

Abstract
Cancer hallmarks allow the complexity and heterogeneity of tumor biology to be better understood, leading to the discovery of various promising targets for cancer therapy. An amorphous iron oxide nanoparticle (NP)-based RNAi strategy is developed to co-target two cancer hallmarks. The NP technology can modulate the glycolysis pathway by silencing MCT4 to induce tumor cell acidosis, and concurrently exacerbate oxidative stress in tumor cells via the Fenton-like reaction. This strategy has the following features for systemic siRNA delivery: 1) siRNA encapsulation within NPs for improving systemic stability; 2) effective endosomal escape through osmotic pressure and/or endosomal membrane oxidation; 3) small size for enhancing tumor tissue penetration; and 4) triple functions (RNAi, Fenton-like reaction, and MRI) for combinatorial therapy and in vivo tracking.
Keywords: cancer hallmarks, iron oxide nanoparticles, MRI, RNAi, tumor penetration
Graphical abstract
A unique amorphous iron oxide nanoparticle (NP)-based RNAi delivery strategy is developed to co-target two cancer hallmarks – aerobic glycolysis and dysregulated redox homeostasis. This NP platform exhibits several appealing features including small size, efficient siRNA loading and gene silencing, high tumor accumulation, deep tumor tissue penetration, and MRI-mediated in vivo tracking of tumor accumulation.
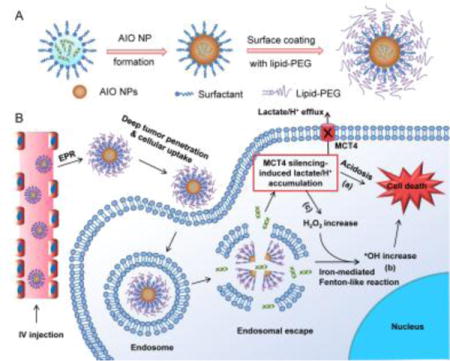
Hypoxia is recognized as a fundamentally important feature of solid tumors and is at the heart of cancer hallmarks.[1] Under hypoxia, metabolic pathways of cancer cells may therefore be rewired in such a way that balances biosynthetic processes with rapid and large amounts of ATP production to support cell proliferation,[2] a phenomenon referred to as aerobic glycolysis or the “Warburg effect” that is uniquely observed in primary and metastatic tumors.[3] The high rate of glycolysis is associated with excessive generation of lactic acid,[4] which leads to the upregulation of monocarboxylate transporters (MCTs),[5] predominantly MCT4, for efflux of lactate/H+ to maintain a stable intracellular pH and induce an acidic tumor microenvironment. The elevated MCT4 expression has been correlated with metastasis, angiogenesis, poor prognosis, and recurrence of many cancers.[6]
In parallel to altering the energy metabolism, hypoxia also triggers more production of reactive oxygen species (ROS) (up to 100 μM) in tumor cells than normal tissues (~20 nM).[7] The elevated ROS (in particular H2O2) could foster tumor growth and malignant progression.[8] On the other hand, due to the high ROS levels, tumor cells are also more vulnerable to further oxidative assault than normal cells.[9] The generation of more reactive and toxic ROS (such as •OH) caused by exogenous agents could disrupt the ROS homeostasis in tumor cells, leading to severe oxidative stress and then cell death. Therefore, we hypothesized that co-targeting of these two cancer hallmarks, aerobic glycolysis and dysregulated redox homeostasis in tumor cells, could represent a novel strategy for efficiently and specifically arresting tumor progression.
Herein, we developed a unique amorphous iron oxide (AIO) RNAi NP platform (Scheme 1) for co-targeting metabolic and ROS homeostasis in tumor cells by simultaneously silencing MCT4 to induce tumor cell acidosis and exacerbating oxidative stress via the Fenton-like reaction. These NPs exhibit multiple appealing features for systemic siRNA delivery. First, different from previous iron oxide RNAi NPs, in which siRNA was simply loaded on NP surface by complexing with cationic materials,[10] our strategy enables siRNA encapsulation within NPs. This could limit enzymatic contact, degradation, and burst release during circulation in the blood. Second, the NPs are coated with lipid-PEG for prolonged blood circulation, and show less prone to the recognition by mononuclear phagocyte system in the liver and spleen. Third, the small size (~14.6 nm) could allow NPs to effectively penetrate deeply in tumor tissues. Fourth, unlike the majority of previous RNAi NPs,[11] which utilize the cationic lipids and/or polymers to facilitate the endosomal escape, the efficient endosomal escape of our system may be attributable to osmotic pressure and/or endosomal membrane oxidation induced by the iron ions released from NPs. By the NP-mediated MCT4 silencing, the efflux of intracellular lactate/H+ can be blocked, leading to acidosis-induced tumor cell death (Scheme 1B).
