

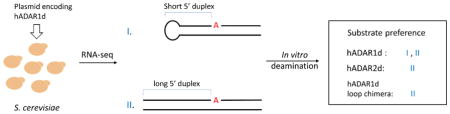
Abstract
Adenosine deamination is one of the most prevalent post-transcriptional modifications in mRNA and is catalyzed by ADAR1 and ADAR2 in humans. ADAR1 and ADAR2 have different substrate selectivity, which is believed to mainly originate from the proteins’ deaminase domains (hADAR1d and hADAR2d, respectively). RNA-seq of the S. cerevisiae transcriptome subjected to ADAR-catalyzed RNA editing identified substrates with common secondary structure features preferentially edited by hADAR1d over hADAR2d. The relatively small size and efficient reaction of one of these substrates suggested it could be useful for further study of the hADAR1d reaction. Indeed, a short hairpin stem from the S. cerevisiae HER1 mRNA was efficiently deaminated by hADAR1d and used to generate an hADAR1d-specific fluorescent reporter of editing activity. Using substrates preferred by either hADAR1d or hADAR2d in vitro, we found that a chimeric protein bearing an RNA binding loop from hADAR2d grafted onto hADAR1d showed ADAR2-like selectivity. Finally, a high-throughput mutagenesis analysis (Sat-FACS-Seq) of conserved residues in an RNA-binding loop of hADAR1d revealed essential amino acids for function and advances our understanding of RNA recognition by this domain.
Graphical Abstract
Introduction
Modified nucleosides are widespread in mRNA and their biological function and regulation have attracted considerable attention recently (1–5). Inosine (I), the deamination product of adenosine (A), is one of the most abundant modified nucleosides in human mRNA. Its formation is catalyzed by adenosine deaminases that act on RNA (ADARs) (6,7). Since A to I conversion changes the Watson-Crick pairing properties of the modified nucleoside and, thus, the coding properties of the mRNA, this reaction is a form of RNA editing. There are three members in the human ADAR family, with ADAR1 and ADAR2 being catalytically active and ADAR3 catalytically inactive (6,8–10). ADARs selectively edit certain adenosines over others in an RNA molecule. ADAR1 and ADAR2 have overlapping yet distinct selectivity (11–13). For instance, in glutamate receptor subunit B, glutamate receptor ionotropic AMPA 2 (GRIA2) mRNA, the Q/R site is specifically edited by ADAR2 while the hotspot 1 is specifically edited by ADAR1 (14,15). The R/G site in the GRIA2 mRNA is edited by both ADAR1 and ADAR2 (15). Several other examples of ADAR-selective editing reactions have been reported (12,16). Recently Tan et al. revealed that most editing events in mammals are caused by ADAR1 with ADAR1 accounting for most editing in repetitive sites while ADAR2 accounts for most editing in non-repetitive sites (17). However, our understanding of the basis for ADAR-specific selectivity is limited. While the N-terminal region of each ADAR, composed of multiple copies of double stranded RNA binding domains (dsRBDs), contributes to substrate binding affinity and selectivity (18,19) , domain swapping experiments suggest that selectivity differences between the ADARs primarily originate from differences in the deaminase domains (20). Substrate recognition by the deaminase domain of human ADAR2 (hADAR2d) has been extensively studied (21–23). Less is known about substrate recognition by the ADAR1 deaminase domain (hADAR1d). The importance of understanding and controlling the selectivity of ADAR deaminase domains has been highlighted by recent reports of directed RNA editing applications that use fusion proteins containing these domains (24–26). Indeed, off targets sites in these applications typically arise from the intrinsic reactivity and selectivity of the deaminase domains used. Here, we used RNA-seq of the S. cerevisiae transcriptome subjected to ADAR-catalyzed RNA editing to identify novel substrates for hADAR1d. We found that a subset of these RNAs are poorly edited by hADAR2d. Results of experiments using a substrate RNA efficiently edited by hADAR1d discovered in this manner allowed us to suggest key features in an RNA substrate that lead to selective recognition by the different ADAR deaminase domains. In addition, we demonstrate the contribution of an RNA-binding loop to the site selectivity of ADAR using substrate RNAs preferentially deaminated by the different ADAR deaminase domains and a chimeric protein with ADAR2 sequence grafted onto hADAR1d. Finally, using a high throughput sequencing method previously reported by us (Sat-FACS-Seq) (21), we identified residues essential for editing and provide insight into the basis for differences in ADAR deaminase domain recognition of RNA.
Materials and Methods
General methods
Components for yeast culture media were purchased from: BTS (biological grade peptone, yeast extracts and biological grade agar); BD (yeast nitrogen base w/o amino acid); Sigma-Aldrich (glucose, glycerol, lactate, galactose, raffinose, amino acids, yeast synthetic dropout supplement medium without uracil and yeast synthetic dropout medium without tryptophan). Phusion hot start DNA polymerase was purchased from Thermo Scientific for high fidelity DNA amplification. Restriction enzymes, T4 ligase and other cloning enzymes were obtained from New England Biolabs. Ligation products were transformed into XL 10-Gold cells. The sequences of plasmids or plasmid libraries were confirmed by Sanger sequencing. Plasmid extraction from E. coli was carried out with QIAprep Spin Miniprep kit (Qiagen). Oligonucleotides were synthesized by Integrated DNA Technology.
RNA-seq
Details for obtaining total RNA from S. cerevisiae expressing wild type hADAR1d or inactive hADAR1d mutant can be found in the Supplementary Information. Strand-specific and barcode-indexed RNA-seq libraries were generated from 2 μg total RNA each after poly-A enrichment using the Kapa Stranded mRNA-seq kit (KapaBiosystems, Cape Town, South Africa) following the instructions of the manufacturer. Libraries were analyzed with a Bioanalyzer 2100 instrument (Agilent, Santa Clara, CA), quantified by fluorometry on a Qubit instrument (LifeTechnologies, Carlsbad, CA) and pooled in equimolar ratios. The pool was quantified by qPCR with a Kapa Library Quant kit (KapaBiosystems) and sequenced on one lane of a HiSeq 3000 flow cell with single-end 50 bp reads, to generate over 135M reads per sample.
Raw, de-multiplexed reads were filtered and trimmed for adapter contamination and low-quality sequences (using the programs Scythe and Sickle, respectively, available at https://github.com/ucdavis-bioinformatics/). The trimmed reads were aligned to the S. cerevisiae S288c genome (NCBI accession GCA_000146045.2_R64, April 2011) with TopHat2 v. 2.1.0, utilizing bowtie2 v. 2.2.6. (27). Variants were called on the aligned reads using freebayes v0.9.18–1-g4233a23. A custom perl script was used to identify genomic locations within transcripts with A-to-G changes, that could be further analyzed as potential editing sites.
