

ABSTRACT
Purpose of Review: This article describes the clinical characteristics, diagnosis, molecular pathogenesis, and treatment of facioscapulohumeral muscular dystrophy (FSHD).
Recent Findings: FSHD comprises two genetically distinct types that converge on a common downstream pathway of the expression of the toxic protein DUX4. Approximately 95% of patients have FSHD type 1 (FSHD1), in which loss of DNA repetitive elements (D4Z4 repeats) in the subtelomeric region of chromosome 4q causes decreased methylation and epigenetic derepression of DUX4, a gene contained within each D4Z4 repeat. FSHD type 2 (FSHD2) occurs through a deletion-independent mechanism but, similar to FSHD1, leads to decreased methylation and epigenetic derepression in the same region of chromosome 4q. Whereas FSHD1 is dominantly inherited, FSHD2 shows digenic inheritance, and about 80% of patients will have a mutation in the SMCHD1 gene. DUX4 lacks a polyadenylation signal, so both FSHD1 and FSHD2 only occur in the presence of permissive 4q polymorphisms, which provide a stabilizing polyadenylation sequence. FSHD is an epigenetic disease, and penetrance and severity are related to both the number of residual D4Z4 units and D4Z4 methylation.
Summary: Recent consensus guidelines outline standards for care for FSHD, and identification of potential therapeutic targets have shifted emphasis in the research community toward drug development and clinical trial planning.
INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies. FSHD has a distinct initial pattern of muscle involvement, often affecting the facial muscles, shoulder girdles, and upper arms, followed by weakness of the trunk, distal lower extremities, and more proximal muscles later in the disease course.1 Patients can show marked side-to-side asymmetry and debilitating truncal weakness. Considerable variability in presentation and rates of progression exist. FSHD can be diagnosed over the full course of a lifespan, from the very young to very old. Although FSHD does not typically shorten the life span, it can result in significant morbidity and loss of the ability to earn a living, and approximately 20% of people older than 50 years of age will require a wheelchair. Two genetically distinct but clinically indistinguishable forms of FSHD occur.2 More than 95% of patients have FSHD type 1 (FSHD1), which is characterized by deletion of large repeated elements on the long arm of chromosome 4q (the D4Z4 region). Healthy individuals have more than 10 repeats, and patients with FSHD1 have between 1 and 10 repeats. A minority of patients have FSHD type 2 (FSHD2), which is caused by a deletion-independent mechanism. Both FSHD1 and FSHD2 have a common downstream mechanism, with loss of methylation in the D4Z4 region and epigenetic derepression of a normally silenced gene, DUX4 (double homeobox 4), which is believed to cause disease through a toxic gain-of-function mechanism. Identification of potential therapeutic targets has shifted the focus of the FSHD research community toward drug development and clinical trial planning. In addition, the recent publication of consensus care guidelines provides many avenues to improve care and quality of life for patients with FSHD.3 This article reviews the clinical features of FSHD, the diagnosis and differential considerations, the pathogenic mechanism, and care guidelines for FSHD.
EPIDEMIOLOGY
Since 1991, studies have shown the worldwide prevalence of FSHD ranging between 2.03 and 6.8 per 100,000 individuals, and the prevalence of FSHD in the United States is commonly quoted as 1 in 15,000, or approximately 21,000 individuals.4 No clear racial or ethnic differences are evident in FSHD but in one of the largest US registries, 95% of participants were white, 4% were Asian, and 0.9% were Native American.5 It is not clear whether this deviation from US population racial distributions is particular to FSHD or represents sampling bias in the registry. FSHD has been described around the world, including in the United States, Europe, and Asia. A study evaluating the ancestral origin for chromosome 4q polymorphisms identified 17 4q haplotypes, which were believed to be present prior to human migration from the African continent.6 This common origin for modern 4q polymorphisms suggests that FSHD may affect most ethnic and racial populations. Penetrance in women is believed to be lower than in men, and, on average, women are diagnosed at an older age and are often less severely affected.7 The vast majority of patients meeting clinical criteria for FSHD will have FSHD1 (approximately 95%). The exact prevalence of FSHD2 is not known, but case series estimate FSHD2 to represent about 5% of patients meeting clinical criteria for FSHD.8
CLINICAL FEATURES
FSHD has a characteristic pattern of muscle involvement and progression but has a high degree of variability both between patients from different families and within an affected family from generation to generation.9 Both FSHD1 and FSHD2 have a similar clinical presentation (Case 8-1). Typically, FSHD will initially affect muscles of the face, shoulder girdle, and upper arms (mimetic muscles, serratus anterior and rhomboid muscles, and biceps and triceps). Ptosis or difficulty swallowing is typically not encountered. Parents of affected children might notice the child’s shoulder blades sticking out or his or her eyes not completely closing when sleeping. The most frequent initial symptom is the inability to lift arms over shoulder height. A high degree of side-to-side asymmetry of muscle involvement can occur. The disease then progresses to involve the lower extremities, typically the distal musculature first (tibialis anterior and gastrocnemius) in a facioscapuloperoneal pattern, then later involving more proximal muscles (quadriceps and hamstrings) and the pelvic girdle. Marked involvement of the abdominal and paraspinal musculature can occur, which can cause exaggerated lumbar lordosis or camptocormia (bent spine syndrome). Unlike other muscular dystrophies, contractures around weak muscles are not common. Pectus excavatum (caved in appearance to chest) occurs more frequently in patients with FSHD than in the general population. Other initial presentations have been described including limb-girdle weakness, isolated limb involvement, severe facial involvement in infancy, and bent spine syndrome (Case 8-2).10–12 Although individual patients may describe an almost relapsing course, where a particular muscle becomes involved and wastes, quickly followed by periods of little or no progression, patients with FSHD show a slow but steady progression of weakness overall. The risk of becoming wheelchair bound is bimodal with a peak in the second decade for patients with a more severe infantile presentation (and often only 1 to 3 residual D4Z4 units on genetic testing), followed by a slow, age-dependent increase in the risk, with approximately 20% of people older than 50 years of age requiring a wheelchair for some portion of the day.5
Case 8-1
A 35-year-old man who worked as a construction worker presented for evaluation because he had been catching his toe and falling multiple times at work. He stated that he had trouble lifting his left foot at his ankle, and while he was not sure when it first started, he felt it progressed fairly quickly over months. He stated that despite his work in construction, he had never been able to lift his right arm above his head and, when playing baseball as a child, he had to throw side arm. He had never been able to whistle and had not been able to perform a sit-up since junior high school. He stated that no one in his family had a similar disorder. On examination, he could not bury his lashes on forced eye closure, and he had a flattened pucker (inability to pucker their lips) affecting the right side of his mouth, loss of the normal clavicular angle, with scapular winging on the right, and active abduction to approximately 80 degrees. He had a positive Beevor sign and weakness of his tibialis anterior that was greater on the left than on the right. His workup included a serum creatine kinase level which was mildly elevated. EMG showed myopathic units only in the biceps and serratus anterior. A genetic test was sent for facioscapulohumeral muscular dystrophy type 1 (FSHD1) and came back normal with a fragment size of 45 kilobases. Subsequent testing for methylation levels showed that methylation was markedly reduced (15%), and follow-up genetic testing identified a mutation in SMCHD1. Testing of remaining family members showed that his father also showed markedly reduced methylation but was clinically unaffected (Figure 8-1).
FIGURE 8-1.