Scheme 1.
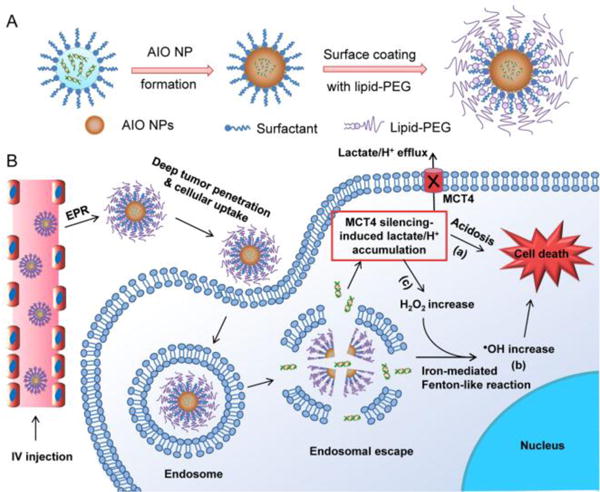
Schematic illustration of (A) the formulation and (B) multiple functions of AIO RNAi NPs.
On the other hand, AIO NPs are responsive to acidic pHs after cellular uptake, and the released iron ions will react with H2O2 to generate highly reactive and toxic •OH via the Fenton-like reaction.[12] In addition to promoting endosomal escape aforementioned, •OH will drastically exacerbate oxidative stress in tumor cells and subsequently induce cell death (Scheme 1B). Notably, the block of intracellular lactate efflux by MCT4 silencing could further stimulate more H2O2 production to amplify the Fenton-like reaction and oxidative damage to tumor cells for effective combinatorial therapy. Moreover, AIO NPs could be useful as a surrogate marker for real-time monitoring of biodistribution and tumor accumulation of siRNA via MRI, which will provide more insights into tumor heterogeneities and the enhanced permeability and retention (EPR) effect to identify cancer patients most likely to benefit from RNAi nanomedicines.[13]
The RNAi NPs were first synthesized by a reversed microemulsion method (Scheme 1A), with a siRNA encapsulation efficiency of ∼50% as determined by inductively coupled plasma-mass spectrometry analysis. The average diameter of NPs was ~14.6 nm by size analysis of random 100 NPs in the transmission electron microscopy (TEM) image (Figure 1A), and the hydrodynamic size was ~45 nm as measured by dynamic light scattering (Figure S1). X-ray diffraction (XRD) confirmed the amorphous structure (Figure 1B), which is presumably composed of Fe2O3 or FeOOH.[14] To assess whether the decomposition of AIO NPs in acidic pHs can trigger •OH production, we used electron spin resonance (ESR) spectroscopy to measure •OH produced by the reaction between NPs and H2O2 at different pHs. The generation of •OH was very low at pH 7.4, as indicated by the low ESR amplitude of paramagnetic adduct DEPMPO-OH (Figure 1C). In contrast, the ESR amplitude of DEPMPO-OH was dramatically increased at pH 6.0, and further enhanced at pH 5.0, suggesting the elevated production of •OH at acidic pHs. Figure S2 also confirms that AIO NPs can be decomposed at acidic pHs, thus contributing to the release of iron ions and accelerated the Fenton-like reaction. Meanwhile, the NP decomposition led to fast siRNA release (Figure S3).
Figure 1.
