanisms across different length scales may allow one to optimally orchestrate the drug presentation. Combining drug–hydrogel interactions with tailored mesh size is one attractive approach. For example, one can use tethered drugs as cross-links in a hydrogel, with the network degradation itself facilitating the drug release. This approach can also minimize the use of excipients like cross-linkers and thus increase the drug loading170. A hetero-bifunctional PEG chain was tethered via the primary amines on BSA, forming a macromer cross-linked PEG hydrogel; by tuning the hydrolysis rate of ester bonds in the network via adjacent charged amino acids, the BSA was released over weeks171.
Composite hydrogels, in which particles are included within the hydrogel network, can also provide multiple mechanisms for controlled drug release. The particles are often used to tether drugs, with a particle size sufficiently large to allow entrapment in hydrogels. Particles used to date include microgels, clay, gold nanoparticles, polymeric particles, liposomes, and micro- and nanocapsules. The drugs are released either by dissociation from the entrapped particles followed by diffusion through the hydrogel, or by release of the particles from the hydrogel. One type of widely used polymeric particle is poly(lactic-co-glycolic acid) (PLGA), which releases the encapsulated drug via hydrolysis. Drug-laden PLGA particles have been incorporated into various hydrogels (for example, alginate and PEO–PPO–PEO) for sustained protein release over several weeks172,173. Clay particles are particularly appealing for binding with drugs, because they typically possess distinct types of drug-binding sites; for example, Laponite, a synthetic smectite, consists of disk-shaped particles of around 25-nm diameter and 1-nm thickness, and possesses a permanent negative surface charge and a pH-dependent edge charge (which is positive if pH < 9)174. In addition, as the clay particles form aggregates in physiological solutions, there exist intra-particle spaces that can retain hydrophobic drugs via van der Waals’ interactions175. Slow release of charged proteins and hydrophobic molecules with incorporation of clay particles into hydrogels has been demonstrated.176 Despite the potential utility of clay nanoparticles, their biodegradation and biological impacts remain largely unexplored.
Interactions between entrapped particles and polymer chains, in addition to allowing control over drug release, can improve the stability of hydrogels by increasing the cross-link density.177,178 This has been shown in the design of hybrid nanogels for the delivery of small interfering RNA; the nanogels contain a rigid silica core to stabilize the pH-responsive hydrogel matrix, which would otherwise be fragile when undergoing a pH transition.179
Release kinetics of hydrogel systems
Since the first description of hydrogel drug delivery systems in the 1950s (REF. 180), a variety of material systems and design principles have been developed to provide different drug release kinetics, such as extended or pulsatile release. Because drug release from hydrogels can involve various chemical, physical and biological interactions, this complicates the theoretical prediction of the drug release profiles. Many systems were originally developed in a trial-and-error manner. However, as substantial experimental data has been collected, a more fundamental understanding of drug release mechanisms has emerged.
Experimental data on the release kinetics is typically presented as the amount of drug released as a function of time (inset of FIG. 5). The general release profiles can be described with an empirical Ritger–Peppas equation181,182:
where Mt is the mass of drug released at time t, M∞ is the total mass of released drug, k is a kinetic constant and n is the diffusional exponent. In this equation, the mass fraction of drug released with time follows a power-law relationship. The power n depends on the type of transport, hydrogel geometry and polymer polydispersity: n = 0.5 if the drug is released by Fickian diffusion; n = 1 when surface erosion dominates the release. In many systems, the value of n is between 0.5 and 1, because more than one mechanism controls the release in a given system. More details about this model have been reviewed elsewhere97,121.
Figure 5. Drug release property chart of hydrogels.
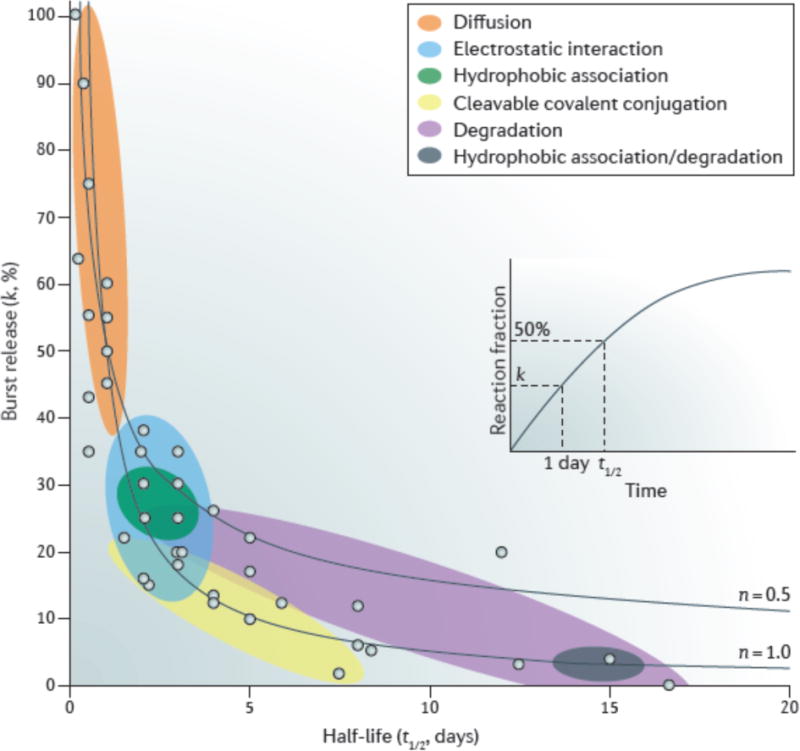
Each dot represents the burst release and half-life of release taken from the literature (see the Supplementary information for the particular reference corresponding to each dot). The inset shows a representative release profile, for which the half-life (t1/2) is determined by the time when the fraction of released drug reaches 50% and the burst release parameter (k) is determined by the drug release fraction at 24 hours. The coloured circles refer to different drug release mechanisms: diffusion-controlled mechanism (red), degradation-controlled mechanism (purple), affinity-controlled mechanisms based on cleavable covalent conjugation (orange), electrostatic interactions (blue), and hydrophobic association (green), as well as a combination of degradation-controlled and hydrophobic association-controlled mechanisms (brown). The two dashed lines were calculated using equation (2) with n = 0.5 and 1.0.
Equation 2 allows us to reanalyse a wide range of experimental data that exists in the literature. To solve the Ritger–Peppas equation, one needs to determine a pair of parameters: (1) The value of k, which is determined by measuring the release fraction over one unit of time (for example, 1 day) and provides an indication of the initial kinetics of drug release (also known as burst release); (2) the half-life time of the release (t1/2), which is defined as the time at which the mass fraction of drug released (Mt/M∞) is equal to 50% and provides an indication of extended drug release in a particular system. These two parameters allow us to compare the drug release properties of different systems.
In FIG. 5, we present a drug release property chart of various hydrogel systems, illustrating the general release behaviour resulting from the differing strategies used to control drug delivery. The overall distribution follows equation (2), because the burst release decreases with increasing half-life, and the experimental data is distributed in the region between the two curves that are calculated by this equation with n = 0.5 and 1.0. As discussed above, the diffusion-controlled mechanism typically yields a relatively short release duration (t1/2 ≈ 1 day) and a large burst release (k > 50%). The release can be slowed via several mechanisms. Electrostatic interactions and hydrophobic association typically lead to a half-life of around 2–3 days, owing to the relatively weak bond strength between the drug and polymer. Slower release rates (up to a week) can be obtained by using cleavable covalent bonds or degradation-controlled mechanisms. A very extended release can be achieved by combining hydrophobic interactions and degradation-controlled mechanisms, as exemplified by a PEO–PPO–PEO hydrogel (Pluronic F127) encapsulating drug-laden PLGA nanoparticles172. There remain many opportunities in developing hydrogel systems for extremely sustained release (with half-lives beyond 20 days). Although FIG. 5 includes only a few representative data, we hope it provides useful insight into the different release mechanisms and can aid in the design of hydrogel delivery systems to meet application-based requirements.
Using hydrogels for cell delivery
Many therapies under development harness the ability of cells (for example, islet cells, stem cells progenitor cells) to actively secrete therapeutic biomolecules such as growth factors and insulin183. Cells are most often delivered while suspended in a solution and typically have very low retention rates in the tissue of interest, rapidly losing viability in the human body within the first few days after delivery184–187. The problem is exemplified with long-term retention rates of mesenchymal stem cells of less than 3% in many injury models (for example, heart, kidney and liver)188. This is attributed to many factors, including exposure of cells to ischemia and inflammation, mechanical washout of cells, and leakage of the cell suspension from the injection site.