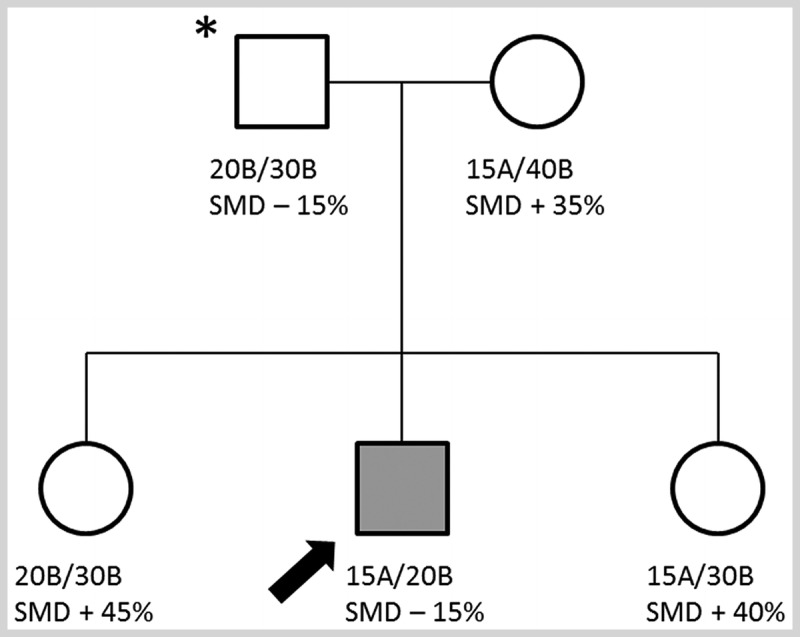
Pedigree of the patient in Case 8-1. The top line indicates the number of D4Z4 repeats on both copies of 4q and whether each has a permissive A or nonpermissive B polymorphism. The bottom line indicates the presence of an SMCHD1 mutation (SMD-) and percent of methylation (normal being greater than 20%) at the D4Z4 repeats. Note that the affected individual (arrow) has an SMCHD1 mutation and a permissive A allele while his father (asterisk) carries the same SMCHD1 mutation and is hypomethylated but is unaffected because he lacks a permissive A polymorphism.
Comment. This case shows the similar presentation between patients with FSHD1 and FSHD2, with characteristic patterns of often asymmetric muscle involvement affecting the face, shoulder, and distal lower extremity. This patient’s FSHD1 testing was negative, but methylation studies showed marked reduction in methylation consistent with FSHD2. This case also demonstrates the digenic inheritance of FSHD2. The patient’s father was also hypomethylated and carried an SMCHD1 mutation (chromosome 18) but was unaffected because he lacked a permissive distal chromosome 4q polymorphism.
Case 8-2
A 17-year-old boy presented because of right shoulder pain and weakness and difficulty lifting his right arm above shoulder level. These symptoms had occurred during a football game when he took a direct hit to the front of his right shoulder from an opponent’s helmet, and he was suspected of having a long thoracic nerve injury. He was otherwise healthy, was a star high school football player, and was aiming for a college scholarship.
On examination, he had no facial weakness. Examination of his shoulders showed straight clavicles with his right shoulder lower than the left at rest, and both shoulders were rounded. On attempted shoulder abduction, prominent winging occurred on the right, and he was unable to abduct beyond approximately 10 degrees from shoulder level. However, his left scapula was unusually far from the midline at rest, and slight winging occurred without limitation in this shoulder’s range of motion. Other than his limitation of right shoulder abduction and forward flexion, his limb muscle strength was completely normal. EMG showed signs of denervation in the right serratus anterior consistent with a long thoracic nerve injury. However, on his left side, his periscapular muscles showed signs of a chronic myopathy. No family history of muscle disease was present. Sporadic facioscapulohumeral muscular dystrophy (FSHD) was suspected, and DNA testing showed a short 4q fragment size of 28 kilobases (approximately 7 repeats) consistent with FSHD type 1.
Comment. This case is an example of sporadic FSHD type 1 and emphasizes the relatively high frequency of patients with FSHD who have de novo mutations. In addition, because of the intrinsic weakness of the shoulder and periscapular muscles, patients with FSHD are more susceptible to overuse musculoskeletal injury or, as in this patient, brachial plexus injury. In fact, chronic shoulder and neck pain is a common symptom among patients with FSHD.
Signs
On neurologic examination, muscle involvement can be symmetric or asymmetric in every region examined, but a number of examination findings, when present in combination, are highly suggestive of FSHD. Numerous signs can be observed on the patient’s face, including decreased brow furrow; an inability to close the eyes or bury the lashes on forced eye closure; a flattened, transverse smile or flattened pucker; and an inability to tense the platysma when asking the patient to growl (Figure 8-2A). The chest wall can show wasting of the pectoral muscles with exaggeration of the axillary crease, flattening of the normal clavicular angle with rounding of the shoulders, and a protuberant abdomen (Figure 8-2B). The deltoid is often preserved, and the trapezius can appear bunched up because of selective involvement of the lower part of the muscle, giving rise the so-called poly-hill appearance of alternating muscle and bony landmarks.13 The biceps and triceps are often wasted with preservation of distal forearm muscles, giving rise to the so-called Popeye arm appearance (Figure 8-2C14). With disease progression, distal upper extremity involvement can be seen, resulting in selective involvement of the finger and wrist extensors. In the shoulders, wasting of the rhomboids can occur, with lateral displacement of the scapula. Both posterior and lateral winging of the scapula can be seen on active shoulder abduction or forward flexion, often with inability to move beyond 90 degrees (Figure 8-2D and Supplemental Digital Content 8-1, http://links.lww.com/CONT/A207). From the front, the tip of the scapula can often be seen above the clavicle during shoulder flexion or abduction. The asymmetric involvement of the abdominal muscles, most typically with the upper abdominals spared initially, can lead to the Beevor sign, which is a fairly specific sign for FSHD in patients with myopathy (Figure 8-2E15 and Supplemental Digital Content 8-2, http://links.lww.com/CONT/A208).16 If, when supine, the patient lifts his or her head off the examining table, tensing the abdominal muscles will cause the umbilicus to be pulled toward the intact muscles (typically up). Hip girdle muscles are often intact early in the disease, but weakness of the tibialis anterior is common.
FIGURE 8-2.