Sequence motif generation
The motif (Figure 1A) was generated by including 10 nt upstream and downstream from the 34 candidate sites identified from RNA-seq using WebLogo 3.
Figure 1.

Sequence motif surrounding candidate editing sites identified from RNA-seq and confirmation of editing by Sanger sequencing. A) Nearest neighbor and next nearest neighbor preference for hADAR1d revealed by yeast candidate substrates. The edited A is labeled with a red star. B) Sanger sequencing confirms in vivo editing on yeast candidate sites by hADAR1d and hADAR2d. The experiment was performed in triplicate. Error bar refers to SD, n≥3. C) Secondary structures of HER1 mRNA (top), GSY1 mRNA (middle), and BDF2 mRNA (bottom) surrounding the edited A (colored in red).
Confirmation of in vivo deamination by Sanger sequencing
The editing of hits from RNA-seq were confirmed by amplifying approximately 200 bp cDNA surrounding the editing sites by access RT-PCR (Promega) from total RNA isolated from S. cerevisiae INVSc1 expressing hADAR1d or hADAR2d. Primers used in the RT-PCR are listed in Table S2. The PCR products were then subjected to Sanger sequencing. The experiments were performed in biological triplicate.
Protein purification
Wild type hADARds, mutants and loop chimera were overexpressed and purified from S. cerevisiae BCY123 strain as described previously (28). Purified hADAR1d, hADAR1d E1008Q, and loop chimera were stored in 50 mM Tris-HCl pH 8.0, 10% glycerol, 0.01% Nonidet P-40, 1 mM dithiothreitol, 5 mM EDTA and 200 mM KCl at −80 °C. Purified hADAR2d and hADAR2d E488Q were stored in 20 mM Tris-HCl pH 8.0, 20% glycerol, 1 mM β-mercaptoethanol and 100 mM NaCl at −80°C.
Preparation of RNA for in vitro deamination
In vitro RNA transcription and purification for preparing the 218 nt HER1 RNA, the BDF2 RNA and the 90 nt Gabra RNA can be found in the Supplementary Information. To prepare the internally labeled HER1 33 mer RNA, a 24 mer RNA strand with sequence 5’-ACCCGCCAAUUCUUCGAGAAUUAGCGGGU-3’, an 18 mer RNA/DNA chimera with sequence 5’-rArGrCrGrGrGrUrCrGdCdCdTdTdGdGdGdTdG-3’ and a 35 mer DNA strand with sequence 5’-CCAAGGCGACCCGCTAATTCTCGAAGAATTGGCGG-3’ were purchased from IDT. The 18 mer RNA/DNA chimera was radiolabeled with [γ-32P] at the 5' end as described previously (18). Splint ligation to ligate the 24 mer RNA and the 18mer RNA/DNA chimera can be found in the Supplementary Information.
In vitro deamination assays
Deamination assays for hADAR1d were performed under single turnover conditions with 15 mM Tris-HCl pH 7.8, 20 mM KCl, 40 mM potassium glutamate, 5 mM NaCl, 4% glycerol, 1.4 mM EDTA, 0.0027% Nonidet P-40, 0.6 mM dithiothreitol, 1.0 μg/ml yeast tRNAPhe, 160 U/mL RNAsin, 10 nM RNA, and specific concentrations of enzyme. Deamination assays for hADAR2d were performed under single turn-over conditions with 17 mM Tris-HCl pH 7.4, 60 mM KCl, 15 mM NaCl, 5% glycerol, 1.5 mM EDTA, 0.003% Nonidet P-40, 0.1 mM β-mercaptoethanol, 0.5 mM dithiothreitol, 1.0 μg/ml yeast tRNAPhe, 160 U/mL RNAsin, 10 nM RNA, and specific concentrations of enzyme. Deamination assays for the loop chimera, hADAR1d E1008Q and hADAR2d E488Q were performed under a low salt conditions with 15 mM Tris-HCl pH 7.8, 6 mM KCl, 4 mM potassium glutamate, 5 mM NaCl, 4% glycerol, 1.5 mM EDTA, 0.003% Nonidet P-40, 0.6 mM dithiothreitol, 1.0 μg/ml yeast tRNAPhe, 160 U/mL RNAsin, and specific concentrations of enzyme and RNA (see figure legends for details). Each solution was incubated at 30 °C for 30 min before adding enzyme and each reaction was allowed to proceed for different time periods at 30 °C prior to stopping with either hot water or hot 1% SDS. Deaminated RNAs were phenol-chloroform extracted and ethanol precipitated.
Processing of deaminated RNA
HER1 218 nt RNA, BDF2 RNA and Gabra3 90 nt RNA from deamination reactions were analyzed with RT-PCR followed by Sanger sequencing (Figure 2). The HER1 33 nt internally labeled RNA from deamination reactions was analyzed with nuclease P1 digestion followed by thin layer chromatography (TLC) (Figure 3). Details of these methods can be found in the Supplementary Information.
Figure 2.

In vitro deamination reveals that HER1 RNA and Gabra3 RNA are hADAR1d preferred substrates. A) Deamination on HER1 RNA and BDF2 RNA by hADAR1d, with 35 nM protein and 10 nM RNA. Error bar indicates SD, n≥3. B) Deamination on HER1 RNA and BDF2 RNA by hADAR2d, with 35 nM protein and 10 nM RNA. Error bar indicates SD, n≥3. There are two off-target sites in the BDF2 RNA other than the one under study; however, the editing at the two sites are very low compared to the main site (Figure S2). ND: non-detectable. C) Local secondary structure of Gabra3 substrate predicted by Mfold. D) In vitro deamination on Gabra3 RNA by hADAR1d and hADAR2d with 35 nM protein and 10 nM RNA. Error bar indicates SD, n≥3. ND: non-detectable.
Figure 3.

Deamination of hADAR1d and hADAR2d on a 33 nt HER1 substrate and construction of a HER1-based reporter. A) Structure of the 33 nt HER1 substrate predicted by Mfold. Positions where are modified from the native HER1 RNA are bolded. B) and C) Editing of the 33 nt HER1 substrate (10 nM) by 150 nM hADAR1d (B) and 150 nM hADAR2d (C) at different time points. D) Plot of kobs for hADAR1d and hADAR2d editing the 33 nt HER1 substrate. Error bar indicates SD, n≥3. ND: non-detectable. E) Components of the HER1 fluorescent reporter. F) Characterization of the HER1 reporter with different ADAR proteins. F/F0 is the ratio of sample fluorescence divided by negative control (inactive mutant) fluorescence. Error bar indicates SD, n≥3.