Compared to simply delivering cells in solution, hydrogel systems may offer a number of significant advantages. First, hydrogels can be designed to protect the cells from attack by the immune system, while remaining permeable for therapeutic, signalling and metabolic biomolecules. Owing to these features, hydrogels are widely used as semipermeable membranes for immunoisolating cells (for example, islet cells) in the treatment of many diseases (for example, diabetes and liver failure)92,189. Numerous successes have been reported in relation to tailoring the permeability and immunoisolation characteristics of hydrogel membranes, such as those made from alginate hydrogels190. The critical design parameters for these applications range from physical properties to geometric factors, such as the thickness and surface/volume ratio92. However, the practical application of immunoisolation techniques has been historically limited by the availability of therapeutic cells. Fortunately, this obstacle can potentially be overcome with advances in embryonic stem cell and induced pluripotent stem cell research and development191,192. A second advantage of hydrogel-based cell delivery is that this approach can concentrate the cell payloads at the target sites, and provide necessary biological or physicochemical cues for cell growth and function193,194. For example, hydrogels used to deliver human mesenchymal stem cells were shown to provide a marked increase in the number of viable cells over time and allow cells to exert therapeutic benefits for extended periods in an infarcted heart or other anatomic sites195,196.
Considerations for cell therapies delivered with hydrogels are often different from those for small and macromolecular drugs. First, because cells are micrometre-sized, they are typically entrapped inside a hydrogel, unless the hydrogel is macro-porous or degradable. Macro-porous hydrogels possess interconnected porous spaces in which cells can adhere and differentiate before and after implantation63,197. Microgels are particularly appealing for cell delivery, owing to a greater inward diffusion of oxygen and the nutrients required to maintain cell viability, rapid release of cell-excreted biomolecules and the capability for sorting193. Second, cell compatibility is crucial, especially during gelation and degradation processes; mild gelation conditions facilitate cell viability after encapsulation31,51,52,198. The use of click chemistry to crosslink hydrogels has attracted much attention in recent years, because it is bio-orthogonal, which enables high cell survival199. Third, tailoring hydrogel–cell interactions may further improve the outcomes of cell therapeutics, in part by tuning the secretions of the cells. Adhesion of cells to the matrix is crucial for their viability, retention and function. Synthetic hydrogels and some natural hydrogels that lack biological recognition sites for supporting cellular activity can be modified with biological moieties to yield bioactive hydrogels, such as a tri-peptide of arginine– glycine–aspartic acid (RGD)62,200–202, and small-molecule functional groups (for example, t-butyl and phosphate groups)203. Physical properties of hydrogels (for example, stiffness204 and stress relaxation205) also play an important role in controlling the fate and behaviour of encapsulated cells and their secretion188,206.
This field is still in its infancy, with a number of issues remaining to be explored. There has been little effort to date to determine or control whether the rate of release of cell-secreted agents is dictated by the release rate from the cells, diffusion through the encapsulating hydrogel, or both. The appropriate degradation rate of the hydrogels (or even if they should degrade at all) is unclear. Coupling the degradation rate of hydrogels with the growth rate of the developing tissue has been shown to improve both the quantity and quality of tissue regeneration with cell transplantation207–209; however, the relevance of these observations to cell bioactivity that is derived from products secreted from the cells needs further investigation.
Clinical translation
Although clinical translation is the main objective of this field, and despite the creation of many new material chemistries and hydrogels, the number of hydrogel drug delivery systems that have entered into the clinic is still limited (TABLE 1). Notably, the most impactful in terms of sales revenue (about US $750 US million) is Medtronic’s INFUSE, which is a collagen gel that releases BMP-2 for bone regeneration. A number of other systems have entered the clinic (for example, VANTAS hydrogel implants for continuous delivery of histrelin for the treatment of prostate cancer)210 and hold great promise in the years to come211.
TABLES.
| Product | Type of hydrogel | Drug | Therapeutic application |
|---|---|---|---|
| INFUSE | Collagen | Recombinant human BMP2 |
• Bone fracture • Oral maxillofacial reconstruction • Spinal fusion |
| MASTERGRAFT | Calcium phosphate and collagen | BMP2 | Spinal fusion |
| OP-1, OP-1 Putty |
Collagen | BMP7 | • Long bone fracture • Spinal fusion |
| VANTAS | Poly(2-hydroxyethyl methacrylate), poly(2-hydroxypropyl methacrylate) |
Histrelin acetate | Subdermal implant for the treatment of prostate cancer |
| SUPPRELIN LA | Poly(2-hydroxyethyl methacrylate) | Histrelin acetate | Subcutaneous implant for the treatment of children’s central precocious puberty |
| AzaSite | Poly(acrylic acid) | Azithromycin | Bacterial conjunctivitis |
| Besivance | Poly(acrylic acid) | Besifloxacin | Bacterial conjunctivitis |
| Cervidil | PEC or urethane polymer | Dinoprostone | Vaginal insert for cervical ripening to induce labour |
| Differin | Carbomer 940 | Adapalene | Topical treatment of acne vulgaris |
| AndroGel | Carbomer 980 | Testosterone | Topical gel for hypogonadism treatment |
| Calamine-zinc gelatin | Gelatin | Calamine zinc oxide | Wound dressing to lessen pain and it ching |
| ALGICELL Ag, Suprasorb A+Ag | Alginate | Silver | Wound dressing with antimicrobial silver |
| Prontosan | Hydroxyethylcellulose | Polyhexanide | Antiseptic wound dressing |
| REGRANEX | Carboxymethylcellulose | Becaplermin, a recombinant human platelet-derived growth factor | Topical gel for the treatment of diabetic foot ulcers |
BMP. bone morphogenetic protein: PEG. poly(ethylene glycol).
Researchers need to overcome many challenges during the translation process, including those related to hydrogel fabrication and storage, regulatory complexity and cost. The high water content can pose challenges, in contrast to dry biodegradable polymers (for example, PLGA microspheres). The hydrated nature of hydrogels can make terminal sterilization difficult, and sterility must therefore be typically validated for all source materials and fabrication processes. If a hydrogel degrades or drug–polymer linkages cleave via hydrolysis, the hydrogel must be dehydrated after fabrication to prevent premature degradation during storage, and such treatment must not change the hydrogel structure or the drug bioactivity. If hydrogels are instead maintained during storage in a hydrated state, the storage conditions (for example, a closed container and temperature-controlled supply chain) should minimize water evaporation and preclude pre-exposure to any medium that causes unwanted drug loss. The effects of the storage conditions (for example, temperature and time) must be carefully examined212. Regulatory concerns and the cost of commercialization often present significant challenges in the development of hydrogel delivery systems. A hydrogel releasing a drug or encapsulating drug-secreting cells is regulated as a combination product, and therefore its regulatory approval process is often longer than a scaffold without any payload. As the duration of patent protection is limited, a longer approval time can limit commercial viability213. The cost to develop hydrogel drug delivery systems from the bench to bedside, as for all drugs, is estimated to be quite high; drug development costs are generally estimated to run between US $50–800 million, which provides a significant impediment to commercialization214.
Conclusions and outlook
For the past several decades, the development of hydrogel drug delivery systems has been aided by advances in material chemistry, polymer physics, fabrication techniques and our fundamental understanding of tissue and cell biology. There exist over 3500 published papers on hydrogel drug delivery systems (according to a search for “hydrogel drug delivery” in PubMed). However, despite the successes, many challenges and unmet clinical needs remain.
Current research focuses in this field are the delivery of multiple drugs from a single system, and on-demand release with a high level of control. Although the release of multiple drugs has been demonstrated, obtaining release of distinct molecules at different rates remains a challenge that is currently met through trial and error. The potential importance of this topic is exemplified by the fact that tissue repair and regeneration involves the sequential signalling of multiple growth factors215–217. Achieving a high level of control with on-demand drug release is also a challenge. An ideal system for on-demand release would have a precisely predictable response of drug release with respect to stimuli, with this response being maintained throughout the lifetime of the system. Many systems to date that demonstrate on-demand release are proofs of concept, with only a few demonstrating low baseline release and the ability to exhibit triggered release repeatedly218. The development of broadly useful systems that provide reliable, robust stimuli responses will likely necessitate new chemical strategies and control principles.