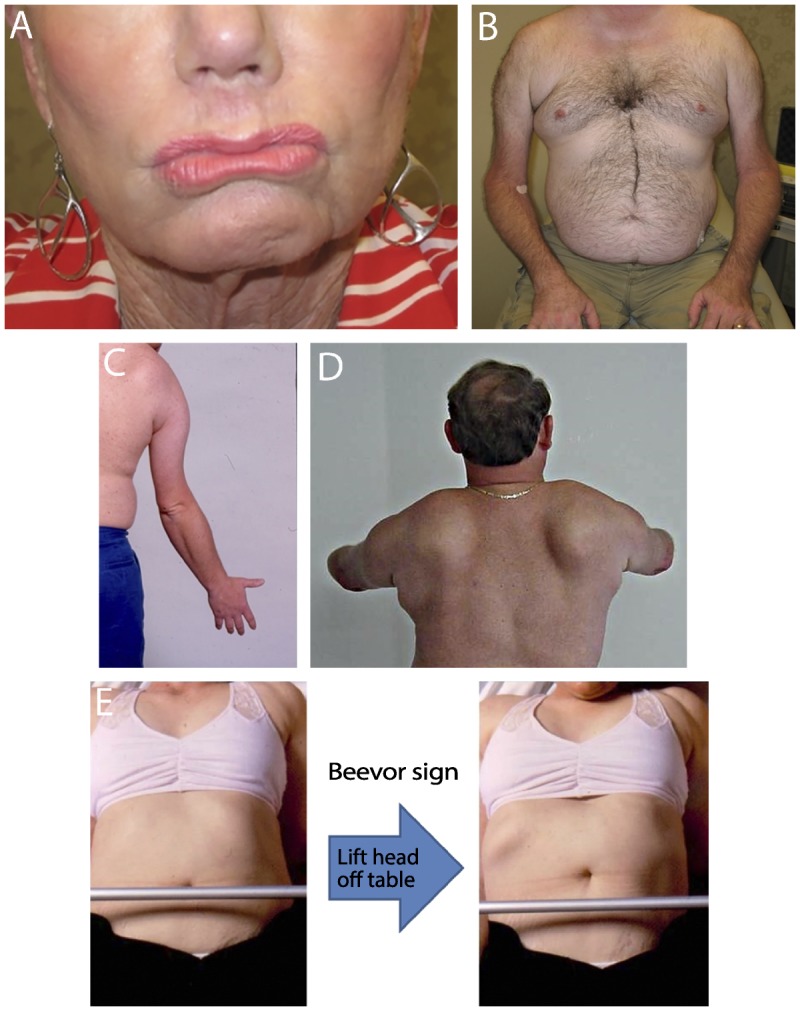
Clinical features of facioscapulohumeral muscular dystrophy. A, Facial weakness with flattened pucker; B, chest wall showing asymmetric wasting of pectoral muscles with prominent axillary fold, upper arm wasting with sparing of forearms, and protruberant abdomen; C, Popeye arm appearance; D, both posterior and lateral winging of the scapula on shoulder forward flexion; E, Beevor sign demonstrating asymmetric abdominal weakness with intact upper abdominals pulling umbilicus upwards when the abdomen is tensed.
Panel C reprinted with permission from Tawil R, Griggs RC, Butterworth-Heinemann.14 © 1997 R Tawil and RC Griggs. Panel E modified with permission from Griggs RC, et al, Muscle Nerve.15 © 1995 John Wiley & Sons, Inc. onlinelibrary.wiley.com/doi/10.1002/mus.880181311/full.
Respiratory Involvement
Restrictive respiratory involvement varies from study to study, with a range of 0% to 13% reported.3 The patterns of involvement for most patients appear to be caused by loss of core/trunk musculature as opposed to diaphragmatic involvement. In FSHD the respiratory complications typically follow the weakness and are more likely in patients with pelvic girdle weakness who are wheelchair bound or who have prominent paraspinal involvement or kyphoscoliosis. The actual incidence of ventilatory support is not known; a Dutch study suggests that approximately 1% of the Dutch population who have FSHD require mechanical ventilation, and a large US registry showed an approximate 8% risk over 6 years of requiring noninvasive ventilator support.5,17
Cardiac Involvement
Structural cardiac involvement (dilated ventricles or reduced cardiac function) is not typically found in FSHD. Largely asymptomatic supraventricular arrhythmias can be found in approximately 5% to 10% of patients.18,19 One study found that approximately one-third of subjects had incomplete right bundle branch block, but when followed for 8 years, no significant progression of ECG changes were found in these subjects.20
Extramuscular Manifestations
A number of characteristic extramuscular manifestations occur in patients with FSHD, but the specific mechanisms of these pleiotropic effects are not known. Interestingly, the more severe clinical manifestations of these extramuscular manifestations are almost entirely found in patients with the smallest number of residual D4Z4 units and have not been described to date in patients with FSHD2. Subtle retinal vascular changes (peripheral telangiectasia) can be found on retinal examination in up to one-fourth of patients with FSHD.21,22 A more severe retinal vasculopathy, known as Coats disease, can be found in less than 1% of patients and is characterized by aneurysmal dilatations, exudation, and can cause retinal detachment or blindness if untreated. While idiopathic Coats disease is typically unilateral and occurs in males, Coats disease in FSHD is often bilateral and found more commonly in females. But perhaps most striking is the association of Coats disease with patients with the smallest number of residual D4Z4 units (1 unit to 3 units remaining).23
Similar to the retinal vascular changes, mild and usually asymptomatic loss of high-frequency hearing can be found in approximately 16% of patients with FSHD, which is not that different than the normal age-related changes in hearing.22,24 Hearing loss is more frequent and can be quite severe (requiring hearing aids) in patients with the smallest number of residual D4Z4 units (1 unit to 4 units remaining).25
Cognitive changes, mental retardation, and even seizures have been described in infants with FSHD and are almost entirely associated with the smallest number of residual D4Z4 units (1 unit to 3 units remaining).
DIAGNOSIS
Supporting features for a diagnosis of FSHD include the following:
Onset of weakness affecting the facial muscles or shoulder girdle musculature
Positive family history
Asymmetric muscle involvement
Abdominal weakness
The presence of retinal vasculopathy or hearing loss in early-onset FSHD
These features supporting a diagnosis of FSHD should occur in the absence of the following:
Ptosis or extraocular muscle involvement
Lingual involvement or difficulty swallowing
Prominent contractures
Cardiomyopathy
Features on EMG or muscle biopsy suggesting an alternative diagnosis
EMG usually shows nonspecific myopathic changes (small, polyphasic motor units), and fibrillations and positive sharp waves are not uncommon. Muscle biopsy typically shows nonspecific myopathic changes (variation in fiber size, internal myonuclei, degenerating/regenerating fibers, increased fibrosis, or fatty replacement). However, approximately 5% to 30% of muscles may show a somewhat characteristic perivascular inflammatory infiltrate, typically CD4 or CD8 cells.26–28 Serum creatine kinase (CK) can be normal or modestly elevated (usually less than 10 times the upper limit of normal). However, current guidelines for the diagnosis of FSHD do not require EMG, serum CK, or muscle biopsy to make the diagnosis. For a patient with a characteristic clinical presentation and findings on examination, the next step is genetic testing. Consensus criteria suggest the following testing order29:
Commercial testing for a D4Z4 contraction using Southern blot after double digestion with EcoRI/BlnI yields a sensitivity of 93% and specificity of 98%.3 Normal individuals have fragments of more than 38 kilobases (kb); patients with FSHD1 have fragments between 10 kb and 38 kb (corresponding to an estimated 1 to 10 residual D4Z4 units).
If Southern blot testing is negative (ie, no contraction of D4Z4 repeats) then determination of the presence of at least one A allele and very low methylation (less than 20%) confirms a diagnosis of FSHD2.
FSHD1 inheritance is autosomal dominant but up to 30% of mutations can be spontaneous (Case 8-2). If two siblings are affected without a known family history, this raises the possibility of either a germ line mutation in one of the parents or a nonmanifesting mosaic parent. For patients with an atypical clinical presentation, confirming the presence of a permissive A polymorphism on the contracted D4Z4 copy decreases the likelihood of a false positive.
Approximately 80% of patients with FSHD2 will have deletions in the structural maintenance of the chromosomal flexible hinge domain containing 1 (SMCHD1) gene.8 While genetic testing for SMCHD1 is becoming widely available, demonstrating the presence of an SMCHD1 mutation alone is not sufficient for a proper genetic confirmation of an SMCHD1-associated FSHD2. As previously noted, FSHD2 is a digenic disease requiring the presence of at least one permissive A polymorphism on allele 4q and profound hypomethylation resulting from a mutation on a hypermethylating gene on another chromosome.