Alanine scanning
The construction of HER1 yeGFP reporter can be found in the Supplementary Information. Alanine mutation was introduced at each of the conserved residues in the hADAR1 putative 5’ binding loop in the hADAR1d E1008Q construct using QuikChange II XL site-directed mutagenesis kit (Agilent) following manufacturer’s protocol. Plasmids for the BDF2 or HER1 yeGFP reporter and expression plasmid of one of the hADAR1d mutant proteins were sequentially transformed into yeast INVSc1 strain using TRP1 and URA3 selection respectively. After transformation, yeast cells were plated onto complete media (CM) – ura – trp + 2% glucose plate for growth. Individual colonies were then harvested and used to inoculate 5 mL CM – ura – trp + 2% glucose media for overnight growth. The resulting culture (0.1 mL) was used to inoculate 20 mL CM – ura – trp + 3% glycerol + 2% lactate media for another growth until the OD600 of the culture reaches 1–2 (approximately 30 h). Galactose was then added to a final concentration of 3% for induction. Cells were harvested after around 10 h of induction and the same number of cells were pelleted, washed twice with PBS and measured for fluorescence in an Optiplate-96 black, black opaque 96-well microplate (PerkinElmer) using a CLARIOstar plate reader (BMG labtech), with excitation at 482/16 nm and emission at 520/10 nm. The fluorescence reading of cells expressing the inactive hADAR1d E912A mutant was subtracted from the absolute fluorescence reading of cells expressing each hADAR1d mutant protein, and the resulting reading for each alanine mutant was normalized to the resulting reading of hADAR1d E1008Q to obtain F/Fref (Figure 6). The experiments were performed in biological triplicate.
Figure 6.

Alanine scan of the ADAR1 5’ binding loop using both HER1 reporter (Figure 3E) and BDF2 reporter (21). The star indicates hADAR1d E1008Q. F/Fref is the ratio of sample fluorescence divided by the parent protein (hADAR1d E1008Q) fluorescence. Error bar indicates SD, n≥3.
Preparation of hADAR1d 5’ binding loop library and fluorescence-activated cell sorting
hADAR1d 5’ binding loop library in hADAR1d E1008Q (in YEpTOP2PGAL1) was prepared by saturation mutagenesis as described previously (21). Oligonucleotide sequences used for saturation mutagenesis can be found in the Supplementary Information (Table S3). The procedures for introducing the hADAR1d library and the reporter plasmid to yeast cells can be found in a previous publication (21). Catalytically inactive mutant of hADAR1d E912A was used as a negative control and processed in the same way. Cells with the hADAR1d loop library and hADAR1d E912A plasmids were diluted in PBS to 20,000 cells/μl and sorted using Beckman Coulter Astrios EQ cell sorter at UC Davis flow cytometry shared resource laboratory. GFP excitation was at 488 nm and emission at 529/28 nm. Cells expressing hADAR1d loop library were collected in a BD falcon polypropylene round-bottom tubes corresponding to each gate (R1–R5) based on the fluorescence level. More than 150,000 cells (in around 2 mL) were collected for each gate and the collected cell samples were each used to inoculate equal volume of CM - ura - trp + 2% glucose media for 24 h growth, after which the whole culture was used to inoculate 13 mL fresh CM - ura - trp + 2% glucose media for another 24 h growth. Cells were then pelleted and plasmids were isolated from each cell pellet. Contour plot creation and data analysis were performed using FlowJo.
Library preparation and Illumina sequencing
hADAR1d sequence spanning amino acids 949–1019 (213 nt) was amplified from the loop plasmid libraries isolated from yeast cells using two-step PCR to prepare Illumina Miseq sequencing samples. The first PCR used primers containing overhang adapter sequence and amplicon-specific sequence. The PCR products were gel purified and used for a second PCR with primers containing a leader sequence (P5, P7), an 8bp barcode specific to each sample, and a sequence overlapping with the adapter sequence in the primers used in the previous PCR. The final PCR products were gel purified and concentrations were determined by nanodrop. PCR products were then pooled in equal amount and sequenced using Illumina Miseq PE250.
Processing of sequencing data
Paired-end reads from Miseq were de-multiplexed according to the sample specific barcodes. Using Trimmomatic, adapter sequences were removed and reads were trimmed on both sides with a quality cutoff of 5. Reads were further filtered with an overall average quality cutoff of 30. The resulting forward reads were long enough to cover the entire 213 nt sequence, thus only the forward reads were utilized from which reads of 213 nt long were selected for further data analysis. For each position of variation, the abundance of each codon was determined by searching 12 nt sequences including the target codon, the positional barcode and six extra nucleotides in the 5’ direction using MobaXterm command lines.
To eliminate bias caused by the difference in the total number of reads among different samples, total number of reads in each sample (input library, R1–R5) were adjusted based on read number in R3. Then, the abundance of amino acids in R1 through R5 were weighed against their abundance in the input library to determine enrichment levels in each gate. Frequencies of amino acids enriched in the R5 gate were calculated from their enrichment levels and plotted using Seq2 Logo 2.0 server. Average fluorescence Fave for each amino acid at each position was calculated using the equation described previously (21) and was used to generate bar graphs for each position using KaleidaGraph.
Results
RNA-seq used to identify novel substrates edited selectively by hADAR1d
We showed previously that RNA-seq of the S. cerevisiae transcriptome after expression of human ADAR2 revealed novel RNA editing substrates useful for biochemical and structural studies (29). We reasoned that such an analysis of yeast RNAs efficiently edited by hADAR1d may provide further insight into ADAR-specific editing selectivity. Here, to study the site selectivity of the isolated ADAR1 deaminase domain, hADAR1d was overexpressed in yeast to induce editing in yeast RNAs. RNA-seq was then performed on mRNA isolated from yeast expressing wild type hADAR1d or an inactive mutant (E912A). A site by site comparison was used to screen for editing sites. Candidate editing sites were identified that 1) had at least 50 reads in both the hADAR1d sample and control sample (deaminase inactive mutant E912A expressed), and 2) the editing level in the hADAR1d sample was >5% and at least 3-fold higher than that in the control sample. Using the same criteria, we also searched for T to C changes in the strand complementary to the RNA transcript. In total, the analysis led to the identification of 34 candidate editing sites. Of note, the Bromodomain Factor 2 (BDF2) mRNA, which was previously identified as an editing site for hADAR2 in this manner, was also identified here. The top 19 sites whose editing levels are ≥10% are listed in Table 1 and sites whose editing levels are between 5% and 10% are listed in Table S1 (34 sites total).