Drug delivery depots will eventually become exhausted of drug and approaches to reload the depot with the same or different drugs could be useful in a number of settings (for example, when it is difficult or impractical to access the anatomic site for replacement of the depot). Although drug–polymer interactions are typically exploited to allow for drug loading and controlled release, they may also be used to reload drug delivery depots that are already present in the body. This concept is dependent on a very high specificity of the drug–polymer bond, which ensures that the drug does not bind indiscriminately to other molecules in the body. Although still at an early stage, both complementary base pairing using DNA molecules and bio-orthogonal click chemistries have been used to reload hydrogels many times in vivo, differentially load two or more hydrogels present in the body with distinct drugs and slow tumour growth219,220.
Theoretical modelling of drug release profiles remains a challenge in the field. Many studies have revealed the basic mechanisms of drug release in different hydrogel systems. However, many parameters in existing models are unknown and remain to be quantified. In particular, modelling of in vivo drug release has been explored to a very limited extent221,222 despite its significance. Integrated understanding of drug release and transport through the local tissue will facilitate the development of drugs and drug delivery systems.
Progress in bioelectronics and emerging areas such as gene editing highlight new opportunities for controlled drug delivery. There have been limited attempts to date using microelectronics and membrane reservoirs for remote-controlled drug delivery223–225. Those devices could be integrated with hydrogel drug delivery systems to create next-generation bioelectronics with therapeutic function. The hybridized systems may someday monitor the body continuously, respond with active control, and regulate the release of therapeutics accordingly. As hydrogels possess attractive features for new bioelectronics, including high stretchability, transparency, and conductivity to ionic currents, they are beginning to allow for the integration of stretchable conductors and drug-delivery channels and reservoirs226. The emergence of gene editing technologies such as CRISPR-Cas9 and zinc-finger nucleases also call for new materials that efficiently target gene modification to diseased cells; hydrogels are likely to be useful systems for the delivery of these molecules.
In conclusion, advances in biomaterials have broadened the repertoire of hydrogels designed for controlled drug delivery. With an expanding arsenal of material systems, target applications, and increasing fundamental understanding, the impact of hydrogel drug delivery systems is expected to increase in importance for years to come. Hydrogel drug delivery systems are likely to further change the scale, efficacy and cost of therapeutics, and to continue to improve human healthcare.
Supplementary Material
One-Sentence Summary.
Hydrogels can provide spatial and temporal control over the release of various therapeutic agents and have found clinical use. This Review presents multiscale mechanisms underlying hydrogel delivery systems and quantitative comparison between them, whilst discussing clinical translation and future opportunities.
Acknowledgments
This work was supported by government support under R01DE0130333 awarded by the National Institute of Dental & Craniofacial Research of the National Institutes of Health, and award A21448 from Novartis Pharmaceuticals Corporation. The authors thank Dr. Luo Gu and Dr. Achim Göpferich for discussion.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Langer R. Drug delivery and targeting. Nature. 1998;392:5–10. [PubMed] [Google Scholar]
- 2.Hoare TR, Kohane DS. Hydrogels in drug delivery: Progress and challenges. Polymer. 2008;49:1993–2007. [Google Scholar]
- 3.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Polymers for drug delivery systems. Ann Rev Chem Biomol Eng. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908–908. doi: 10.1126/science.270.5238.908a. [DOI] [PubMed] [Google Scholar]
- 5.Florence AT, Jani PU. Novel oral drug formulations. Drug Safety. 1994;10:233–266. doi: 10.2165/00002018-199410030-00005. [DOI] [PubMed] [Google Scholar]
- 6.Ashley GW, Henise J, Reid R, Santi DV. Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc Natl Acad Sci USA. 2013;110:2318–2323. doi: 10.1073/pnas.1215498110. This study features cleavable covalent linkages with tunable half-lives over a wide range, and demonstrates different drug release kinetics by orchestrating the rates of bulk erosion and linkage cleavage independently. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiwari G, et al. Drug delivery systems: An updated review. Int J Pharm Investig. 2012;2:2–11. doi: 10.4103/2230-973X.96920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tibbitt MW, Dahlman JE, Langer R. Emerging frontiers in drug delivery. J Am Chem Soc. 2016;138:704–717. doi: 10.1021/jacs.5b09974. [DOI] [PubMed] [Google Scholar]
- 9.Calvert P. Hydrogels for Soft Machines. Adv Mater. 2009;21:743–756. [Google Scholar]
- 10.Arakaki K, et al. Artificial cartilage made from a novel double-network hydrogel: In vivo effects on the normal cartilage and ex vivo evaluation of the friction property. J Biomed Mater Res Part A. 2010;93A:1160–1168. doi: 10.1002/jbm.a.32613. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Illeperuma WR, Suo Z, Vlassak JJ. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Letters. 2014;3:520–523. doi: 10.1021/mz5002355. [DOI] [PubMed] [Google Scholar]
- 12.Bodugoz-Senturk H, Macias CE, Kung JH, Muratoglu OK. Poly(vinyl alcohol)–acrylamide hydrogels as load-bearing cartilage substitute. Biomaterials. 2009;30:589–596. doi: 10.1016/j.biomaterials.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Su J, Hu BH, Lowe WL, Kaufman DB, Messersmith PB. Anti-inflammatory peptide-functionalized hydrogels for insulin-secreting cell encapsulation. Biomaterials. 2010;31:308–314. doi: 10.1016/j.biomaterials.2009.09.045. This study demonstrates a synergy between adhesion ligands and cytokine-suppressive peptides in improving viability of insulin secreting cells in the presence of pro-inflammatory cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reichert JM. Trends in development and approval times for new therapeutics in the United States. Nat Rev Drug Discov. 2003;2:695–702. doi: 10.1038/nrd1178. [DOI] [PubMed] [Google Scholar]
- 15.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 16.Khan TA, Peh KK, Ch’ng HS. Mechanical, bioadhesive strength and biological evaluations of chitosan films for wound dressing. J Pharm Pharmaceut Sci. 2000;3:303–311. [PubMed] [Google Scholar]
- 17.Mahdavi A, et al. A biodegradable and biocompatible gecko-inspired tissue adhesive. Proc Natl Acad Sci USA. 2008;105:2307–2312. doi: 10.1073/pnas.0712117105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di J, et al. Stretch-triggered drug delivery from wearable elastomer films containing therapeutic depots. ACS Nano. 2015;9:9407–9415. doi: 10.1021/acsnano.5b03975. [DOI] [PubMed] [Google Scholar]
- 19.Bessa PC, Casal M, Reis R. Bone morphogenetic proteins in tissue engineering: the road from laboratory to clinic, part II (BMP delivery) J Tissue Eng Regen Med. 2008;2:81–96. doi: 10.1002/term.74. [DOI] [PubMed] [Google Scholar]
- 20.Thorn R, Greeman J, Austin A. An in vitro study of antimicrobial activity and efficacy of iodine-generating hydrogel dressings. J Wound Care. 2006;15:305. doi: 10.12968/jowc.2006.15.7.26929. [DOI] [PubMed] [Google Scholar]
- 21.Momoh FU, Boateng JS, Richardson SC, Chowdhry BZ, Mitchell JC. Development and functional characterization of alginate dressing as potential protein delivery system for wound healing. Int J Biol Macromolec. 2015;81:137–150. doi: 10.1016/j.ijbiomac.2015.07.037. [DOI] [PubMed] [Google Scholar]
- 22.Pandit A, Ashar R, Feldman D. The effect of TGF-β delivered through a collagen scaffold on wound healing. J Invest Surg. 1999;12:89–100. doi: 10.1080/089419399272647. [DOI] [PubMed] [Google Scholar]