The list of other neuromuscular conditions that present with a similar clinical picture is short and includes: certain limb-girdle muscular dystrophies (eg, limb-girdle muscular dystrophy type 2A, calpain 3 deficiency), scapuloperoneal syndromes (eg, Davidenkow syndrome), mitochondrial myopathy, inclusion body myositis (sporadic/hereditary), polymyositis, and acid maltase deficiency (Pompe disease). Most of these can be easily distinguished from FSHD based on clinical history, examination findings, or on subsequent workup (EMG, muscle biopsy).
PATHOLOGIC MECHANISM
Both FSHD1 and FSHD2 converge on a common downstream pathway, which leads to the derepression of a presumed retrogene, DUX4, that is believed to cause disease by a toxic gain of function when expressed in adult tissue (Figure 8-330).31
FIGURE 8-3.
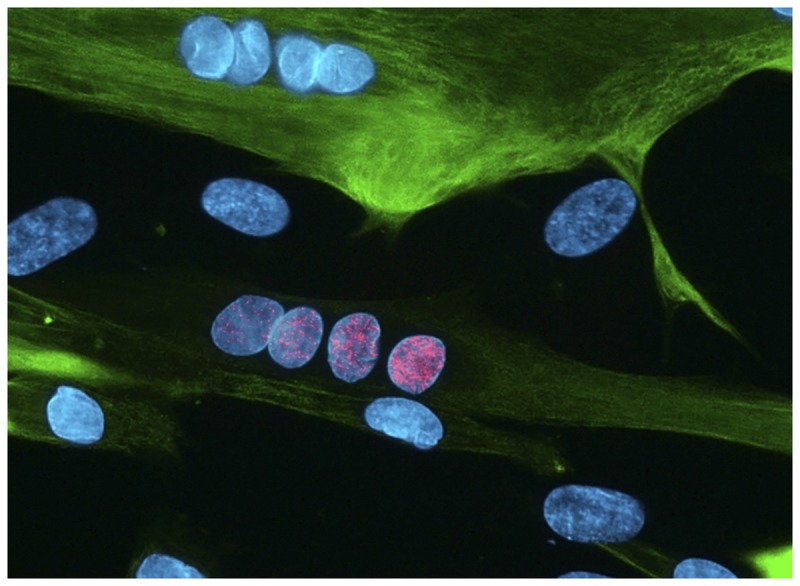
Muscle culture from a patient with facioscapulohumeral muscular dystrophy type 1 (FSHD1) showing a myotube (green) with a row of nuclei (blue) showing bursts of DUX4 expression (pink). Muscle tissue from patients with both FSHD1 and FSHD2 will show bursts of DUX4 expression.
Reprinted with permission from Cell. Photograph by Linda Geng.30 © 2012 Elsevier, Inc. www.cell.com/cell/fulltext/S0092-8674(12)00160-2.
Patients with FSHD1 have loss of D4Z4 macrosatellite repeated units (each more than 3.3 kb long) in the subtelomeric region of the long arm of chromosome 4q.32,33 Healthy individuals have more than 10 repeated units. Patients with FSHD1 have at least 1 D4Z4 unit, as monosomy of chromosome 4q does not cause FSHD1.34 This region of chromosome 4q is normally highly methylated and epigenetically silenced. In FSHD1 loss of D4Z4 units to between 1 and 10 repeats causes decreased methylation and opening of the chromatin structure in the D4Z4 region.35,36 Contained within each D4Z4 repeat is an open reading frame coding for a DUX4 gene.37 However DUX4 lacks a polyadenylation sequence, so even with an open chromatin structure in the D4Z4 region, DUX4 messenger RNA is normally broken down by the cell. Certain 4q polymorphisms (the so-called A polymorphism) contain a polyadenylation sequence distal to the last D4Z4 repeat, which can be added to nascent DUX4 transcripts, stabilizing them and leading to translation of DUX4 protein.31
Whereas in patients with FSHD1 only the contracted D4Z4 copy is selectively hypomethylated, in patients with FSHD2, both noncontracted D4Z4 copies are profoundly hypomethylated. In approximately 80% of patients with FSHD2, the cause of this profound hypomethylation is a deletion in another gene, SMCHD1, on chromosome 18.8 SMCHD1 is a hypermethylating enzyme with a normal role in X chromosome inactivation. As in FSHD1, in FSHD2 in order for the hypomethylated D4Z4 repeats to transcribe viable DUX4 messenger RNA, at least one of the two 4q alleles should have a permissive A polymorphism just distal to the last repeat. Thus, whereas FSHD1 is a dominantly inherited disease resulting from a contraction in the number of D4Z4 repeats on one copy of 4q in the presence of a distal A polymorphism, FSHD2 is a digenic disease requiring at least one permissive A polymorphism on 4q and a mutation in a methylating gene (eg, SMCHD1) on another chromosome.
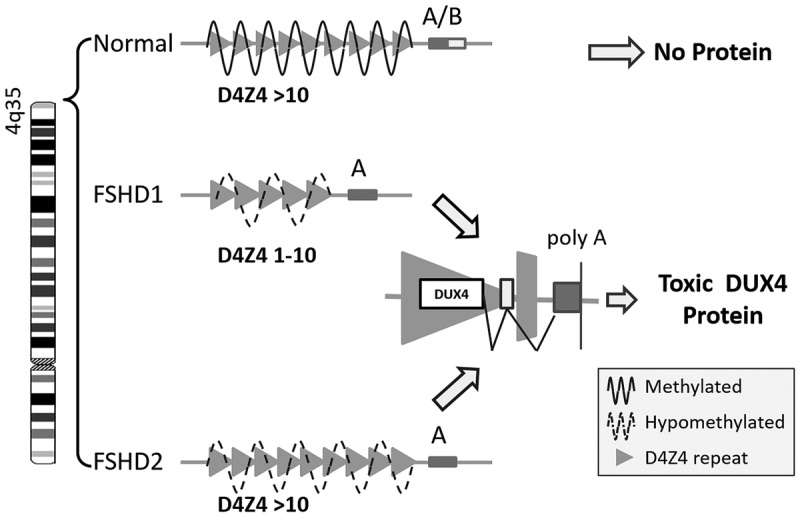
The unified genetic model for FSHD posits that the minimum necessary requirements for disease includes loss of methylation in the D4Z4 region on chromosome 4q, opening of the chromatin structure enabling transcription, and a permissive 4q polymorphism providing a stabilizing polyadenylation signal, resulting in the expression of DUX4 (Figure 8-438).
FIGURE 8-4.
Unified genetic model of facioscapulohumeral muscular dystrophy (FSHD). In FSHD, loss of methylation in the D4Z4 region on the long arm of chromosome 4q (due to loss of D4Z4 repeats in FSHD1 and due to mutations in other genes involved in methylation in FSHD2) causes epigenetic derepression, opening of the chromosome structure, and certain permissive polymorphisms that provide a polyadenylation sequence (poly A), resulting in the expression of DUX4.
Reprinted with permission from Statland J, Tawil R, Curr Opin Neurol.38 © 2011 Lippincott Williams & Wilkins, Inc. journals.lww.com/co-neurology/Abstract/2011/10000/Facioscapulohumeral_muscular_dystrophy___molecular.3.aspx.