Table 1.
Top 19 sites from RNA-seq whose editing levels are ≥10%
| Position | Edited region | Editing level | transcript |
|---|---|---|---|
| ChrXV: 766200 |
|
22% | HER1 |
| ChrXV: 94295 |
|
19% | MIP1 |
| ChrIV: 1282492 |
|
17% | PDR15 |
| ChrVII: 724023 |
|
15% | SPT6 |
| ChrII: 204592 |
|
15% | SCT1 |
| ChrX: 287154 |
|
15% | SCP160 |
| ChrIV: 1437562 |
|
15% | RSM28 |
| ChrXV: 965732 |
|
14% | RPA190 |
| ChrVI: 175014 |
|
14% | GSY1 |
| ChrXIV: 715206 |
|
13% | LYS9 |
| ChrXIII: 782664 |
|
13% | YMR258C |
| ChrIV: 469105 |
|
11% | SNQ2 |
| ChrV: 116713 |
|
11% | URA3 |
| ChrXII: 812027 |
|
11% | FKS1 |
| ChrXI: 104771 |
|
11% | FAS1 |
| ChrVIII: 497886 |
|
10% | AIM46 |
| ChrXII: 650120 |
|
10% | HAP1 |
| ChrVII: 407681 |
|
10% | TIF4632 |
| ChrXI: 251826 |
|
10% | HSL1 |
The editing sites are colored in red.
The sequences surrounding all 34 hADAR1d editing sites in yeast identified by RNA-seq were analyzed for preferred nucleotides. A preference for a U 5’ to the editing site, a G 3’ to the editing site and a U two nucleotides 5’ to the editing site was revealed (Figure 1A), in agreement with previously observed nearest neighbor and next nearest neighbor preference for hADAR1d (12).
We confirmed editing on the top nine candidate substrates from the RNA-seq analysis, as well as for the BDF2 site, using Sanger sequencing of cDNA generated from total RNA isolated from yeast expressing hADAR1d (Figure 1B). In order to determine if any of these yeast targets were preferred by hADAR1d, total RNA was also isolated from yeast expressing hADAR2d and editing on these same sites was quantified. Other than the MIP1 site, all sites tested were confirmed to be edited. The Sanger sequencing data showed a similar trend of editing but generally higher editing levels compared to that determined by RNA-seq, likely due to differences in sample processing between the two techniques. Two of the sites identified as hADAR1d editing sites, located in mRNAs of HER1 (required for Hmg2p-induced ER remodeling) and GSY1 (glycogen synthase), were edited to 25–45% by ADAR1d, but to below 10% by hADAR2d (Sanger sequencing data) (Figure 1B), indicating that they are hADAR1d preferred sites. This is not due to an expression difference between hADAR2d and hADAR1d since hADAR2d showed a higher expression level than hADAR1d in yeast (Figure S6A). We predicted the secondary structures surrounding the editing sites using Mfold (Figure 1C and Figure S1). Interestingly, the secondary structures surrounding the HER1 site and the GSY1 site are strikingly similar with both comprising a five bp A•U rich duplex with a five or six nucleotide (nt) terminal loop on the 5’ side of the editing site (Figure 1C).
As an additional test of selectivity, we measured the deamination rates for hADAR1d and hADAR2d editing using an RNA corresponding to a 218 nt HER1 transcript with the editing site in the center. The two proteins were assayed with the same protein concentration (35 nM) and under respective assay conditions previously optimized for each ADAR (i.e. glutamate ion was used as an anion source in addition to Cl− in the ADAR1 assay buffer for better activity of ADAR1d in vitro (30)). Considering the possible influence of different assay conditions on enzymatic activities, we used a transcribed RNA containing the BDF2 site as a control (Figure 1C), which was shown previously to be an excellent substrate for both isolated deaminase domains (29,31). Deaminated RNAs were analyzed with RT-PCR followed by Sanger sequencing to determine editing efficiencies. In agreement with the editing patterns observed in RNAs isolated from yeast expressing the proteins, hADAR1d was able to edit the HER1 site more efficiently than the BDF2 site (Figure 2A). In contrast, hADAR2d demonstrated minimal editing on the HER1 site although it maintained excellent editing activity on the BDF2 site (Figure 2B). These results confirm the HER1 derived RNA is more efficiently edited by hADAR1d than by hADAR2d. Importantly, these results suggested the HER1 substrate RNA could be useful for the study of ADAR selectivity since it is efficiently edited by hADAR1d and poorly edited by hADAR2d.
A feature of the HER1 RNA that could be responsible for the low editing efficiency of hADAR2d is the relatively short (five bp) hairpin stem on the 5’ side of the editing site. Human GABAA receptor 3 subunit transcript (Gabra3), which is a substrate of both full length ADAR1 and ADAR2 (32,33) , has a similar hairpin stem structure on the 5’ side of the editing site as seen in the HER1 RNA (Figure 2C). We carried out deamination reactions on a 90 nt Gabra3-derived RNA with the editing site in the center, and found that this RNA is also edited substantially more efficiently by hADAR1d than hADAR2d, although it exhibited lower absolute editing efficiencies compared to the HER1 RNA (Figure 2D).
A minimal substrate for hADAR1d-selective editing
Short oligonucleotide substrate RNAs are useful for biochemical, biophysical and structural studies of ADARs (22,29,34). We wished to determine whether an RNA comprising only the predicted secondary structure surrounding the HER1 site could support editing by hADAR1d in vitro. Thus, we prepared a 33 nt HER1 RNA with the editing site specifically 32P labeled at its 5’ phosphate. This RNA is derived from the natural HER1 sequence with some modifications– the G•U wobble pair on the 3’ side of the editing site was converted to a G-C base pair and two additional G-C pairs were added to increase the stability of the folded RNA (Figure 3A). Deaminated RNAs were analyzed with nuclease P1 digestion followed by thin layer chromatography (TLC) (18) (Figure 3B, C). The 33 nt HER1 RNA is edited by hADAR1d with a kobs = 0.091± 0.023 min−1. Importantly, little editing is observed for hADAR2d with this RNA under these conditions (Figure 3D). To eliminate the possibility that the absence of editing of hADAR2d on this 33 nt HER1 RNA is a result of assay condition used, we also measured its editing on this RNA under the same assay conditions as hADAR1d and still no editing was observed (data not shown). Thus, hADAR1d-selective editing requires only the RNA present on the minimal 33 nt HER1 substrate RNA. This RNA is short enough to allow for routine incorporation of nucleoside analogs such as 2-aminopurine ribonucleoside (35) and 8-azanebularine (34) where needed for biochemical and biophysical studies of hADAR1-RNA complexes.
