

Abstract
We previously discovered and validated a class of piperidinyl ureas that regulate defective in cullin neddylation 1 (DCN1)-dependent neddylation of cullins. Here, we report preliminary structure-activity relationship studies aimed at advancing our high-throughput screen hit into a tractable tool compound for dissecting the effects of acute DCN1-UBE2M inhibition on the NEDD8/cullin pathway. Structure-enabled optimization led to a 100-fold increase in biochemical potency and modestly increased solubility and permeability as compared to our initial hit. The optimized compounds inhibit the DCN1-UBE2M protein-protein interaction in our TR-FRET binding assay and inhibit cullin neddylation in our pulse-chase NEDD8 transfer assay. The optimized compounds bind to DCN1 and selectively reduce steady-state levels of neddylated CUL1 and CUL3 in a squamous cell carcinoma cell line. Ultimately, we anticipate that these studies will identify early lead compounds for clinical development for the treatment of lung squamous cell carcinomas and other cancers.
TOC image

Introduction
Squamous cell carcinomas (SCCs) of mucosal origin, including those of the lung, head, and neck, account for more than 200,000 cancer-related deaths annually in the United States. The five-year survival rate of SCC patients (16% for lung) is worse than that for patients with other high-morbidity cancers such as colon (65%), breast (90%), and prostate (>99%).2 Studies have identified defective in cullin neddylation 1 (DCN1) as an oncogene within a recurrent 3q26.3 chromosomal amplification present in many SCCs; this amplification is associated with poorer survival outcome.3–7 DCN1 is also known as DCUN1D1, DCNL1, or SCCRO (squamous cell carcinoma-related oncogene); we use “DCN1” hereafter.8
DCN1 is a co-E3 ligase in the pathway regulating conjugation of NEDD8 to cullin-RING ligases (CRLs), ultimately controlling the full activation of CRLs as ubiquitin E3 ligases (Figure 1A).9 DCN1 binds the acetylated N-terminus of UBE2M (an E2 enzyme for the ubiquitin-like protein NEDD8) and the cullin family of proteins, acting as a co-E3 promoting NEDD8 modification of cullins (Figure 1B).10 Once activated by neddylation, the cullins go on to form CRLs, the largest family of ubiquitin E3 ligase enzymes and broad regulators of ubiquitin conjugation, ultimately controlling many cellular phenomena, especially protein homeostasis.11–12 Thus, we would expect inhibition of the DCN1-UBE2M interaction to reduce cullin neddylation.
Figure 1.
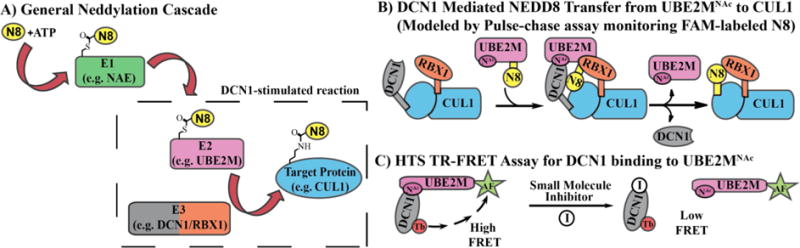
A) Scheme of general neddylation tri-enzyme cascades. B) Cartoon of DCN1/RBX1-stimulated transfer of NEDD8 (“N8” in cartoon) from UBE2M to a cullin (CUL1), which can be monitored in vitro using a pulse-chase assay. Desired inhibitors will block transfer of Nedd8 C) TR-FRET assay for DCN1 binding to UBE2MNAc. Desired inhibitors will possess low FRET signal.
Inhibiting post-translational modification by ubiquitin and ubiquitin-like proteins, such as NEDD8, is gaining increasing attention due to the clinical successes of proteasome inhibitors (e.g., Bortezomib or Carfilzomib).13–20 One approach is the inhibition of neddylation that subsequently decreases the activity of CRLs, affecting only a subset of ubiquitin-dependent proteasomal degradation.21–25 The NEDD8 E1 (NAE) inhibitor Pevonedistat (MLN4924) completely blocks all neddylation, inhibits all CRL function, reduces total cellular ubiquitination by 10-20%, and stabilizes hundreds of CRL substrates.21, 26 Pevonedistat treatment elicits broad cellular effects including checkpoint activation, DNA re-replication, and induction of apoptosis.27–33 Pevonedistat is currently in Phase II clinical trials and validates the neddylation pathway as an oncology target.
Several other approaches have been taken to block the NEDD8 pathway. Virtual screens have identified a natural product-like flavonoid derivative 6,6″-biapigenin34 and 1-benzyl-N-(2,4-dichlorophenethyl) piperidin-4-amine, a selective reversible small molecule NAE inhibitor, that is efficacious in multiple cancer lines.35 Metal-based NAE inhibitors have been described as viable alternatives to organic small-molecule-based drugs.36–37 These molecules effectively inhibit NAE activity and exhibit anti-inflammatory activity in a murine model of inflammatory bowel disease. Recently, an inhibitor of the COP9 signalosome, responsible for deneddylation of the CRLs, has been reported and displays anti-tumor activity.38
Other studies have demonstrated that selective inhibition of UB E3 ligases is possible.39–41 Some of the more well-characterized of these inhibitors include Thalidomide,42 Nutlin-3a,43 SMER3,44, Heclin,45 and VH29846. We investigated whether we could inhibit a subset of the NEDD8 pathway by selectively targeting the co-E3 ligase DCN1, which is essential for SCC transformation and maintenance,7 and thought to regulate only a subset of CRLs.47–48
DCN1 is one of five paralogues in the human genome (DCN1-5) that function in the neddylation pathway.49 Since DCN1 is the most commonly dysregulated paralogue in SCCs and the other DCNs do not appear to contribute to malignancy, a selective DCN1 inhibitor might be as effective as Pevonedistat while having fewer side effects.50 However, compounds that are more promiscuous may have significant ancillary affects that contribute to efficacy. Therefore, it is unclear whether selective or pan inhibition of the DCN isoforms will provide the most efficacious therapeutics in vivo.49, 51–52 Thus, we set out to identify and optimize inhibitors of DCN1 activity with sufficient potency, selectivity, and bioavailability to be used to examine the consequences of inhibiting the DCN1 interaction with UBE2M in cells and animals. These compounds would be useful in dissecting the effects of acute DCN1-UBE2M inhibition on downstream NEDD8/cullin pathways in normal and SCC cells.1, 53 We particularly focused on developing isoform-specific inhibitors in order to study the consequences of differential inhibition of certain isoforms. We previously reported the discovery of a class of piperidinyl ureas that regulate DCN1-dependent cullin neddylation.1 Here, we delineate preliminary structure-activity relationships (SARs) and structure-property relationships (SPRs) that we developed during optimization of these inhibitors. Our optimization strategy focused on identifying the minimum pharmacophore and understanding the structural determinants of affinity and potency.
Our optimization studies followed a recursive process of analog design, synthesis, testing, and design refinement. We designed analogs based on the combination of empirically derived SAR and SPR data with structural data obtained by examination of co-crystal structures of our compounds bound to DCN1 (Figure 2). We tested the biochemical potency of all new compounds using an assay measuring the time-resolved fluorescence resonance energy transfer (TR-FRET) signal between a biotinylated version of DCN1, recognized by terbium-linked streptavidin, and a stapled peptide corresponding to N-terminally acetylated UBE2M labeled at its C-terminus with AlexaFluor 488. We previously showed that this system faithfully recapitulates the interactions and affinity of longer constructs of UBE2M (Figure 1C).10, 54 We then tested compounds with significantly improved inhibitory potency, relative to those in prior rounds, for their ability to inhibit DCN1 mediated transfer of [FAM]-NEDD8 to cullin targets using our previously reported pulse-chase assay (Figure 1B).10, 49 In each round of optimization, we obtained X-ray co-structures of representative inhibitors bound to DCN1. Finally, we determined cellular activity and selectivity using in vitro proliferation assays with BJ cells (normal fibroblasts) and a lung cancer cell line with amplified DCN1 expression (HCC95). These studies identified compound 52, which is 100-fold more potent in our biochemical TR-FRET assay and is modestly more soluble and permeable as compared to our initial high-throughput screen (HTS) hit. Compound 52 engages DCN1 and reduces steady-state levels of cullin neddylation in HCC95 cells.
Figure 2.
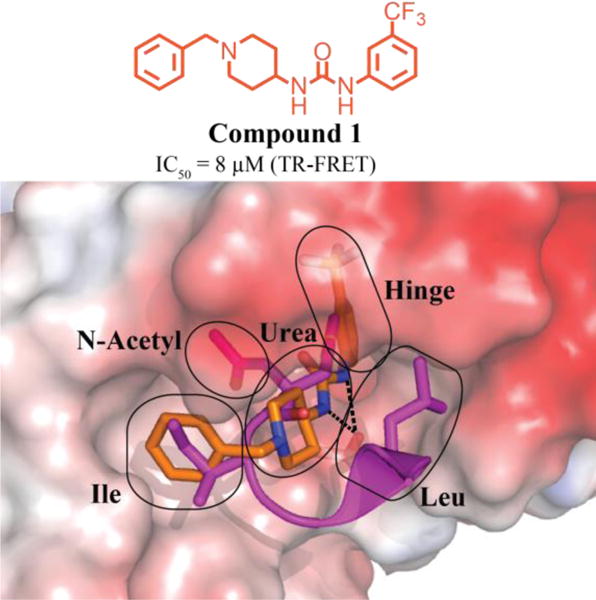
Chemical structure of the HTS hit (1) and overlay of 1(orange):DCN1(electrostatic potential surface) and UBE2MNAc(magenta): DCN1 X-ray co-crystal structures (PDB 5V83 and 3TDU), highlighting the key regions targeted for optimization. Hashed black lines represented key hydrogen-bonding interaction between the urea and backbone amide of Gln114 (3.0 and 2.7 Å, respectively).1
Results
We designed our initial analogs based on the data generated from roughly 300 structurally related compounds present in the original screening collection, as well as examination of the 1:DCN1 and DCN1-UBE2MNAc X-ray crystal co-structures (Figure 2).1 For these initial analogs, we focused on validating hypotheses concerning binding mode and minimum pharmacophore. Based on the 1:DCN1 X-ray co-structure, we identified five sub-pockets within the DCN1-UBE2M binding site to target with optimized compounds: the Ile, urea, hinge, Leu, and N-acetyl pockets (Figure 2).