- 23.Jayakumar R, Prabaharan M, Kumar PS, Nair S, Tamura H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv. 2011;29:322–337. doi: 10.1016/j.biotechadv.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Lee KY, Mooney DJ. Alginate: Properties and biomedical applications. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tellechea A, et al. Alginate and DNA gels are suitable delivery systems for diabetic wound healing. Inter Lower Extremity Wounds. 2015;14:146–53. doi: 10.1177/1534734615580018. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Chen J, Han C. A multicenter clinical trial of recombinant human GM-CSF hydrogel for the treatment of deep second-degree burns. Wound Repair Regen. 2009;17:685–689. doi: 10.1111/j.1524-475X.2009.00526.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Griffith M, Fengfu L. Alginate microsphere-collagen composite hydrogel for ocular drug delivery and implantation. J Mater Sci Mater Med. 2008;19:3365–3371. doi: 10.1007/s10856-008-3486-2. [DOI] [PubMed] [Google Scholar]
- 28.Dash A, Cudworth G. Therapeutic applications of implantable drug delivery systems. J Pharmacol Toxicol Methods. 1998;40:1–12. doi: 10.1016/s1056-8719(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 29.Yu L, Ding J. Injectable hydrogels as unique biomedical materials. Chem Soc Rev. 2008;37:1473–1481. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 30.Silva EA, Mooney DJ. Spatiotemporal control of vascular endothelial growth factor delivery from injectable hydrogels enhances angiogenesis. J Thromb Haemost. 2007;5:590–598. doi: 10.1111/j.1538-7836.2007.02386.x. This study demonstrates the ability of needle-injectable alginate hydrogels to regulate the temporal and spatial presentation of VEGF for the treatment of ischemic diseases in a rodent model. [DOI] [PubMed] [Google Scholar]
- 31.Silva EA, Kim ES, Kong HJ, Mooney DJ. Material-based deployment enhances efficacy of endothelial progenitor cells. Proc Natl Acad Sci USA. 2008;105:14347–14352. doi: 10.1073/pnas.0803873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiemstra C, et al. In vitro and in vivo protein delivery from in situ forming poly(ethylene glycol)–poly(lactide) hydrogels. J Control Release. 2007;119:320–327. doi: 10.1016/j.jconrel.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Jin R, et al. Synthesis and characterization of hyaluronic acid–poly(ethylene glycol) hydrogels via Michael addition: An injectable biomaterial for cartilage repair. Acta Biomater. 2010;6:1968–1977. doi: 10.1016/j.actbio.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Lim DW, Nettles DL, Setton LA, Chilkoti A. Rapid cross-linking of elastin-like polypeptides with (hydroxymethyl) phosphines in aqueous solution. Biomacromolecules. 2007;8:1463–1470. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wieduwild R, et al. Minimal peptide motif for non-covalent peptide–heparin hydrogels. J Am Chem Soc. 2013;135:2919–2922. doi: 10.1021/ja312022u. [DOI] [PubMed] [Google Scholar]
- 36.Kiick KL. Peptide-and protein-mediated assembly of heparinized hydrogels. Soft Matter. 2008;4:29–37. doi: 10.1039/b711319f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishii S, Kaneko J, Nagasaki Y. Development of a long-acting, protein-loaded, redox-active, injectable gel formed by a polyion complex for local protein therapeutics. Biomaterials. 2016;84:210–218. doi: 10.1016/j.biomaterials.2016.01.029. [DOI] [PubMed] [Google Scholar]
- 38.Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS. Versatile click alginate hydrogels crosslinked via tetrazine–norbornene chemistry. Biomaterials. 2015;50:30–37. doi: 10.1016/j.biomaterials.2015.01.048. [DOI] [PubMed] [Google Scholar]
- 39.Jewett JC, Bertozzi CR. Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeForest CA, Anseth KS. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. This study demonstrates the combination of bio-orthogonal click chemistries and photoreactions to synthesize light-responsive hydrogels, which enable photoconjugation of peptides and cell encapsulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao Y, et al. Poly(N-isopropylacrylamide)–chitosan as thermosensitive in situ gel-forming system for ocular drug delivery. J Control Release. 2007;120:186–194. doi: 10.1016/j.jconrel.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Mortensen K, Pedersen JS. Structural study on the micelle formation of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer in aqueous solution. Macromolecules. 1993;26:805–812. [Google Scholar]
- 43.Kwon DY, et al. Synergistic anti-tumor activity through combinational intratumoral injection of an in-situ injectable drug depot. Biomaterials. 2016;85:232–245. doi: 10.1016/j.biomaterials.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 44.DAVIDORF FH, et al. Ocular toxicity of vitreal pluronic polyol F-127. Retina. 1990;10:297–300. doi: 10.1097/00006982-199010000-00013. [DOI] [PubMed] [Google Scholar]
- 45.Censi R, et al. Photopolymerized thermosensitive hydrogels for tailorable diffusion-controlled protein delivery. J Control Release. 2009;140:230–236. doi: 10.1016/j.jconrel.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 46.van de Wetering P, Metters AT, Schoenmakers RG, Hubbell JA. Poly(ethylene glycol) hydrogels formed by conjugate addition with controllable swelling, degradation, and release of pharmaceutically active proteins. J Control Release. 2005;102:619–627. doi: 10.1016/j.jconrel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 47.Guvendiren M, Lu HD, Burdick JA. Shear-thinning hydrogels for biomedical applications. Soft Matter. 2012;8:260–272. [Google Scholar]
- 48.Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ. Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials. 2011;32:5906–5914. doi: 10.1016/j.biomaterials.2011.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajagopal K, Schneider JP. Self-assembling peptides and proteins for nanotechnological applications. Curr Opin Struct Biol. 2004;14:480–486. doi: 10.1016/j.sbi.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 50.Haines-Butterick L, et al. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc Natl Acad Sci USA. 2007;104:7791–7796. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan C, et al. Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter. 2010;6:5143–5156. doi: 10.1039/C0SM00642D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haines-Butterick LA, Salick DA, Pochan DJ, Schneider JP. In vitro assessment of the pro-inflammatory potential of β-hairpin peptide hydrogels. Biomaterials. 2008;29:4164–4169. doi: 10.1016/j.biomaterials.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Micklitsch CM, et al. Zinc-triggered hydrogelation of a self-assembling β-hairpin peptide. Angew Chem. 2011;123:1615–1617. doi: 10.1002/anie.201006652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rowan SJ, Cantrill SJ, Cousins GR, Sanders JK, Stoddart JF. Dynamic covalent chemistry. Angew Chem Int Ed. 2002;41:898–952. doi: 10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 55.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Bis-aliphatic hydrazone-linked hydrogels form most rapidly at physiological pH: identifying the origin of hydrogel properties with small molecule kinetic studies. Chem Mater. 2014;26:2382–2387. [Google Scholar]
- 56.Jin Y, Yu C, Denman RJ, Zhang W. Recent advances in dynamic covalent chemistry. Chem Soc Rev. 2013;42:6634–6654. doi: 10.1039/c3cs60044k. [DOI] [PubMed] [Google Scholar]
- 57.Yesilyurt V, et al. Injectable self-healing glucose-responsive hydrogels with pH-regulated mechanical properties. Adv Mater. 2016;28:86–91. doi: 10.1002/adma.201502902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plieva FM, Galaev IY, Noppe W, Mattiasson B. Cryogel applications in microbiology. Trends Microbiol. 2008;16:543–551. doi: 10.1016/j.tim.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Sheridan M, Shea L, Peters M, Mooney D. Bioabsorbable polymer scaffolds for tissue engineering capable of sustained growth factor delivery. J Control Release. 2000;64:91–102. doi: 10.1016/s0168-3659(99)00138-8. [DOI] [PubMed] [Google Scholar]
- 60.Zhou S, Bismarck A, Steinke JH. Ion-responsive alginate based macroporous injectable hydrogel scaffolds prepared by emulsion templating. J Mater Chem B. 2013;1:4736–4745. doi: 10.1039/c3tb20888e. [DOI] [PubMed] [Google Scholar]
- 61.Hassan CM, Peppas NA. Structure and morphology of freeze/thawed PVA hydrogels. Macromolecules. 2000;33:2472–2479. [Google Scholar]
- 62.Huebsch N, et al. Matrix elasticity of void-forming hydrogels controls transplanted-stem-cell-mediated bone formation. Nat Mater. 2015;14:1269–1277. doi: 10.1038/nmat4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bencherif SA, et al. Injectable preformed scaffolds with shape-memory properties. Proc Natl Acad Sci USA. 2012;109:19590–19595. doi: 10.1073/pnas.1211516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bencherif SA, et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Comm. 2015;6:7556. doi: 10.1038/ncomms8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitragotri S, Lahann J. Physical approaches to biomaterial design. Nat Mater. 2009;8:15–23. doi: 10.1038/nmat2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Euliss LE, DuPont JA, Gratton S, DeSimone J. Imparting size, shape, and composition control of materials for nanomedicine. Chem Soc Rev. 2006;35:1095–1104. doi: 10.1039/b600913c. [DOI] [PubMed] [Google Scholar]