DUX4 is a transcription factor whose role in normal germ cell development is unknown. It is found in the spermatogonia in adult male testes. Expression of DUX4 in adult tissue leads to activation of a number of canonical pathways not normally expressed in adult tissues, including cancer testes antigen, genes involved in innate immunity, and genes involved in protein degradation and muscle atrophy (Figure 8-5).39,40 Possible pathogenic mechanisms include induction of mitotic pathway in a postmitotic cell leading to apoptosis or expression of germ line antigens inducing an immune response. The forced expression of DUX4 in muscle tissue is highly toxic, leading to apoptosis, oxidative stress, and interference with myogenic differentiation.41–43
FIGURE 8-5.
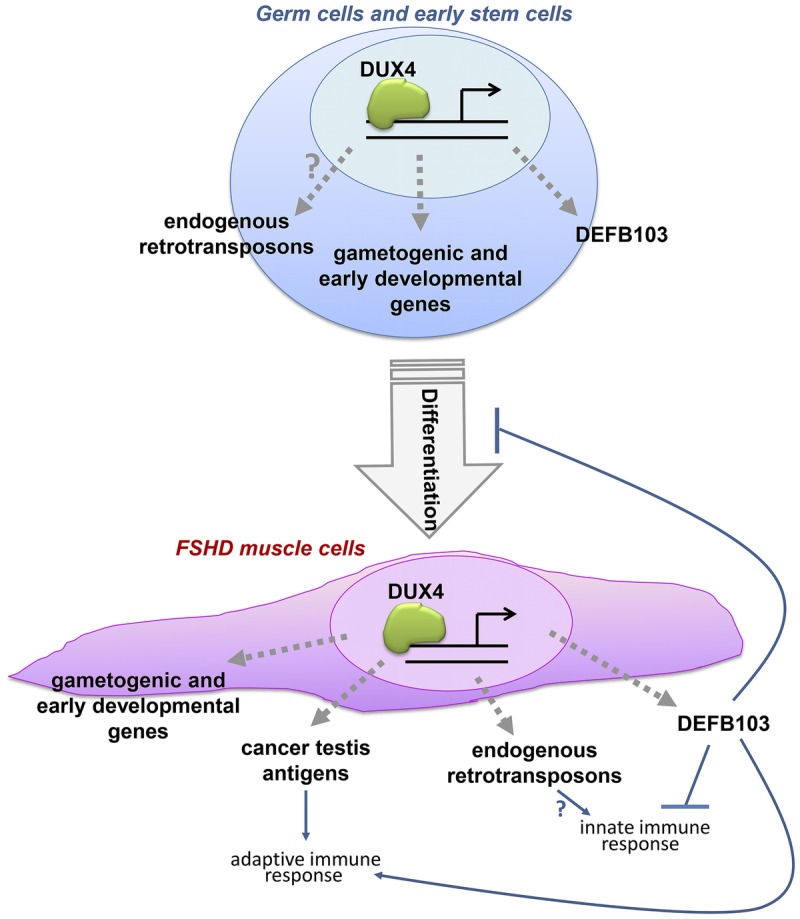
DUX4 is a transcription factor that activates a number of genetic cascades not normally expressed in adult tissue.
FSHD = facioscapulohumeral muscular dystrophy.
Reprinted with permission from Geng LN, et al, Dev Cell.39 © 2012 Elsevier, Inc. www.cell.com/developmental-cell/fulltext/S1534-5807(11)00523-5.
Genotype-Phenotype Associations
For patients with between 1 and 6 residual D4Z4 units, a fairly linear association occurs with the number of D4Z4 units, methylation in the D4Z4 region, and clinical severity (Figure 8-6).44,45 Patients with between 1 and 3 remaining D4Z4 units may have additional disease complications related to the size of the deletion; for example, extramuscular complications are almost exclusively found in this group and are not seen to date in patients with FSHD2. For patients with 7 to 10 remaining D4Z4 units, no clear association exists between the number of D4Z4 units and clinical severity, with lower penetrance in this group.46 In Japan, patients with 7 to 10 remaining D4Z4 units do not acquire FSHD.47 One study in a mostly European population showed that penetrance in this group could be tied to lower observed methylation than would be expected by the number of remaining D4Z4 units alone, suggesting a role for other genetic or environmental modifiers of the disease. Indeed, a study of families with mild disease and 9 D4Z4 units identified family members who were more severely affected who had lower methylation than would be predicted, and who were found to have mutations in SMCHD1, making this not only a causative gene in FSHD2, but a modifying gene in FSHD1.48
FIGURE 8-6.
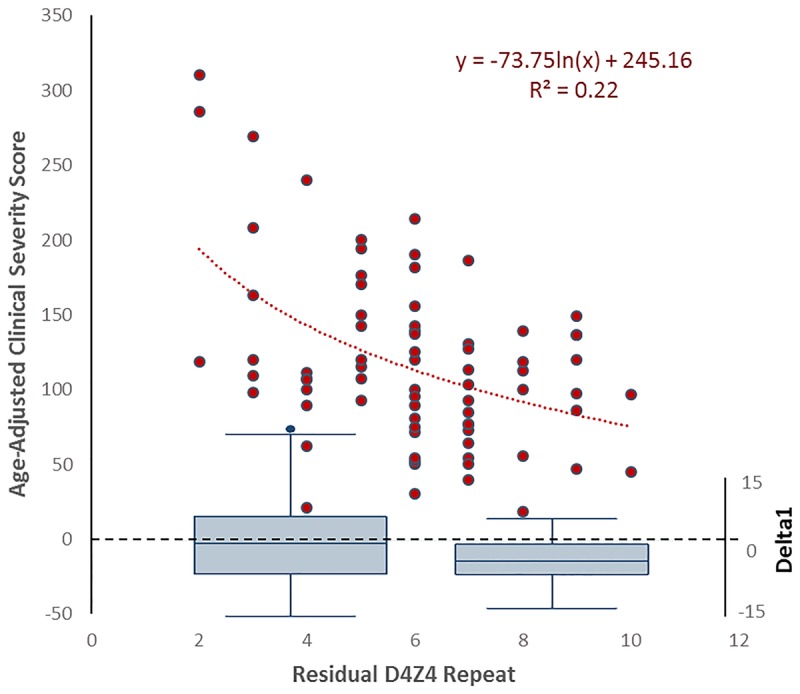
The relationship of the number of residual D4Z4 units to age-adjusted clinical severity and methylation. The relationship of age-adjusted clinical severity to D4Z4 repeat size is not linear (left y axis, n = 74). While the relationship of severity to repeat size is linear from 1 to 6 repeats, some other factor aside from repeat size appears to determine severity for 7 to 10 repeats. The bottom of the graph shows box plots of the difference between observed methylation values and expected methylation values (right y axis) for 1 to 6 repeats (left) and 7 to 10 repeats (right). Note the negative difference in methylation values for 7 to 10 repeats, suggesting that some other factor, other than he number of D4Z4 units, is affecting disease severity for this group.
Reprintedwith permission fromStatland JM, et al, Neurology.45 © 2015 American Academy of Neurology. www.neurology.org/content/85/24/2147.short.