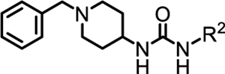
Compound 1 accesses the Ile, urea, and hinge pockets. The Ile pocket is primarily solvent exposed, and the preliminary SAR data suggested that both alkyl and aryl substitutions were well tolerated. The piperidine core, which occupies the urea pocket, was critical to compound activity; decreasing the ring size, exchanging for the 1,3-substituted piperidine, or replacing with aromatic rings all resulted in a complete loss in activity. Our examination of the 1:DCN1 X-ray crystal co-structure also revealed an H-bond within the urea pocket between the urea N-H protons and the backbone carbonyl of Gln114 of DCN1. We hypothesized that this H-bonding interaction was required to maintain compound potency. The initial SAR set supported our hypothesis as replacement of the urea with an amide led to retained potency whereas removal of the hydrogen bond donor by either introduction of alkyl linkers or alkylating the nitrogen resulted in loss of activity. The SAR data indicated that the substituent occupying the hinge pocket offered the most restrictive SAR. Adding bulky substituents (i.e., t-Bu and aryl) or alternative phenyl ring substitution patterns (ortho- and para-) caused substantial decreases in activity. Replacement of the aromatic ring with either smaller aromatic or saturated rings caused a complete loss of activity. The dense packing of hydrophobic residues around the hinge region substituent, as observed in the crystal structure, supported this SAR data.
Our examination of the UBE2MNAc:DCN1 co-structure revealed two additional pockets, the N-acetyl pocket and the Leu pocket, that were unoccupied by compound 1 (Figure 2). As these pockets offered us the potential to increase binding affinity, we targeted them for further study. We hypothesized that by accessing the N-acetyl pocket, we could benefit from additional electrostatic interactions with the water-mediated H-bonding network that interacts with the hydrophilic N-terminally acetylated methionine of the native substrate. In prior studies using peptide inhibitors, we showed that occupying this pocket with an acetyl group can increase affinity by more than ten-fold.10 We envisioned that accessing the predominately hydrophobic Leu pocket would afford additional van der Waals contacts.
Our initial analysis of the SAR data and 1:DCN1 X-ray crystal co-structures suggested a minimal pharmacophore consisting of three key structural drivers: (1) the saturated amine linker that orients the two ends of the molecule into the hydrophobic Ile and hinge sub-pockets, (2) a hydrogen bond between the aryl N-H of the urea and Gln114 that anchors the inhibitor into the DCN1-UBE2MNAc binding site, and (3) the tight steric packing of the hinge pocket around the m-CF3-substituted phenyl ring.
In order to support further SAR studies aimed at fully understanding the drivers of binding to the targeted site on DCN1, we developed the general synthetic routes shown in Scheme 1. In route i, reductive amination of a boc-protected 4-amino piperidine (2) yielded a common protected amine (3) with good yield (70 – 99%). Subsequent acid-mediated deprotection and condensation of the resulting amine with isocyanates furnished the desired disubstituted urea analogs (4) in moderate yields over the two steps (40 – 75%).55–58 Synthesis of the tri-substituted ureas utilized two separate routes (ii and iii) that permitted the late-stage introduction of either the Ile or the hinge pocket substitutions. Starting from 4-piperidone (5), alkylation to give 6 and reductive amination afforded an amine common intermediate 7 that was coupled with isocyanates to give alkylurea analogs targeting the hinge pocket 8. In general, the reductive amination and isocyanate coupling reactions proceeded with good yields (70 – 99%), while the alkylation reactions were less efficient (30-50%). Alternatively, starting from 1-boc-4-piperidone (9), reductive amination gave 10, followed by coupling to an isocyanate to introduce the hinge substitution of intermediate 11. Deprotection of 11 afforded the free piperidine intermediate that was functionalized by reductive amination (12) in good yields over the sequence (40 – 95%, three steps), permitting exploration of the Ile pocket.
Scheme 1.
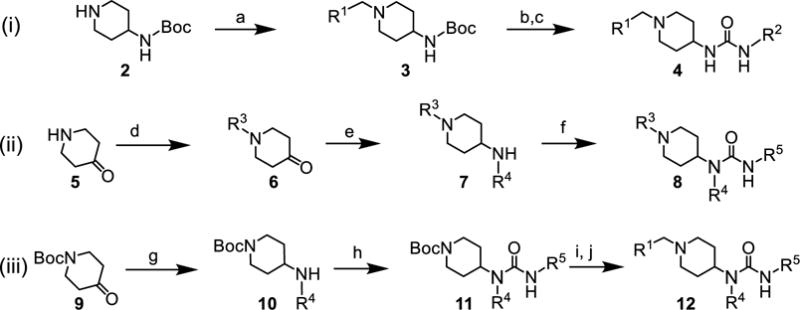
General synthetic routes for the generation of analogs. (a) R1-CHO, sodium triacetoxyborohydride, acetic acid, dichloromethane, 70-99%; (b) trifluoroacetic acid, dichloromethane, 0 °C – rt, 16 h; (c) R2-isocyanate, N,N-diisopropylethylamine, dichloromethane, rt, 40-75% (two steps); (d) R3-1, potassium carbonate, acetonitrile, 30-50%; (e) R4-NH2, sodium triacetoxyborohydride, acetic acid, dichloromethane, 70-99%; (f) R5-isocyanate, N,N-diisopropylethylamine, dichloromethane, rt 70-99%; (g) R4-NH2, sodium triacetoxyborohydride, acetic acid, dichloromethane, 70-99%; (h) R5-isocyanate, N,N-diisopropylethylamine, dichloromethane, rt, 65-95%; (i) trifluoroacetic acid, dichloromethane, 0 °C – rt, h; (j) R1-CHO, sodium triacetoxyborohydride, acetic acid, dichloromethane, 75-99% (two steps).
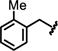
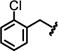
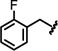
We initially focused on expanding the SAR data set around the Ile and hinge pockets (Table 1). We probed the Ile pocket with a variety of benzyl-substituted piperidines. We independently introduced methyl, chloro, and fluoro substituents in the ortho-, meta-, and para- positions to probe the steric and electronic requirements of the Ile pocket (13-21). While introduction of the methyl group at the ortho position (13) had little effect on potency, the electronegative chlorine and the fluorine each decreased potency (14 or 15). This suggested that, while the Ile pocket was relatively forgiving of steric bulk, electronegative substituents are disfavored. We observed this general trend for the meta and para positions (16-18) as well. This result suggested that maintaining the basicity of the piperidine amine was critical to retaining potency. One possible explanation is that the protonated piperidine ring is engaged in a water-mediated hydrogen bonding network within the N-acetyl pocket. This trend was reinforced by the observation that introduction of the benzoyl piperidine substitution drastically reduced potency (22).
Table 1. Optimization of the Hinge and Ile regions.
Values for the TR-FRET, solubility, and permeability experiments are represented as means plus or minus standard deviation calculated from at least one experiment, run in triplicate. ND indicates values were not determined.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| No | R1 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) | No | R2 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) |
| 1 |
|
7.6 ±1.0 | 32 ± 1.6 | 670 ± 100 | 23 |
|
>67 | 68 ± 3.4 | 1400 ± 250 |
| 13 |
|
4.0 ±0.18 | 44 ± 3.4 | 1200 ± 35 | 24 |
|
23 ±4.7 | ND | ND |
| 14 |
|
28 ±1.9 | 0.3 ±0.2 | 830 ± 1 | 25 |
|
>67 | 2.4 ± 0.1 | >2200 |
| 15 |
|
13 ±1.7 | 44 ± 1.4 | 1500 ± 1 | 26 |
|
44 ± 6.2 | ND | ND |
| 16 |
|
6.6 ± 0.32 | 35 ±8 | >2200 | 27 |
|
>67 | ND | ND |
| 17 |
|
33 ±0.11 | 0.7 ±0.6 | 1700 ± 1 | 28 |
|
>67 | ND | ND |
| 18 |
|
16 ±1.5 | 50 ± 2.7 | >2200 | 29 |
|
>67 | ND | ND |
| 19 |
|
19 ±1.9 | 14 ±0.7 | >2200 | 30 |
|
>67 | ND | ND |
| 20 |
|
25 ± 2.7 | 0.8 ±0.4 | 1300 ± 43 | 31 |
|
>67 | 1.0 ±0.6 | 930 ± 190 |
| 21 |
|
14 ±0.5 | 19 ± 1.2 | >2200 | 32 |
|
>67 | 78 ± 5.5 | >2200 |
| 22 |
|
>67 | 2.8 ± 0.8 | 910 ±290 | 33 |
|
>67 | 75 ± 0.6 | 220 ± 120 |
Next, we focused on optimizing substituents binding to the hinge pocket (Table 1). The 1:DCN1 co-structure (Figure 2) and initial SAR data set suggested that the hinge pocket is tightly packed. Thus, we designed analogs to be closely isosteric substitutions that maintain packing interactions, while testing whether incorporating heteroatoms could establish new electrostatic interactions with Thr113, Gln114, Glu116, or Glu121 (within 5-Å of hinge) (24-30). In general, any major deviation from the meta-trifluoromethyl substitution caused a sharp reduction in potency. Of note, even the meta-methyl-substituted phenyl ring (26) was practically inactive in the TR-FRET assay. Introduction of five- or six-membered heterocyclic rings also reduced potency (31- 33). These results supported our initial hypothesis that the hinge pocket afforded the sharpest SAR and that the tight steric packing of the hinge pocket around the m-CF3-substituted phenyl ring is crucial to compound binding. Our results also suggest that there is little room for further compound optimization in the hinge pocket. Thus, the remainder of our studies utilized the meta-CF3-substituted phenyl ring.