- 68.Gratton SE, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci USA. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merkel TJ, et al. The effect of particle size on the biodistribution of low-modulus hydrogel PRINT particles. J Control Release. 2012;162:37–44. doi: 10.1016/j.jconrel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012–an update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 71.Peer D, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 72.Vinogradov SV, Bronich TK, Kabanov AV. Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. Adv Drug Deliv Rev. 2002;54:135–147. doi: 10.1016/s0169-409x(01)00245-9. [DOI] [PubMed] [Google Scholar]
- 73.Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24:39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 74.Li J, et al. Self-assembly of DNA nanohydrogels with controllable size and stimuli-responsive property for targeted gene regulation therapy. J Am Chem Soc. 2015;137:1412–1415. doi: 10.1021/ja512293f. A modular design of DNA nanogels for gene therapy was presented which can incorporate different functional elements to target specific cells and release therapeutic genes inside cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. The development of microgels/nanogels for drug delivery applications. Prog Polym Sci. 2008;33:448–477. [Google Scholar]
- 76.Rolland JP, et al. Direct fabrication and harvesting of monodisperse, shape-specific nanobiomaterials. J Am Chem Soc. 2005;127:10096–10100. doi: 10.1021/ja051977c. This study presents a versatile “top-down” technique for the fabrication of nanogels and microgels, which provides finely control over particle size and shape, and is compatible with various therapeutic agents. [DOI] [PubMed] [Google Scholar]
- 77.Perry JL, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012;12:5304–5310. doi: 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dunn SS, et al. Reductively responsive siRNA-conjugated hydrogel nanoparticles for gene silencing. J Am Chem Soc. 2012;134:7423–7430. doi: 10.1021/ja300174v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peppas NA, Sahlin JJ. Hydrogels as mucoadhesive and bioadhesive materials: a review. Biomaterials. 1996;17:1553–1561. doi: 10.1016/0142-9612(95)00307-x. [DOI] [PubMed] [Google Scholar]
- 80.Chaturvedi M, Kumar M, Pathak K. A review on mucoadhesive polymer used in nasal drug delivery system. J Adv Pharm Technol Res. 2011;2:215. doi: 10.4103/2231-4040.90876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reece TB, Maxey TS, Kron IL. A prospectus on tissue adhesives. Am J Surg. 2001;182:S40–S44. doi: 10.1016/s0002-9610(01)00742-5. [DOI] [PubMed] [Google Scholar]
- 82.Xu J, Strandman S, Zhu JX, Barralet J, Cerruti M. Genipin-crosslinked catechol-chitosan mucoadhesive hydrogels for buccal drug delivery. Biomaterials. 2015;37:395–404. doi: 10.1016/j.biomaterials.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 83.Nho YC, Park JS, Lim YM. Preparation of poly(acrylic acid) hydrogel by radiation crosslinking and its application for mucoadhesives. Polymers. 2014;6:890–898. [Google Scholar]
- 84.Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2010;62:83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 85.Ponchel G, Irache JM. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Adv Drug Deliv Rev. 1998;34:191–219. doi: 10.1016/s0169-409x(98)00040-4. [DOI] [PubMed] [Google Scholar]
- 86.Shojaei AH, Paulson J, Honary S. Evaluation of poly(acrylic acid-co-ethylhexyl acrylate) films for mucoadhesive transbuccal drug delivery: factors affecting the force of mucoadhesion. J Control Release. 2000;67:223–232. doi: 10.1016/s0168-3659(00)00216-9. [DOI] [PubMed] [Google Scholar]
- 87.Das Neves J, Bahia M. Gels as vaginal drug delivery systems. Int J Pharm. 2006;318:1–14. doi: 10.1016/j.ijpharm.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 88.Luppi B, et al. Novel mucoadhesive nasal inserts based on chitosan/hyaluronate polyelectrolyte complexes for peptide and protein delivery. J Pharm Pharmacol. 2009;61:151–157. doi: 10.1211/jpp/61.02.0003. [DOI] [PubMed] [Google Scholar]
- 89.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Mussel-inspired adhesives and coatings. Ann Rev Mater Res. 2011;41:99. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brubaker CE, Kissler H, Wang LJ, Kaufman DB, Messersmith PB. Biological performance of mussel-inspired adhesive in extrahepatic islet transplantation. Biomaterials. 2010;31:420–427. doi: 10.1016/j.biomaterials.2009.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nafea E, Marson A, Poole-Warren L, Martens P. Immunoisolating semi-permeable membranes for cell encapsulation: focus on hydrogels. J Control Release. 2011;154:110–122. doi: 10.1016/j.jconrel.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 93.Lake GJ, Thomas AG. Strength of highly elastic materials. Proc R Soc A. 1967;300:108–119. [Google Scholar]
- 94.Kong HJ, Wong E, Mooney DJ. Independent control of rigidity and toughness of polymeric hydrogels. Macromolecules. 2003;36:4582–4588. [Google Scholar]
- 95.Gong JP, Katsuyama Y, Kurokawa T, Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv Mater. 2003;15:1155–1158. [Google Scholar]
- 96.Sun JY, et al. Highly stretchable and tough hydrogels. Nature. 2012;489:133–136. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin CC, Metters AT. Hydrogels in controlled release formulations: network design and mathematical modeling. Adv Drug Deliv Rev. 2006;58:1379–1408. doi: 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 98.Burczak K, Fujisato T, Hatada M, Ikada Y. Protein permeation through poly(vinyl alcohol) hydrogel membranes. Biomaterials. 1994;15:231–238. doi: 10.1016/0142-9612(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 99.Dubrovskii SA, Rakova GV. Elastic and osmotic behavior and network imperfections of nonionic and weakly ionized acrylamide-based hydrogels. Macromolecules. 1997;30:7478–7486. [Google Scholar]
- 100.Sakai T, et al. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules. 2008;41:5379–5384. [Google Scholar]
- 101.Lee KY, Mooney DJ. Hydrogels for tissue engineering. Chem Rev. 2001;101:1869–1880. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 102.Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853–2888. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 103.Young M, Carroad P, Bell R. Estimation of diffusion coefficients of proteins. Biotechnol Bioeng. 1980;22:947–955. [Google Scholar]
- 104.Brazel CS, Peppas NA. Modeling of drug release from swellable polymers. Eur J Pharm Biopharm. 2000;49:47–58. doi: 10.1016/s0939-6411(99)00058-2. [DOI] [PubMed] [Google Scholar]
- 105.Lin YH, Liang HF, Chung CK, Chen MC, Sung HW. Physically crosslinked alginate/N,O-carboxymethyl chitosan hydrogels with calcium for oral delivery of protein drugs. Biomaterials. 2005;26:2105–2113. doi: 10.1016/j.biomaterials.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 106.Amsden B. Solute diffusion within hydrogels. Mechanisms and models. Macromolecules. 1998;31:8382–8395. [Google Scholar]
- 107.Kong HJ, Kaigler D, Kim K, Mooney DJ. Controlling rigidity and degradation of alginate hydrogels via molecular weight distribution. Biomacromolecules. 2004;5:1720–1727. doi: 10.1021/bm049879r. [DOI] [PubMed] [Google Scholar]
- 108.Boontheekul T, Kong HJ, Mooney DJ. Controlling alginate gel degradation utilizing partial oxidation and bimodal molecular weight distribution. Biomaterials. 2005;26:2455–2465. doi: 10.1016/j.biomaterials.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 109.O’Shea TM, Aimetti AA, Kim E, Yesilyurt V, Langer R. Synthesis and characterization of a library of in‐situ curing, nonswelling ethoxylated polyol thiol‐ ene hydrogels for tailorable macromolecule delivery. Adv Mater. 2015;27:65–72. doi: 10.1002/adma.201403724. [DOI] [PubMed] [Google Scholar]
- 110.Ishihara M, et al. Controlled release of fibroblast growth factors and heparin from photocrosslinked chitosan hydrogels and subsequent effect on in vivo vascularization. J Biomed Mater Res A. 2003;64:551–559. doi: 10.1002/jbm.a.10427. [DOI] [PubMed] [Google Scholar]