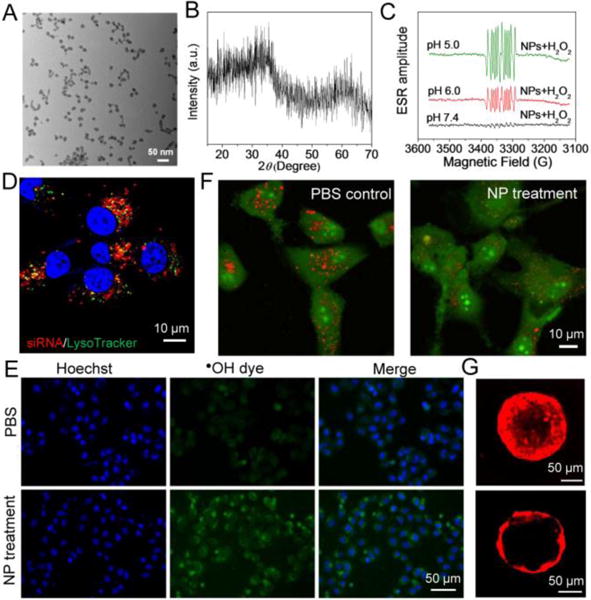
(A) TEM image and (B) XRD spectrum of AIO RNAi NPs. (C) ESR spectra of AIO RNAi NPs in the presence of H2O2 at different pHs. 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO) was used as the spin trap agent. (D) Fluorescent image of HeLa cells treated with Cy5.5-siRNA-loaded AIO NPs for 4 h. (E) Confocal imaging of intracellular •OH production and (F) AO staining of PC3 cells treated with PBS vs. AIO NPs. The reduced red dots in the NP-treated cells (F) indicate the loss of membrane integrity of endo/lysosomes. (G) Tissue penetration of (top) AIO NPs and (down) polymer NPs loaded with Cy5.5-siRNA in a 3D spheroid model after 4 h incubation.
The endosomal escape ability of AIO NPs was tested by staining endosomes with LysoTracker Green. Figure 1D shows the effective cytosolic transport of the internalized siRNA from endosomes. Notably, the endosomal escape mechanism associated with AIO NPs may be attributable to two potential processes: (i) after internalization, AIO NPs will be dissolved at acidic endosomal pHs to release iron ions and thus increase osmotic pressure in endosomes, leading to endosomal swelling and rupture; and (ii) the released iron ions can also react with intracellular H2O2 to generate highly reactive •OH via the Fenton-like reaction, as identified by •OH staining using aminophenyl fluorescein (APF) (Figure 1E). •OH could then contribute to the endosomal membrane oxidation and rupture, as confirmed by staining the endosomal membrane integrity with acridine orange (AO) (Figure 1F).
Since small size NPs have been suggested to diffuse through the tumor tissue more efficiently than large size NPs,[15] we proceeded to evaluate the tissue penetration of AIO NPs using a 3D spheroid tumor model. As a proof-of-concept test, polymer NPs with similar surface coating but with a particle size ∼100 nm were used as a reference (Figure S4), given the challenge of making large AIO NPs with the reversed microemulsion method adopted in this work. Results showed that red fluorescence was observed only in the periphery of the spheroid treated with large polymer RNAi NPs, while much better tissue penentration can be seen for the group treated with AIO RNAi NPs (Figure 1G).
Next, prostate cancer (PCa), characterized by upregulation of MCT4 and overproduction of ROS,[16] was used as a model to examine the combinatorial therapy of AIO RNAi NPs. The overexpression of MCT4 and the increased ROS (mainly H2O2) has been correlated with the cell proliferation, drug resistance, invasion, and metastasis of PCa.[6] However, to the best of our knowledge, no study has thus far explored systemic delivery of siRNA targeting MCT4 (siMCT4) to PCa. To do this, we first examined the MCT4 expression and ROS production in three PCa cell lines including DU145, PC3, and LNCaP. All the cells have high expression of MCT4, while the ROS production in PC3 and LNCaP cells was higher than that in DU145 cells (Figure S5). Correspondingly, the cell death in PC3 and LNCaP cells caused by AIO NP-mediated oxidative damage was more obvious than that in DU145 cells (Figure S6). Thus, PC3 cells, with high ROS production and MCT4 expression, were used for the following experiments unless otherwise specified. Western blot and immunofluorescence analysis demonstrated that NP(siMCT4) treatment significantly suppressed the expression of MCT4 in PC3 cells (Figure 2A,B). As a result, a significant increase of the intracellular lactate was observed (Figure 2C). Moreover, we found that the intracellular H2O2 production in PC3 cells was dramatically enhanced after MCT4 silencing (Figure 2D). This may be presumably attributed to the MCT4 silencing-induced lactate accumulation, which can activate mitochondrial biogenesis via upregulation of PGC1α and subsequently increase the intracellular H2O2.[17] The produced H2O2 could further react with iron ions released from AIO NPs to generate more •OH and thus amplify the oxidative damage to tumor cells. Figure 2E shows that NP(siMCT4) were most effective in suppressing cell proliferation compared to the NP(siControl) or non-treated group, primarily due to the drastic apoptosis caused by MCT4 silencing and the amplified oxidative damage (Figure 2F-H).
