

Summary
Fetal haemoglobin (HbF, α2γ2) induction has long been an area of investigation, as it is known to ameliorate the clinical complications of sickle cell disease (SCD). Progress in identifying novel HbF-inducing strategies has been stymied by limited understanding of gamma (γ)–globin regulation. Genome-wide association studies (GWAS) have identified variants in BCL11A and HBS1L-MYB that are associated with HbF levels. Functional studies have established the roles of BCL11A, MYB, and KLF1 in γ–globin regulation, but this information has not yielded new pharmacological agents. Several drugs are under investigation in clinical trials as HbF-inducing agents, but hydroxycarbamide remains the only widely used pharmacologic therapy for SCD. Autologous transplant of edited haematopoietic stem cells holds promise as a cure for SCD, either through HbF induction or correction of the causative mutation, but several technical and safety hurdles must be overcome before this therapy can be offered widely, and pharmacological therapies are still needed.
Keywords: sickle cell disease, hydroxycarbamide, fetal haemoglobin, gene therapy, globin genes
Haemoglobin structure
Haemoglobin (Hb) is a tetramer protein composed of two alpha (α)-like globin chains and two beta (β)-like globin chains. In humans, the β-like globin locus, found on chromosome 11, is composed of five different genes (HBE1 [ε], HGB2 [γG], HBG1 [γA], HBD [δ] and HBB [β]). The α-like globin locus, found on chromosome 16, is composed of three different genes (HBZ [ζ,] HBA1 [α1] and HBA2 [α2]). In early fetal development, the embryonic globin (ζ2ε2) transitions to fetal haemoglobin (α2γ2, HbF) and then, near birth, fetal globin (α2γ2) begins to be replaced by adult globin (α2β2) (Stamatoyannopoulos, 2005). In normal individuals, HbF comprises <5% of the total Hb at 6 months, and reaches adult levels by 2 years of age (Thein & Menzel, 2009). In normal adults, the major Hb is HbA (α2β2), with 2·5–3·5% HbA2 (α2δ2) and, usually, <1% HbF (α2γ2). Interestingly, this residual HbF is not generally distributed evenly and is concentrated in a small number of erythrocytes termed F-cells. An increase in the number of F-cells is observed during fetal development (peaks at 18–22 weeks of gestation) and after bone marrow (BM) suppression caused by various myelosuppressive therapies (Popat et al, 1977; Eridani & Mosca, 2011).
Globin switching regulation
The β-like globin genes are linearly arranged in the order in which they are expressed during development (HBE1, HBG1/HBG2, HBD/HBB). The 16-kb-long locus control region (LCR) consists of four erythroid specific DNase I hypersensitive sites (DHS) that are made up of clusters of binding sites for transcription activators. The LCR is located 40–60 kb upstream of the β-like genes cluster, and regulates the expression of the β-like genes by looping and direct interaction with the β-like globin promoters (Kim & Dean, 2012). A complex combination of factors, including GATA1, TAL1, E2A, LMO2 and LDB1, is thought to mediate formation of the loop between the LCR and the globin promoters (Breda et al, 2016). β-like globin gene expression is regulated by gene-autonomous control, competition between the globin genes for the LCR (Enver et al, 1990), and repressive elements, prominently trans-acting factors binding to the γ-δ intergenic region, such as BCL11A (Sankaran et al, 2011a).
Genome-wide association (GWAS) and functional follow up studies have identified BCL11A (B-cell lymphoma/leukaemia 11A) as a key negative regulator of HBG (HBG1/HBG2) expression (Uda et al, 2008). The BCL11A protein, a zincfinger transcriptional repressor, occupies critical sites within the β-like globin gene cluster and promotes long-range physical interactions between the LCR and the HBB promoter at the expense of the HBG promoter. It is thought that BCL11A exerts this role by interacting with the erythroid master regulators GATA1, SOX6, ZFPM1/FOG1 and the NuRD repressor complex, which includes HDAC1 and HDAC2 (Bradner et al, 2010). In the absence of BCL11A, upstream enhancers of the β-globin gene cluster, the LCR, co-locate with the transcriptionally activated HBG (Basak & Sankaran, 2016), and γ–globin is expressed. Inhibition or reduction of BCL11A, therefore, is an excellent strategy for HbF induction. However, BCL11A has multiple functions outside of HbF regulation in non-erythroid cells. Indeed, it is essential that the erythroid-specific BCL11A enhancer be used as a target for HbF induction (Bauer et al, 2013, Canver et al, 2015).
Genomic studies have also identified common variants within the intergenic region between GTP-binding elongation factor HBS1L and myeloblastosis oncogene MYB on chromosome 6q that are associated with elevated HbF levels (Thein et al, 2007). The MYB transcription factor is a key regulator of haematopoiesis, erythropoiesis, and HbF levels, and modulates the erythroid traits via two mechanisms: first, directly via activation of KLF1 and through other repressors of HBG, such as the nuclear receptors TR2/TR4; and second, indirectly through alteration of the kinetics of erythroid differentiation. Low MYB levels accelerate erythroid differentiation, leading to release of early erythroid progenitor cells that are larger, and still predominantly express γ-globin (Stadhouders et al, 2014).
KLF1 (Kruppel-like factor 1) is an essential erythroid-specific transcription factor that plays a key role in erythropoiesis. KLF1 influences haemoglobin switching both by directly activating β-globin gene expression in the adult stage through interacting with the HBB promoter, and by increasing the expression of the γ-globin silencer BCL11A by occupying the BCL11A promoter. Naturally occurring heterozygous KLF1 mutations are reported to cause hereditary persistence of fetal haemoglobin (HPFH), and lentiviral knockdown of KLF1 in cultured adult erythroblasts results in increased γ-globin and decreased BCL11A. It is, therefore, possible that pharmacological or gene manipulation of KLF1 levels may induce γ-globin, and be a practical therapy for sickle cell disease (SCD) (Borg et al, 2010; Zhou et al, 2010; Vinjamur et al, 2016). Several other transcription factors have been implicated in HBE/HBG silencing. These include GATA1 in association with FOG1 and the NuRD complex, NF-E4 (Zhou et al, 2004), LRF/ZBTB7A (Masuda et al, 2016), the TR2/TR4/DRED complex, and Ikaros in association with the PYR co-regulatory complex (Bank, 2006).
Developmental haemoglobin switching is also regulated epigenetically, including alterations to higher-order chromatin structure, histone modifications, and DNA methylation. In adult erythroid cells, HBG is associated with increased cytosine methylation, loss of surrounding active histone modifications (such as H3K36me3, H3K27ac), and a decrease in chromatin accessibility compared with fetal erythroid cells (Forsberg et al, 2000; Mabaera et al, 2007; Yin et al, 2007). Elevation of γ-globin expression is observed following genetic or chemical inhibition of DNA methylation (DeSimone et al, 1982) by the methyl-cytosine binding protein MBD2 (Rupon et al, 2006), the histone arginine methyltransferase PRMT5 (Zhao et al, 2009), the histone methyltransferases EHMT1 and EHMT2 (Renneville et al, 2015) and histone deacetylases (HDACs). Furthermore, chromatin regulators are observed to occupy the HBG promoter in the form of repressive complexes including the DNA methylating enzyme DNMT3A, the lysine methyltransferase SUV4-20 h1, the serine/threonine kinase CK2alpha, and components of NuRD (Zhao et al, 2009; Rank et al, 2010).
Lysine-specific demethylases (LSDs) are also involved in HBG silencing. LSD1 demethylates lysine 4 and lysine 9 in histone H3K4 and H3K9 respectively, and diminished HBG promoter DNA methylation (Cui et al, 2015; Rivers et al, 2016). In addition, LSD1, along with several other co-repressors, is involved in TR2/TR4/DRED critical role in silencing HBG (Cui et al, 2011). Inhibition of LSD1 results in increased γ-globin gene expression in transgenic mice, primate models, and cultured primary human erythroid cells. However, since LSD1 is required for normal erythroid maturation, its inhibition can cause adverse effects, such as neutropenia (Cui et al, 2011, 2015; Xu et al, 2013).