- 111.Lutolf M, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci USA. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Um SH, et al. Enzyme-catalysed assembly of DNA hydrogel. Nat Mater. 2006;5:797–801. doi: 10.1038/nmat1741. [DOI] [PubMed] [Google Scholar]
- 113.Purcell BP, et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater. 2014;13:653. doi: 10.1038/nmat3922. This study features a biomolecule-responsive hydrogel that can degrade in response to matrix metalloproteinase, and release drugs for the treatment of myocardial infarction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fischel-Ghodsian F, Brown L, Mathiowitz E, Brandenburg D, Langer R. Enzymatically controlled drug delivery. Proc Natl Acad Sci USA. 1988;85:2403–2406. doi: 10.1073/pnas.85.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Podual K, Doyle FJ, Peppas NA. Glucose-sensitivity of glucose oxidase-containing cationic copolymer hydrogels having poly(ethylene glycol) grafts. J Control Release. 2000;67:9–17. doi: 10.1016/s0168-3659(00)00195-4. [DOI] [PubMed] [Google Scholar]
- 116.Maitz MF, et al. Bio-responsive polymer hydrogels homeostatically regulate blood coagulation. Nat Comm. 2013;4:2168. doi: 10.1038/ncomms3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin KY, Lo JH, Consul N, Kwong GA, Bhatia SN. Self-titrating anticoagulant nanocomplexes that restore homeostatic regulation of the coagulation cascade. ACS Nano. 2014;8:8776–8785. doi: 10.1021/nn501129q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y, Wang R, Hua Y, Baumgartner R, Cheng J. Trigger-responsive poly(β-amino ester) hydrogels. ACS Macro Lett. 2014;3:693–697. doi: 10.1021/mz500277j. [DOI] [PubMed] [Google Scholar]
- 119.Tibbitt MW, Han BW, Kloxin AM, Anseth KS. Synthesis and application of photodegradable microspheres for spatiotemporal control of protein delivery. J Biomed Mater Res A. 2012;100:1647. doi: 10.1002/jbm.a.34107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yan B, Boyer JC, Habault D, Branda NR, Zhao Y. Near infrared light triggered release of biomacromolecules from hydrogels loaded with upconversion nanoparticles. J Am Chem Soc. 2012;134:16558–16561. doi: 10.1021/ja308876j. [DOI] [PubMed] [Google Scholar]
- 121.Siepmann J, Göpferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Deliv Rev. 2001;48:229–247. doi: 10.1016/s0169-409x(01)00116-8. [DOI] [PubMed] [Google Scholar]
- 122.Yu H, Lu J, Xiao C. Preparation and properties of novel hydrogels from oxidized konjac glucomannan cross linked chitosan for in vitro drug delivery. Macromol Biosci. 2007;7:1100–1111. doi: 10.1002/mabi.200700035. [DOI] [PubMed] [Google Scholar]
- 123.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(α-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581–587. [Google Scholar]
- 124.Ma G, Miao B, Song C. Thermosensitive PCL-PEG-PCL hydrogels: Synthesis, characterization, and delivery of proteins. J Appl Polym Sci. 2010;116:1985–1993. [Google Scholar]
- 125.van de Manakker F, et al. Protein release behavior of self assembled PEG–β-cyclodextrin/PEG–cholesterol hydrogels. Adv Func Mater. 2009;19:2992–3001. [Google Scholar]
- 126.Brannonpeppas L, Peppas NA. Equilibrium swelling behavior of pH-sensitive hydrogels. Chem Eng Sci. 1991;46:715–722. [Google Scholar]
- 127.Hong W, Zhao X, Zhou J, Suo Z. A theory of coupled diffusion and large deformation in polymeric gels. J Mech Phys Solids. 2008;56:1779–1793. [Google Scholar]
- 128.Hirokawa Y, Tanaka T. Volume phase-transition in a nonionic gel. J Chem Phys. 1984;81:6379–6380. [Google Scholar]
- 129.Obaidat AA, Park K. Characterization of protein release through glucose-sensitive hydrogel membranes. Biomaterials. 1997;18:801–806. doi: 10.1016/s0142-9612(96)00198-6. [DOI] [PubMed] [Google Scholar]
- 130.Kokufata E, Zhang YQ, Tanaka T. Saccharide-sensitive phase transition of a lectin-loaded gel. Nature. 1991;351:302–304. [Google Scholar]
- 131.Zhang S, et al. A pH-responsive supramolecular polymer gel as an enteric elastomer for use in gastric devices. Nat Mater. 2015 doi: 10.1038/nmat4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ohmine I, Tanaka T. Salt effects on the phase-transition of ionic gels. J Chem Phys. 1982;77:5725–5729. [Google Scholar]
- 133.Murdan S. Electro-responsive drug delivery from hydrogels. J Control Release. 2003;92:1–17. doi: 10.1016/s0168-3659(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 134.Mumper RJ, Huffman AS, Puolakkainen PA, Bouchard LS, Gombotz WR. Calcium-alginate beads for the oral delivery of transforming growth factor-β1 (TGF-β1): stabilization of TGF-β1 by the addition of polyacrylic acid within acid-treated beads. J Control Release. 1994;30:241–251. [Google Scholar]
- 135.Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials. 2016;85:152–167. doi: 10.1016/j.biomaterials.2016.01.061. [DOI] [PubMed] [Google Scholar]
- 136.Shirakura T, Kelson TJ, Ray A, Malyarenko AE, Kopelman R. Hydrogel nanoparticles with thermally controlled drug release. ACS Macro Lett. 2014;3:602–606. doi: 10.1021/mz500231e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ankareddi I, Brazel CS. Synthesis and characterization of grafted thermosensitive hydrogels for heating activated controlled release. Int J Pharm. 2007;336:241–247. doi: 10.1016/j.ijpharm.2006.11.065. [DOI] [PubMed] [Google Scholar]
- 138.Huebsch N, et al. Ultrasound-triggered disruption and self-healing of reversibly cross-linked hydrogels for drug delivery and enhanced chemotherapy. Proc Natl Acad Sci USA. 2014;111:9762–9767. doi: 10.1073/pnas.1405469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brudno Y, Mooney DJ. On-demand drug delivery from local depots. J Control Release. 2015;219:8–17. doi: 10.1016/j.jconrel.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 140.Lee KY, Peters MC, Anderson KW, Mooney DJ. Controlled growth factor release from synthetic extracellular matrices. Nature. 2000;408:998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 141.Liu TY, Hu SH, Liu TY, Liu DM, Chen SY. Magnetic-sensitive behavior of intelligent ferrogels for controlled release of drug. Langmuir. 2006;22:5974–5978. doi: 10.1021/la060371e. [DOI] [PubMed] [Google Scholar]
- 142.Hu SH, Liu TY, Liu DM, Chen SY. Nano-ferrosponges for controlled drug release. J Control Release. 2007;121:181–189. doi: 10.1016/j.jconrel.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 143.Zhao X, et al. Active scaffolds for on-demand drug and cell delivery. Proc Natl Acad Sci USA. 2011;108:67–72. doi: 10.1073/pnas.1007862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mitragotri S. Healing sound: the use of ultrasound in drug delivery and other therapeutic applications. Nat Rev Drug Dis. 2005;4:255–260. doi: 10.1038/nrd1662. [DOI] [PubMed] [Google Scholar]
- 145.Mitragotri S, Blankschtein D, Langer R. Ultrasound-mediated transdermal protein delivery. Science. 1995;269:850–853. doi: 10.1126/science.7638603. [DOI] [PubMed] [Google Scholar]
- 146.Mann BK, Schmedlen RH, West JL. Tethered-TGF-β increases extracellular matrix production of vascular smooth muscle cells. Biomaterials. 2001;22:439–444. doi: 10.1016/s0142-9612(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 147.Kolate A, et al. PEG—a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81. doi: 10.1016/j.jconrel.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 148.Ehrbar M, et al. Cell-demanded liberation of VEGF121 from fibrin implants induces local and controlled blood vessel growth. Circ Res. 2004;94:1124–1132. doi: 10.1161/01.RES.0000126411.29641.08. [DOI] [PubMed] [Google Scholar]
- 149.Traub S, et al. The promotion of endothelial cell attachment and spreading using FNIII10 fused to VEGF-A 165. Biomaterials. 2013;34:5958–5968. doi: 10.1016/j.biomaterials.2013.04.050. [DOI] [PubMed] [Google Scholar]
- 150.Van Hove AH, Beltejar MJG, Benoit DS. Development and in vitro assessment of enzymatically-responsive poly(ethylene glycol) hydrogels for the delivery of therapeutic peptides. Biomaterials. 2014;35:9719–9730. doi: 10.1016/j.biomaterials.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Greenwald RB, et al. Controlled release of proteins from their poly(ethylene glycol) conjugates: drug delivery systems employing 1, 6-elimination. Bioconjugate Chem. 2003;14:395–403. doi: 10.1021/bc025652m. [DOI] [PubMed] [Google Scholar]
- 152.Schneider EL, Henise J, Reid R, Ashley GW, Santi DV. Hydrogel drug delivery system using self-cleaving covalent linkers for once-a-week administration of exenatide. Bioconjugate Chem. 2016 doi: 10.1021/acs.bioconjchem.5b00690. [DOI] [PubMed] [Google Scholar]