An hADAR1d-specific fluorescent reporter
To gain more insight into the substrate recognition by ADAR1, we developed a fluorescence-based editing reporter assay using the minimal HER1 substrate RNA (Figure 3E). With this assay, yeast cells generate a fluorescence signal when an editing event converts a stop codon into a tryptophan codon in an eGFP reporter transcript within the yeast cells, thus allowing an evaluation of editing activity quantitatively. This new reporter system was then characterized with different ADARs. Consistent with the specificity pattern observed in vitro, the reporter generated fluorescence with hADAR1d, but not with hADAR2d (Figure 3F). However, the fluorescence generated with hADAR1d was only approximately 4-fold higher than the negative control. To increase the sensitivity of the assay, we optimized the hADAR1d coding sequence for better expression in yeast (Figure S6B) and introduced the E1008Q mutation in hADAR1d. With this construct, the HER1 reporter produced approximately 30 times higher fluorescence than the negative control, while still no fluorescence was produced with hADAR2d E488Q mutant, showing that the HER1 reporter is hADAR1d specific (Figure 3F).
The 5’ RNA binding loops contribute to ADAR selectivity
Recently, crystal structures of hADAR2d bound to RNA substrates revealed a protein loop that contacts the RNA substrate on the 5’ side of the editing site (i.e. the 5’ binding loop) (Figure 4A, B) (22). Sequence alignment between ADAR1 and ADAR2 in this region revealed the 5’ binding loops of the two proteins are substantially different (Figure 4C). These observations suggest this loop could contribute to specificity differences between the two ADARs. Our discovery of RNA substrates that are efficiently and selectively deaminated by the different ADAR deaminase domains in vitro enabled experiments to test the importance of the 5’ RNA binding loops in determining substrate preferences for ADAR deaminase domains. For these experiments, we generated hADAR1d mutants bearing the hADAR2 5’ binding loop. Initially, we replaced the 5’ binding loop (aa969-aa999) of ADAR1 with the ADAR2 5’ binding loop (aa454-aa479) in a protein that otherwise comprises the human ADAR1 deaminase domain. The resulting chimeric protein was tested with a colorimetric screening assay described by us previously (31,36) and showed no activity. By examining crystal structures of hADAR2-RNA complexes, we found that the first three amino acids of the loop are located close to the active site and may be involved in interactions essential for the respective protein. Thus, we used a shorter region from the ADAR2 5’ binding loop (aa457-aa479) to replace a corresponding region in the ADAR1 5’ binding loop (aa972-aa999) (Figure 5A). We also included the E1008Q mutation shown previously to enhance editing activity (31). The resulting construct was tested with the colorimetric assay and shown to be active, although its activity is significantly lower than the wild-type hADAR1d based on the time taken to show a positive phenotype (Figure S3).
Figure 4.

The ADAR 5’ binding loop (22). A) Crystal structure of hADAR2 deaminase domain bound to dsRNA substrate (22). The editing site nucleotide is flipped out of the RNA duplex. The 5’ RNA binding loop is highlighted in green. B) Close-up view of interactions between the ADAR2 5’ binding loop and the RNA (21,22). Residues F457 (corresponding to F972 in ADAR1), D469, H471, P472, R474, and R477 are highlighted. C) Sequence alignment around the 5’ binding loops of ADAR1s (A1) and ADAR2s (A2) from different organisms. The conservation patterns are labeled in different colors. Purple: conserved in both ADAR1 and ADAR2; red: conserved in ADAR1; green: conserved in ADAR2; black: not conserved.
Figure 5.

A loop swapping experiment shows that the 5’ RNA binding loops contribute to ADAR selectivity. A) Construction of the loop chimera protein. Residues in the 5' binding loop regions are shown. B) hADAR1d E1008Q (2 nM) acting on the HER1 and BDF2 substrates (10 nM). C) hADAR2d E488Q (2 nM) acting on the HER1 and BDF2 substrates (10 nM). D) Loop chimera protein (10 nM) acting on the HER1 and BDF2 substrates (10 nM). Error bar indicates SD, n≥3.
The loop chimera protein was purified and tested in in vitro deamination assays. The activity of this protein was compared to that of hADAR1d E1008Q and hADAR2d E488Q (a counterpart of ADAR1d E1008Q (37)) acting on both the 218 nt HER1 substrate and the BDF2 substrate previously shown to be efficiently deaminated by hADAR2d. Low salt concentration was used in the assay to further increase editing activity of the chimeric protein in vitro (see Materials and Methods section). For direct comparisons, all proteins were assayed under the same conditions (See Materials and Methods section). In order to compare specificities among these proteins, the protein concentrations and reaction time were controlled so that early stages of the reactions were monitored. Deaminated RNAs were analyzed with RT-PCR followed by Sanger sequencing to determine editing efficiencies. Consistent with the editing patterns of the corresponding wild-type proteins, hADAR1d E1008Q prefers the HER1 site over the BDF2 site, while hADAR2d E488Q prefers the BDF2 site over the HER1 site (Figure 5B, C and Figure S5A, B). Importantly, the chimeric protein has a preference for the BDF2 site over the HER1 site (Figure 5D and Figure S5C), indicating that the chimera lost the selectivity of hADAR1d and gained the selectivity of hADAR2d by incorporating the ADAR2 5’ binding loop into the ADAR1 deaminase domain. These results clearly showed a shift of the substrate preference with loop swapping. The change in preference is not likely simply a result of differences in catalytic activities for the two proteins (the chimera is substantially less active) as both wild type ADAR1d and the E1008Q mutant have different catalytic activities but exhibit the same preference for the HER1 site over the BDF2 site (Figure 2A, B and Figure 5A, B).