MicroRNAs (miRNAs) have also been implicated in globin gene switching through interactions with known γ–globin regulators and through unknown mechanisms. Lin28B proteins and its known target let-7 miRNA family are involved in fetal to adult erythroid development; at least part via the inhibitory effect of LIN28B on BCL11A expression (Lee et al, 2013). MIR486-1-3p has been shown to bind to the BCL11A mRNA and down regulate its expression concomitant with upregulation of γ-globin expression in cultured human erythroid cells (Lulli et al, 2013). Sankaran et al (2011b) also have shown that miR-15a and miR-16-1 can elevate HbF expression by acting via MYB. Several other miRNAs such as miR-221/222, miR-26b, miR-146a and miR-96, are implicated in the regulation of γ-globin gene expression and globin gene switching, although their mechanism of action may not be well known. Well-described molecular mechanisms of HbF regulation are summarized in Fig 1.
Fig 1.
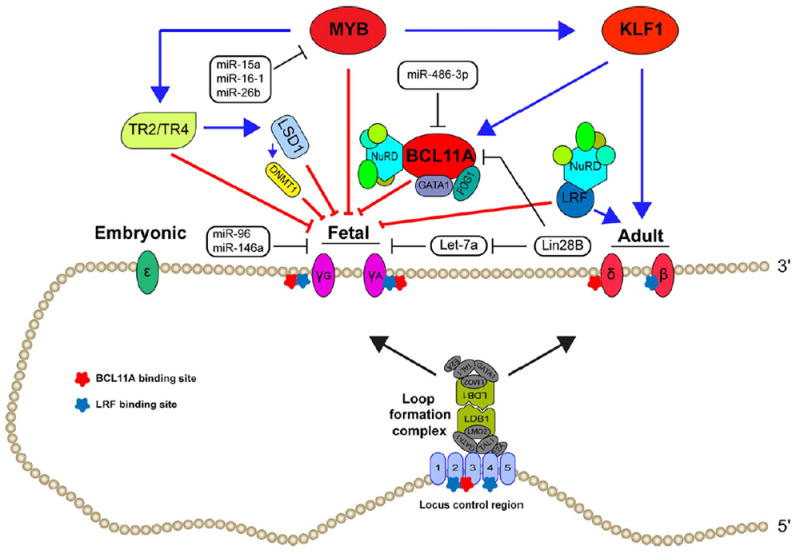
Regulation of fetal haemoglobin (HbF) production. Globin switching cis and trans regulators and chromatin loop formation complex.
Importance of HbF in pathophysiology of SCD
Sickle cell anaemia is a prototypical monogenic disorder, caused by the autosomal recessive inheritance of a single base substitution (A-T) in the first exon of HBB. This substitution results in the replacement of negatively charged, hydrophilic glutamic acid by a hydrophobic amino acid, valine, at position 6 (HBB; glu(E)6 val(A); GAG-GTG; rs334), which leads to defective haemoglobin tetramers that polymerize and aggregate upon deoxygenation, changing flexible, soft discoid red blood cells into stiff, sickle-shaped cells. Biochemical studies have demonstrated that the presence of HbF profoundly delays the polymerization and increases the solubility of HbS under deoxygenated conditions. HbF is even more potent than HbA in terms of polymer interference. X-ray crystallography shows that the sickle haemoglobin polymer is stabilized by the hydrophobic β 6 Val of sickle haemoglobin on one strand binding to a hydrophobic patch at β 85–88 on the adjacent strand. γ-globin has a glutamine rather than a threonine at position 87, which makes this hydrophobic interaction weaker (Nagel et al, 1979). Consequently, HbF tetramers containing γ-chains have a much lower probability of co-polymerizing with the sickle haemoglobin tetramers containing two βS peptides.
Clinically, the importance of HbF levels in SCD was theorized by a paediatrician, Janet Watson, in 1948, well before the mechanistic work summarized above, when she noted that SCD clinical complications were rare before the age of 1 year of age, while HbF levels were still elevated (Watson, 1948). The role of HbF was confirmed by studies describing essentially asymptomatic patients with SCD who co-inherited a HPFH phenotype marked by a substantially elevated HbF level in adulthood. HPFH can be caused by cis-acting factors, such as deletions in the β-globin gene cluster, leading to compensatory increases in γ-globin synthesis in response to decreased or absent γ-globin synthesis, and mutations in the HBG promoter regions or inheritance of HbF modulating quantitative trait loci (QTL), such as HBS1L-MYB intergenic region (6q23) and BCL11A (2p16) (Galarneau et al, 2010; Akinsheye et al, 2011).
As a representation of patients’ ethnic background or geographical area of origin, five major SCD haplotypes have been described, namely Benin (BEN), Bantu or Central African Republic (CAR), Cameroon (CAM), Arab-Indian (ARAB) and Senegal (SEN), in addition to atypical haplotypes (Pagnier et al, 1984; Labie et al, 1985; Srinivas et al, 1988; Lapoumeroulie et al, 1992). Each haplotype is associated with a characteristic average level of HbF (Arab-Indian>Senegal>Benin>Bantu); however, HbF levels varied among patients homozygous for any haplotype. For example, in carriers of Senegal and Saudi-Indian haplotypes, Xmn1 CT restriction site polymorphism (158 bps upstream of HBG2) is associated with high HbF and Gγ-globin (HBG2) levels (Green et al, 1993; Chang et al, 1995; Pandey et al, 2012).
Although the pathophysiology of β-thalassaemia, insufficient or absent production of β-globin chains, is different from SCD, induction of HbF can ameliorate the symptoms at least by alleviating imbalanced non-α to α-globin chain synthesis and its consequent ineffective erythropoiesis and haemolysis caused by precipitation of unpaired α-globin chains. More mechanistic details and studies related to HbF induction in β-thalassaemia can be found in other reviews (Sripichai & Fucharoen, 2016; Thein, 2017).
Pharmacological induction
Hydroxycarbamide
Hydroxycarbamide (also known as hydroxyurea) is the only pharmacological HbF inducer approved for use by the US Food and Drug Administration (in 1998), and the European Medicines Agency (in 2007). Despite recent National Heart, Lung, and Blood Institute (NHLBI) recommendations to offer hydroxycarbamide to all sickle cell anaemia patients over the age of 9 months (Yawn et al, 2014), these approvals apply only to adult SCD patients and patients over 2 years of age, respectively. With its favourable side effect profile, daily oral dosing, and proven efficacy, hydroxycarbamide appeared to be an ideal drug (Charache et al, 1996; Kinney et al, 1999). Several clinical trials have established the efficacy of hydroxycarbamide in preventing pain events, acute chest syndrome, need for blood transfusions and hospitalizations (Wykes & Rees, 2011). A recent paediatric trial demonstrated non-inferiority to chronic blood transfusions for combined hydroxycarbamide and phlebotomy (Ware et al, 2016).
However, long term observation of the Multicenter Study of Hydroxyurea in Patients with Sickle Cell Anemia (MSH) participants found that up to half of adult patients did not exhibit clinical improvement on hydroxycarbamide (Steinberg et al, 2010). While hydroxycarbamide has many beneficial effects, including reduction in red cell adhesion and white blood cell count (Ferster et al, 1996), HbF levels are considered strongly associated with clinical response (Strouse et al, 2008). A recent report demonstrated that HbF response to hydroxycarbamide may decline over time, and leave patients vulnerable to worsening disease complications and increased hospitalizations (Green et al, 2016). A better understanding of this decline in HbF response to hydroxycarbamide in some patients is needed, with clarity on whether this an age-related issue, or related to the duration of hydroxycarbamide use. This question was explored in vitro by Baliga et al (2000). Further investigation into not only the effects of hydroxycarbamide on erythroid progenitors, but also the stem cell niche, may also be relevant.