Figure 2.
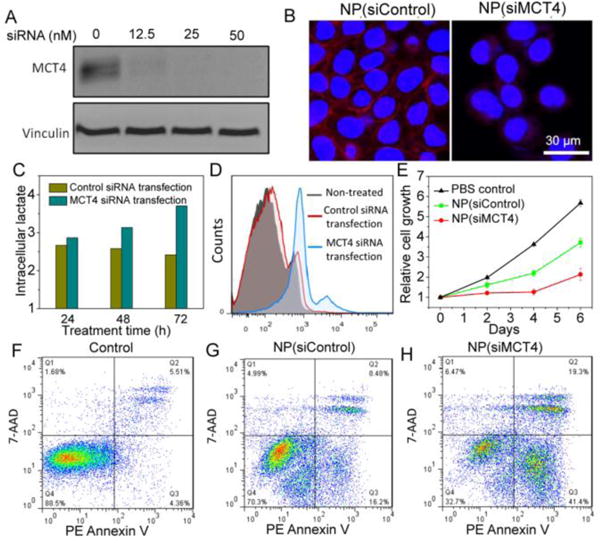
(A) Western blot and (B) immunofluorescence analysis of MCT4 expression in PC3 cells treated with NP(siControl) vs. NP(siMCT4). (C) Time-dependent intracellular lactate change in PC3 cells with vs. without MCT4 silencing. (D) ROS production of PC3 cells with vs. without MCT4 silencing. Note that Lipofectamine 2000 was used for siRNA transfection in this experiment, as it will not induce Fenton-like reaction. (E) Proliferation of PC3 cells treated with PBS, NP(siControl), or NP(siMCT4). (F-H) Flow cytometry analysis of PC3 cell apoptosis post treatment of (F) PBS, (G) NP(siControl), and (H) NP(siMCT4).
Given these promising in vitro results, we proceeded to examine the in vivo performance of AIO NPs. The pharmacokinetics and biodistribution studies were first carried out by intravenous (iv) injection of Cy5.5-siRNA-loaded AIO NPs. The AIO NPs demonstrated long circulation in the blood (Figure 3A), effective accumulation in the tumor, and less uptake by the liver and spleen (Figure 3B,C). In addition, we also examined the in vivo tumor-penetrating ability of AIO NPs by immunofluorescence analysis. As shown in Figure 3D, bright red fluorescence was visualized in the blood vessels and the extravascular tumor parenchyma, suggesting that AIO NPs could efficiently extravasate from the leaky tumor vasculature and transport deeply into the tumor tissue.
Figure 3.
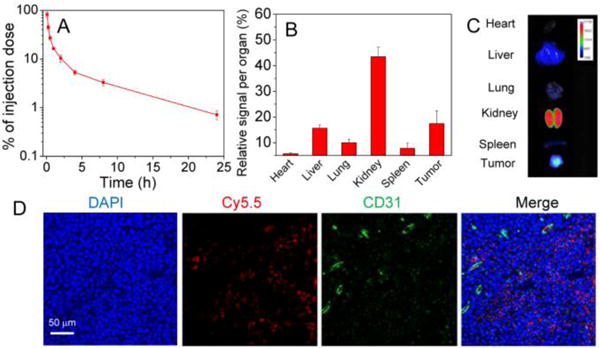
(A) Pharmacokinetic profile and (B) biodistribution of Cy5.5-siRNA-loaded AIO NPs. (C) Fluorescent imaging of organs from PC3 tumor-bearing mouse at 24 h after iv injection of Cy5.5-siRNA-loaded AIO NPs. (D) Fluorescent images of the tumor sections of the PC3 tumor-bearing mouse sacrificed at 4 h post-injection of Cy5.5-siRNA-loaded AIO NPs.