TREATMENT
There are no registered therapies approved for FSHD. Although many drugs have been tried in clinical trials (prednisone diltiazem, albuterol, and a myostatin inhibitor), none showed a clear benefit.49–54 The mainstay of treatment for FSHD is multidisciplinary care, and recent evidence-based guidelines for care have been published by the American Academy of Neurology (AAN) (Supplemental Digital Content 8-3, http://links.lww.com/CONT/A209).3 The guideline conclusions are based on a modified Grading of Recommendations Assessment, Development, and Evaluation (GRADE) process, and recommendations are made by a modified Delphi consensus process that includes consideration of the evidence.3
The authors commonly recommend the following in their clinical practice:
It can benefit patients to be assessed of their need for assistive devices through physical/occupational therapy. Stretching and range of motion exercises are also recommended.
Baseline pulmonary function testing should be obtained at diagnosis, and then routine yearly testing should occur once patients develop proximal lower extremity weakness. Patients experiencing difficulty breathing despite normal pulmonary function testing should be sent to pulmonary medicine for possible sleep study/overnight oximetry.
Cardiac screening should be performed in patients on a symptomatic basis.
Baseline dilated eye examination is recommended in all patients newly diagnosed with FSHD to screen for potentially reversible retinal vascular involvement. Patients with small numbers of residual D4Z4 units should be followed closer by an ophthalmologist with repeat testing as needed.
All children with FSHD should be screened for hearing deficits as this can affect learning and language development. Adults should be screened if symptomatic.
Patients should routinely be asked about pain, and referrals should be made to physical therapy or recommendations should be given regarding the use of nonsteroidal antiinflammatory drugs for acute pain or anticonvulsants or antidepressants for chronic pain. Patients with chronic pain may benefit from a referral to a pain clinic.
Scapular Fixation
Limited range of motion at the shoulders due to shoulder weakness is not only one of the first presenting symptoms of FSHD, but is a major functional limitation. Scapular fixation uses surgical techniques to fix the scapula to the chest wall (typically a combination of wires and bone graft) and can improve function (eg, dressing, washing hair) in patients with otherwise preserved upper extremity strength. A simple bedside procedure where the physician manually fixes the scapula to the chest wall can be used to screen for potential benefit with surgery (Figure 8-7). This needs to be weighed against the possible complications of surgery, which include hemothorax/pneumothorax, pain, infection, and a possible decrease in respiratory reserve. Many case series have documented a possible benefit in function for this approach.3,55
FIGURE 8-7.
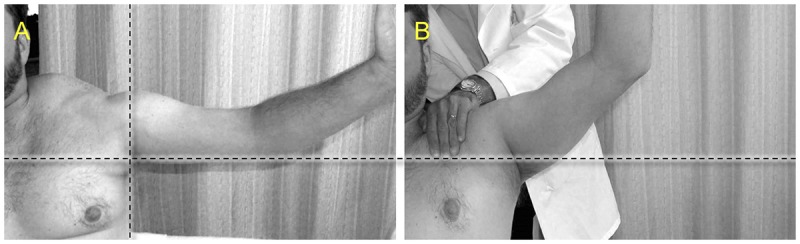
Manual fixation of scapula in the clinic. A, In this patient with facioscapulohumeral muscular dystrophy, abduction of shoulder is only to approximately 90 degrees. B, The physician manually pulls the shoulder back while pushing against the scapula with the other hand, fixing the scapula to the chest wall, and abduction increases beyond 90 degrees. This would be a positive test and may support the notion that scapular fixation surgery could improve function for this patient.
Reprinted with permission from Tawil R, Van DerMaarel SM, Muscle Nerve.9 © 2006 John Wiley & Sons, Inc. onlinelibrary.wiley.com/doi/10.1002/mus.20522/full.
Exercise
Aerobic exercise appears to be safe and possibly beneficial for improving function or quality of life in patients with FSHD.56,57 One study that analyzed the effects of strength training in patients with FSHD did not show worsening with strength training. Patients interested in strength training should work with a physical therapist to design a program with the general precept of lower resistance/increased repetitions, and therapists should take into account the patient’s physical limitations.52
FUTURE THERAPEUTIC DIRECTIONS
FSHD1 and FSHD2 are genetically distinct but converge on a common downstream mechanism of aberrant expression of DUX4, which causes disease in a toxic gain-of-function fashion. This has identified many potential disease-directed therapeutic targets for FSHD, including increasing methylation in the D4Z4 region, knocking down expression of DUX4, or targeting downstream targets of DUX4. Emphasis in the research community has turned to clinical trial planning, and many animal or cell models for testing new therapeutics have been developed.58–61 Proof of concept for the use of antisense oligonucleotide therapies to target DUX4 are being provided in a variety of other neuromuscular diseases where antisense therapies are entering human studies, including spinal muscular atrophy, Duchenne muscular dystrophy, and myotonic dystrophy. Other possible therapies for FSHD might include drugs that are not disease specific but either target inflammation (a dose-escalating study of an immunomodulatory drug is underway62), muscle bulk, or muscle function. Evidence of this shift in focus in the research community can be seen in international workshops geared toward outcome measure development.63,64 Ultimately, effective treatments for FSHD will require the dissemination of standards of care, along with international cooperation to prepare for possible future therapeutic trials.
CONCLUSION
FSHD is one of the most prevalent muscular dystrophies. Patients experience progressive weakness commonly first affecting the face and shoulder muscles, followed by muscles of the trunk and lower extremity. Considerable variability occurs in clinical presentation and morbidity associated with FSHD, with approximately 20% of patients older than the age of 50 eventually requiring use of a wheelchair. Recent advances in our molecular understanding of FSHD have revealed a common downstream pathway for patients with FSHD1 and FSHD2. In both FSHD1 and FSHD2, epigenetic changes lead to the derepression of the gene DUX4, which causes disease by a toxic gain of function. This common downstream pathway raises the possibility for future disease-targeted therapies. The current best practice for treatment for FSHD is supportive care, and AAN care guidelines for FSHD were published in 2015.3
VIDEO LEGENDS
Supplemental Digital Content 8-1
Scapular winging. When asked to abduct her arms over her head, this 65-year-old woman with facioscapulohumeral muscular dystrophy is only able to abduct to about 70 degrees with prominent posterior winging of her scapula. Her deltoid muscles are preserved.inks.
http://links.lww.com/CONT/A207
© 2016 American Academy of Neurology.
Supplemental Digital Content 8-2
Beevor sign. A 50-year-old man with facioscapulohumeral muscular dystrophy lies on his back with his head to the left. When he lifts his head, the asymmetric involvement of his abdominal musculature causes the umbilicus to be pulled in the direction of the unaffected muscles.
http://links.lww.com/CONT/A208
© 2016 American Academy of Neurology.
USEFUL WEBSITES
ClinicalTrials.gov
Friends of FSH Research
FSH Society
GeneReviews
Muscular Dystrophy Association
mdausa.org
National Registry for Myotonic Dystrophy and Facioscapulohumeral Dystrophy
The Richard Fields Center for FSHD Research
KEY POINTS
Facioscapulohumeral muscular dystrophy types 1 and 2 converge on a common pathologic pathway and present with a similar pattern of weakness.
Facioscapulohumeral muscular dystrophy is one of the most common muscular dystrophies with approximately 21,000 affected individuals in the United States.
Facioscapulohumeral muscular dystrophy has a characteristic descending pattern of weakness, first affecting the face and shoulder followed by the distal lower extremity and the proximal hip girdle muscles, but many variations in presentation can occur.
Facioscapulohumeral muscular dystrophy can show marked side-to-side asymmetry of muscle involvement.