Despite being a mainstay of SCD therapy in high to moderate resource countries for over 20 years, much is still not known about why some patients produce several times more HbF in response to the drug than others. Hydroxycarbamide is a ribonucleotide reductase inhibitor, causing a reduction in the available intracellular deoxynucleotide triphosphates needed to synthesize DNA in the cell. However, the mechanism by which hydroxycarbamide induces HbF is still not known. Many theories have been proposed; perhaps the most widely accepted theory is that hydroxycarbamide causes a “stress erythropoiesis” response (Galanello et al, 1985; Stamatoyannopoulos et al, 1985). Briefly, this theory postulates that altering erythroid differentiation kinetics causes a more immature erythroid progenitor, with HbF turned on, to exit the BM. This hypothesis is supported by the fact that other cytotoxic agents also induce HbF, such as DNA methyltransferase (DNMT) inhibitors, 50azacytodine and HDAC inhibitors. Another proposed mechanism for HbF induction by hydroxycarbamide involves induction of the nitric oxide (NO)-cyclic guanosine monophosphate (cGMP) pathway (Cokic et al, 2003; Lou et al, 2009). In this model, hydroxycarbamide is converted to NO, NO stimulates intracellular soluble guanylate cyclase which in turn, increases cGMP and induces HbF via cGMP-dependent protein kinase (PKG) (Almeida et al, 2012). Individuals with variants in BCL11A have been reported to have a higher HbF response to hydroxycarbamide (Friedrisch et al, 2016), and hydroxycarbamide has been shown to decrease BCL11A expression in erythroid cells (Grieco et al, 2015), suggesting a role for BCL11A in HbF induction by hydroxycarbamide.
DNA methyltransferase (DNMT) inhibitors
In vertebrates, DNA methylation occurs at cytosine residues that precede guanine (CpG dinucleotides) (Saunthararajah & DeSimone, 2004). Three DNMTs have been identified in mammalian cells, the genes of which are all expressed in haematopoietic cells, and while DNMT1 is responsible for maintenance of existing methylation patterns, DNMT3A and DNMT3B can methylate de novo previously unmethylated sequences (Bestor, 2000).
Direct modification of the DNA by methylation of its cytidine residues is associated with transcriptional repression or silencing of genes (van der Ploeg & Flavell, 1980). Indeed, it has been shown that the overall methylation patterns of the HBG and HBB promoter regions are inversely related to gene expression and are near the known transcription factor binding sites (Mabaera et al, 2007). These are consistent with the findings that DNMT inhibition, either through knockdown of DNMT1 in human erythroid precursors, knockout in transgenic mice, or chemical inhibitors, results in reactivation of fetal haemoglobin expression (Charache et al, 1983; Xu et al, 2013).
The DNMT inhibitors, azacytidine (5-Aza) and decitabine (20deoxy-5-azacytidine), are analogues of cytidine that incorporate into DNA and inactivate the DNMT enzymes that methylate DNA (Gnyszka et al, 2013). 5-Aza was one of the first HbF-inducing agents investigated, first in anaemic baboons, then in patients with haemoglobinopathies. However, concerns for 5-Aza’s carcinogenic potential and increased interest in hydroxycarbamide ended further clinical investigation. Decitabine is incorporated specifically into DNA, unlike 5-Aza, which is incorporated into both DNA and RNA (Lavelle, 2004). It is a more potent inhibitor of DNMT, in addition to having a more favourable safety profile than 5-Aza.
Although DNMT inhibitors have shown efficacy in HbF induction, they are not widely used. Decitabine is limited by its inability to be intestinally absorbed without suppression of cytidine deaminase. Oral administration of tetrahydrouridine (THU) one hour before decitabine can overcome this limitation, and this combined therapy is the subject of a phase I trial (NCT01685515). There is also concern that chronic decitabine usage will lead to an increased risk of cancer; the mutagenic effect of decitabine has been demonstrated in cell lines like L5178Y mouse lymphoma cells, colonic cells from mice transgenic for Escherichia coli lacI, and Salmonella typhimurium tester strain TA100 (Marquardt & Marquardt, 1977). In addition, there are concerns about cytogenetic instability secondary to DNA hypomethylation, given that profound DNA hypomethylation, produced by knocking out DNMT genes, causes chromosome instability and carcinogenesis in mouse models. Finally, decitabine treatment could reactivate genes that may favour cellular transformation. However, in practice, decitabine has been noted to reactivate expression of a range of tumour-suppressor genes (Leone et al, 2003; Karpf et al, 2004).
Histone deacetylase inhibitors
The zinc-dependent HDACs are a group of enzymes that remove acetyl groups, primarily from histone lysine residues; acetylated chromatin is more accessible to interacting proteins (Tse et al, 1998). In this context, site-specific acetylation or deacetylation leads to locally restricted activation or repression of transcription, respectively. Inhibition of HDAC activity results in elevated histone acetylation which has been associated with increased chromatin accessibility and gene expression (Forsberg et al, 2000; Cao et al, 2004). A well-characterized example is provided by the beta-globin locus, which reveals broad acetylation throughout domains with defined boundaries as a function of transcriptional competence. In vitro studies in an erythroleukaemia cell line (K562) and in HSCs have shown that HDAC inhibitors can induce HbF. The most likely mechanism for HbF induction by HDAC inhibitors is thought to be increased acetylation of the HBG promoter, leading to increased transcription.
Butyrate was the earliest HDAC inhibitor investigated as a HbF inducer. Butyrate increases the transcription rate of the HBG genes, as well as the translation of HBG1/HBG2 mRNA (Weinberg et al, 2005). In clinical trials, intravenous infusions of arginine butyrate and oral administration of sodium phenylbutyrate increased HbF in patients with SCD and β-thalassaemia; however, oral butyrate compounds only produced a modest elevation of HbF and compliance was poor (Sher et al, 1995). Intravenous administration required pulse treatments and HbF response was variable. Panobinostat is a pan-HDAC inhibitor reported to have greater potency than sodium butyrate (Bradner et al, 2010); it is listed in clinicaltrials. gov as an active study, not currently recruiting patients (Table I).
Table I.
Clinical trials involving HbF induction in SCD currently posted in Clinicaltrials.gov.
| Trial | NCT identifier | Status | Reported results |
|---|---|---|---|
| Study of decitabine and THU in patients with SCD | NCT01685515 | Active, not recruiting | N/A |
| Gum arabic as a HbF agent in SCA | NCT02467257 | Completed | None |
| Hydroxyurea and erythropoietin to treat SCA | NCT00270478 | Completed | None |
| Study to determine the maximum tolerated dose, safety and effectiveness of pomalidomide for patients with SCD | NCT01522547 | Completed | None |
| Effects of HQK-1001 in patients with SCD | NCT01601340 | Completed | None |
| Decitabine for high-risk SCD | NCT01375608 | Completed | 4·8% average HbF increase |
| Effectiveness of arginine as a treatment for SCA | NCT00513617 | Completed | None for HbF induction |
| Fetal haemoglobin induction treatment with metformin | NCT02981329 | Recruiting | N/A |
| Gene transfer for patients with SCD | NCT02186418 | Recruiting | N/A |
| Study of panobinostat in patients with SCD | NCT01245179 | Recruiting | N/A |
| A study to evaluate safety, pharmacokinetic, and biological activity of INCB059872 | NCT03132324 | Recruiting | N/A |
| Efficacy of vorinostat to induce HbF in SCD | NCT01000155 | Terminated | None; closed for slow accrual |
HbF, fetal haemoglobin; N/A, not applicable; NCT, National Clinical Trial registry (https://clinicaltrials.gov/); None, closed or completed without reported results; SCA, sickle cell anaemia; SCD, sickle cell disease; THU, tetrahydrouridine.
Like butyrate and panobinostat, a variety of non-selective HDAC inhibitors have been used successfully to induce HbF in preclinical studies (Witt et al, 2003; Cao & Stamatoyannopoulos, 2006). Although these agents induce γ-globin expression in culture, many are ineffective in vivo. In addition, the use of non-selective HDAC inhibitors in the clinical setting has been associated with significant toxicity and adverse events (Subramanian et al, 2010; Guha, 2015). Therefore, development of potent and selective HDAC1/2 inhibitors leading to HbF induction with less toxicity would represent a promising therapeutic approach for the treatment of SCD and β-thalassaemia.