Alanine scan and Sat-FACS-Seq analysis of the ADAR1 5’ binding loop
With efficient fluorescent reporters for hADAR1d activity in hand, we chose to carry out mutagenesis analysis of its 5’ binding loop. Initially we carried out an alanine scan for sixteen conserved residues in the 5’ loop of hADAR1d (E1008Q) (Figure 6). The resulting alanine mutants were each tested with the HER1 fluorescent reporter and the fluorescence intensities were measured and normalized to the fluorescence generated by the parent construct (hADAR1d E1008Q). The alanine scan revealed that F972, D973, F992, K996 and K999 in this loop are particularly important for editing activity (Figure 6), as the corresponding alanine mutants led to a reduction of more than 50% in fluorescence intensity relative to the parent protein. Alanine mutation of residues K974 and H988 also led to obvious decrease in editing activity to around 50–80% of the parent protein, suggesting that they also play a role in editing activity. The alanine mutants were also tested with the previously described reporter that uses a BDF2-derived substrate RNA (Figure 6) (21). Interestingly, the alanine scan produced similar results with the two different reporters, indicating that the 5’ binding loop of hADAR1 interacts with the HER1 and BDF2 substrates in similar ways, possibly by contacting a surface shared by the two substrates.
In the ADAR2 5’ binding loop, residue D469 contacts residue R477 via ionic interactions and residue F457 provides a platform onto which the guanidinium group of R477 stacks in an apparent cation-π interaction (Figure 4B). These interactions stabilize the loop structure in the conformation necessary to contact RNA (21,22). In analogy to the conformation of ADAR2 5’ binding loop, it is intriguing to speculate that the ADAR1 5’ binding loop is also stabilized in a similar manner, possibly through interactions between the negatively charged residue D973 and one or both of the two positively charged lysine residues, K996 and K999. It is also tempting to suggest that the phenylalanine residues, F972 or F992, may engage in cation-π interactions with the lysine to stabilize the conformation. To test these ideas further, we carried out a Sat-FACS-Seq analysis of the 5’ binding loop of the ADAR1 deaminase domain (21). This approach involves saturation mutagenesis at specific codons for amino acids of interest (Sat), fluorescence-activated cell sorting to fractionate yeast cells with varying levels of signal from a fluorescent reporter of editing activity (FACS) and next-generation sequencing to identify and quantify the ADAR mutants with varying levels of activity (Seq). We generated saturation mutagenesis libraries for hADAR1d (E1008Q) corresponding to residues G969, A970, L971, F972, D973, K974, S975, C976, S977, H988, P990, F992, E993, N994, K996, Q997, G998 and K999. These 18 residues include all of the 16 residues mutated in the alanine scan along with A970 and L971. We screened these libraries using a fluorescent reporter containing the BDF2-derived substrate RNA so that a direct comparison could be made to results of a similar experiment for the hADAR2 5’ binding loop (21).
Yeast cells after induction of expression of hADAR1d and reporter were analyzed by FACS (Figure 7). Cells expressing inactive hADAR1d E912A mutant were used to define background fluorescence, and five gates, R1 through R5, were used for separating cells with various fluorescence intensities. R1 contained cells with background fluorescence and R2 through R5 had cells with above-background fluorescence, in an order of increasing fluorescence intensity from R2 to R5. Cells expressing the inactive E912A mutant mostly fell into the R1 gate (Figure 7A). In contrast, cells expressing the loop library were found in each gate and were collected and cultured, followed by plasmid isolation to obtain R1 through R5 libraries (Figure 7B).
Figure 7.

Fluorescence activated cell sorting for hADAR1d 5’ binding loop library. A) Top: cells expressing the yeGFP reporter and hADAR1d E912A were used to define background fluorescence and above-background fluorescence categories. Bottom: parameters corresponding to each sorting gate. B) Top: cells expressing the yeGFP reporter and hADAR1d loop library were sorted into different categories based on different levels of fluorescence. Bottom: parameters corresponding to each sorting gate.
To analyze the selection for different amino acids at each position, we used barcoded PCR and sequencing with Illumina Miseq (21). The resulting data were decoded for the sample-specific barcode and the internal positional barcode to obtain the abundance of each amino acid for each position in each sample (Table S4). The abundance of variants in R1 through R5 were weighted against their abundance in the input library and the resulting data were used to analyze the distributions of the variants in each fluorescence category (Table S4).
Wild type amino acids are highly enriched in the R5 gate but not in other gates (R5/input library ratios are around 2–4). Using the enrichment levels of amino acids across the 5 gates and the median fluorescence value of populations falling into each gate, we calculated an overall average fluorescence value (Fave) for each amino acid at each position (Table S4). The wild type residue Fave value at each location was normalized to 1 and the other values were adjusted accordingly providing an assessment of the impact of all 20 common amino acids at each of 18 locations in the ADAR1 5’ binding loop (Figure 8). For an integrated picture of the selection at each position within the loop, the distribution of amino acids in R5, which is highly correlated with the Fave values, was expressed as a logo plot (Figure 9A). We set a cutoff for R5/input ratio to be 1 to include variants of highest editing activity. The residues under study show a high level of conservation among ADAR1 sequences from different organisms (Figure 4C). However, the screening results showed that only positions F972 and D973 prefer the wild type amino acid over all others. For the 973 position, aspartic acid (D) is the only amino acid found in the most active clones whereas only aromatic amino acids are tolerated at position 972 (F, W, and Y). At position 996, the wild type residue (K) showed equal activity with the positively charged K996R mutant with all other amino acids excluded at this position (Figure 8, Figure 9A). Thus, the Sat-FACS-Seq analysis clearly demonstrates the importance of F972, D973 and K996 consistent with these residues playing roles similar to F457, D469 and R477 in ADAR2. Interestingly, when the same Sat-FACS-Seq analysis was carried out for the conserved residues in the 5’ binding loop of the ADAR2 deaminase domain, six wild type residues were found to be essential for editing the reporter RNA (F457, D469 and R477 along with H471, P472, and R474) (Figure 9B) (21). H471, P472 and R474 are important for ADAR2 deaminase domain binding to the phosphodiester backbone of the edited strand approximately 10 nt on the 5’ side of the editing site (Figure 4B) (21,22,38). The absence of a similar cluster of critical residues for ADAR1’s 5’ binding loop suggests its deaminase domain lacks this contact to the reporter RNA.
Figure 8.

Normalized average fluorescence corresponding to each of the twenty common amino acids at 18 positions in the hADAR1 5’ binding loop. The star refers to stop codon. For the Q997 position, no codon sequence for cysteine was identified in either the input library or the active clones.
Figure 9.

Logo plots summarizing results of Sat-FACS-Seq analyses of residues present in A) the 5’ binding loop of the ADAR1 deaminase domain and B) the 5’ binding loop of the ADAR2 deaminase domain (21). Positions where the wild type residue is highly preferred are labeled with red stars. Arrows indicate residues that are likely to be functionally equivalent.