- 153.Shah NJ, et al. Adaptive growth factor delivery from a polyelectrolyte coating promotes synergistic bone tissue repair and reconstruction. Proc Natl Acad Sci USA. 2014;111:12847–12852. doi: 10.1073/pnas.1408035111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Macdonald ML, et al. Tissue integration of growth factor-eluting layer-by-layer polyelectrolyte multilayer coated implants. Biomaterials. 2011;32:1446–1453. doi: 10.1016/j.biomaterials.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Silva EA, Mooney DJ. Effects of VEGF temporal and spatial presentation on angiogenesis. Biomaterials. 2010;31:1235–1241. doi: 10.1016/j.biomaterials.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kolambkar YM, et al. An alginate-based hybrid system for growth factor delivery in the functional repair of large bone defects. Biomaterials. 2011;32:65–74. doi: 10.1016/j.biomaterials.2010.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Martino MM, et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science. 2014;343:885–888. doi: 10.1126/science.1247663. Growth factors were engineered to bind strongly to the ECM, which led to superior tissue repair and decreased side effects in the treatment of diabetic wounds, compared to the wild-type proteins with low affinity to the ECM. [DOI] [PubMed] [Google Scholar]
- 158.Pike DB, et al. Heparin-regulated release of growth factors in vitro and angiogenic response in vivo to implanted hyaluronan hydrogels containing VEGF and bFGF. Biomaterials. 2006;27:5242–5251. doi: 10.1016/j.biomaterials.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 159.Freeman I, Kedem A, Cohen S. The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials. 2008;29:3260–3268. doi: 10.1016/j.biomaterials.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 160.Freudenberg U, et al. Heparin desulfation modulates VEGF release and angiogenesis in diabetic wounds. J Control Release. 2015;220:79–88. doi: 10.1016/j.jconrel.2015.10.028. [DOI] [PubMed] [Google Scholar]
- 161.Thatiparti TR, Shoffstall AJ, von Recum HA. Cyclodextrin-based device coatings for affinity-based release of antibiotics. Biomaterials. 2010;31:2335–2347. doi: 10.1016/j.biomaterials.2009.11.087. [DOI] [PubMed] [Google Scholar]
- 162.Zhang P, Cheetham AG, Lin Y-a, Cui H. Self-assembled Tat nanofibers as effective drug carrier and transporter. ACS Nano. 2013;7:5965–5977. doi: 10.1021/nn401667z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Soukasene S, et al. Antitumor activity of peptide amphiphile nanofiber-encapsulated camptothecin. ACS Nano. 2011;5:9113–9121. doi: 10.1021/nn203343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jensen BE, Dávila I, Zelikin AN. Poly(vinyl alcohol) physical hydrogels: Matrix-mediated drug delivery using spontaneously eroding substrate. J Phys Chem B. 2016 doi: 10.1021/acs.jpcb.6b01381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mateen R, Hoare T. Injectable, in situ gelling, cyclodextrin–dextran hydrogels for the partitioning-driven release of hydrophobic drugs. J Mater Chem B. 2014;2:5157–5167. doi: 10.1039/c4tb00631c. [DOI] [PubMed] [Google Scholar]
- 166.Kearney CJ, Mooney DJ. Macroscale delivery systems for molecular and cellular payloads. Nat Mater. 2013;12:1004–1017. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 167.Alconcel SN, Baas AS, Maynard HD. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym Chem. 2011;2:1442–1448. [Google Scholar]
- 168.Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97:4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 169.Lee S, Greenwald RB, McGuire J, Yang K, Shi C. Drug delivery systems employing 1, 6-elimination: releasable poly(ethylene glycol) conjugates of proteins. Bioconjugate Chem. 2001;12:163–169. doi: 10.1021/bc000064z. [DOI] [PubMed] [Google Scholar]
- 170.Cheetham AG, Ou YC, Zhang P, Cui H. Linker-determined drug release mechanism of free camptothecin from self-assembling drug amphiphiles. Chem Comm. 2014;50:6039–6042. doi: 10.1039/c3cc49453e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jo YS, Gantz J, Hubbell JA, Lutolf MP. Tailoring hydrogel degradation and drug release via neighboring amino acid controlled ester hydrolysis. Soft Matter. 2009;5:440–446. [Google Scholar]
- 172.Geng H, Song H, Qi J, Cui D. Sustained release of VEGF from PLGA nanoparticles embedded thermo-sensitive hydrogel in full-thickness porcine bladder acellular matrix. Nanoscale Res Lett. 2011;6:1–8. doi: 10.1186/1556-276X-6-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee J, Lee KY. Injectable microsphere/hydrogel combination systems for localized protein delivery. Macromol Biosci. 2009;9:671–676. doi: 10.1002/mabi.200800317. [DOI] [PubMed] [Google Scholar]
- 174.Johnston CT, Premachandra GS, Szabo T, Lok J, Schoonheydt RA. Interaction of biological molecules with clay minerals: A combined spectroscopic and sorption study of lysozyme on saponite. Langmuir. 2011;28:611–619. doi: 10.1021/la203161n. [DOI] [PubMed] [Google Scholar]
- 175.Dawson JI, Oreffo RO. Clay: new opportunities for tissue regeneration and biomaterial design. Adv Mater. 2013;25:4069–4086. doi: 10.1002/adma.201301034. [DOI] [PubMed] [Google Scholar]
- 176.Takahashi T, Yamada Y, Kataoka K, Nagasaki Y. Preparation of a novel PEG–clay hybrid as a DDS material: Dispersion stability and sustained release profiles. J Control Release. 2005;107:408–416. doi: 10.1016/j.jconrel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 177.Abdurrahmanoglu S, Okay O. Rheological behavior of polymer-clay nanocomposite hydrogels: Effect of nanoscale interactions. J Appl Polym Sci. 2010;116:2328–2335. [Google Scholar]
- 178.Appel EA, et al. Exploiting electrostatic interactions in polymer–nanoparticle Hydrogels. ACS Macro Lett. 2015;4:848–852. doi: 10.1021/acsmacrolett.5b00416. [DOI] [PubMed] [Google Scholar]
- 179.Khaled SZ, et al. One-pot synthesis of pH-responsive hybrid nanogel particles for the intracellular delivery of small interfering RNA. Biomaterials. 2016;87:57–68. doi: 10.1016/j.biomaterials.2016.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wichterle O, Lim D. Hydrophilic gels for biological use. Nature. 1960;185:117–118. [Google Scholar]
- 181.Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5:23–36. [PubMed] [Google Scholar]
- 182.Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5:37–42. [PubMed] [Google Scholar]
- 183.Schmidt JJ, Rowley J, Kong HJ. Hydrogels used for cell-based drug delivery. J Biomed Mater Res A. 2008;87:1113–1122. doi: 10.1002/jbm.a.32287. [DOI] [PubMed] [Google Scholar]
- 184.Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med. 2013;5:179ps177–179ps177. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 186.Ballios BG, et al. A hyaluronan-based injectable hydrogel improves the survival and integration of stem cell progeny following transplantation. Stem Cell Reports. 2015;4:1031–1045. doi: 10.1016/j.stemcr.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Robey TE, Saiget MK, Reinecke H, Murry CE. Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol. 2008;45:567–581. doi: 10.1016/j.yjmcc.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rustad KC, et al. Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials. 2012;33:80–90. doi: 10.1016/j.biomaterials.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lim F, Sun AM. Microencapsulated islets as bioartificial endocrine pancreas. Science. 1980;210:908–910. doi: 10.1126/science.6776628. [DOI] [PubMed] [Google Scholar]
- 190.Trivedi N, et al. Islets in alginate macrobeads reverse diabetes despite minimal acute insulin secretory responses. Transplantation. 2001;71:203–211. doi: 10.1097/00007890-200101270-00006. [DOI] [PubMed] [Google Scholar]
- 191.Wang N, Adams G, Buttery L, Falcone FH, Stolnik S. Alginate encapsulation technology supports embryonic stem cells differentiation into insulin-producing cells. J Biotechnol. 2009;144:304–312. doi: 10.1016/j.jbiotec.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 192.Liras A. Future research and therapeutic applications of human stem cells: general, regulatory, and bioethical aspects. J Transl Med. 2010;8:131. doi: 10.1186/1479-5876-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ma M, et al. Core–shell hydrogel microcapsules for improved islets encapsulation. Adv Healthc Mater. 2013;2:667–672. doi: 10.1002/adhm.201200341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Parisi-Amon A, Mulyasasmita W, Chung C, Heilshorn SC. Protein-engineered injectable hydrogel to improve retention of transplanted adipose-derived stem cells. Adv Healthc Mater. 2013;2:428–432. doi: 10.1002/adhm.201200293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Roche ET, et al. Comparison of biomaterial delivery vehicles for improving acute retention of stem cells in the infarcted heart. Biomaterials. 2014;35:6850–6858. doi: 10.1016/j.biomaterials.2014.04.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Levit RD, et al. Cellular encapsulation enhances cardiac repair. J Am Heart Assoc. 2013;2:e000367. doi: 10.1161/JAHA.113.000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Newland B, et al. Tackling cell transplantation anoikis: An injectable, shape memory cryogel microcarrier platform material for stem cell and neuronal cell growth. Small. 2015;11:5047–5053. doi: 10.1002/smll.201500898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Alsberg E, Anderson K, Albeiruti A, Franceschi R, Mooney D. Cell-interactive alginate hydrogels for bone tissue engineering. J Dental Res. 2001;80:2025–2029. doi: 10.1177/00220345010800111501. [DOI] [PubMed] [Google Scholar]