Thalidomide derivatives
Pomalidomide and lenalidomide (derivatives of thalidomide) are antiangiogenic and immunomodulatory agents used in relapsed and refractory multiple myeloma; pomalidomidetreated multiple myeloma patients exhibit an increase in γ-globin production (Dulmovits et al, 2016). In a humanized mouse model of SCD, pomalidomide increased the level of HbF production comparable with hydroxycarbamide treatment but without myelosuppressive effects (Meiler et al, 2011). However, when pomalidomide was given in combination with hydroxycarbamide, the HbF-inducing activity of both drugs was lost. In addition, treatment of human HSCs with pomalidomide and lenalidomide in a two-phase erythroid differentiation liquid culture significantly stimulated the proliferation of HSCs and induced HbF by modulating the transcription of HBB and HBG (Moutouh-de Parseval et al, 2008).
A recent publication confirmed pomalidomide can induce a fetal-like erythroid differentiation programme, leading to a reversion of HBG silencing in adult human erythroblasts by selectively and differentially affecting the main regulators of γ- and β-globin synthesis, namely BCL11A, SOX6, IKZF1, KLF1, and LSD1 (Dulmovits et al, 2016). However, given the toxicity profile of pomalidomide, including potential neuropathy, it is unlikely it will become a widely used therapy for individuals with haemoglobinopathies.
Metformin
Another emerging HbF-inducing agent is metformin. Whole exome sequencing and analysis of rare variants in patients with SCD suggested that FOXO3 was a positive regulator of HbF. Functional studies in human HSCs confirmed the association (Sheehan, 2013), and lead to the identification of metformin, a FOXO3 inducer, as a potential HbF-inducing agent. It is now under investigation in a pilot clinical trial in patients with haemoglobinopathies (NCT02981329). The mechanism by which metformin induces HbF remains to be elucidated but may be related to metabolic manipulation in the erythroid progenitor. The possible role of metabolic manipulation in haemoglobin switching was suggested by the observation made by Perrine et al (1985) over 30 years ago, that the infants of diabetic mothers showed a delay in fetal haemoglobin switch.
Gene therapy
The only curative therapy for SCD is a haematopoietic stem cell transplant (HSCT), typically from a matched related donor, which is available to only about 15% of patients (Mentzer et al, 1994). Morbidity and mortality from HSCT increase significantly when using matched but unrelated or haploidentical donors. A recent prospective study of unrelated donor HSCT in SCD concluded that, without modifications to existing regimens, this therapy is not safe for widespread adoption (Shenoy et al, 2016). Gene therapy using autologous HSCs would avoid the limitation of needing a matched related donor, eliminate the need for immunosuppressive drugs, and avoid all risk of graft-versus-host disease.
There are several possible gene editing strategies to ameliorate SCD: (i) correction of the causative A-T point mutation in HBB, (ii) induction of HbF through introduction of a HPFH deletion (Traxler et al, 2016), down regulation of BCL11A, or double strand breaks, and (iii) gene addition of HBB, HBG, or anti-sickling β-globin cassette (Urbinati et al, 2015), among which correction of the A-T mutation or producing high enough levels of HbF could be curative (Renneville et al, 2015; Masuda et al, 2016). Another question is what technology to use; lentiviral vectors have been used extensively in clinical trials, and the first SCD patient reported to have successfully been treated with gene therapy received an ex-vivo lentivirus-altered autologous transplant on the HGB-205 study (Ribeil et al, 2017). Two other active clinical trials use a lentiviral vector, one to express γ–globin, the other to introduce a β-globin transgene that contains three anti-sickling mutations (NCT02186418, NCT02151526). Other approaches include zinc finger nucleases (ZFNs), transcription activator like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeat (CRISPR) associated nuclease Cas9.
Lentivirus-based gene editing to induce HbF in SCD
As mentioned above, two of the three lentiviral gene therapy strategies for SCD under active investigation do not attempt to increase γ–globin, but instead seek to correct the HBB sickle mutation, or produce a unique, “anti-sickling” beta globin. All three use a modified LCR to express genes in an erythroid-specific manner, and the HBB promoter to express the various β-globin-like genes. The non γ-globin approaches do have the advantage of being easily distinguished from endogenous β-globin production; HbF may be elevated due to stress erythropoiesis, obscuring the contribution of the γ–globin based editing strategy, but these strategies are outside of the scope of this review. Cincinnati Children’s Medical Center has an open protocol to perform autologous stem cell transplants of ex-vivo edited HSCs following reduced intensity conditioning with melphalan, but no results have been reported to date (NCT02186418). This protocol uses ex-vivo lentiviral delivery of HBG. In order to avoid excess unpaired β-like globin chains, they seek to preferentially enrol HbSβ0 patients. Proof of principle of lentiviral vector delivery of HBG under control of the HBB promoter was shown in the Berkeley SCD mouse (Perumbeti et al, 2009).
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (CRISPR/Cas9)
With the advancement of CRISPR/Cas9 technology (Hsu et al, 2013), effective gene correction of sickling mutation in HBB of SCD patient-derived HSCs using CRISPR/Cas9 has been demonstrated (Dever et al, 2016; DeWitt et al, 2016). But significant challenges in bringing this technology to a human clinical trial remain - some common to all CRISPR Cas9 applications, and some SCD-specific. First, off-target effects remain a major issue. Of concern is the fact that no published studies performed unbiased genome- wide off-target analysis of CRISPR/Cas9 edited HSCs to rigorously identify all genomic changes. Second, the efficacy of gene editing for treating SCD needs to be increased significantly, as only a low-level of engraftment of gene-edited SCD HSCs without selection has been shown (DeWitt et al, 2016). Finally, a particular challenge in using CRISPR/Cas9 gene editing in SCD is the source of autologous CD34+ cells. Patients with SCD cannot be mobilized with granulocyte colony-stimulating factor, limiting the number of available patient CD34+ cells to what can be obtained through BM harvest. The number of CD34+ cells needed is also greater than for an allogeneic transplant, as ex vivo gene editing is associated with cell death, and edited cells show lower levels of engraftment. Addressing these issues will be essential to translating the gene-correction based treatment of SCD into clinical practice.
Several strategies for HbF induction are possible using CRISPR-Cas9. Traxler et al (2016) deleted a 13-nucleotide sequence present in the promoters of the HBG1 and HBG2 genes in HSCs from SCD patients via microhomologymediated end-joining (MMEJ), recapitulating a known mutation that causes HPFH. This resulted in production of red blood cells (RBCs) with increased HbF levels sufficient to inhibit the pathological hypoxia-induced RBC morphology found in SCD (Traxler et al, 2016).
CRISPR-Cas9 targeting of HbF regulators, such as KLF1, can lead to inhibition of the γ- to β-globin switching process (Shariati et al, 2016). However, there is concern that these transcription factors have essential roles in native erythropoiesis and/or non-erythroid contexts. For example, BCL11A, a potent HbF silencer, has a significant role in lymphoid and neuronal development, and BCL11A knockout mice are perinatal lethal (Liu et al, 2003; John et al, 2012). In this regard, instead of complete knockout of BCL11A, approaches focusing on the deletion of BCL11A-binding sequences in the proximity of HBG are promising. Another approach is repression of erythroid-specific BCL11A (Bauer et al, 2013) as tried successfully by Canver et al (2015). In order to interrupt the BCL11A effect on fetal haemoglobin silencing, they used CRISPR-Cas9 technology in HUDEP-2 cells to delete a 12-kb BCL11A enhancer, located in the first intron of BCL11A, by introduction of a pair of chimeric single guide RNAs (sgRNAs). This site is targeted by GATA1 and is essential for BCL11A expression only in erythroid cells. The result was a significant increase in expression of γ-globin and HbF protein, suggesting that perturbation of defined critical sequences within the BCL11A enhancer may result in HbF levels exceeding a clinical threshold required to ameliorate the SCD (Canver et al, 2015).