Preliminary optimization of the Ile and hinge regions, although informative concerning determinants of binding, did not lead to the significant improvements in potency. In an effort to gain potency, we turned our attention towards the currently unoccupied N-acetyl and Leu pockets. Based on the 1:DCN1 co-structure (Figure 2), we envisaged that substitution of the 3-position of the piperidine ring could provide access to the N-acetyl pocket. Modifications of published procedures allowed diastereoselective synthesis of the trans-3-acetyl piperidine 34.59–65 In contrast to expectations, 34 did not possess improved potency compared to our initial HTS hit 1. Analysis of the 34:DCN1 X-ray co-structure (Figure 3) provided an explanation: the acetate did not fill the intended N-acetyl pocket but instead the molecule had flipped to place this substituent into the Leu pocket, suggesting that fulfilling this hydrophobic interaction was more important than fulfilling those in the N-acetyl pocket. To test this hypothesis, we focused on analogs that would target the Leu pocket.
Figure 3.
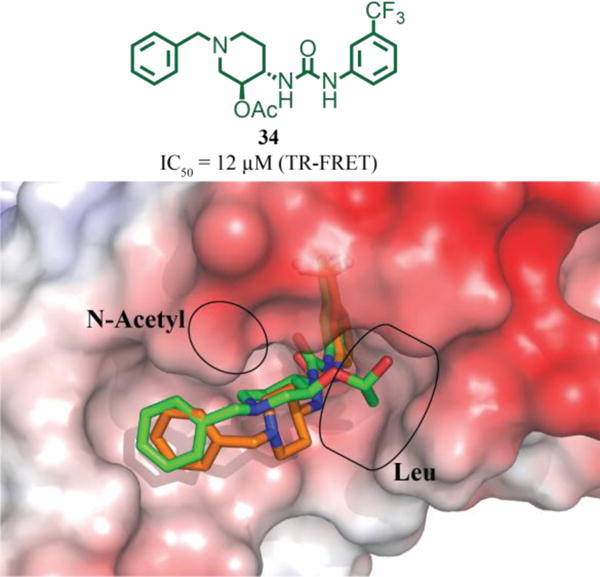
Chemical structure of 34 and overlay of 34(green):DCN1(electrostatic potential surface) and 1(orange):DCN1 X-ray co-crystal structures (PDB 6BG3 and 5V83), highlighting that the acetate substitution, which targeted the N-acetyl pocket flipped into the Leu pocket.
The HTS set contained a small number of analogs possessing a tri-substituted urea in which the alkyl nitrogen of the urea was substituted with a furfuryl group. Given the placement of the alkyl-substituted nitrogen of the urea in the 1:DCN1 co-structure and the requirement for only one H-bond donor (urea NH), we hypothesized that this furfuryl substitution occupied the targeted Leu pocket. Based on this precedent we developed a short synthetic sequence that provided rapid access to such tri-substituted ureas (Scheme 1).
In contrast to the unsubstituted urea subseries, introduction of the alkyl urea substituent targeting the Leu sub-pocket switched preferences in the originally forgiving Ile pocket to favor alkyl over aryl substitutions. Accommodating this observation, we fixed the hinge targeting substituent as the m-CF3-phenyl and the Ile-targeting substituent as a propyl group, and then varied the moiety targeting the Leu pocket (Table 2). Alkyl groups were generally well-tolerated (35-38), but introduction of the linear ether (39) reduced potency, suggesting electrostatic repulsion. The butyl (36) and methyl cyclohexyl (40) substitutions afforded the most potent compounds among the alkyl series. Exploring aromatic substituents, the furfuryl substitution (41) provided the first sub-micromolar potency compound. Further exploration revealed that a variety of differentially substituted six-membered aryl and heteroaryl rings also produced potent inhibitors (42-46). Overall, both alkyl and benzyl substituents were well tolerated. We hypothesized that the increase in potency came from the urea substituent accessing the targeted Leu pocket, while maintaining the urea pocket hydrogen bond with Gln114 and the tight steric fit in the hinge pocket. This model was supported by the X-ray co-structure of 45 bound to DCN1 (Figure 4), which showed that all of the hypothesized interactions were fulfilled. Surprisingly, introduction of the third substituent on the urea led to modest improvements in solubility and permeability, suggesting some self-organization in solvent if the full hydrogen-bonding capability of the ureas is maintained. Given the equivalent potencies of the various Leu substituents, we chose the benzyl group for final optimization due to easy access to a wide range of related analogs and ease of synthesis.
Table 2. Optimization of the Leu pocket.
Values for the TR-FRET, solubility, and permeability experiments are represented as means plus or minus standard deviation calculated from at least one experiment, run in triplicate. ND indicates values were not determined.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| No | R1 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) | No | R1 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) |
| 35 |
|
10 ± 1.2 | 61 ± 3.1 | 1600 ±170 | 41 |
|
0.75 ± 0.26 | 69 ± 1.4 | >2200 |
| 36 |
|
1.4 ±0.23 | 70 ± 2.3 | >2200 | 42 |
|
0.82 ± 0.36 | 39 ± 2.9 | 1800 ± 1 |
| 37 |
|
5.8 ± 1.3 | 96 ± 9.7 | 1900±200 | 43 |
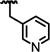
|
1.3 ±0.25 | <0.1 | 1300 ±79 |
| 38 |
|
2.7 ± 0.29 | 61 ± 3.7 | 1500 ±5.4 | 44 |
|
4.2 ±0.38 | 85 ± 2.7 | 700 ± 73 |
| 39 |
|
21 ±3.8 | 64 ± 7.6 | 1600 ± 1 | 45 |
|
> 1.1 ±0.12 | 66 ± 4.2 | >2200 |
| 40 |
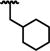
|
2.4 ±2.4 | 90 ± 12 | >2200 | 46 |
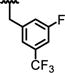
|
1.0 ±0.53 | 37 ± 2.4 | >2200 |
Figure 4.
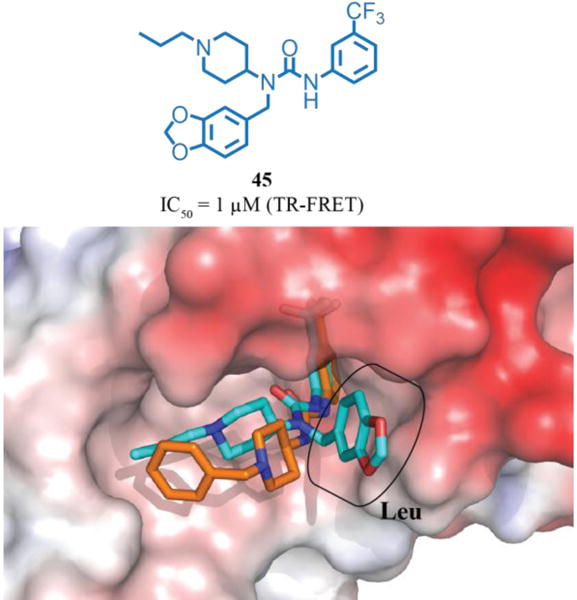
Chemical structure of 45 and overlay of 45(green):DCN1(electrostatic potential surface) and 1(orange):DCN1 X-ray co-crystal structures, highlighting successful targeting of the Leu pocket (PDB 6BG5 and 5V83).
One of our key observations in these studies was that the contributions of the Ile-targeting substituent could be non-additive depending upon the identity of the other substituents. Therefore, for the final round of optimization we fixed the substituent targeting the hinge region as the m-CF3–phenyl and that targeting the Leu region as an unsubstituted benzyl. We then readdressed optimization of the substituent targeting the Ile pocket (Table 3). Removal of N-alkyl group from the piperidine reduced activity (47). Careful exploration of the nature of the alkyl chain revealed a preference for an n-butyl group (47-51) and that branching groups on the alkyl chain improved potency (52-56). While a variety of patterns was tolerated, the 2-pentyl group (52) produced the most potent analog synthesized to date. This alkyl chain, which represents a constrained version of the native Ile side chain of UBE2M, most efficiently occupies the sub-pocket. Substitution of the piperidine with cyclic five- or six-membered alkyl rings led to slight decreases in potency (57 and 58). Consistent with the previous studies described above, completely removing the ionizable nitrogen by introduction of an amide caused a significant drop in potency (59 and 60). One explanation for the observed loss of potency is that the reduction in basicity substantially reduced protonation of the piperidine nitrogen, inhibiting its ability to partake in the previously hypothesized hydrogen-bonding network. To test this hypothesis, we introduced a sulfonamide substitution. Our specific structural hypothesis was that while reducing the basicity of the piperidine nitrogen, the sulfonamide could correctly orient one of its oxygen atoms to participate in the hydrogen-bonding network. The sulfonamides possessed superior potency, supporting our hypothesis (61 and 62). Next, we explored introducing aromatic and heteroaromatic substituents, all of which possessed reduced potency (63-70). Notably, the dihydrobenzo[1,4]dioxine analog (71) retained potency but at significant detriment to solubility. These results show that switching to a substituted urea to efficiently target the Leu sub-pocket leads to significantly more stringent steric requirements in the Ile-targeting substituent, as compared to the di-substituted ureas.
Table 3. Final optimization of the Ile pocket.
Values for the TR-FRET, solubility, and permeability experiments are represented as means plus or minus standard deviation calculated from at least one experiment, run in triplicate. ND indicates values were not determined.