In vivo gene silencing of AIO RNAi NPs was then examined after iv injection of the NPs into PC3 tumor-bearing mice for three consecutive injections at a 900 μg siRNA/kg dose. Compared to the NP(siControl)-treated group, the MCT4 expression was significantly suppressed in mice treated with NP(siMCT4) (Figure 4A and Figure S7). Next, the in vivo antitumor effect of NPs was evaluated. AIO RNAi NPs were iv injected into PC3 tumor-bearing mice for four consecutive injections at a 900 μg siRNA/kg dose and the tumor growth was monitored. NP(siControl) treatment reduced the tumor growth compared to the control group due to the Fenton-like reaction-induced oxidative damage (Figure 4B,C). More impressively, a further suppression of tumor growth was observed for the NP(siMCT4)-treated group, without causing noticeable influence on the body weight (Figure S8).
Figure 4.
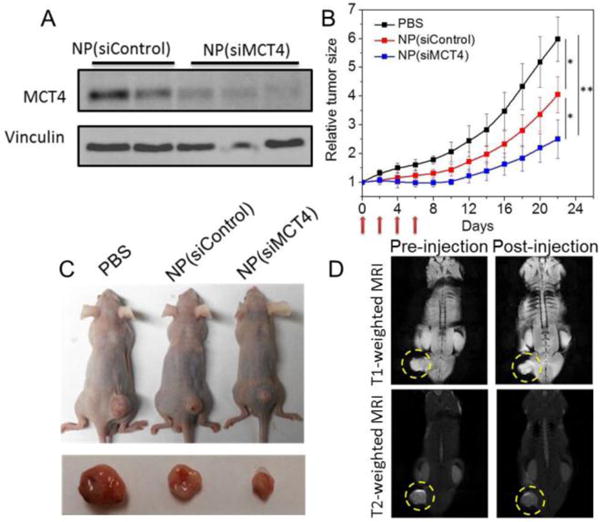
(A) Western blot analysis of the MCT4 expression in PC3 tumor-bearing mice after iv injection of NP(siControl) vs. NP(siMCT4). (B) Tumor growth of PC3 xenograft mice after treatment with PBS, NP(siControl), or NP(siMCT4). (*p<0.05 and **p<0.01 vs. PBS control). (C) Representative picture of tumor-bearing mice and tumor tissues from three different groups. (D) Whole body T1- and T2-weighted MRI images of the PC3 tumor-bearing mouse after iv injection of AIO RNAi NPs.
In addition to combinatorial cancer therapy, noninvasive visualization of the tumor accumulation of AIO RNAi NPs could be achieved via MRI, as iron oxide NPs are well-known as MRI contrast agents.[18] Notably, unlike commercial iron oxide NPs that are generally T2-weighted MRI contrast agents, our AIO NPs showed both T1 and T2 contrast effects (Figure S9). The dual-model imaging effect may be presumably due to the amorphous structure and small size of AIO NPs, while further studies will still be needed to reveal the imaging mechanisms. Our preliminary data also demonstrated that the tumor accumulation of AIO NPs via the EPR effect could be visualized in both T1- and T2-weighted MRI images after iv injection (Figure 4D), suggesting the potential use of AIO NPs for MRI-guided siRNA delivery and the selection of patients with high EPR effect.[13]
Finally, the potential side effects of NPs were evaluated. Systemic administration of AIO NPs did not induce obvious changes in the serum levels of multiple hematological parameters (Figure S10) and cytokines (Figure S11). Moreover, histological analysis shows no noticeable inflammatory response or tissue injury in the NP-treated mice, as compared to the control group (Figure S12). All these results suggest the good biocompatibility of AIO NPs.
In conclusion, we developed an innovative amorphous iron oxide NP platform for effective systemic siRNA delivery, and for concurrently targeting two distinct cancer hallmarks for combinatorial therapy. The NPs exhibit several promising features, such as small size, efficient gene silencing, high tumor accumulation, deep tumor tissue penetration, good biocompatibility, and MRI-mediated tracking of tumor accumulation. We expect this unique platform to become a valuable tool for theranostic treatment of advanced cancers.