Transcription activator-like effector nucleases (TALEN)
A TALEN monomer comprises a DNA-binding domain (DBD) formed by tandem repeats of about 34 amino acids and each repeat recognizes one nucleotide. The DBD is fused to FokI nuclease and a TALEN pair is required for the formation of a site-specific double-strand break within the spacer region. While the therapeutic potential of TALEN in SCD has been investigated in targeted genetic correction of the SCD mutation, Humbert and Kiem (2015) recruited TALENs to perform double-strand DNA break-induced mutagenesis and inactivate BCL11A function in inducible HSCs. They reported up to 30% gene editing efficiency in human and nonhuman primate CD34+ cells by TALEN mRNAs electroporation targeting exon 2 of BCL11A in vitro. While colony-forming efficiency was slightly lower in BCL11A-edited CD34+ cells, lineage differentiation potential was unchanged. Erythroid differentiation of CD34+ cells in culture showed an increased γ- to β-globin ratio in both human and primate Bcl11a-modified cells as compared to control cells, and sustained HbF elevation at almost a year post-transplant was achieved (Humbert & Kiem, 2015).
Zinc finger nucleases (ZFN)
ZFN was the first gene editing technology used to correct the single point mutation in HBB. This system has been used to correct the sickle mutation in patient-derived induced pluripotent stem cells and CD34+ HSCs by separate groups. Chang et al (2017) used zinc finger nucleases system to disrupt BCL11A in human HSCs, either within exon 2 or at the GATAA motif in the intronic erythroid-specific enhancer. Both targeting strategies upregulated γ-globin expression in erythroid cells to the levels predicted to inhibit HbS polymerization. However, complete inactivation of BCL11A resulting from bi-allelic frameshift mutations in BCL11A exon 2 adversely affected erythroid enucleation. In contrast, bi-allelic disruption of the GATAA motif in the erythroid enhancer of BCL11A did not negatively affect enucleation. Furthermore, BCL11A exon 2-edited BM CD34+ cells demonstrated a significantly reduced engraftment potential in immunodeficient mice. Such an adverse effect on HSC function was not observed upon BCL11A erythroid-enhancer GATAA motif editing because enhancer-edited CD34+ cells achieved robust long-term engraftment and gave rise to erythroid cells with elevated levels of HbF when chimeric BM was cultured ex vivo (Chang et al, 2017).
HbF induction by artificial transcription factors
Artificial transcription factors (ATFs) are synthetic proteins designed to modulate endogenous gene expression by interacting with specific DNA sequence. For example, programmable nucleases have the potential to be used as transcription factors if attached to appropriate effector domains (activation domains, epigenetic modifiers, etc.) and can correct selected DNA sequences. Graslund et al (2005) designed a zinc fingerbased protein called gg1-VP64, consisting of a DBD, recognizing a specific sequence proximal to the −117 position of the HBG promoter, and a transcriptional activator domain VP64. Delivery of gg1-VP64 to K562 cells resulted in up to a 16-fold increase of HbF compared to the native K562 cell line. To demonstrate that the regulation of γ-globin expression by gg1- VP64 was direct, they used gg1-KRAB, a designed repressor bearing the same DNA recognition domain, which resulted in near complete repression of γ-globin expression upon delivery of the repressor to K562 cells, providing strong evidence for direct target HBG regulation as opposed to non-specific binding of the transcription factor that might lead to erythroid differentiation of K562 cells and subsequent indirect upregulation of γ-globin expression (Graslund et al, 2005). Gg1-VP64 was then tested in cytokine-mobilized peripheral blood CD34+ cells, which increased HbF levels from 2 to 20% of the total haemoglobin (Wilber et al, 2010). Later, in vivo investigation directed by Costa et al (2012) showed five-fold induction of HbF in gg1-VP64 β-YAC double-transgenic mice.
Chromatin structural alterations
For a long time, DNA organization and its effects on how cells access, read, and interpret genetic information have been an area of interest. The three-dimensional organization of chromatin to create chromosomal contacts are critical in the regulation of gene expression (Dekker et al, 2013), and there is increasing data suggesting that the folding of chromatin within the nucleus are key determinants of several gene expression programs (Krivega & Dean, 2016). These concepts have been most thoroughly demonstrated at the human β-like globin gene LCR, which regulates the expression of the distant β-like globin genes through formation of a long-range chromatin loop (Kim & Dean, 2012). Using lentiviral vectors expressing an artificial zinc finger (ZF) protein directed against the γ-globin promoter and fused to the looping factor LDB1, Deng et al (2014) developed an interesting approach for repressing γ-globin expression which forced the formation of a loop between the LCR and the HBG promoters. The result was re-activation of endogenous γ-globin expression and concomitant reduction of sickle β-globin levels in primary erythroid cells derived from adult HSCs, enough to reduce the proportion of sickling RBCs derived from human SCD HSCs in vitro (Deng et al, 2014). Breda et al (2016) used a similar strategy and reported that treatment with ZF-Ldb1 raised HbF in adult sickle cells to a higher level than pharmacological inducers. They showed that even relatively low expression level of ZF-Ldb1 was enough to activate the HBG genes and allow sustained HbF production. The short ankyrin promoter driving ZF-LDB1 expression will probably enable the production of a relatively high-titre lentiviral vector. This may represent a considerable advantage over lentiviral vectors based on a classic additive gene-therapy approach, like those carrying curative HBB/HBG, which require additional large genomic elements to achieve therapeutic expression levels. The therapeutic implication of this strategy needs more preclinical in vivo studies to address safety and specificity (Breda et al, 2016).
Ongoing clinical trials and future directions
A survey of clinical trials with the key words “sickle cell disease” and “fetal haemoglobin”, omitting trials designed to optimize hydroxycarbamide use, are summarized in Table I. All but one are studying pharmacological agents; one is a gene therapy trial. There are other gene therapy trials underway, but their intent is not to induce HbF. Identifying novel HbFinducing agents is challenging; there are few in vitro systems in which to screen compounds, and animal models have limited utility. The agents listed in Table I have made the leap to a clinical trial; that is already a significant achievement. However, notably, there are many closed studies with no published or reported results, either because there was no significant effect, or the study was unable to be completed. Until the recent approval of L-glutamine as an FDA approved agent, there have been no other agents approved by the FDA for the treatment of SCD since hydroxycarbamide. The results of the phase III clinical trial (NCT01179217) that lead to L-glutamine’s approval have not been published at the time of this article’s submission, and its impact on HbF, if any, has not been released, although the effect of L-glutamine on HbF was a secondary endpoint of the trial.
Genomic sequencing revealed the roles of BCL11A and HBS1L-MYB in γ–globin regulation. Further large scale whole genome sequencing of thousands of patients with SCD is underway as part of the Transcriptomics in Precision Medicine (TOPMed) NHLBI initiative. It is very possible that this work will identify other regulators of γ–globin, more easily “druggable” than BCL11A, or new targets for gene therapy. Gene therapy is a very promising strategy, and may lead to a cure for individuals in high resource countries in the next 5–10 years, provided there are no unexpected setbacks. Despite our hope for autologous stem cell transplant of edited HSC as a cure, we must not neglect the search for inexpensive, well tolerated pharmacological agents that can be used in low resource countries where SCD is endemic. It is essential that we have several therapeutics and strategies to offer our patients so that they can make choices that fit their health status, environment, pharmacogenomic status and personal preferences.
Acknowledgments
VAS is supported by the National Heart, Lung and Blood Institute, 1K08DK110448-01 award.
Footnotes
Author contribution
AP and VAS reviewed the literature, wrote, and edited the paper.
The authors declare no competing interests.