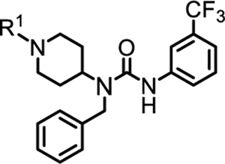
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| No | R1 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) | No | R1 | IC50 (μM) (TR-FRET) | Avg. Sol (μM) | Pampa (×10−6 cm/s) |
| 47 |
|
21 ±6.1 | 71 ±0.9 | >2200 | 61 |
|
0.74 ± 0.03 | 0.5 ± 0.6 | <1 |
| 42 |
|
0.82 ± 0.36 | 39 ±2.9 | 1800 ± 1 | |||||
| 48 |
|
23 ± 3.8 | 2.3 ± 1.5 | 440 ± 1 | 62 |
|
0.14 ±0.11 | 1.0 ±0.1 | <1 |
| 49 |
|
0.26 ±0.13 | 70 ± 1.4 | >2200 | 63 |
|
> 67 | 6.3 ± 1.4 | 310 ±540 |
| 50 |
|
0.66 ±0.10 | 58 ± 1.4 | 1400 ± 540 | |||||
| 51 |
|
1.1 ±0.09 | ND | ND | 64 |
|
5.9 ± 2.2 | 110 ±2.4 | 50 ± 1.2 |
| 52 |
|
0.062 ± 0.01 | 57 ± 1.4 | 1400 ±39 | 65 |
|
2.4 ±2.3 | 54 ± 3.2 | >2200 |
| 53 |
|
0.21 ± 0.02 | 75 ± 1.8 | >2200 | 66 |
|
3.2 ± 1.5 | 11 ±0.3 | 300 ± 460 |
| 54 |
|
0.96 ± 0.08 | ND | ND | 67 |
|
20 ± 12 | 30 ± 1.1 | >2200 |
| 55 |
|
0.79 ± 0.05 | 60 ± 0.7 | >2200 | |||||
| 56 |
|
0.17 ±0.03 | 5.8 ± 0.6 | >2200 | 68 |
|
10 ±0.69 | 0.9 ±0.1 | >2200 |
| 57 |
|
0.37 ± 0.06 | 48 ± 0.3 | 990±110 | 69 |
|
> 67 | 0.4 ±0.1 | 110 ±190 |
| 58 |
|
0.62 ± 0.06 | 1.2 ±0.1 | <1 | |||||
| 59 |
|
36 ± 7.2 | ND | ND | 70 |
|
12 ±4.2 | <0.1 | >2200 |
| 60 |
|
36 ± 15 | <0.1 | >2200 | 71 |
|
0.36 ± 0.21 | 1.0 ± 1.3 | >2200 |
Having improved potency by 100-fold without detriment to solubility or permeability, we focused on evaluating the best compounds in more stringent assays (Table 4). We measured equilibrium binding constants using isothermal titration calorimetry. Generally, the binding affinity tracked well with the previously measured inhibitory potency. This finding suggests that the observed SAR trends derive from the varying affinity of the compounds for DCN1. Next, we tested the ability of the compounds to inhibit DCN1-mediated cullin neddylation of the C-terminal domain of Cul2 as representative of cullins 1-5 based on prior data, using a variation of our published assay (Figure 1B).1, 49 The potency for inhibiting neddylation activity correlated closely with the ability to prevent formation of the DCN1-UBE2M complex.
Table 4. Evaluation of top compounds for equilibrium binding, biochemical inhibition of cullin neddylation, and proliferation inhibition.
Values for the TR-FRET, proliferation, solubility, and permeability experiments are represented as means plus or minus standard deviation calculated from at least one experiment, run in triplicate. Values for the pulse-chase and isothermal titration calorimetry (ITC) experiments are from a single experiment. BJ cells are normal fibroblasts lacking DCN1 amplification. HCC95 cells are a squamous cell carcinoma line containing DCN1 amplification. The greater than symbol indicates that at the highest concentration tested less than 50% growth inhibition was observed. ND indicates values were not determined.
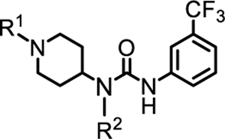
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| No | R1 | R2 | IC50 (μM) (TR-FRET) |
Kd (μM) (ITC) |
EC50 (μM) (Pulse-Chase) |
Proliferation EC50 (μM)
|
Avg. Sol (μM) |
Pampa (×10−6 cm/s) |
|
| BJ cells | HCC95 cells | ||||||||
| 1 |
|
|
7.6 ± 1.0 | ND | 4.2 | >24 | >32 | 32 ± 1.6 | 670±100 |
| 41 |
|
|
0.74 ± 0.26 | 0.70 | 0.57 | >23 | >23 | 69 ± 1.4 | >2200 |
| 42 |
|
|
0.82 ± 0.36 | 0.86 | 0.40 | >23 | >40 | 39 ± 2.9 | 1800 ± 1 |
| 43 |
|
|
1.3 ± 0.25 | ND | 1.7 | >23 | >23 | <0.1 | 1300 ±79 |
| 45 |
|
|
1.1 ± 0.12 | ND | 0.49 | >23 | >40 | 66 ± 4.2 | >2200 |
| 46 |
|
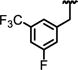
|
1.0 ± 0.53 | ND | 0.18 | >23 | 21 ± 1.0 | 37 ± 2.4 | >2200 |
| 49 |
|
|
0.26 ± 0.13 | 0.70 | 0.14 | >40 | >40 | 70 ± 1.4 | >2200 |
| 52 |
|
|
0.062 ± 0.01 | 0.14 | 0.079 | >40 | 47 ± 2.6 | 57 ± 1.4 | 1400 ± 39 |
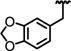
| 72 |
|
|
0.36 ± 0.21 | ND | 0.11 | >40 | 46 ± 5.6 | 71 ± 4.4 | >2200 |
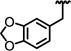
| 73 |
|
|
0.31 ± 0.05 | ND | 0.37 | >40 | >40 | 75 ± 3.1 | >2200 |
Since DCN1 is one of five paralogues in the human genome (DCN1-5) that all function in the neddylation pathway, we elected to evaluate the isoform selectivity of our most potent compound 52 (Figure 5) at concentrations similar to the ones used for our cell-based studies. Compound 52 was highly selective for inhibiting neddylation activity of DCN1 and DCN2, exhibiting only very moderate inhibition of DCN3-5 (slight activity at 50 μM). Dose-response isoform selectivity studies for compound 42 (Supplementary Figure 2) and our previously published studies1 suggest selective and approximately equipotent inhibition of DCN1/2 over DCN3/4/5 is a general feature of the optimized scaffold. These results reflect the high sequence identity between DCN1 and DCN2 (identity = 82%), including identical residues comprising the 52 binding pocket on DCN1. DCN3-5 exhibit lower sequence identity (< 40%). The high specificity of 52 amongst the DCN family members bodes well for its selectivity against other less-related binding pockets.
Figure 5.
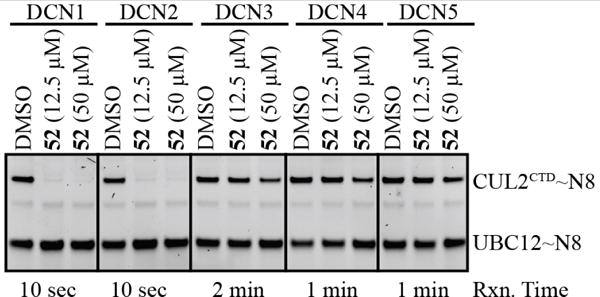
Pulse-chase assay monitoring transfer of NEDD8 from UBE2M to CUL2 with the indicated concentrations of 52 as tested against all five of the DCN family members.
Next, we evaluated the potential of our compounds to inhibit related enzymes within the NEDD8 pathway. First, we tested whether our compounds inhibit the NEDD8 E1 enzyme. Compounds 49 and 52 do not inhibit the E1 activating enzyme for NEDD8, even at concentrations as high as 50 μM (Supplementary Figure 3). Next, we investigated whether our compounds inhibit UBE2F mediated cullin neddylation. Previous studies have shown that both NEDD8 E2 enzymes, UBE2M and UBE2F, mediate cullin neddylation and are stimulated by the presence of a DCN family member.49 As expected, both compounds 49 and 52 inhibit DCN1 but not DCN3 stimulated NEDD8 transfer from UBE2F to the cullins (Supplementary Figure 4). Finally, we demonstrated that our top compounds (46, 49, 52, and 73) do not inhibit the slower, DCN1-independent neddylation of cullins, catalyzed solely by the NEDD8 co-E3 enzyme RBX1. (Supplementary Figure 5). Satisfied that the best compounds were selectively and effectively inhibiting both the protein-protein interaction and cullin neddylation, we examined their activity in cell-based studies.
First, we assessed non-specific inhibition of cell growth using a normal fibroblast (BJ) cell line lacking DCN1 amplification; none of the compounds were toxic or growth inhibitory (Table 4). Next, we assessed growth inhibition using a SCC line that contains significant DCN1 amplification (HCC95 cells); the compounds were slightly more toxic in this cell line, but only at high doses nearing their maximum solubility (Table 4). All of the lead compounds tested exhibited reasonable to good solubility (> 30 μM in pH = 7.4 buffer) and permeability (> 1000 ×10−6 cm/s), suggesting that they would be active against cultured cells and that oral dosing may be possible in animal models (Table 4).
Next, we used the cellular thermal shift assay (CETSA)66 to demonstrate that compounds 49 and 52 bind to DCN1 in HCC95 cells (Figure 6). The DCN1 protein is largely degraded at 53 °C in HCC95 cells treated with either DMSO or a previously reported negative control (NAcM-NEG)1. However, in the presence of 10 μM of compound 49, 52, or the known DCN1/2 inhibitor NAcM-OPT1, the thermal stability of the DCN1 protein is clearly enhanced. This suggests that both compounds 49 and 52 engage DCN1 in cells.
Figure 6.