When present in combination, the following key signs are highly suggestive of facioscapulohumeral muscular dystrophy: weakness on forced eye closure, transverse smile, pectoral wasting, scapular winging, poly-hill sign, Beevor sign, and ankle weakness that occurs before hip girdle weakness.
Restrictive respiratory involvement is found more frequently in patients who are severely affected by facioscapulohumeral muscular dystrophy, and cardiac involvement, when present, is typically not structural but most commonly manifests as asymptomatic supraventricular arrhythmias.
While extramuscular manifestations such as retinal vascular changes and hearing loss can be found in one-fourth to one-half of patients with facioscapulohumeral muscular dystrophy, only a minority of patients, typically with a small number of residual D4Z4 units (1 unit to 3 units), ever have symptomatic involvement.
Diagnosis of facioscapulohumeral muscular dystrophy is based on family history and clinical presentations and is confirmed by genetic testing.
For patients with typical clinical features of facioscapulohumeral muscular dystrophy, commercial testing for a D4Z4 contraction shows 93% sensitivity and 98% specificity; for patients with negative testing, methylation studies can confirm a diagnosis of facioscapulohumeral muscular dystrophy type 2.
Facioscapulohumeral muscular dystrophy type 1 is due to the deletion of repeated elements in the D4Z4 region of chromosome 4q; healthy individuals have more than 10 units, and patients with facioscapulohumeral muscular dystrophy have between 1 unit and 10 units.
Facioscapulohumeral muscular dystrophy type 2 occurs through a contraction-independent mechanism, and about 80% of patients will turn out to have a mutation in the gene SMCHD1 on chromosome 18.
Both facioscapulohumeral muscular dystrophy types 1 and 2 have the following characteristics: decreased methylation on chromosome 4q and opening of the chromatin structure, a permissive polymorphism that provides a polyadenylation sequence, and expression of the normally repressed DUX4 gene.
DUX4 is a transcription factor that activates many genetic pathways and is believed to cause disease in a toxic gain-of-function manner.
Patients with facioscapulohumeral muscular dystrophy type 1 with 1 to 3 remaining D4Z4 units tend to have more severe disease presentations with earlier onset and increased frequency of extramuscular manifestations. Patients with 7 to 10 remaining D4Z4 units have a milder disease with decreased penetrance.
The American Academy of Neurology has recently published evidence-based guidelines for the diagnosis and management of facioscapulohumeral muscular dystrophy. Care is largely supportive, with regular evaluations assessing the patient’s need for assistive devices or pain management as well as monitoring for respiratory complications or extramuscular manifestations.
Scapular fixation may improve arm range of motion and function in some individuals with facioscapulohumeral muscular dystrophy, and aerobic exercise may improve quality of life or function.
The identification of a unified genetic hypothesis has identified potential future disease-directed targets for therapy of facioscapulohumeral muscular dystrophy.
Acknowlegment
This work was supported by grants from the National Institutes of Health (U01 NS061795-04, 1U54AR065139, 1P01 NS069539).
REFERENCES
- 1.Statland J, Tawil R. Facioscapulohumeral muscular dystrophy. Neurol Clin 2014;32(3):721–728, ix. doi:10.1016/j.ncl.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med 2011;17(5):252–258. doi:10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tawil R, Kissel JT, Heatwole C, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015;85(4):357–364. doi:10.1212/WNL.0000000000001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deenen JCW, Horlings CGC, Verschuuren JJGM, et al. The epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J Neuromuscul Dis 2015;2:73–85. doi:10.3233/JND-140045. [PubMed] [Google Scholar]
- 5.Statland JM, Tawil R. Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve 2014;49(4):520–527. doi:10.1002/mus.23949. [DOI] [PubMed] [Google Scholar]
- 6.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet 2010;86(3):364–377. doi:10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonini MM, Passos-Bueno MR, Cerqueira A, et al. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord 2004;14(1):33–38. doi:10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012;44(12):1370–1374. doi:10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006;34(1):1–15. doi:10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 10.Jordan B, Eger K, Koesling S, Zierz S. Camptocormia phenotype of FSHD: a clinical and MRI study on six patients. J Neurol 2011;258(5):866–873. doi:10.1007/s00415-010-5858-z. [DOI] [PubMed] [Google Scholar]
- 11.Kottlors M, Kress W, Meng G, Glocker FX. Facioscapulohumeral muscular dystrophy presenting with isolated axial myopathy and bent spine syndrome. Muscle Nerve 2010;42(2):273–275. doi:10.1002/mus.21722. [DOI] [PubMed] [Google Scholar]
- 12.Krasnianski M, Eger K, Neudecker S, et al. Atypical phenotypes in patients with facioscapulohumeral muscular dystrophy 4q35 deletion. Arch Neurol 2003;60(10):1421–1425. doi:10.1001/archneur.60.10.1421. [DOI] [PubMed] [Google Scholar]
- 13.Pradhan S. Poly-Hill sign in facioscapulohumeral dystrophy. Muscle Nerve 2002;25(5):754–755. doi:10.1002/mus.10108. [DOI] [PubMed] [Google Scholar]
- 14.Tawil R, Griggs RC. Facioscapulohumeral muscular dystrophy. In: Rosenberg RN, Prusiner SB, DiMauro S, Barch RL, editors. The molecular and genetic basis of neurological disease. 2nd ed Oxford, UK: Butterworth-Heinemann, 1997. [Google Scholar]
- 15.Griggs RC, Tawil R, McDermott M, et al. Monozygotic twins with facioscapulohumeral dystrophy (FSHD): implications for genotype/phenotype correlation. Muscle Nerve 1995;18(suppl 13):S50–S55. doi:10.1002/mus.880181311. [PubMed] [Google Scholar]
- 16.Shahrizaila N, Wills AJ. Significance of Beevor’s sign in facioscapulohumeral dystrophy and other neuromuscular diseases. J Neurol Neurosurg Psychiatry 2005;76(6):869–870. doi:10.1136/jnnp.2004.052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wohlgemuth M, van der Kooi EL, van Kesteren RG, et al. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 2004;63(1):176–178. doi:10.1212/01.WNL.0000133126.86377.E8 1526-632X. [DOI] [PubMed] [Google Scholar]
- 18.Laforêt P, de Toma C, Eymard B, et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 1998;51(5):1454–1456. doi:10.1212/WNL.51.5.1454. [DOI] [PubMed] [Google Scholar]
- 19.Trevisan CP, Pastorello E, Armani M, et al. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol 2006;56(1):1–5. doi:10.1159/000094248. [DOI] [PubMed] [Google Scholar]
- 20.van Dijk GP, van der Kooi E, Behin A, et al. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct Neurol 2014;29(3):159–165. [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain 1987;110(pt 3):631–648. doi:10.1093/brain/110.3.631. [DOI] [PubMed] [Google Scholar]
- 22.Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve Suppl 1995;(2):S73–S80. doi:10.1002/mus.880181314. [PubMed] [Google Scholar]
- 23.Statland JM, Sacconi S, Farmakidis C, et al. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology 2013;80(13):1247–1250. doi:10.1212/WNL.0b013e3182897116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trevisan CP, Pastorello E, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy: hearing loss and other atypical features of patients with large 4q35 deletions. Eur J Neurol 2008;15(12):1353–1358. doi:10.1111/j.1468-1331.2008.02314.x. [DOI] [PubMed] [Google Scholar]