References
- Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, Chui DH, Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, Conran N, Frenette PS. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood. 2012;120:2879–2888. doi: 10.1182/blood-2012-02-409524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga BS, Pace BS, Chen HH, Shah AK, Yang YM. Mechanism for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. American Journal of Hematology. 2000;65:227–233. doi: 10.1002/1096-8652(200011)65:3<227::aid-ajh9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Bank A. Regulation of human fetal hemoglobin: new players, new complexities. Blood. 2006;107:435–443. doi: 10.1182/blood-2005-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A, Sankaran VG. Regulation of the fetal hemoglobin silencing factor BCL11A. Annals of the New York Academy of Sciences. 2016;1368:25–30. doi: 10.1111/nyas.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Human Molecular Genetics. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, Cassar W, Galdies R, van Ijcken W, Ozgur Z, Gillemans N, Hou J, Bugeja M, Grosveld FG, von Lindern M, Felice AE, Patrinos GP, Philipsen S. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nature Genetics. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, Lin K, Shen JP, Kwiatkowski NP, Gheldof N, Dekker J, DeAngelo DJ, Carr SA, Schreiber SL, Golub TR, Ebert BL. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12617–12622. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breda L, Motta I, Lourenco S, Gemmo C, Deng W, Rupon JW, Abdulmalik OY, Manwani D, Blobel GA, Rivella S. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 2016;128:1139–1143. doi: 10.1182/blood-2016-01-691089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, Luc S, Kurita R, Nakamura Y, Fujiwara Y, Maeda T, Yuan GC, Zhang F, Orkin SH, Bauer DE. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Stamatoyannopoulos G. Histone deacetylase inhibitor FK228 is a potent inducer of human fetal hemoglobin. American Journal of Hematology. 2006;81:981–983. doi: 10.1002/ajh.20676. [DOI] [PubMed] [Google Scholar]
- Cao H, Stamatoyannopoulos G, Jung M. Induction of human gamma globin gene expression by histone deacetylase inhibitors. Blood. 2004;103:701–709. doi: 10.1182/blood-2003-02-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Smith KD, Moore RD, Serjeant GR, Dover GJ. An analysis of fetal hemoglobin variation in sickle cell disease: the relative contributions of the X-linked factor, beta-globin haplotypes, alpha-globin gene number, gender, and age. Blood. 1995;85:1111–1117. [PubMed] [Google Scholar]
- Chang KH, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, Liu M, Liu Y, Vieira BF, Sun C, Hong VP, Zhang M, Yang X, Reik A, Urnov FD, Rebar EJ, Holmes MC, Danos O, Jiang H, Tan S. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow- derived CD34+ hematopoietic stem and progenitor cells. Molecular Therapy Methods & Clinical Development. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Dover G, Smith K, Talbot CC, Jr, Moyer M, Boyer S. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:4842–4846. doi: 10.1073/pnas.80.15.4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Barton FB, Moore RD, Terrin ML, Steinberg MH, Dover GJ, Ballas SK, McMahon RP, Castro O, Orringer EP. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–326. doi: 10.1097/00005792-199611000-00002. [DOI] [PubMed] [Google Scholar]
- Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, Schechter AN. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. The Journal of Clinical Investigation. 2003;111:231–239. doi: 10.1172/JCI16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa FC, Fedosyuk H, Neades R, de Los Rios JB, Barbas CF, 3rd, Peterson KR. Induction of fetal hemoglobin in vivo mediated by a synthetic gamma-globin zinc finger activator. Anemia. 2012;2012:507894. doi: 10.1155/2012/507894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Kolodziej KE, Obara N, Amaral-Psarris A, Demmers J, Shi L, Engel JD, Grosveld F, Strouboulis J, Tanabe O. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic beta-type globin promoters in differentiated adult erythroid cells. Molecular and Cellular Biology. 2011;31:3298–3311. doi: 10.1128/MCB.05310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Lim KC, Shi L, Lee M, Jearawiriyapaisarn N, Myers G, Campbell A, Harro D, Iwase S, Trievel RC, Rivers A, DeSimone J, Lavelle D, Saunthararajah Y, Engel JD. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood. 2015;126:386–396. doi: 10.1182/blood-2015-02-626259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, Blobel GA. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone J, Heller P, Hall L, Zwiers D. 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:4428–4431. doi: 10.1073/pnas.79.14.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg KI, Porteus MH. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Muñoz DP, Boffelli D, Kohn DB, Walters MC, Carroll D, Martin DI, Corn JE. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulmovits BM, Appiah-Kubi AO, Papoin J, Hale J, He M, Al-Abed Y, Didier S, Gould M, Husain-Krautter S, Singh SA, Chan KW, Vlachos A, Allen SL, Taylor N, Marambaud P, An X, Gallagher PG, Mohandas N, Lipton JM, Liu JM, Blanc L. Pomalidomide reverses gamma-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood. 2016;127:1481–1492. doi: 10.1182/blood-2015-09-667923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enver T, Raich N, Ebens AJ, Papayannopoulou T, Costantini F, Stamatoyannopoulos G. Developmental regulation of human fetal-to-adult globin gene switching in transgenic mice. Nature. 1990;344:309–313. doi: 10.1038/344309a0. [DOI] [PubMed] [Google Scholar]
- Eridani S, Mosca A. Fetal hemoglobin reactivation and cell engineering in the treatment of sickle cell anemia. Journal of Blood Medicine. 2011;2:23–30. doi: 10.2147/JBM.S14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, Devalck C, Fondu P, Toppet M, Sariban E. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood. 1996;88:1960–1964. [PubMed] [Google Scholar]
- Forsberg EC, Downs KM, Christensen HM, Im H, Nuzzi PA, Bresnick EH. Developmentally dynamic histone acetylation pattern of a tissue-specific chromatin domain. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14494–14499. doi: 10.1073/pnas.97.26.14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrisch JR, Sheehan V, Flanagan JM, Baldan A, Summarell CC, Bittar CM, Friedrisch BK, Wilke II, Ribeiro CB, Daudt LE, da Rocha Silla LM. The role of BCL11A and HMIP-2 polymorphisms on endogenous and hydroxyurea induced levels of fetal hemoglobin in sickle cell anemia patients from southern Brazil. Blood Cells, Molecules, & Diseases. 2016;62:32–37. doi: 10.1016/j.bcmd.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanello R, Veith R, Papayannopoulou T, Stamatoyannopoulos G. Pharmacologic stimulation of Hb F in patients with sickle cell anemia. Progress in Clinical and Biological Research. 1985;191:433–445. [PubMed] [Google Scholar]
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Finemapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nature Genetics. 2010;42:1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Research. 2013;33:2989–2996. [PubMed] [Google Scholar]
- Graslund T, Li X, Magnenat L, Popkov M, Barbas CF., 3rd Exploring strategies for the design of artificial transcription factors: targeting sites proximal to known regulatory regions for the induction of gamma-globin expression and the treatment of sickle cell disease. Journal of Biological Chemistry. 2005;280:3707–3714. doi: 10.1074/jbc.M406809200. [DOI] [PubMed] [Google Scholar]
- Green NS, Fabry ME, Kaptue-Noche L, Nagel RL. Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia. American Journal of Hematology. 1993;44:145–146. doi: 10.1002/ajh.2830440214. [DOI] [PubMed] [Google Scholar]