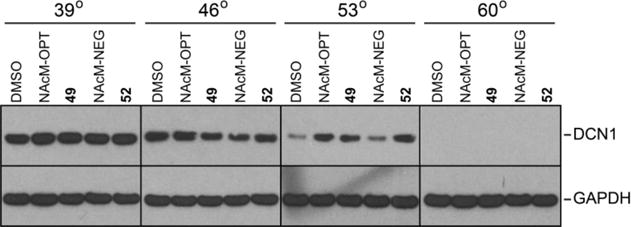
Enhancement of DCN1 thermal stability by compounds 49, 52, and NAcM-OPT (positive control) but not by NAcM-NEG (negative control) or DMSO. HCC95 cells were treated with either DMSO or 10 μM of the indicated compound for 1 h, heated at the indicated temperature for 3 minutes, lysed, and blotted with the indicated antibodies. Note: NAcM-OPT and NAcM-NEG correspond to previously published DCN1/2 inhibitors.1
Finally, we evaluated our top compounds’ efficacy at inhibiting cullin neddylation in HCC95 cells.67,1 While we observed minimal cellular activity at a concentration matching the biochemical IC50 of 0.060 μM, increasing the concentration of our inhibitors to 20 μM led to a significant decrease in cullin neddylation (Figure 7, Supplementary Figure 6). Although compounds 49 and 52 inhibited neddylation at 20 μM, this concentration was only minimally growth inhibitory for monolayer cultures (Table 4). The concentration required to suppress cullin neddylation in cells represents the biochemical IC95 of our inhibitors and suggests that cellular activity requires complete inhibition of DCN1 activity. This hypothesis is further supported by previous studies with the NEDD8 E1 inhibitor Pevonedistat, which exhibits complete inhibition of cellular neddylation at concentrations well above its biochemical IC50 value of 5 nM (Pevonedistat: biochemical IC50 = 5 nM, cell-based studies routinely conducted at 0.3-1 μM).26
Figure 7.
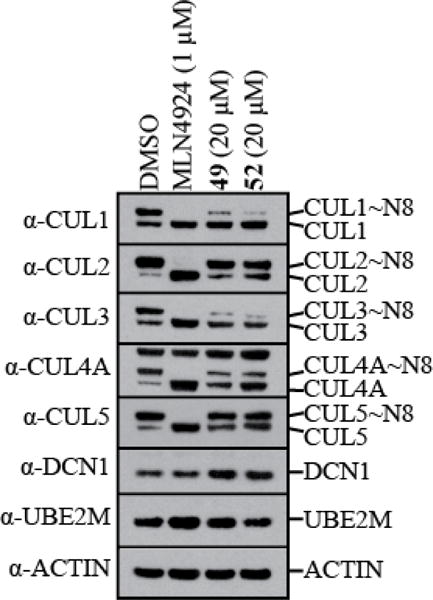
Western blot for inhibition of cellular neddylation at 48 hours by DMSO, MLN4924 (positive control, 1 μM, single dose at T = 0), 49 (20 μM, dosing at T = 0, and T = 24 hr), and 52 (20 μM, dosing at T = 0, and T = 24 hr).
Discussion and Conclusions
In summary, structure-enabled optimization of our initial HTS hit 1, has led to the discovery of 52, which is a 100-fold more potent inhibitor of the interaction between UBE2M and DCN1, is ten-fold more tightly bound to DCN1, blocks DCN1-mediated neddylation in biochemical assays with potency equivalent to that for inhibiting the protein interaction, and possesses modestly increased solubility and permeability (Figure 8). A ten-fold increase in potency is attributed to the benzyl group accessing the Leu pocket as confirmed by X-ray crystallography of 45 bound to DCN1. Another ten-fold increase in potency is due to the optimization of interactions in the Ile pocket, again confirmed by X-ray crystallography. The combination of multiple X-ray co-structures of our inhibitors bound to DCN1, the HTS data set, and preliminary synthetic efforts provide a detailed model of the binding mode of this series of compounds, enabling us to refine our minimum pharmacophore to include four key drivers for potency: (1) the piperidine linker that orients the ends of the molecule into the hydrophobic Ile and hinge sub-pockets, (2) a hydrogen bond between the urea aryl N-H and Gln114 that anchors the inhibitor into the binding site, (3) the tight steric packing of the hinge pocket around the substituted phenyl ring, and (4) the hydrophobic interactions of the benzyl substituent within the Leu region (Figure 8B).
Figure 8.
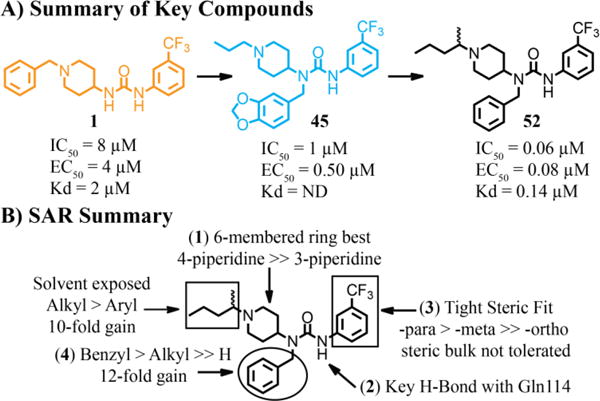
Summary of optimization. A) Chemical structure of key compounds B) Structure-activity relationship summary for HTS hits and newly synthesized inhibitors. ND indicates values were not determined.
Compound 52 and its closely related analogs (Table 4) are the first set of compounds to exhibit stronger binding affinities for DCN1 than the native UBE2MNAc substrate (Kd ~ 1 μM)49. Importantly, these compounds have been sufficiently optimized to bind to DCN1 and inhibit cullin neddylation in cells (Figure 6, 7). The highly modular and short synthetic routes outlined in Scheme 1 enabled rapid access to analogs as well as the preparation of our top compound, 52, on gram scale. Future efforts will focus on further improving potency by accessing both the Leu and currently unoccupied N-acetyl pockets and establishing the pharmacokinetic profile of our top compounds in vivo.
The cellular effects of compound 52 on the steady-state level of cullin neddylation differ substantially from treatments with Pevonedistat, which completely inhibits all cullin neddylation. In HCC95 cells, treatment with either compound 49 or 52 selectively decreases the steady-state level of cullin neddylation for CUL1 and CUL3, while CUL2, CUL4A, and CUL5 are relatively unaffected (Figure 7). In the cellular context, it is currently unknown why neddylation of a particular Cullin family member would require DCN1 activity or not. This requirement likely varies from cell type to cell type. In lower eukaryotes (C. elegans and S. cerevisiae) that express only a single DCN family member, elimination of DCN1 leads to a 90% decrease in the steady-state level of neddylated cullins.68 However in mammalian cell lines, which contain five DCN isoforms, DCN1 knockdown or knockout has relatively subtle effects on steady-state levels of cullin neddylation.48, 69–72 Potential factors dictating this selectivity include, but are not limited to, the expression of other DCN family members, cellular localization, expression levels of neddylation components, expression levels of Cullin binding modulators, and substrate demand.
We, and others, have recently reported the discovery of inhibitors of DCN1.1, 53, 73 In concordance with our previous studies,1 acute pharmacological inhibition of DCN1-mediated cullin neddylation with compounds 49 and 52 failed to cause gross accumulation of several well-recognized proteasomal targets as measured by immunoblot analysis (Supplementary Figure 6). The lack of gross substrate stabilization is likely why our compounds do not exhibit the potent cytotoxicity of Pevonedistat and related NAE inhibitors. A more thorough biochemical and cellular characterization of an optimized variant of the scaffold presented herein was recently published.1 Experiments with the optimized DCN1/2 selective inhibitors in numerous cell lines illustrate the Cullin effects are cell line dependent and suggest multiple factors, which are likely cell-type specific, contribute to the dependence on DCN1 for levels of neddylation.1 Future experiments, outside the scope of this work, will focus on understanding the selectivity and the phenotypic consequences of acute DCN inhibition in cells and murine models.
Experimental Section
General Synthetic procedures
All moisture sensitive reactions were performed using syringe-septum techniques under an atmosphere of dry N2 unless otherwise noted. All glassware was dried in an oven at 140 °C for a minimum of 6 h or flame-dried under an atmosphere of dry nitrogen prior to use. Methylene chloride and acetonitrile were dried using an aluminum oxide column. Deuterated chloroform was stored over anhydrous potassium carbonate. Reactions were monitored by TLC analysis (pre-coated silica gel 60 F254 plates, 250 μm layer thickness) and visualized by using UV lamp (254 nm) or by staining with either Vaughn’s reagent (4.8 g of (NH4)6Mo7O24·4H2O and 0.2 g of Ce(SO4)2 in 100 mL of a 3.5 N H2SO4) or a potassium permanganate solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of a 0.1% NaOH solution). Unless otherwise specified, commercially available reagents were used as received. Flash column chromatography was performed using a Biotage Isolera one and Biotage KP-SIL SNAP cartridges. Melting points were acquired on Buchi Melting Point B-545. All NMR data was collected at room temperature in CDCl3 or (CD3)2SO on a 400 or 500 MHz Bruker instrument. Chemical shifts (δ) are reported in parts per million (ppm) with internal CHCl3 (δ 7.26 ppm for 1H and 77.00 ppm for 13C), internal DMSO (δ 2.50 ppm for 1H and 39.52 ppm for 13C), or internal TMS (δ 0.0 ppm for 1H and 0.0 ppm for 13C) as the reference. 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, p = pentet, sext = sextet, sep = septet, m= multiplet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, qd = quartet of doublets), coupling constant(s) (J) in Hertz (Hz), and integration. Purity was assessed by LC/MS/UV/ELSD using a Waters Acquity UPLC-MS with the purity being assigned as the average determined by UV/ELSD. All compounds were confirmed to ≥95% purity. Identity and purity were further confirmed by NMR.
General reductive amination procedure
To a stirred solution of Boc-4-aminopiperidine (5.00 g, 25.0 mmol, 1 equiv) and benzaldehyde (2.54 mL, 25.0 mmol, 1 equiv) in dry CH2Cl2 (60 mL) was added acetic acid (1.72 mL, 25.0 mmol, 1.2 equiv). The resulting cloudy mixture was stirred at room temperature for 1 hr and sodium triacetoxyborohydride (10.6 g, 50.0 mmol, 2 equiv) was added portion wise. The resulting heterogeneous mixture was stirred at room temperature overnight (ca. 16 h), quenched with a saturated aqueous solution of NaHCO3, the organic layer was separated, and the remaining aqueous layer was extracted with EtOAc (×2). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The crude mixture was purified by chromatography on SiO2 (MeOH/CH2Cl2, 1:20 to 1:9) to give 5.63 g (78% yield) of the desired product as a colorless oil.