Supplementary Material
Acknowledgments
Financial support by NIH/NCI R01CA200900 and the Prostate Cancer Foundation Young Investigator Award.
References
- 1.Wilson WR, Hay MP. Nat Rev Cancer. 2011;11:393. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 2.Denko NC. Nat Rev Cancer. 2008;8:705. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Nat Rev Clin Oncol. 2017;14:113. doi: 10.1038/nrclinonc.2017.1. [DOI] [PubMed] [Google Scholar]
- 4.Cairns RA, Harris IS, Mak TW. Nat Rev Cancer. 2011;11:85. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 5.Halestrap AP. IUBMB Life. 2012;64:1. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- 6.Choi SY, Xue H, Wu R, Fazli L, Lin D, Collins CC, Gleave ME, Gout PW, Wang Y. Clin Cancer Res. 2016;22:2721. doi: 10.1158/1078-0432.CCR-15-1624. [DOI] [PubMed] [Google Scholar]
- 7.a) Sabharwal SS, Schumacker PT. Nat Rev Cancer. 2014;14:709. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhai S, Hu X, Hu Y, Wu B, Xing D. Biomaterials. 2017;121:41. doi: 10.1016/j.biomaterials.2017.01.002. [DOI] [PubMed] [Google Scholar]; c) Halliwell B, Clement MV, Long LH. FEBS Lett. 2000;486:10. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 8.Panieri E, Santoro MM. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey MR, Sharpless NE. Nat Cell Biol. 2006;8:1213. doi: 10.1038/ncb1106-1213. [DOI] [PubMed] [Google Scholar]
- 10.a) Jiang S, Eltoukhy AA, Love KT, Langer R, Anderson DG. Nano Lett. 2013;13:1059. doi: 10.1021/nl304287a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu G, Xie J, Zhang F, Wang Z, Luo K, Zhu L, Quan Q, Niu G, Lee S, Ai H, Chen X. Small. 2011;7:2742. doi: 10.1002/smll.201100825. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Park JW, Bae KH, Kim C, Park TG. Biomacromolecules. 2011;12:457. doi: 10.1021/bm101244j. [DOI] [PubMed] [Google Scholar]
- 11.a) Kanasty R, Dorkin JR, Vegas A, Anderson D. Nat Mater. 2013;12:967. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]; b) Liu Y, Gunda V, Zhu X, Xu X, Wu J, Askhatova D, Farokhzad OC, Parangi S, Shi J. Proc Natl Acad Sci U S A. 2016;113:7750. doi: 10.1073/pnas.1605841113. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zuckerman JE, Davis ME. Nat Rev Drug Discov. 2015;14:843. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]; d) Lee J, Saw PE, Gujrati V, Lee Y, Kim H, Kang S, Choi M, Kim JI, Jon S. Theranostics. 2016;6:192. doi: 10.7150/thno.13657. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wang Y, Miao L, Satterlee A, Huang L. Adv Drug Deliv Rev. 2015;87:68. doi: 10.1016/j.addr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Foy SP, Labhasetwar V. Biomaterials. 2011;32:9155. doi: 10.1016/j.biomaterials.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pham AL, Doyle FM, Sedlak DL. Water Res. 2012;46:6454. doi: 10.1016/j.watres.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Nat Rev Cancer. 2017;17:20. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leidinger P, Treptow J, Hagens K, Eich J, Zehethofer N, Schwudke D, Oehlmann W, Lunsdorf H, Goldmann O, Schaible UE, Dittmar KE, Feldmann C. Angew Chem Int Ed. 2015;54:12597. doi: 10.1002/anie.201505493. [DOI] [PubMed] [Google Scholar]
- 15.Barua S, Mitragotri S. Nano today. 2014;9:223. doi: 10.1016/j.nantod.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marignol L, Rivera-Figueroa K, Lynch T, Hollywood D. Nat Rev Urology. 2013;10:405. doi: 10.1038/nrurol.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto T, Hussien R, Oommen S, Gohil K, Brooks GA. FASEB J. 2007;21:2602. doi: 10.1096/fj.07-8174com. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Jon S. Theranostics. 2012;2:122. doi: 10.7150/thno.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.