- Green NS, Manwani D, Qureshi M, Ireland K, Sinha A, Smaldone AM. Decreased fetal hemoglobin over time among youth with sickle cell disease on hydroxyurea is associated with higher urgent hospital use. Pediatric Blood & Cancer. 2016;63:2146–2153. doi: 10.1002/pbc.26161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco AJ, Billett HH, Green NS, Driscoll MC, Bouhassira EE. Variation in gamma-globin expression before and after induction with hydroxyurea associated with BCL11A, KLF1 and TAL1. PLoS One. 2015;10:e0129431. doi: 10.1371/journal.pone.0129431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M. HDAC inhibitors still need a home run, despite recent approval. Nature Reviews Drug Discovery. 2015;14:225–226. doi: 10.1038/nrd4583. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert O, Kiem H-P. Long-term increase in fetal hemoglobin expression in nonhuman primates following transplantation of autologous Bcl11a nuclease-edited HSCs. Blood. 2015;126:2035–2035. [Google Scholar]
- John A, Brylka H, Wiegreffe C, Simon R, Liu P, Juttner R, Crenshaw EB, 3rd, Luyten FP, Jenkins NA, Copeland NG, Birchmeier C, Britsch S. Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development. 2012;139:1831–1841. doi: 10.1242/dev.072850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine. Molecular Pharmacology. 2004;65:18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- Kim A, Dean A. Chromatin loop formation in the beta-globin locus and its role in globin gene transcription. Molecules and Cells. 2012;34:1–5. doi: 10.1007/s10059-012-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney TR, Helms RW, O’Branski EE, Ohene-Frempong K, Wang W, Daeschner C, Vichinsky E, Redding-Lallinger R, Gee B, Platt OS, Ware RE. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Pediatric Hydroxyurea Group Blood. 1999;94:1550–1554. [PubMed] [Google Scholar]
- Krivega I, Dean A. Chromatin looping as a target for altering erythroid gene expression. Annals of the New York Academy of Sciences. 2016;1368:31–39. doi: 10.1111/nyas.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labie D, Pagnier J, Lapoumeroulie C, Rouabhi F, Dunda-Belkhodja O, Chardin P, Beldjord C, Wajcman H, Fabry ME, Nagel RL. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:2111–2114. doi: 10.1073/pnas.82.7.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapoumeroulie C, Dunda O, Ducrocq R, Trabuchet G, Mony-Lobe M, Bodo JM, Carnevale P, Labie D, Elion J, Krishnamoorthy R. A novel sickle cell mutation of yet another origin in Africa: the Cameroon type. Human Genetics. 1992;89:333–337. doi: 10.1007/BF00220553. [DOI] [PubMed] [Google Scholar]
- Lavelle DE. The molecular mechanism of fetal hemoglobin reactivation. Seminars in Hematology. 2004;41:3–10. doi: 10.1053/j.seminhematol.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Lee YT, de Vasconcellos JF, Yuan J, Byrnes C, Noh SJ, Meier ER, Kim KS, Rabel A, Kaushal M, Muljo SA, Miller JL. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122:1034–1041. doi: 10.1182/blood-2012-12-472308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, Voso MT, Teofili L, Lubbert M. Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Clinical Immunology. 2003;109:89–102. doi: 10.1016/s1521-6616(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Bcl11a is essential for normal lymphoid development. Nature Immunology. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- Lou TF, Singh M, Mackie A, Li W, Pace BS. Hydroxyurea generates nitric oxide in human erythroid cells: mechanisms for gamma-globin gene activation. Experimental Biology and Medicine (Maywood, N J) 2009;234:1374–1382. doi: 10.3181/0811-RM-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulli V, Romania P, Morsilli O, Cianciulli P, Gabbianelli M, Testa U, Giuliani A, Marziali G. MicroRNA-486-3p regulates gamma-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One. 2013;8:e60436. doi: 10.1371/journal.pone.0060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabaera R, Richardson CA, Johnson K, Hsu M, Fiering S, Lowrey CH. Developmental- and differentiation-specific patterns of human gamma- and beta-globin promoter DNA methylation. Blood. 2007;110:1343–1352. doi: 10.1182/blood-2007-01-068635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt H, Marquardt H. Induction of malignant transformation and mutagenesis in cell cultures by cancer chemotherapeutic agents. Cancer. 1977;40:1930–1934. doi: 10.1002/1097-0142(197710)40:4+<1930::aid-cncr2820400826>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, Fisher C, Suciu M, Martyn GE, Norton LJ, Zhu C, Kurita R, Nakamura Y, Xu J, Higgs DR, Crossley M, Bauer DE, Orkin SH, Kharchenko PV, Maeda T. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler SE, Wade M, Kutlar F, Yerigenahally SD, Xue Y, Moutouh-de Parseval LA, Corral LG, Swerdlow PS, Kutlar A. Pomalidomide augments fetal hemoglobin production without the myelosuppressive effects of hydroxyurea in transgenic sickle cell mice. Blood. 2011;118:1109–1112. doi: 10.1182/blood-2010-11-319137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzer WC, Heller S, Pearle PR, Hackney E, Vichinsky E. Availability of related donors for bone marrow transplantation in sickle cell anemia. The American Journal of Pediatric Hematology/Oncology. 1994;16:27–29. [PubMed] [Google Scholar]
- Moutouh-de Parseval LA, Verhelle D, Glezer E, Jensen-Pergakes K, Ferguson GD, Corral LG, Morris CL, Muller G, Brady H, Chan K. Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. The Journal of Clinical Investigation. 2008;118:248–258. doi: 10.1172/JCI32322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel RL, Bookchin RM, Johnson J, Labie D, Wajcman H, Isaac-Sodeye WA, Honig GR, Schiliro G, Crookston JH, Matsutomo K. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:670–672. doi: 10.1073/pnas.76.2.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer-Rego KE, Beldjord C, Nagel RL, Labie D. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:1771–1773. doi: 10.1073/pnas.81.6.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Pandey S, Mishra RM, Saxena R. Modulating effect of the -158 gamma (C–>T) Xmn1 polymorphism in Indian sickle cell patients. Mediterranean Journal of Hematology and Infectious Diseases. 2012;4:e2012001. doi: 10.4084/MJHID.2012.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrine SP, Greene MF, Faller DV. Delay in the fetal globin switch in infants of diabetic mothers. New England Journal of Medicine. 1985;312:334–338. doi: 10.1056/NEJM198502073120602. [DOI] [PubMed] [Google Scholar]
- Perumbeti A, Higashimoto T, Urbinati F, Franco R, Meiselman HJ, Witte D, Malik P. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114:1174–1185. doi: 10.1182/blood-2009-01-201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg LH, Flavell RA. DNA methylation in the human gamma delta betaglobin locus in erythroid and nonerythroid tissues. Cell. 1980;19:947–958. doi: 10.1016/0092-8674(80)90086-0. [DOI] [PubMed] [Google Scholar]
- Popat N, Wood WG, Weatherall DJ, Turnbull AC. Pattern of maternal F-cell production during pregnancy. Lancet. 1977;2:377–379. doi: 10.1016/s0140-6736(77)90305-1. [DOI] [PubMed] [Google Scholar]
- Rank G, Cerruti L, Simpson RJ, Moritz RL, Jane SM, Zhao Q. Identification of a PRMT5-dependent repressor complex linked to silencing of human fetal globin gene expression. Blood. 2010;116:1585–1592. doi: 10.1182/blood-2009-10-251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renneville A, Van Galen P, Canver MC, McConkey M, Krill-Burger JM, Dorfman DM, Holson EB, Bernstein BE, Orkin SH, Bauer DE, Ebert BL. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood. 2015;126:1930–1939. doi: 10.1182/blood-2015-06-649087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P, Cavazzana M. Gene therapy in a patient with sickle cell disease. New England Journal of Medicine. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- Rivers A, Vaitkus K, Ibanez V, Ruiz MA, Jagadeeswaran R, Saunthararajah Y, Cui S, Engel JD, DeSimone J, Lavelle D. The LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis) Haematologica. 2016;101:688–697. doi: 10.3324/haematol.2015.140749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupon JW, Wang SZ, Gaensler K, Lloyd J, Ginder GD. Methyl binding domain protein 2 mediates gamma-globin gene silencing in adult human betaYAC transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6617–6622. doi: 10.1073/pnas.0509322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, Sabath DE, Groudine M, Orkin SH, Premawardhena A, Bender MA. A functional element necessary for fetal hemoglobin silencing. New England Journal of Medicine. 2011a;365:807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Scepanovic D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proceedings of the National Academy of Sciences of the United States of America. 2011b;108:1519–1524. doi: 10.1073/pnas.1018384108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunthararajah Y, DeSimone J. Clinical studies with fetal hemoglobin-enhancing agents in sickle cell disease. Seminars in Hematology. 2004;41:11–16. doi: 10.1053/j.seminhematol.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Shariati L, Khanahmad H, Salehi M, Hejazi Z, Rahimmanesh I, Tabatabaiefar MA, Modarressi MH. Genetic disruption of the KLF1 gene to overexpress the gamma-globin gene using the CRISPR/Cas9 system. The Journal of Gene Medicine. 2016;18:294–301. doi: 10.1002/jgm.2928. [DOI] [PubMed] [Google Scholar]