General Boc deprotection procedure
To a stirred solution of tert-butyl (1-benzylpiperidin-4-yl) carbamate (1.00 g, 3.44 mmol, 1 equiv) in CH2Cl2 (20 mL) at 0 °C was slowly added trifluoroacetic acid (1.32 mL, 17.2 mmol, 5 equiv). The reaction mixture was slowly warmed to room temperature, stirred at room temperature overnight (ca. 16 h), and concentrated under reduced pressure. The crude reaction mixture was directly carried onto the next reaction without purification.
General isocyanate addition
To a stirred solution of 1-benzylpiperidin-4-amine (0.250 g, 1.314 mmol, 1.2 equiv) in CH2Cl2 (6 mL) was added 1-isocyanato-3-(trifluoromethyl)benzene (1.53 mL, 1.10 mmol, 1 equiv) and N,N-diisopropylethylamine (0.286 mL, 1.64 mmol, 1.5 equiv). The resulting mixture was stirred at room temperature overnight (ca. 16 hr), concentrated under reduced pressure, and purified by chromatography on SiO2.
Spectral characterization of key compounds
1-(1-benzylpiperidin-4-yl)-3-(3-(trifluoromethyl)phenyl)urea (1)
Prepared according to general procedure C. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.96 (d, J = 2.2 Hz, 1H), 7.50 – 7.38 (m, 2H), 7.35 – 7.27 (m, 4H), 7.27 – 7.19 (m, 2H), 6.24 (d, J = 7.6 Hz, 1H), 3.56 – 3.40 (m, 3H), 2.71 (d, J = 11.4 Hz, 2H), 2.07 (t, J = 11.0 Hz, 2H), 1.80 (dd, J = 16.0, 4.0 Hz, 2H), 1.41 (qd, J = 16.0, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 154.3, 141.3, 138.6, 129.7, 129.4 (q, J = 30.3 Hz, 1C), 128.8, 128.1, 126.9, 124.3 (q, J = 273.3 Hz, 1C), 121.0, 117.1 (q, J = 4.0 Hz, 1C), 113.4 (q, J = 4.0 Hz, 1C), 62.2, 51.7, 46.3, 32.0. m/z (APCI-pos) M+1 = 378.5. HRMS (ESI+) m/z calcd for C20H23F3N3O [M+H]+ 378.1793, found 378.1790. Note: Spectra can be found in a previous publication.1
(3S,4S)-1-benzyl-4-(3-(3-(trifluoromethyl)phenyl)ureido)piperidin-3-yl acetate (34)
Prepared as a racemic mixture according based on published procedures (ref59–65). 1H NMR (400 MHz, Acetone-d6) 8.30 (s, 1H), 8.05 (s, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.38 – 7.28 (m, 4H), 7.28 – 7.18 (m, 2H), 5.80 (d, J = 8.1 Hz, 1H), 4.79 (td, J = 10.5, 5.5 Hz, 1H), 3.86 – 3.70 (m, 1H), 3.57 (s, 2H), 3.04-2.83 (m, 3H), 2.29 – 2.09 (m, 2H), 1.94 (2, 3H), 1.59 (q, J = 12.3 Hz, 1H). 13C NMR (101 MHz, Acetone-d6) δ 170.9, 155.4, 142.4, 139.1, 131.3 (q, J = 31.9 Hz), 130.4, 129.7, 129.1, 128.0, 125.4 (q, J = 271.4 Hz) 122.0, 118.46 (q, J = 3.8 Hz), 115.06 (q, J = 4.0 Hz), 72.4, 62.7, 56.8, 52.5, 52.3, 31.9, 21.0. m/z (APCI-pos) M+1 = 436.3 HRMS (ESI+) m/z calcd for C22H25F3N3O3 [M+H]+ 436.1844, found 436.1848. Note: Spectra are reproduced in supplemental information.
1-(benzo[d][1,3]dioxol-5-ylmethyl)-1-(1-propylpiperidin-4-yl)-3-(3-(trifluoromethyl)phenyl)urea (45)
Prepared according to general procedure C. 1H NMR (400 MHz, Chloroform-d) δ 7.53 (s, 1H), 7.27 – 7.10 (m, 3H), 6.78 – 6.68 (m, 3H), 6.44 (s, 1H), 5.91 (s, 2H), 4.34 (s, 3H), 2.97 (d, J = 15.0 Hz, 2H), 2.34 – 2.20 (m, 2H), 2.12 – 1.97 (m, 2H), 1.80 – 1.66 (m, 4H), 1.44 (sext, J = 10.0 Hz, 2H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.5, 148.7, 147.4, 139.6, 131.2, 131.0 (q, J = 31.5 Hz, 1C), 120.1, 123.9 (q, J = 273.7 Hz, 1C), 122.6, 121.1, 119.4 (q, J = 3.8 Hz, 1C), 119.1, 116.3 (q, J = 3.8 Hz, 1C), 108.7, 106.6, 101.3, 60.4, 53.0, 52.5, 45.9, 29.8, 20.0, 11.8. m/z (APCI-pos) M+1 = 464.4 HRMS (ESI+) m/z calcd for C24H29F3N3O3 [M+H]+, 464.2161 found 464.2159. Note: Spectra are reproduced in supplemental information.
1-benzyl-1-(1-butylpiperidin-4-yl)-3-(3-(trifluoromethyl)phenyl)urea (49)
Prepared according to general procedure C. 1H NMR (500 MHz, Chloroform-d) δ 7.60 (s, 1H), 7.37 (dd, J = 8.5, 6.5 Hz, 2H), 7.34 – 7.26 (m, 3H), 7.25-7.21 (m, 2H), 7.20-7.17 (m, 1H), 6.67 (s, 1H), 4.51 (s, 2H), 4.38 (tt, J = 10.6, 5.0 Hz, 1H), 2.96 (dt, J = 12.4, 2.8 Hz, 2H), 2.35 – 2.21 (m, 2H), 1.99 (td, J = 11.4, 3.4 Hz, 2H), 1.80 – 1.63 (m, 4H), 1.43 (ddd, J = 15.4, 8.9, 6.1 Hz, 2H), 1.30 (sext, J = 7.3 Hz, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 155.4, 139.5, 137.5, 130.8 (q, J = 31.5 Hz, 1C), 128.9, 128.8, 127.6, 125.9, 123.8 (q, J = 272.2 Hz, 1C), 122.7, 119.1 (q, J = 3.8 Hz, 1C), 116.4 (q, J = 3.8 Hz, 1C), 58.1, 52.9, 52.8, 45.8, 29.9, 29.1, 20.6, 13.8. m/z (APCI-pos) M+1 = 434.4. HRMS (ESI+) m/z calcd for C24H31F3N3O [M+H]+ 434.2419, found 434.2418. Note: Spectra are reproduced in supplemental information.
1-benzyl-1-(1-(pentan-2-yl)piperidin-4-yl)-3-(3-(trifluoromethyl)phenyl)urea (52)
Prepared according to general procedure C. 1H NMR (500 MHz, Chloroform-d) δ 7.57 (s, 1H), 7.42 – 7.36 (m, 2H), 7.34-7.30 (m, 3H), 7.29 – 7.17 (m, 4H), 6.50 (s, 1H), 4.53 (s, 2H), 4.37 (tt, J = 12.1, 4.2 Hz, 1H), 2.84-2.80 (m, 2H), 2.57 (sext, J = 7.0 Hz, 1H), 2.42 (td, J = 11.6, 2.5 Hz, 1H), 2.27 (td, J = 11.6, 2.3 Hz, 1H), 1.83 – 1.76 (m, 2H), 1.69 (dqd, J = 24.1, 12.0, 3.8 Hz, 2H), 1.47 (dq, J = 12.5, 5.2 Hz, 1H), 1.41 – 1.13 (m, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 155.4, 139.6, 137.5, 130.8 (q, J = 31.5 Hz, 1C), 129.1, 129.0, 127.1, 126.0, 123.9 (q, J = 273.4 Hz, 1C), 122.6, 119.2 (q, J = 3.8 Hz, 1C), 116.3 (q, J = 3.8 Hz, 1C), 58.8, 53.2, 49.5, 46.1, 46.0, 35.7, 30.6, 30.4, 20.1, 14.1, 14.0. m/z (APCI-pos) M+1 = 448.2. HRMS (ESI+) m/z calcd for C25H33F3N3O [M+H]+, 448.2575 found 448.2575. Note: Spectra are reproduced in supplemental information.
TR-FRET Assay.1
The TR-FRET assay was carried out in black 384-well microtiter plates at a final volume of 20 μL per well. To screen library compounds, the assay cocktail was prepared as a mixture of 50 nM Biotin-DCN1, 20 nM Ac-UBE2M12-AlexaFluor488, 2.5 nM Tb-Streptavidin (ThermoFisher) in assay buffer (25 mM HEPES, 100 mM NaCl, 0.1% Triton X-100, 0.5 mM DTT, pH 7.5). Then the assay cocktail was incubated for 1 hr at room temperature and distributed using a WellMate instrument (Matrix). Compounds to be screened were added to assay plates from DMSO stock solutions by pin transfer using 50SS pins (V&P Scientific). The assay mixture was incubated for 1 hr at room temperature prior to measuring the TR-FRET signal with a PHERAstar FS plate reader (BMG Labtech) equipped with modules for excitation at 337 nm and emissions at 490 and 520 nm. The integration start was set to 100 μs and the integration time to 200 μs. The number of flashes was fixed at 100. The ratio of 520/490 was used as TR-FRET signal in calculations. Assay endpoints were normalized from 0% (DMSO only) to 100% inhibition (unlabeled competitor peptide) for hit selection and for curve fitting. All compounds were tested in at least triplicate.