- 199.Lin CC, Raza A, Shih H. PEG hydrogels formed by thiol-ene photo-click chemistry and their effect on the formation and recovery of insulin-secreting cell spheroids. Biomaterials. 2011;32:9685–9695. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rowley JA, Mooney DJ. Alginate type and RGD density control myoblast phenotype. J Biomed Mater Res. 2002;60:217–223. doi: 10.1002/jbm.1287. [DOI] [PubMed] [Google Scholar]
- 201.Bidarra SJ, et al. Injectable in situ crosslinkable RGD-modified alginate matrix for endothelial cells delivery. Biomaterials. 2011;32:7897–7904. doi: 10.1016/j.biomaterials.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 202.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 203.Benoit DS, Schwartz MP, Durney AR, Anseth KS. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat Mater. 2008;7:816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 205.Chaudhuri O, et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat Mater. 2015 doi: 10.1038/nmat4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shin JW, Mooney DJ. Improving Stem Cell Therapeutics with Mechanobiology. Cell Stem Cell. 2016;18:16–19. doi: 10.1016/j.stem.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Alsberg E, et al. Regulating bone formation via controlled scaffold degradation. J Dental Res. 2003;82:903–908. doi: 10.1177/154405910308201111. [DOI] [PubMed] [Google Scholar]
- 208.Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nat Mater. 2015;14:737–744. doi: 10.1038/nmat4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Stevens KR, Miller JS, Blakely BL, Chen CS, Bhatia SN. Degradable hydrogels derived from PEG-diacrylamide for hepatic tissue engineering. J Biomed Mater Res A. 2015;103:3331–3338. doi: 10.1002/jbm.a.35478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schlegel PN, Group, H. S Efficacy and safety of histrelin subdermal implant in patients with advanced prostate cancer. J Urology. 2006;175:1353–1358. doi: 10.1016/S0022-5347(05)00649-X. [DOI] [PubMed] [Google Scholar]
- 211.Jaklenec A, Stamp A, Deweerd E, Sherwin A, Langer R. Progress in the tissue engineering and stem cell industry “are we there yet?”. Tissue Eng Part B Rev. 2012;18:155–166. doi: 10.1089/ten.TEB.2011.0553. [DOI] [PubMed] [Google Scholar]
- 212.Wurm A, Nogler M, Ammann CG, Coraça-Huber DC. Effect of storage temperature and antibiotic impregnation on the quantity of bone morphogenetic protein seven in human bone grafts. Int Orthop. 2014;38:1513–1517. doi: 10.1007/s00264-014-2349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Spiller KL, Vunjak-Novakovic G. Clinical translation of controlled protein delivery systems for tissue engineering. Drug Deliv Transl Res. 2015;5:101–115. doi: 10.1007/s13346-013-0135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hunziker E, et al. Translation from research to applications. Tissue Eng. 2006;12:3341–3364. doi: 10.1089/ten.2006.12.3341. [DOI] [PubMed] [Google Scholar]
- 215.Chen RR, Silva EA, Yuen WW, Mooney DJ. Spatio–temporal VEGF and PDGF delivery patterns blood vessel formation and maturation. Pharm Res. 2007;24:258–264. doi: 10.1007/s11095-006-9173-4. [DOI] [PubMed] [Google Scholar]
- 216.Kanczler JM, et al. The effect of the delivery of vascular endothelial growth factor and bone morphogenic protein-2 to osteoprogenitor cell populations on bone formation. Biomaterials. 2010;31:1242–1250. doi: 10.1016/j.biomaterials.2009.10.059. [DOI] [PubMed] [Google Scholar]
- 217.Basmanav FB, Kose GT, Hasirci V. Sequential growth factor delivery from complexed microspheres for bone tissue engineering. Biomaterials. 2008;29:4195–4204. doi: 10.1016/j.biomaterials.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 218.Kearney CJ, et al. Switchable release of entrapped nanoparticles from alginate hydrogels. Adv Healthc Mater. 2015;4:1634–1639. doi: 10.1002/adhm.201500254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brudno Y, et al. Refilling drug delivery depots through the blood. Proc Natl Acad Sci USA. 2014;111:12722–12727. doi: 10.1073/pnas.1413027111. A new paradigm of refilling hydrogel drug depots that are already present in the body was presented, and the utility of highly specific drug-polymer interactions for this application was demonstrated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Brudno Y, et al. In vivo targeting through click chemistry. ChemMedChem. 2015;10:617–620. doi: 10.1002/cmdc.201402527. [DOI] [PubMed] [Google Scholar]
- 221.Saltzman WM, Radomsky ML. Drugs released from polymers: diffusion and elimination in brain tissue. Chem Eng Sci. 1991;46:2429–2444. [Google Scholar]
- 222.Weiser JR, Saltzman WM. Controlled release for local delivery of drugs: barriers and models. J Control Release. 2014;190:664–673. doi: 10.1016/j.jconrel.2014.04.048. This review provides a comprehensive overview of mathematical models for controlled drug release, highlighting the effect of tissue barriers on drug transport in the body. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Santini JT, Cima MJ, Langer R. A controlled-release microchip. Nature. 1999;397:335–338. doi: 10.1038/16898. [DOI] [PubMed] [Google Scholar]
- 224.Grayson ACR, et al. Multi-pulse drug delivery from a resorbable polymeric microchip device. Nat Mater. 2003;2:767–772. doi: 10.1038/nmat998. [DOI] [PubMed] [Google Scholar]
- 225.Santini JT, Jr, Richards AC, Scheidt R, Cima MJ, Langer R. Microchips as controlled drug-delivery devices. Angew Chem Int Ed. 2000;39:2396–2407. [PubMed] [Google Scholar]
- 226.Lin S, et al. Stretchable hydrogel electronics and devices. Adv Mater. 2015 doi: 10.1002/adma.201504152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Flory PJ, Rehner J. Statistical mechanics of cross-linked polymer networks II Swelling. J Chem Phys. 1943;11:521–526. [Google Scholar]
- 228.Kuijpers A, et al. Characterization of the network structure of carbodiimide cross-linked gelatin gels. Macromolecules. 1999;32:3325–3333. [Google Scholar]
- 229.Anseth KS, Bowman CN, Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996;17:1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 230.Li JY, Hu YH, Vlassak JJ, Suo ZG. Experimental determination of equations of state for ideal elastomeric gels. Soft Matter. 2012;8:8121–8128. [Google Scholar]
- 231.Hu YH, Zhao XH, Vlassak JJ, Suo ZG. Using indentation to characterize the poroelasticity of gels. Appl Phys Lett. 2010;96:121904. [Google Scholar]
- 232.Drury JL, Dennis RG, Mooney DJ. The tensile properties of alginate hydrogels. Biomaterials. 2004;25:3187–3199. doi: 10.1016/j.biomaterials.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 233.Koshy ST, et al. Click crosslinked injectable gelatin hydrogels. Adv Healthc Mater. 2016;5:541–547. doi: 10.1002/adhm.201500757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Adhikari B, Banerjee A. Short peptide based hydrogels: incorporation of graphene into the hydrogel. Soft Matter. 2011;7:9259–9266. [Google Scholar]
- 235.Waters DJ, et al. Morphology of photopolymerized end-linked poly(ethylene glycol) hydrogels by small-angle X-ray scattering. Macromolecules. 2010;43:6861–6870. doi: 10.1021/ma101070s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Krogstad DV, et al. Small angle neutron scattering study of complex coacervate micelles and hydrogels formed from ionic diblock and triblock copolymers. J Phys Chem B. 2014;118:13011–13018. doi: 10.1021/jp509175a. [DOI] [PubMed] [Google Scholar]
- 237.Zhang X, Hansing J, Netz RR, DeRouchey JE. Particle transport through hydrogels is charge asymmetric. Biophys J. 2015;108:530–539. doi: 10.1016/j.bpj.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fatin-Rouge N, Starchev K, Buffle J. Size effects on diffusion processes within agarose gels. Biophys J. 2004;86:2710–2719. doi: 10.1016/S0006-3495(04)74325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.