- Sheehan VA. Pediatric Blood & Cancer; Abstracts of the American Society of Pediatric Hematology/Oncology (ASPHO) 26th Annual Meeting; Miami, Florida, USA. April 24-27, 2013; 2013. pp. S1–S105. [DOI] [PubMed] [Google Scholar]
- Shenoy S, Eapen M, Panepinto JA, Logan BR, Wu J, Abraham A, Brochstein J, Chaudhury S, Godder K, Haight AE, Kasow KA, Leung K, Andreansky M, Bhatia M, Dalal J, Haines H, Jaroscak J, Lazarus HM, Levine JE, Krishnamurti L, Margolis D, Megason GC, Yu LC, Pulsipher MA, Gersten I, DiFronzo N, Horowitz MM, Walters MC, Kamani N. A BMT CTN phase II trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128:2561–2567. doi: 10.1182/blood-2016-05-715870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher GD, Ginder GD, Little J, Yang S, Dover GJ, Olivieri NF. Extended therapy with intravenous arginine butyrate in patients with beta-hemoglobinopathies. New England Journal of Medicine. 1995;332:1606–1610. doi: 10.1056/NEJM199506153322404. [DOI] [PubMed] [Google Scholar]
- Srinivas R, Dunda O, Krishnamoorthy R, Fabry ME, Georges A, Labie D, Nagel RL. Atypical haplotypes linked to the beta S gene in Africa are likely to be the product of recombination. American Journal of Hematology. 1988;29:60–62. doi: 10.1002/ajh.2830290117. [DOI] [PubMed] [Google Scholar]
- Sripichai O, Fucharoen S. Fetal hemoglobin regulation in beta-thalassemia: heterogeneity, modifiers and therapeutic approaches. Expert Review of Hematology. 2016;9:1129–1137. doi: 10.1080/17474086.2016.1255142. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, Lenhard B, Rooks H, Best S, Menzel S, Grosveld F, Thein SL, Soler E. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. The Journal of Clinical Investigation. 2014;124:1699–1710. doi: 10.1172/JCI71520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Experimental Hematology. 2005;33:259–271. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos G, Veith R, Galanello R, Papayannopoulou T. Hb F production in stressed erythropoiesis: observations and kinetic models. Annals of the New York Academy of Sciences. 1985;445:188–197. doi: 10.1111/j.1749-6632.1985.tb17188.x. [DOI] [PubMed] [Google Scholar]
- Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, Swerdlow P, Kutlar A, DeCastro L, Waclawiw MA. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. American Journal of Hematology. 2010;85:403–408. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strouse JJ, Lanzkron S, Beach MC, Haywood C, Park H, Witkop C, Wilson RF, Bass EB, Segal JB. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics. 2008;122:1332–1342. doi: 10.1542/peds.2008-0441. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel) 2010;3:2751–2767. doi: 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein SL. Molecular basis of beta thalassemia and potential therapeutic targets. Blood Cells, Molecules, & Diseases. 2017:30210–30213. doi: 10.1016/j.bcmd.2017.06.001. S1079-9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. British Journal of Haematology. 2009;145:455–467. doi: 10.1111/j.1365-2141.2009.07650.x. [DOI] [PubMed] [Google Scholar]
- Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, Silver N, Gerovasilli A, Ping C, Yamaguchi M, Wahlberg K, Ulug P, Spector TD, Garner C, Matsuda F, Farrall M, Lathrop M. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11346–11351. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, Weiss MJ. A genome-editing strategy to treat betahemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nature Medicine. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Molecular and Cellular Biology. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, Piras MG, Sestu N, Lai S, Dei M, Mulas A, Crisponi L, Naitza S, Asunis I, Deiana M, Nagaraja R, Perseu L, Satta S, Cipollina MD, Sollaino C, Moi P, Hirschhorn JN, Orkin SH, Abecasis GR, Schlessinger D, Cao A. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbinati F, Hargrove PW, Geiger S, Romero Z, Wherley J, Kaufman ML, Hollis RP, Chambers CB, Persons DA, Kohn DB, Wilber A. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34+ cells. Experimental Hematology. 2015;43:346–351. doi: 10.1016/j.exphem.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinjamur DS, Alhashem YN, Mohamad SF, Amin P, Williams DC, Jr, Lloyd JA. Kruppel-like transcription factor KLF1 is required for optimal gamma- and beta-globin expression in human fetal erythroblasts. PLoS One. 2016;11:e0146802. doi: 10.1371/journal.pone.0146802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, Odame I, Fuh B, George A, Owen W, Luchtman-Jones L, Rogers ZR, Hilliard L, Gauger C, Piccone C, Lee MT, Kwiatkowski JL, Jackson S, Miller ST, Roberts C, Heeney MM, Kalfa TA, Nelson S, Imran H, Nottage K, Alvarez O, Rhodes M, Thompson AA, Rothman JA, Helton KJ, Roberts D, Coleman J, Bonner MJ, Kutlar A, Patel N, Wood J, Piller L, Wei P, Luden J, Mortier NA, Stuber SE, Luban NL, Cohen AR, Pressel S, Adams RJ. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387:661–670. doi: 10.1016/S0140-6736(15)01041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. American Journal of the Medical Sciences. 1948;215:419–423. doi: 10.1097/00000441-194804000-00008. [DOI] [PubMed] [Google Scholar]
- Weinberg RS, Ji X, Sutton M, Perrine S, Galperin Y, Li Q, Liebhaber SA, Stamatoyannopoulos G, Atweh GF. Butyrate increases the efficiency of translation of gammaglobin mRNA. Blood. 2005;105:1807–1809. doi: 10.1182/blood-2004-02-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber A, Tschulena U, Hargrove PW, Kim YS, Persons DA, Barbas CF, 3rd, Nienhuis AW. A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood. 2010;115:3033–3041. doi: 10.1182/blood-2009-08-240556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt O, Monkemeyer S, Ronndahl G, Erdlenbruch B, Reinhardt D, Kanbach K, Pekrun A. Induction of fetal hemoglobin expression by the histone deacetylase inhibitor apicidin. Blood. 2003;101:2001–2007. doi: 10.1182/blood-2002-08-2617. [DOI] [PubMed] [Google Scholar]
- Wykes C, Rees DC. The safety and efficacy of hydroxycarbamide in infants with sickle cell anemia. Expert Review of Hematology. 2011;4:407–409. doi: 10.1586/ehm.11.40. [DOI] [PubMed] [Google Scholar]
- Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, Yao H, Trowbridge JJ, Mandel G, Orkin SH. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6518–6523. doi: 10.1073/pnas.1303976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- Yin W, Barkess G, Fang X, Xiang P, Cao H, Stamatoyannopoulos G, Li Q. Histone acetylation at the human beta-globin locus changes with developmental age. Blood. 2007;110:4101–4107. doi: 10.1182/blood-2007-05-091256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, Cunningham JM, Jane SM. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nature Structural & Molecular Biology. 2009;16:304–311. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Zhao Q, Sutton R, Cumming H, Wang X, Cerruti L, Hall M, Wu R, Cunningham JM, Jane SM. The role of p22 NF-E4 in human globin gene switching. Journal of Biological Chemistry. 2004;279:26227–26232. doi: 10.1074/jbc.M402191200. [DOI] [PubMed] [Google Scholar]
- Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nature Genetics. 2010;42:742–744. doi: 10.1038/ng.637. [DOI] [PubMed] [Google Scholar]