Cellular thermal shift assay
The cellular thermal shift assay (CETSA) was performed according to the previously published procedure.66 Briefly, 500,000 HCC95 cells were aliquoted in 100 μL of PBS and DMSO or the indicated compound (10 μM) was added (2% final DMSO). Mixtures were incubated on ice for 1 h and the cells were washed three times with PBS buffer. Cell pellets were resuspended in 50 μL of PBS and heated for 3 minutes at the indicated temperature in a thermocycler. Cells were lysed by three rounds of freezing in liquid nitrogen and thawing on ice. After pelleting at 14,000 rpm for 15 minutes, equal amounts of supernatant were removed and blotted with the indicated antibodies.
Supplementary Material
Acknowledgments
We acknowledge the High Throughput Biosciences Center, Medicinal Chemistry Center, Compound Management, and High Throughput Analytical Chemistry Centers in Chemical Biology and Therapeutics; Hartwell Center for use of their personnel and facilities. We thank the staff at the BL8.2.1 and 22-ID beamlines at the Advanced Light Source and Advanced Photon Source. This study was funded through B.A.S., HHMI, and NIH R37GM069530, P30CA021765; J.T.H., NIH F32GM113310; American Syrian Lebanese Associated Charities, and St Jude Children’s Research Hospital.
Abbreviations Used
- DCN1
defective in cullin neddylation 1
- UBE2M
NEDD8-conjugating enzyme Ubc12
- SCC
Squamous cell carcinoma
- CRL
cullin ring ligases
- NEDD8
Neural Precursor Cell Expressed, Developmentally Down-Regulated 8
- SAR
structure-activity relationship
- SPR
structure-property relationship
- TR-FRET
time-resolved fluorescence resonance energy transfer
- HTS
high-throughput screen
- t-Bu
tert-butyl
- NAE
NEDD8-activating enzyme
- RBX1
Ring-Box 1, E3 Ubiquitin Protein Ligase
- CUL1
cullin 1
- AF
AlexaFluor 488
Footnotes
Supporting Information:
Characterization details for the other synthetic compounds as well as additional information regarding the X-ray crystallography and biological studies are described in Supporting Information.
PDB ID Codes:
PDB: 5V83, 3TDU, 6BG3, and 6BG5
Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions:
All authors contributed to specific parts of the manuscript. J.T.H and R.K.G. assume responsibility for the manuscript in its entirety. All authors have given approval to the final version of the manuscript.
References
- 1.Scott DC, Hammill JT, Min J, Rhee DY, Connelly M, Sviderskiy VO, Bhasin D, Chen Y, Ong SS, Chai SC, Goktug AN, Huang G, Monda JK, Low J, Kim HS, Paulo JA, Cannon JR, Shelat AA, Chen T, Kelsall IR, Alpi AF, Pagala V, Wang X, Peng J, Singh B, Harper JW, Schulman BA, Guy RK. Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat Chem Biol. 2017;13(8):850–857. doi: 10.1038/nchembio.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute; Bethesda, MD: https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April 2017, accessed Aug 8, 2017. [Google Scholar]
- 3.Estilo CL, P OC, Ngai I, Patel SG, Reddy PG, Dao S, Shaha AR, Kraus DH, Boyle JO, Wong RJ, Pfister DG, Huryn JM, Zlotolow IM, Shah JP, Singh B. The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110alpha in squamous cell carcinoma of the oral tongue. Clin Cancer Res. 2003;9(6):2300–2306. [PubMed] [Google Scholar]
- 4.Broderick SR, Golas BJ, Pham D, Towe CW, Talbot SG, Kaufman A, Bains S, Huryn LA, Yonekawa Y, Carlson D, Hambardzumyan D, Ramanathan Y, Singh B. SCCRO promotes glioma formation and malignant progression in mice. Neoplasia. 2010;12(6):476–484. doi: 10.1593/neo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.P OC, Sarkaria I, Talbot SG, Reddy P, Dao S, Ngai I, Shaha A, Kraus D, Shah J, Rusch V, Ramanathan Y, Singh B. SCCRO (DCUN1D1) induces extracellular matrix invasion by activating matrix metalloproteinase 2. Clin Cancer Res. 2008;14(21):6780–6789. doi: 10.1158/1078-0432.CCR-08-0719. [DOI] [PubMed] [Google Scholar]
- 6.Sarkaria IS, Pham D, Ghossein RA, Talbot SG, Hezel M, Dudas ME, Ebright MI, Chuai S, Memoli N, Venkatraman ES, Miller VA, Kris MG, Zakowski MF, Rusch VW, Singh B. SCCRO expression correlates with invasive progression in bronchioloalveolar carcinoma. The Annals of thoracic surgery. 2004;78(5):1734–1741. doi: 10.1016/j.athoracsur.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 7.Sarkaria I, P OC, Talbot SG, Reddy PG, Ngai I, Maghami E, Patel KN, Lee B, Yonekawa Y, Dudas M, Kaufman A, Ryan R, Ghossein R, Rao PH, Stoffel A, Ramanathan Y, Singh B. Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 2006;66(19):9437–9444. doi: 10.1158/0008-5472.CAN-06-2074. [DOI] [PubMed] [Google Scholar]
- 8.Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, Lancet D. The genecards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. 2016;130(30):54. 1–1, 33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 9.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334(6056):674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 12.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 13.Curran MP, McKeage K. Bortezomib: a review of its use in patients with multiple myeloma. Drugs. 2009;69(7):859–888. doi: 10.2165/00003495-200969070-00006. [DOI] [PubMed] [Google Scholar]
- 14.Jagannath S, Vij R, Stewart AK, Trudel S, Jakubowiak AJ, Reiman T, Somlo G, Bahlis N, Lonial S, Kunkel LA, Wong A, Orlowski RZ, Siegel DS. An open-label single-arm pilot phase II study (PX-171-003-A0) of low-dose, single-agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin Lymphoma, Myeloma Leuk. 2012;12(5):310–318. doi: 10.1016/j.clml.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Vij R, Wang M, Kaufman JL, Lonial S, Jakubowiak AJ, Stewart AK, Kukreti V, Jagannath S, McDonagh KT, Alsina M, Bahlis NJ, Reu FJ, Gabrail NY, Belch A, Matous JV, Lee P, Rosen P, Sebag M, Vesole DH, Kunkel LA, Wear SM, Wong AF, Orlowski RZ, Siegel DS. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661–5670. doi: 10.1182/blood-2012-03-414359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Wu P, Hu Y. Clinical and marketed proteasome inhibitors for cancer treatment. Curr Med Chem. 2013;20(20):2537–2551. doi: 10.2174/09298673113209990122. [DOI] [PubMed] [Google Scholar]
- 17.Stintzing S, Lenz HJ. Molecular pathways: turning proteasomal protein degradation into a unique treatment approach. Clin Cancer Res. 2014;20(12):3064–3070. doi: 10.1158/1078-0432.CCR-13-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 19.Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system – implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta. 2014;1843(1):150–162. doi: 10.1016/j.bbamcr.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Silva SR, Paiva SL, Lukkarila JL, Gunning PT. Exploring a new frontier in cancer treatment: targeting the ubiquitin and ubiquitin-like activating enzymes. J Med Chem. 2013;56(6):2165–2177. doi: 10.1021/jm301420b. [DOI] [PubMed] [Google Scholar]
- 21.Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15(12):3912–3916. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- 22.Gao Q, Yu GY, Shi JY, Li LH, Zhang WJ, Wang ZC, Yang LX, Duan M, Zhao H, Wang XY, Zhou J, Qiu SJ, Jeong LS, Jia LJ, Fan J. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget. 2014;5(17):7820–7832. doi: 10.18632/oncotarget.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes & cancer. 2010;1(7):708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 26.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 27.Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr -Relat Cancer. 2015;22(1):T55–70. doi: 10.1530/ERC-14-0315. [DOI] [PubMed] [Google Scholar]
- 28.Rulina AV, Mittler F, Obeid P, Gerbaud S, Guyon L, Sulpice E, Kermarrec F, Assard N, Dolega ME, Gidrol X, Balakirev MY. Distinct outcomes of CRL-Nedd8 pathway inhibition reveal cancer cell plasticity. Cell Death Dis. 2016;7(12):e2505. doi: 10.1038/cddis.2016.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS, Bence NF, Bolen JB, Brownell J, Dick LR, Loke HK, McDonald AA, Ma J, Manfredi MG, Sells TB, Sintchak MD, Yang X, Xu Q, Koenig EM, Gavin JM, Smith PG. Treatment-emergent mutations in NAEbeta confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21(3):388–401. doi: 10.1016/j.ccr.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U, Milhollen MA, Lightcap ES. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73(1):225–234. doi: 10.1158/0008-5472.CAN-12-1729. [DOI] [PubMed] [Google Scholar]
- 31.Lan H, Tang Z, Jin H, Sun Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep. 2016;6:24218. doi: 10.1038/srep24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310–10320. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116(9):1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 34.Leung CH, Chan DS, Yang H, Abagyan R, Lee SM, Zhu GY, Fong WF, Ma DL. A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun. 2011;47(9):2511–2513. doi: 10.1039/c0cc04927a. [DOI] [PubMed] [Google Scholar]
- 35.Lu P, Liu X, Yuan X, He M, Wang Y, Zhang Q, Ouyang PK. Discovery of a novel NEDD8 Activating Enzyme Inhibitor with Piperidin-4-amine Scaffold by Structure-Based Virtual Screening. ACS Chem Biol. 2016;11(7):1901–1907. doi: 10.1021/acschembio.6b00159. [DOI] [PubMed] [Google Scholar]
- 36.Zhong HJ, Wang W, Kang TS, Yan H, Yang Y, Xu L, Wang Y, Ma DL, Leung CH. A Rhodium(III) complex as an inhibitor of neural precursor cell expressed, developmentally down-regulated 8-activating enzyme with in vivo activity against inflammatory bowel disease. J Med Chem. 2017;60(1):497–503. doi: 10.1021/acs.jmedchem.6b00250. [DOI] [PubMed] [Google Scholar]