Discussion
The importance of the ADAR double stranded RNA binding domains (dsRBDs) in controlling editing efficiency and site selectivity has been studied for many years (12,18–20). For instance, early work from Samuel and coworkers demonstrated that dsRBDs in ADAR1 could be replaced by those from the RNA-dependent protein kinase (PKR) and editing activity was retained for a synthetic duplex RNA substrate but was dramatically reduced for the natural RNA editing substrates tested (19). However, factors that determine the selectivity differences between ADAR1 and ADAR2 have not been fully explored (39,40). Interestingly, early domain swapping experiments suggested that the site selectivity differences between ADAR1 and ADAR2 primarily originate from differences in the deaminase domains (20). How might the ADAR1 and ADAR2 deaminase domains recognize substrate RNAs differently given their high degree of similarity (39% identical, 59% similar over the 400 aa deaminase domains (41)? Of the three main RNA contacting surfaces in the ADAR deaminase domain revealed by hADAR2d-RNA crystal structures, the editing site contact residues and 3’ binding loops are highly similar for ADAR1 and ADAR2 (38). However, their 5’ binding loops are substantially different, suggesting that the 5’ binding loops may dictate selectivity differences between ADAR deaminase domains (Figure 4C) (12,19,22,42). Indeed, the loop swapping experiment described here clearly shows substrate preference tracking with loop identity such that an ADAR1 deaminase domain with an ADAR2 5’ binding loop shows ADAR2 deaminase domain selectivity. The ADAR2 5’ binding loop recognizes the duplex RNA structure 5’ to the editing site by contacting the phosphodiester backbone of each strand of the duplex across a major groove (Figure 4A, B). Formation of this RNA contact surface requires ~10 additional base pairs 5’ from the editing site. How the ADAR1 5’ binding loop binds RNA remains to be established. Since hADAR1d clearly requires less duplex 5’ to the editing site than does hADAR2d, we suggest here that the hADAR1 5’ binding loop adopts a structure that primarily contacts the non-edited strand in this location of the duplex (strand colored blue in Figure 4A, B) instead of using a contact surface that spans the major groove and contacts both strands as does the 5’ binding loop of hADAR2. In support of this hypothesis, our Sat-FACS-Seq results failed to reveal a cluster of ADAR1 residues essential for editing corresponding to H471, P472 and R474 found in the ADAR2 5’ binding loop (Figure 4B). The lack of such a contact would make the hADAR1d reaction less dependent on extension of the duplex in the 5’ direction. However, without a high resolution structure of the ADAR1 deaminase domain bound to RNA, a detailed understanding of how this loop binds RNA remains unknown. In this regard, we expect substrates derived from the HER1 sequence to be useful for future biochemical and structural studies that will define the ADAR1 5’ binding loop-RNA interactions.
The residues varied in our Sat-FACS-Seq analysis are conserved among ADAR1 sequences from different organisms (Figure 4C) yet only a relatively small number require the wild type amino acid or an amino acid with very similar physicochemical properties (e.g. G969, A970, L971, F972, D973, K996 and G998) (Figure 8, Figure 9A). It is possible that the evolutionary conservation of these residues is a result of their role in a function not evaluated in our simple screen for A to I editing in the reporter RNA by the isolated deaminase domain. Such functions could include protein-protein contacts, either to other ADAR1 domains or to other proteins. It is also possible these residues are used for interaction with substrate RNAs that differ in their structure from that found in the reporter used here. In this regard, it would be illustrative to carry out the Sat-FACS-Seq screen with the HER1-based reporter bearing the short 5’ duplex.
Interestingly, for most of the positions varied in our screen, the wild type residue corresponded to the highest or near highest Fave value observed (Figure 8). An exception to this in the H988C mutant that gave a Fave value approximately 50% higher than that of the parent sequence (Figure 8). At this time, we do not know the mechanistic basis for the hyperactivity of the H988C mutant but it could arise from disulfide stabilization of the 5’ binding loop by reaction with nearby C976. Further experiments are necessary to test this hypothesis. It is also worth noting that the K999N mutation is observed in the human disease Aicardi-Goutières Syndrome (43). The mutation of K999 to an alanine led to a decrease in editing activity and the normalized Fave value from the Sat-FACS-Seq analysis of the K999N mutant was a relatively low value of 0.35, indicating that the K999N mutation reduces editing activity in the deaminase domain.
It should be noted that the minimal substrate for hADAR1d described here, represented by the 33 nt HER1 RNA (Figure 3A), is similar to that reported by Herbert et al. (44). These authors showed, using cell-based assays, that a 15 bp RNA duplex with a single base mismatch at the editing site and seven base pairs on each side was efficiently edited by the ADAR1 deaminase domain (44). Such a minimal substrate is not efficiently edited by hADAR2d because the duplex region 5’ to the editing site is too short for full contact (22). Earlier studies also demonstrated that full length ADAR1 preferred editing adenosines close to the 5’ termini of the dsRNA substrates over adenosines close to the 3’ termini, suggesting that a long 3’ duplex is more advantageous for productive editing than a long 5’ duplex (45). The preference for a long 3’ duplex likely arises from binding of dsRBDs present in the full length enzyme. With this understanding of ADAR selectivity, we can re-examine substrates that are known to be selectively edited by ADAR1. For instance, examples are found on the well-studied 5-HT2cR RNA substrate (12). Interestingly, three specific editing sites on this substrate are located 3–5 bp from the nearest 5’ internal or terminal loops that interrupt duplex structures that could be bound by ADARs. These sites are each edited by ADAR1 and ADAR1d, but not by ADAR2 or ADAR2d (12). It was shown that, at these positions, the selectivity of the full length protein was the same as the isolated deaminase domain indicating the deaminase domains alone dictate the selectivity (12). In addition, the ADAR1-selective editing of the NEIL1 mRNA (16,46) , whose editing site is six base pairs from a 5’ terminal loop, could also be explained by superior interaction of the ADAR1d with substrates containing a short 5’ duplex. Also, we found adenosines edited efficiently by ADAR1d but poorly by ADAR2d in our studies of a model RNA editing substrate mimicking the natural GRIA2 mRNA. The hADAR1d-selective sites are located close to a 5’ terminus in this RNA (sites 4 and 5, Figure S7). Overall, these observations together with the other findings in this study suggest that a short stem on the 5’ side of an editing site distinguishes it as a substrate for hADAR1d (and not hADAR2d). In cases where full length ADAR2 editing takes place on substrates with short 5’ duplexes, such as for the GRIA2 R/G site or the Gabra3 site, dsRBD binding to duplex on the 3’ side of the editing site likely compensates for imperfect contacts to the ADAR2 deaminase domain (15,32).