- 25.Lutz KL, Holte L, Kliethermes SA, et al. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013;81(16):1374–1377. doi:10.1212/WNL.0b013e3182a84140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arahata K, Ishihara T, Fukunaga H, et al. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve Suppl 1995;(2):S56–S66. doi:10.1002/mus.880181312. [PubMed] [Google Scholar]
- 27.Carpenter SKG. Pathology of skeletal muscle. 2nd ed New York: Oxford University Press, 2001. [Google Scholar]
- 28.Statland JM, Shah B, Henderson D, et al. Muscle pathology grade for facioscapulohumeral muscular dystrophy biopsies. Muscle Nerve 2015;52(4):521–526. doi:10.1002/mus.24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemmers RJ, O’Shea S, Padberg GW, et al. Best practice guidelines on genetic diagnostics of facioscapulohumeral muscular dystrophy: workshop 9th June 2010, LUMC, Leiden, The Netherlands. Neuromuscul Disord 2012;22(5):463–470. doi:10.1016/j.nmd.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Leading edge select: muscle. Cell 2012;148(4):629–631. doi:http://dx.doi.org/10.1016/j.cell.2012.02.003. [Google Scholar]
- 31.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329(5999):1650–1653. doi:10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Upadhyaya M, Lunt PW, Sarfarazi M, et al. DNA marker applicable to presymptomatic and prenatal diagnosis of facioscapulohumeral disease. Lancet 1990;336(8726):1320–1321. doi:10.1016/0140-6736(90)93005-A. [DOI] [PubMed] [Google Scholar]
- 33.Wijmenga C, Frants RR, Brouwer OF, et al. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990;336(8716):651–653. doi:10.1016/0140-6736(90)92148-B. [DOI] [PubMed] [Google Scholar]
- 34.Tupler R, Berardinelli A, Barbierato L, et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet 1996;33(5):366–370. doi:10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Overveld PG, Enthoven L, Ricci E, et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol 2005;58(4):569–576. doi:10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 36.Zeng W, de Greef JC, Chen YY, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 2009;5(7):e1000559 doi:10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hewitt JE, Lyle R, Clark LN, et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet 1994;3(8):1287–1295. doi:10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 38.Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy: molecular pathological advances and future directions. Curr Opin Neurol 2011;24(5):423–428. doi:10.1097/WCO.0b013e32834959af. [DOI] [PubMed] [Google Scholar]
- 39.Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 2012;22(11):38–51. doi:10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Z, Snider L, Balog J, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet 2014;23(20):5342–5352. doi:10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord 2007;17(8):611–623. doi:10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Snider L, Asawachaicharn A, Tyler AE, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 2009;18(13):2414–2430. doi:10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol 2010;3(4):386–400. [PMC free article] [PubMed] [Google Scholar]
- 44.Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet 2015;24(3):659–669. doi:10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Statland JM, Donlin-Smith CM, Tapscott SJ, et al. Milder phenotype in facioscapulohumeral dystrophy with 7-10 residual D4Z4 repeats. Neurology 2015;85(24):2147–2150. doi:10.1212/WNL.0000000000002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricci G, Scionti I, Sera F, et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 2013;136(pt 11):3408–3417. doi:10.1093/brain/awt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa M, Matsuzaki T, Higuchi I, et al. Facioscapulohumeral muscular dystrophy: clinical diversity and genetic abnormalities in Japanese patients. Intern Med 1997;36(5):333–339. doi:10.2169/internalmedicine.36.333. [DOI] [PubMed] [Google Scholar]
- 48.Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 2013;93(4):744–751. doi:10.1016/j.ajhg.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kissel JT, McDermott MP, Mendell JR, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology 2001;57(8):1434–1440. doi:10.1212/WNL.57.8.1434. [DOI] [PubMed] [Google Scholar]
- 50.Payan CA, Hogrel JY, Hammouda EH, et al. Periodic salbutamol in facioscapulohumeral muscular dystrophy: a randomized controlled trial. Arch Phys Med Rehabil 2009;90(7):1094–1101. doi:10.1016/j.apmr.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 51.Tawil R, McDermott MP, Pandya S, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology 1997;48(1):46–49. doi:10.1212/WNL.48.1.46. [DOI] [PubMed] [Google Scholar]
- 52.van der Kooi EL, Vogels OJ, van Asseldonk RJ, et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology 2004;63(4):702–708. doi:10.1212/01.WNL.0000134660.30793.1F. [DOI] [PubMed] [Google Scholar]
- 53.Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 2008;63(5):561–571. doi:10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 54.Lefkowitz DL, Lefkowitz SS. Facioscapulohumeral muscular dystrophy: a progressive degenerative disease that responds to diltiazem. Med Hypotheses 2005;65(4):716–721. doi:10.1016/j.mehy.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 55.Orrell RW, Copeland S, Rose MR. Scapular fixation in muscular dystrophy. Cochrane Database Syst Rev 2010:CD003278 doi:10.1002/14651858.CD003278.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olsen DB, Ørngreen MC, Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology 2005;64(6):1064–1066. doi:10.1212/01.WNL.0000150584.45055.27. [DOI] [PubMed] [Google Scholar]
- 57.Voet N, Bleijenberg G, Hendriks J, et al. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in FSHD: an RCT. Neurology 2014;83(21):1914–1922. doi:10.1212/WNL.0000000000001008. [DOI] [PubMed] [Google Scholar]
- 58.Ansseau E, Domire JS, Wallace LM, et al. Aberrant splicing in transgenes containing introns, exons, and V5 epitopes: lessons from developing an FSHD mouse model expressing a D4Z4 repeat with flanking genomic sequences. PLoS One 2015;10(3):e0118813 doi:10.1371/journal.pone.0118813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dandapat A, Bosnakovski D, Hartweck LM, et al. Dominant lethal pathologies in male mice engineered to contain an X-linked DUX4 transgene. Cell Rep 2014;8(5):1484–1496. doi:10.1016/j.celrep.2014.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krom YD, Dumonceaux J, Mamchaoui K, et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a Mosaic patient: a cellular model for FSHD. Am J Pathol 2012;181(4):1387–1401. doi:10.1016/j.ajpath.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krom YD, Thijssen PE, Young JM, et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet 2013;9(4):e1003415 doi:10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.PR Newsire Association. aTyr Pharma announces encouraging phase 1b/2 results for ResolarisTM in its first rare myopathy trial. www.prnewswire.com/news-releases/atyr-pharma-announces-encouraging-phase-1b2-results-for-resolaris-in-its-first-rare-myopathy-trial-300243634.html. Published March 30, 2016. Accessed October 12, 2016.
- 63.Tawil R, Padberg GW, Shaw DW, et al. FSHD Workshop Participants. Clinical trial preparedness in facioscapulohumeral muscular dystrophy: clinical, tissue, and imaging outcome measures 29–30 May 2015, Rochester, New York. Neuromuscular Disorders 2015. pii: S0960-8966(15)30028-6. doi:10.1016/j.nmd.2015.10.005. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 64.Tawil R, Shaw DW, van der Maarel SM, Tapscott SJ. Clinical trial preparedness in facioscapulohumeral dystrophy: outcome measures and patient access: 8–9 April 2013, Leiden, The Netherlands. Neuromuscul Disord 2014;24(1):79–85. doi:10.1016/j.nmd.2013.07.009. [DOI] [PubMed] [Google Scholar]