- 37.Zhong HJ, Yang H, Chan DS, Leung CH, Wang HM, Ma DL. A metal-based inhibitor of NEDD8-activating enzyme. PloS one. 2012;7(11):e49574. doi: 10.1371/journal.pone.0049574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, Jones M, Hassiepen U, Schaefer M, Kiffe M, Weiss A, Wiesmann C, Sedrani R, Eder J, Martoglio B. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. 2016;7:13166. doi: 10.1038/ncomms13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discovery. 2014;13(12):889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi D, Grossman SR. Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther. 2010;10(8):737–747. doi: 10.4161/cbt.10.8.13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bielskiene K, Bagdoniene L, Mozuraitiene J, Kazbariene B, Janulionis E. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina (Kaunas) 2015;51(1):1–9. doi: 10.1016/j.medici.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL, Thoma NH. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 44.Aghajan M, Jonai N, Flick K, Fu F, Luo M, Cai X, Ouni I, Pierce N, Tang X, Lomenick B, Damoiseaux R, Hao R, Del Moral PM, Verma R, Li Y, Li C, Houk KN, Jung ME, Zheng N, Huang L, Deshaies RJ, Kaiser P, Huang J. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol. 2010;28(7):738–742. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mund T, Lewis MJ, Maslen S, Pelham HR. Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc Natl Acad Sci US A. 2014;111(47):16736–16741. doi: 10.1073/pnas.1412152111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frost J, Galdeano C, Soares P, Gadd MS, Grzes KM, Ellis L, Epemolu O, Shimamura S, Bantscheff M, Grandi P, Read KD, Cantrell DA, Rocha S, Ciulli A. Potent and selective chemical probe of hypoxic signalling downstream of HIF-alpha hydroxylation via VHL inhibition. Nat Commun. 2016;7:13312. doi: 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurz T, Chou YC, Willems AR, Meyer-Schaller N, Hecht ML, Tyers M, Peter M, Sicheri F. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell. 2008;29(1):23–35. doi: 10.1016/j.molcel.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Keuss MJ, Thomas Y, McArthur R, Wood NT, Knebel A, Kurz T. Characterization of the mammalian family of DCN-type NEDD8 E3 ligases. J Cell Sci. 2016;129(7):1441–1454. doi: 10.1242/jcs.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monda Julie K, Scott Daniel C, Miller Darcie J, Lydeard J, King D, Harper JW, Bennett Eric J, Schulman Brenda A. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure. 2013;21(1):42–53. doi: 10.1016/j.str.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang G, Singh B. Coamplification and cooperation: toward identifying biologically relevant oncogenes. Clin Cancer Res. 2013;19(20):5549–5551. doi: 10.1158/1078-0432.CCR-13-2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bommelje CC, Weeda VB, Huang G, Shah K, Bains S, Buss E, Shaha M, Gonen M, Ghossein R, Ramanathan SY, Singh B. Oncogenic function of SCCRO5/DCUN1D5 requires its neddylation E3 activity and nuclear localization. Clin Cancer Res. 2014;20(2):372–381. doi: 10.1158/1078-0432.CCR-13-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma T, Shi T, Huang J, Wu L, Hu F, He P, Deng W, Gao P, Zhang Y, Song Q, Ma D, Qiu X. DCUN1D3, a novel UVC-responsive gene that is involved in cell cycle progression and cell growth. Cancer Sci. 2008;99(11):2128–2135. doi: 10.1111/j.1349-7006.2008.00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Lu J, Liu L, Bernard D, Yang CY, Fernandez-Salas E, Chinnaswamy K, Layton S, Stuckey J, Yu Q, Zhou W, Pan Z, Sun Y, Wang S. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat Commun. 2017;8(1):1150. doi: 10.1038/s41467-017-01243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, Schulman BA. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157(7):1671–1684. doi: 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carling RW, Moore KW, Moyes CR, Jones EA, Bonner K, Emms F, Marwood R, Patel S, Patel S, Fletcher AE, Beer M, Sohal B, Pike A, Leeson P. D. 1-(3-Cyanobenzylpiperidin-4-yl)-5-methyl-4-phenyl-1, 3-dihydroimidazol-2-one: a selective high-affinity antagonist for the human dopamine D(4) receptor with excellent selectivity over ion channels. J Med Chem. 1999;42(14):2706–2715. doi: 10.1021/jm991029k. [DOI] [PubMed] [Google Scholar]
- 56.Archibald JL, Beardsley DR, Bisset GM, Fairbrother P, Jackson JL, Opalko A, Ward TJ. Antihypertensive ureidopiperidines. J Med Chem. 1980;23(8):857–861. doi: 10.1021/jm00182a009. [DOI] [PubMed] [Google Scholar]
- 57.McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC. Potent and selective inhibition of human cytochrome P450 3A4 by seco-pancratistatin structural analogs. BioorgMed Chem Lett. 2010;20(7):2335–2339. doi: 10.1016/j.bmcl.2010.01.157. [DOI] [PubMed] [Google Scholar]
- 58.Seifert K, Buttner A, Rigol S, Eilert N, Wandel E, Giannis A. Potent small molecule Hedgehog agonists induce VEGF expression in vitro. Bioorg Med Chem. 2012;20(21):6465–6481. doi: 10.1016/j.bmc.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 59.Miles TJ, Axten JM, Barfoot C, Brooks G, Brown P, Chen D, Dabbs S, Davies DT, Downie DL, Eyrisch S, Gallagher T, Giordano I, Gwynn MN, Hennessy A, Hoover J, Huang J, Jones G, Markwell R, Miller WH, Minthorn EA, Rittenhouse S, Seefeld M, Pearson N. Novel amino-piperidines as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg Med Chem Lett. 2011;21(24):7489–7495. doi: 10.1016/j.bmcl.2011.09.117. [DOI] [PubMed] [Google Scholar]
- 60.Di Bussolo V, Fiasella A, Favero L, Bertolini F, Crotti P. Stereoselective synthesis of 4-amino-2,3-unsaturated-N-Cbz-imino-O-glycosides via new diastereoisomeric N-Cbz-imino glycal-derived allyl N-nosyl aziridines. Org Lett. 2009;11(12):2675–2678. doi: 10.1021/ol900742p. [DOI] [PubMed] [Google Scholar]
- 61.Danieli E, Proietti D, Brogioni G, Romano MR, Cappelletti E, Tontini M, Berti F, Lay L, Costantino P, Adamo R. Synthesis of Staphylococcus aureus type 5 capsular polysaccharide repeating unit using novel L-FucNAc and D-FucNAc synthons and immunochemical evaluation. Bioorg Med Chem. 2012;20(21):6403–6415. doi: 10.1016/j.bmc.2012.08.048. [DOI] [PubMed] [Google Scholar]
- 62.Bagal SK, Davies SG, Lee JA, Roberts PM, Scott PM, Thomson JE. Syntheses of the enantiomers of 1-deoxynojirimycin and 1-deoxyaltronojirimycin via chemo- and diastereoselective olefinic oxidation of unsaturated amines. J Org Chem. 2010;75(23):8133–8146. doi: 10.1021/jo101756g. [DOI] [PubMed] [Google Scholar]
- 63.Cherney RJ, Nelson DJ, Lo YC, Yang G, Scherle PA, Jezak H, Solomon KA, Carter PH, Decicco CP. Synthesis and evaluation of cis-3,4-disubstituted piperidines as potent CC chemokine receptor 2 (CCR2) antagonists. Bioorg Med Chem Lett. 2008;18(18):5063–5065. doi: 10.1016/j.bmcl.2008.07.123. [DOI] [PubMed] [Google Scholar]
- 64.Wang W, Cui J, Lu X, Padakanti PK, Xu J, Parsons SM, Luedtke RR, Rath NP, Tu Z. Synthesis and in vitro biological evaluation of carbonyl group-containing analogues for sigma1 receptors. J Med Chem. 2011;54(15):5362–5372. doi: 10.1021/jm200203f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheunemann M, Hennig L, Funke U, Steinbach J. High regiocontrol in the nucleophilic ring opening of 1-aralkyl-3,4-epoxypiperidines with amines—a short-step synthesis of 4-fluorobenzyltrozamicol and novel anilidopiperidines. Tetrahedron. 2011;67(19):3448–3456. [Google Scholar]
- 66.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 67.Cell were graciously provided by Bhuvanesh Singh’s lab at Memorial Sloan Kettering Cancer Center
- 68.Kurz T, Ozlu N, Rudolf F, O’Rourke SM, Luke B, Hofmann K, Hyman AA, Bowerman B, Peter M. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature. 2005;435(7046):1257–1261. doi: 10.1038/nature03662. [DOI] [PubMed] [Google Scholar]
- 69.Kim AY, Bommelje CC, Lee BE, Yonekawa Y, Choi L, Morris LG, Huang G, Kaufman A, Ryan RJ, Hao B, Ramanathan Y, Singh B. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J Biol Chem. 2008;283(48):33211–33220. doi: 10.1074/jbc.M804440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang G, Kaufman AJ, Ramanathan Y, Singh B. SCCRO (DCUN1D1) promotes nuclear translocation and assembly of the neddylation E3 complex. J Biol Chem. 2011;286(12):10297–10304. doi: 10.1074/jbc.M110.203729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meyer-Schaller N, Chou YC, Sumara I, Martin DD, Kurz T, Katheder N, Hofmann K, Berthiaume LG, Sicheri F, Peter M. The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proc Natl Acad Sci US A. 2009;106(30):12365–12370. doi: 10.1073/pnas.0812528106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang G, Kaufman AJ, Xu K, Manova K, Singh B. Squamous cell carcinoma-related oncogene (SCCRO) neddylates Cul3 protein to selectively promote midbody localization and activity of Cul3KLHL21 protein complex during abscission. J Biol Chem. 2017;292(37):15254–15265. doi: 10.1074/jbc.M117.778530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou H, Zhou W, Zhou B, Liu L, Chern TR, Chinnaswamy K, Lu J, Bernard D, Yang CY, Li S, Wang M, Stuckey J, Sun Y, Wang S. High-affinity peptidomimetic inhibitors of the DCN1-UBC12 protein-protein interaction. J Med Chem. 2018;61(5):1934–1950. doi: 10.1021/acs.jmedchem.7b01455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.