The results described here are important for investigators designing reagents for directed editing applications that use ADAR deaminase domains (24–26). Typically, these systems employ a fusion protein with an ADAR deaminase domain linked to an RNA-targeting element with a guide strand capable of forming a duplex at the target editing site. The design of guide strands in these applications should take into account the longer 5’ duplex needed for efficient reaction by the ADAR2 deaminase domain. If fusion proteins containing the ADAR1 deaminase domain are used, a guide strand forming a shorter 5’ duplex is likely to be effective whereas a longer 5’ duplex duplex would be needed for ADAR2 deaminase domain fusions. In addition, we have identified locations within the ADAR1 5’ binding loop where extensive mutagenesis is tolerated. New deaminase domain constructs with novel RNA-binding elements inserted at this site could produce proteins with unique editing properties.
Supplementary Material
Acknowledgments
Funding
National Institutes of Health (NIH) [GM061115 to P.A.B.]
The authors acknowledge UC Davis DNA Technologies Core and Monica Britton in the UC Davis Bioinformatics Core for technical support with the next-generation sequencing and data analysis and Bridget McLaughlin in the UC Davis Flow Cytometry Shared Resource Laboratory for technical assistance with FACS. We also thank Justin M Thomas for helpful discussion and Anna I Scott for the hADAR2d protein sample.
Footnotes
The authors declare no competing financial interest.
References
- 1.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: form, distribution, and function. Science. 2016;352:1048–1412. doi: 10.1126/science.aad8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol. 2016;12:311–316. doi: 10.1038/nchembio.2040. [DOI] [PubMed] [Google Scholar]
- 4.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. Reversible methylation of m6Am in the 5' cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Zheng Y, Beal PA. Adenosine Deaminases That Act on RNA (ADARs) The Enzymes. 2017;41:215–268. doi: 10.1016/bs.enz.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Cho D, Wang Q, Lai F, Carter K, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6:755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O'Connell MA, Samuel CE, Herbert A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 10.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. Communication -- RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 11.Kallman AM, Sahlin M, Ohman M. ADAR2 A->I editing: site selectivity and editing efficiency are separate events. Nucleic Acids Res. 2003;31:4874–4881. doi: 10.1093/nar/gkg681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 14.Maas S, Melcher T, Herb A, Seeburg PH, Keller W, Krause S, Higuchi M, O'Connell MA. Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J Biol Chem. 1996;271:12221–12226. doi: 10.1074/jbc.271.21.12221. [DOI] [PubMed] [Google Scholar]
- 15.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 16.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2's dsRBMs contributes to RNA-editing selectivity. Chem Biol. 2004;11:1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Lei M, Samuel CE. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc Natl Acad Sci U S A. 2000;97:12541–12546. doi: 10.1073/pnas.97.23.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong S, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–858. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Beal PA. Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res. 2016;44:9872–9880. doi: 10.1093/nar/gkw799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol. 2016;23:426–433. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. RNA editing with CRISPR-Cas13. Science. 2017 doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montiel-González MF, Vallecillo-Viejo IC, Rosenthal Joshua JC. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016;44:e157–e157. doi: 10.1093/nar/gkw738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wettengel J, Reautschnig P, Geisler S, Kahle PJ, Stafforst T. Harnessing human ADAR2 for RNA repair – Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017;45:2797–2808. doi: 10.1093/nar/gkw911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions, and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macbeth MR, Bass BL. Large-scale overexpression and purification of ADARs from Saccharomyces cerevisiae for biophysical and biochemical studies. Methods Enzymol. 2007;424:319–331. doi: 10.1016/S0076-6879(07)24015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eifler T, Pokharel S, Beal PA. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry. 2013;52:7857–7869. doi: 10.1021/bi4006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palavicini JP, Correa-Rojas RA, Rosenthal JJ. Extra double-stranded RNA binding domain (dsRBD) in a squid RNA editing enzyme confers resistance to high salt environment. J Biol Chem. 2012;287:17754–17764. doi: 10.1074/jbc.M112.366005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Havel J, Beal PA. A phenotypic screen for functional mutants of human adenosine deaminase acting on RNA 1. ACS Chem Biol. 2015;10:2512–2519. doi: 10.1021/acschembio.5b00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohlson J, Pedersen JS, Haussler D, Öhman M. Editing modifies the GABA(A) receptor subunit α3. RNA. 2007;13:698–703. doi: 10.1261/rna.349107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gumireddy K, Li A, Kossenkov AV, Sakurai M, Yan J, Li Y, Xu H, Wang J, Zhang PJ, Zhang L, et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat Commun. 2016;7:10715. doi: 10.1038/ncomms10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ, Beal PA. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 2015;43:1123–1132. doi: 10.1093/nar/gku1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephens OM, Yi-Brunozzi HY, Beal PA. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000;39:12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 36.Pokharel S, Beal PA. High-throughput screening for functional adenosine to inosine RNA editing systems. ACS Chem Biol. 2006;1:761–765. doi: 10.1021/cb6003838. [DOI] [PubMed] [Google Scholar]
- 37.Kuttan A, Bass BL. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci U S A. 2012;109:E3295–3304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas J, Beal PA. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays. 2017;39:1600187. doi: 10.1002/bies.201600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deffit SN, Hundley HA. To edit or not to edit: regulation of ADAR editing specificity and efficiency. Wiley Interdiscip Rev RNA. 2016;7:113–127. doi: 10.1002/wrna.1319. [DOI] [PubMed] [Google Scholar]
- 40.Wahlstedt H, Ohman M. Site-selective versus promiscuous A-to-I editing. Wiley Interdiscip Rev RNA. 2011;2:761–771. doi: 10.1002/wrna.89. [DOI] [PubMed] [Google Scholar]
- 41.Fisher AJ, Beal PA. Effects of Aicardi-Goutières syndrome mutations predicted from ADAR-RNA structures. RNA Biol. 2016;14:164–170. doi: 10.1080/15476286.2016.1267097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keegan LP, Leroy A, Sproul D, O'Connell MA. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004;5:209. doi: 10.1186/gb-2004-5-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herbert A, Rich A. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc Natl Acad Sci U S A. 2001;98:12132–12137. doi: 10.1073/pnas.211419898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Li Y, Lin CH, Chan THM, Chow RKK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.